

Abstract
Large-conductance voltage- and Ca2+-gated K+ channels are negative-feedback regulators of excitability in many cell types. They are complexes of α subunits and of one of four types of modulatory β subunits. These have intracellular N- and C-terminal tails and two transmembrane (TM) helices, TM1 and TM2, connected by an ∼100-residue extracellular loop. Based on endogenous disulfide formation between engineered cysteines (Cys), we found that in β2 and β3, as in β1 and β4, TM1 is closest to αS1 and αS2 and TM2 is closest to αS0. Mouse β3 (mβ3) has seven Cys in its loop, one of which is free, and this Cys readily forms disulfides with Cys substituted in the extracellular flanks of each of αS0–αS6. We identified by elimination mβ3-loop Cys152 as the only free Cys. We inferred the disulfide-bonding pattern of the other six Cys. Using directed proteolysis and fragment sizing, we determined this pattern first among the four loop Cys in β1. These are conserved in β2–β4, which have four additional Cys (eight in total), except that mβ3 has one fewer. In β1, disulfides form between Cys at aligned positions 1 and 8 and between Cys at aligned positions 5 and 6. In mβ3, the free Cys is at position 7; position 2 lacks a Cys present in all other β2–β4; and the disulfide pattern is 1–8, 3–4, and 5–6. Presumably, Cys 2 cross-links to Cys 7 in all other β2–β4. Cross-linking of mβ3 Cys152 to Cys substituted in the flanks of αS0–S5 attenuated the protection against iberiotoxin (IbTX); cross-linking of Cys152 to K296C in the αS6 flank and close to the pore enhanced protection against IbTX. In no case was N-type inactivation by the N-terminal tail of mβ3 perturbed. Although the mβ3 loop can move, its position with Cys152 near αK296, in which it blocks IbTX binding, is likely favored.
INTRODUCTION
The large-conductance voltage- and Ca2+-gated K+ (BK) channel consists of a tetramer of α subunits (Butler et al., 1993) and cell-specific modulatory β subunits (Knaus et al., 1994b; Wallner et al., 1999; Xia et al., 1999; Brenner et al., 2000; Uebele et al., 2000). BK α contains a short extracellular N-terminal segment, a unique transmembrane (TM) helix, S0, and, in common with all other voltage-gated K+ channels, six TM helices, S1–S6, with a reentrant pore loop between S5 and S6 (Fig. 1 A). The C-terminal two thirds of α are intracellular and contain Ca2+-binding and other regulatory domains (Wallner et al., 1996; Meera et al., 1997; Schreiber and Salkoff, 1997). Previously, using the extent of endogenous disulfide bond formation between cysteines (Cys) substituted for residues just flanking and within the first helical turns of the extracellular ends of the TM helices, we found that S0 is next to S4 (Liu et al., 2008a, 2010) outside of the S1–S4 four-helix bundle (Liu et al., 2010).
Figure 1.

Mouse and human BK α and β subunits. (A–C) Membrane topology of BK α, β2, and β3 showing the residues mutated to Cys in the extracellular flanks of the TM helices. The native extracellular Cys residues of BK α, Cys14, and Cys141 were mutated to Ala to create the pWT α background. Extracellular Cys residues in the extracellular loop of β2 and β3 are shown. The four Cys residues within the extracellular loop that are conserved in all four β-subunit genes are marked with an asterisk. (D) Sequences of the predicted extracellular loops of human and mouse BK β subunits aligned with ClustalW. The Cys in each of the β-subunit genes are highlighted in white text on a black background, and the four conserved Cys are marked with an asterisk. β4-subunit residues that have been implicated in mediating resistance to ChTX (Gan et al., 2008) are highlighted in green. β2-subunit residues that have been implicated in mediating resistance to ChTX and imparting rectification to α (Chen et al., 2008) are highlighted in red. Lys69 in the β1 subunit, which cross-links to ChTX (Knaus et al., 1994a; Munujos et al., 1995), is highlighted in gray. Gain-of-function and loss-of-function β1 polymorphisms, E65K (Fernández-Fernández et al., 2004) and R140W (Seibold et al., 2008), are highlighted in blue. Residues in β1 implicated in promoting voltage-sensor activation and reducing intrinsic gating (Gruslova et al., 2012) are highlighted in purple. The numbering of Cys from 1–8 is based upon the Cys in β2, hβ3, and β4.
The four types of BK β subunits, β1–β4, are similar in structure: the N and C termini are intracellular, and the two TM helices, TM1 and TM2, are connected by an extracellular loop containing ∼100 residues (Fig. 1, B and C). All β types modulate channel function but with different, albeit overlapping, repertoires. Among other effects, β1 and β4 stabilize the activated state of BK relative to the deactivated state and slow activation and deactivation (Brenner et al., 2000; Wang et al., 2006), and β2 and β3 cause N-type channel inactivation (Xia et al., 1999; Uebele et al., 2000). All four types to a greater or lesser extent retard the binding of the channel-blocking scorpion toxins, charybdotoxin (ChTX) and iberiotoxin (IbTX), to the extracellular end of the pore (Kaczorowski et al., 1996; Xia et al., 1999; Behrens et al., 2000; Meera et al., 2000; Lippiat et al., 2003).
This protection against ChTX and IbTX is a function of the extracellular loop, which is likely to cover at least partially the toxin-binding site, itself close to the mouth of the pore. There is additional evidence that the extracellular loop reaches close to the mouth of the pore. For example, the loops of β2 and β3 are responsible for outward rectification (Zeng et al., 2003; Chen et al., 2008). Furthermore, the loops have other functionally significant interactions with α. In β1, a gain-of-function loop mutation (E65K) is associated with protection from diastolic hypertension (Fernández-Fernández et al., 2004), and a loss-of-function loop mutation (R140W) is associated with exacerbation of asthma (Seibold et al., 2008). Ala substitutions of several other residues within the loop reduced the β1-induced hyperpolarizing shift in the G-V curve (Gruslova et al., 2012).
The loops of the β subunits contain multiple Cys. β1 has four Cys in its loop, and these form two disulfides (Hanner et al., 1998). The four Cys are conserved in all β subunits. Most species of β2, β3, and β4 have four additional aligned Cys in their loops, presumably also paired in disulfide bonds (Fig. 1 D). In β3, reduction of the loop disulfide bonds leads to diminished rectification and increased sensitivity to blockade of BK currents by IbTX (Zeng et al., 2003). Mouse β3a (mβ3a) and rat β3a are unique in having only three additional loop Cys, i.e., seven loop Cys in total (Zeng et al., 2008). At least one of these Cys must be free. A free loop Cys presents us with an opportunity to investigate both the interaction of the β loop with the extracellular face of BK α and the pattern of native disulfide bonding of the remaining loop Cys.
Previously, we had used endogenous disulfide bond formation between Cys substituted for residues flanking the TM helices of α and β to determine the relative positions of the extracellular ends of these helices. We found with both β1 and β4 that TM1 is closest to α S1 and S2 and TM2 is closest to α S0 (Liu et al., 2008b, 2010; Wu et al., 2009). Initially, our extension of this approach to β3a was confounded by the weak heterologous expression of human β3a in HEK cells and by the endogenous cross-linking of WT mβ3a to Cys-substituted α. After identifying and mutating the free Cys in the mβ3a loop, we were able to determine the positions of β3a TM1 and TM2 relative to α S0–S6. In the course of the identification of the free loop Cys, we determined first the disulfide cross-linking pattern of the four conserved Cys in the mouse β1 loop and inferred the cross-linking pattern of the six disulfide-bonded Cys in the mβ3a loop.
MATERIALS AND METHODS
Constructs
Mutants of mouse BK α subunit (mSlo1, KCNMA1; available from GenBank under accession no. NM_010610) were generated in a pseudo-WT (pWT) α background, in which the two extracellular Cys, Cys14 and Cys141, were mutated to Ala (Fig. 1 A). Cys14 and Cys141 are disulfide cross-linked in WT BK α (Liu et al., 2008a). Mutations in the human β1 subunit (KCNMB1; available from GenBank under accession no. NM_004137; 191 residues) were made in a pWT β1 background with Ala substitutions of Cys18 and Cys26 in TM1. HA-HisG-pWT β1-FLAG contained an N-terminal six–His-Gly tag preceding the HA-epitope tag and a C-terminal FLAG tag. pWT human β2 (hβ2) contained Ala substitutions of Cys57 in TM1 and Cys205 in TM2 of the BK hβ2 (KCNMB2; available from GenBank under accession no. bc017825; Open Biosystems catalog no. MHS1011-7509571; 235 residues; molecular weight, 27,100) (Fig. 1, B and D). mβ3a subunit cDNA was provided by C. Lingle (Washington University, St. Louis, MO). Cys substitutions in mβ3a (KCNMB3; available from GenBank under accession no. NM_001195074; 239 residues; molecular weight, 26,484) were made in a pWT background in which Cys202 was mutated to Ala (Fig. 1, C and D). FLAG-pWT-mβ3a had a 3X FLAG epitope ligated to the C terminus of pWT mβ3a. pWT2 mβ3a contained the mutation C152A in the background of pWT mβ3a.
Expression, surface biotinylation, and cell lysis
HEK293 cells were cultured and transfected as described previously (Liu et al., 2008a,b). To ensure excess β-subunit expression, hβ2 and mβ3a were transfected with five times as much DNA as pWT α. To determine the extent of cross-linking, we surface-biotinylated the cells with 1 mM sulfosuccinimidyl-6-(biotinamido) hexanoate (sulfoNHS-LC-biotin; Thermo Fisher Scientific) in Dulbecco’s PBS (DPBS), pH 7.4, and solubilized the cells in lysis buffer containing 1% Triton X-100, 150 mM NaCl, 50 mM Tris, 2 mM N-ethylmaleimide, 1 mM EDTA, and protease inhibitors (Liu et al., 2008a,b). The lysates were mixed with Ultralink Immobilized NeutrAvidin Plus beads (Thermo Fisher Scientific), washed extensively, and eluted in 4 M urea in 2% SDS at 100°C. The samples were electrophoresed, transferred to nitrocellulose, and immunoblotted with either anti-BK α antibody (BD) or anti-FLAG antibody (Sigma-Aldrich) for detecting FLAG-tagged mβ3.
Reduction and reformation of disulfides
HEK293 cells, adherent to the tissue culture dish, were washed, and surface proteins were biotinylated with 4 mM sulfoNHS-LC-biotin solution. The cells were washed with DPBS, and a solution of 137 mM NaCl, 2.7 mM KCl, 0.1 mM CaCl2, 0.1 mM MgCl2, and 40 mM HEPES, pH 8.0, was added to the dish. Dithiothreitol (DTT) was added to a final concentration of 20 mM. The cells were washed with DPBS, collected, and lysed as detailed above. In experiments to test the susceptibility of two Cys on the cell surface to reform a disulfide after reduction, intact cells were suspended in a solution of 137 mM NaCl, 2.7 mM KCl, 0.9 mM CaCl2, 0.49 mM MgCl2, and 10 mM MOPS, pH 7.2. The doubly charged oxidant 4,4′-(azodicarbonyl)-bis-[1,1-dimethylpiperazinium, diiodide] (quaternary piperaziniumdiamide [QPD]) (Kosower et al., 1974; Liu et al., 2008a) was added to a final concentration of 40 µM for 10 min. Cells were washed and lysed as detailed above.
Cross-linking of α and β subunits
The extent of cross-linking between Cys-substituted pWT α and either Cys-substituted pWT β2 or Cys-substituted mβ3a was determined as described previously (Liu et al., 2008b, 2010; Wu et al., 2009). We calculated the extent of α–β cross-linking from the integrated luminescence from the band at apparent mass of ∼160 kD, divided by the sum of the integrated luminescence of the bands at ∼130 and ∼160 kD.
Determining disulfide cross-linking pattern of native Cys in β1 extracellular loop
HEK cells were transfected with pWT α and the mutant HA-6XHis-Gly-pWT β1 E13Q, E50Q, E143Q-FLAG. The cells were solubilized in 1% Triton X-100, 200 mM NaCl, 20 mM Tris, 2 mM N-ethylmaleimide, and 20 mM imidazole, pH 8.0, with complete protease inhibitors (Roche). The lysate was mixed for 1 h at room temperature with 50 µl Ni-NTA beads (GE Healthcare). The beads were washed four times with the same buffer. Proteins were eluted and simultaneously denatured by mixing the beads with 20 mM EDTA, 0.2% SDS, and 20 mM HEPES, pH 7.0; after elution, the eluate was held at 95°C for 4 min. Half of the eluate was deglycosylated with PNGase F (New England Biolabs, Inc.) for 3 h at 37°C. To half of each sample, GluC endoproteinase (Roche) was added and mixed at 37°C for 18 h. The samples were then split again. To one half of each sample, 10 mM DTT was added, and to the other half, water was added; all aliquots were held at 50°C for 20 min. Samples were electrophoresed on a 16% Tris-glycine gel, transferred to nitrocellulose, and blotted with either horseradish peroxidase (HRP)-conjugated anti-FLAG antibody or anti-HA antibody (Covance) and HRP-conjugated secondary antibody.
Electrophysiology
Macroscopic currents were recorded from HEK293 cells in the outside-out patch-clamp configuration at 22–24°C (Liu et al., 2008a,b). For the measurement of conductance as a function of membrane potential (G-V curves), currents were activated by depolarizing steps, in 20-mV increments from a holding potential of −120 mV, and deactivated by repolarization to the holding potential, at which tail currents were measured. The bath solution was 150 mM KCl, 1 mM MgCl2, and 5 mM TES, pH 7.4, and the pipette solution was 150 mM KCl, 5 mM TES, pH 7.0, 1 mM HEDTA, and 10 µM free Ca2+. The free Ca2+ concentration was calculated using the MaxChelator program. The extent of inhibition of BK current by 100 nM IbTX (Tocris Bioscience) was measured after its addition to the bath from the current invoked by depolarizing steps from −120 to +100 mV every 5 s. The membrane potential was held at 0 mV between steps. IbTX was delivered to patch using the SF-77B Perfusion system (Warner Instruments).
Statistical analysis
A one-way ANOVA was used for multiple comparisons, followed by Tukey’s post-hoc test if the null hypothesis was rejected. Differences were considered statistically significant at P < 0.05. All statistical analysis was performed using Prism 6 (GraphPad Software).
RESULTS
Disulfide cross-linking of pWT mβ3a to Cys-substituted α
Initially, we tried to determine the proximities of the extracellular flanks of β2 and β3 TM1 and TM2 to α S0–S6 by the same approach we had applied to β1 and β4 (Liu et al., 2008a,b; Wu et al., 2009). We started with hβ3a, but this expressed poorly in HEK293 cells, as determined both by immunoblotting and by patch-clamp functional analysis. We turned to mβ3a. This expressed well, but our analysis of disulfide cross-linking between the Cys-substituted flanks of α S0 and mβ3a TM1 and TM2 was confounded by the considerable cross-linking of the Cys-substituted α with pWT mβ3a. For example, a Cys substituted in the extracellular flank of α S0, R17C, readily cross-linked to pWT mβ3a (Fig. 2). Previously, neither pWT β1 nor pWT β4 cross-linked to any Cys substituted in α (Liu et al., 2008a,b; Wu et al., 2009). Mouse and rat β3 subunits have seven Cys in their extracellular loops (Zeng et al., 2008), unlike all sequenced β3 subunits from other species and unlike all sequenced β2 and β4 subunits, which have eight Cys. One of the seven Cys in the mβ3a loop is free. We set out to determine which one is free so that we could remove it.
Figure 2.
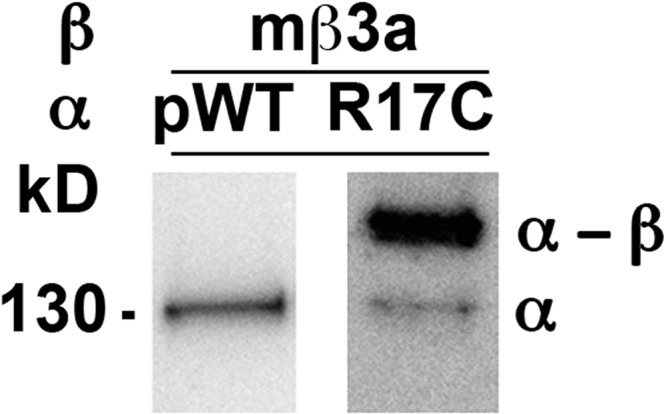
Endogenous disulfide cross-linking between α R17C and a native Cys in pWT mβ3a. Either pWTα or R17C α was coexpressed with pWT mβ3a in HEK cells, and the channel complexes were extracted and denatured in SDS. The blots were developed with anti-BK α antibody, which reacts with both α (130 kD) and the cross-linked mβ3a dimer (∼160 kD).
Disulfide cross-linking pattern of the conserved Cys in the β1 extracellular loop
From the alignment of the sequences of all β subunits, it is evident that the β1 loop contains only four Cys and that these are conserved in all other β subunits (Fig. 1 D). Because these four form two disulfides in β1, it seemed likely that they would also form disulfides in the homologous loops of the other β subunits. By determining the disulfide bond pattern in β1 and by determining which Cys was free in the mβ3a loop, we could infer the complete disulfide-bonding pattern of mβ3a. Further, assuming conservation of the disulfide-bonding pattern among β2, β3, and β4, we could infer the bonding pattern of all the β subunits with eight Cys in their loops.
Our strategy to determine the disulfide-bonding pattern in β1 was to fragment β1 between Cys and to determine the apparent molecular masses of the fragments with the disulfides intact and after they were reduced. Because limited amounts of β1 are available, we used SDS-PAGE to estimate the mass of the fragments and immunoblotting to identify fragments that contained N-terminal tandem HA and His6Gly epitopes and fragments that contained a C-terminal FLAG epitope.
We determined that after mutation of three Glu to Gln (E13Q, E50Q, and E143Q) (Fig. 3 A), cleavage at the remaining Glu with GluC endoproteinase would give different sets of fragments for the three possible disulfide-bonding patterns (Fig. 3 B).
Figure 3.

Determination of disulfide cross-linking pattern of Cys in the β1 extracellular loop. (A) Locations of Cys, Glu, Glu mutated to Gln, and the two N-glycosylation sites (*) in HA-His6Gly-β1-FLAG are labeled above and below the thick line representing the sequence. Above the sequence are the molecular weights of the fragments between the remaining Glu, labeled A–F; these would result from complete cleavage by GluC endoproteinase of the fully reduced and deglycosylated subunit. (B) The predicted and observed molecular weights for the GluC proteolytic fragments detectable either with antibody against the N-terminal HA epitope or with antibody against the C-terminal FLAG epitope, for the three possible combinations of disulfides. These N- and C-terminal fragments are arranged by whether or not they are deglycosylated. To the right are the molecular weights of the N- and C-terminal GluC fragments of the fully reduced subunit. Predicted molecular weights of N-and C-terminal fragments were calculated based on the molecular weights of the fragments in A; each of two glycosylation sites was presumed to add ∼4.2 kD. The observed molecular weights are the mean of at least two independent experiments and are entered below the most closely matching predicted molecular weights. (C) Immunoblot (IB) with anti-HA antibody showing subunit fragments containing the N terminus of the subunit. The treatments of the samples match those in the table in B. *, single glycosylation site; **, two glycosylation sites. (D) Immunoblot with anti-FLAG antibody showing subunit fragments containing the C terminus. (E) Schematic of β1 depicting the only disulfide cross-linking pattern consistent with the observed fragment molecular weights.
The β1 loop is N-glycosylated at Asn80 and Asn142. Deglycosylation of fully reduced β1 with PNGase F decreased the molecular mass by ∼8.4 kD per subunit and by ∼4.6 kD for the GluC-cleaved, FLAG-tagged fragment F (Fig. 3, B–D). Therefore, we accounted for the glycosylation of the two sites in β1 by adding 8.4 kD to the predicted molecular mass of the intact mutant β1 and 4.2 kD to the predicted molecular mass of fragment F (Fig. 3, A and B). Knaus et al. (1994a) reported that each glycosylation site adds ∼5 kD to the apparent molecular mass of β1. The apparent masses of the GluC fragments before and after deglycosylation provided one criterion for the correct disulfide-bonding pattern. In particular, if the pattern were Cys53 to Cys103 and Cys76 to Cys135, deglycosylation should have had no effect on the molecular weight of the N-terminal GluC fragment. Obviously, that was not the case. Only the disulfide-bonding pattern of Cys53 (position 1) to Cys135 (position 8) and Cys76 (position 5) to Cys103 (position 6) is consistent with all of the observed fragment masses (Fig. 3 E).
Identifying the free Cys and disulfide cross-linking in the mβ3a extracellular loop
If there were only one free Cys in the mβ3a loop that formed a disulfide with Cys in α, then mutation of that β-loop Cys to Ala should eliminate the cross-linking. The mutation C152A (position 7) completely blocked formation of α R17C-mβ3a dimer and of larger complexes (Fig. 4 A). With Cys152 in place, mβ3a cross-linked to α R17C to the extent of 90%. The complete elimination of this cross-linking was not caused by lack of assembly of β3a C152A with α, because the channels resulting from their coexpression showed normal inactivation (Fig. 5, I and J).
Figure 4.

Cys152 in the extracellular loop of mβ3a cross-links with α S0 R17C. (A) α S0 R17C was coexpressed with FLAG-tagged pWTβ3 or FLAG-tagged mutant β3a in which one or more of the native Cys in the extracellular loop were mutated to Ala. The intact cells were surface biotinylated, proteins were extracted, and surface proteins were purified using NeutrAvidin. The proteins were denatured in SDS and either reduced with DTT or not. The blots were developed with anti-BK α (top and middle) and anti-FLAG (bottom) antibodies. The four conserved Cys residues are indicated. Representative of more than four similar experiments. The extents of cross-linking were calculated from the relative integrated density of an ∼160-kD band divided by integrated densities of ∼160- and ∼130-kD bands. (B) Schematic of mβ3a depicting disulfide cross-linking between Cys in the extracellular loop.
Figure 5.

Inactivation and resistance to IbTX block and conserved Cys in the mβ3a extracellular loop. (A–I) Macroscopic current conducted by pWT BK α with or without pWT or mutant mβ3a in response to step depolarizations from −80 to +140 mV. The recordings were made in outside-out macropatches with 10 µM Ca2+ inside the pipette. (J) Fraction of current remaining at the end of a 500-ms depolarizing step to +160 mV. Mean ± SEM; n = 4–6. (K) Normalized current conducted by either pWT BK α alone (no mβ3a) or pWT BK α plus pWT mβ3a before and after 100 nM IbTX. Recordings were made in outside-out macropatches. Data are means ± SEM; n ≥ 3. (L and M) Normalized current conducted by pWT BK α plus either pWT mβ3a or mutant mβ3a before and after 100 nM IbTX. Recordings were made in outside-out macropatches. Mean ± SEM; n ≥ 3.
The results of substitution of the other six Cys by Ala were consistent with each of these Cys normally participating in a disulfide. The mutation to Ala of each of the Cys at positions 1, 5, 6, and 8 (Fig. 1 D) decreased the extent of dimer formed with α R17C. In all cases, judging from the blots for α and for β after reduction, both α and β expressed and were transported to the cell surface. However, none of these β mutants coexpressed with α resulted in normal N-type inactivation (Fig. 5, A–F and J), and none protected against IbTX block (Fig. 5, K and L). These functional deficits were likely caused by the disruption of the β3a loop structure and of the assembly of β with α. These results are consistent with these Cys forming structurally and functionally significant conserved disulfides.
The β3a mutants C105A (at position 3) or C109A (at position 4) did cross-link to α R17C, yielding increased relative amounts of the α–β 160-kD band and of higher molecular weight bands, possibly oligomers of α–β (Fig. 4 A, top blot). The double mutant, C105A/C109A, eliminated the higher molecular weight bands, whereas the α–β dimer band (∼160 kD) was stronger (Fig. 4 A). This result is consistent with Cys105 and Cys109 normally being paired in a disulfide (Fig. 4 B). In contrast to the effects of mutating any one of the conserved Cys, mutating either Cys105 or Cys109 had no effect on the mβ3a-induced inactivation of pWT α (Fig. 5, G, H, and J). These mutations did, however, decrease the protection by mβ3a against IbTX block (Fig. 5, K and M), likely as a result of the perturbation of the loop structure caused by the loss of this disulfide.
Collectively, these results imply that Cys152 is the one unpaired Cys in the mβ3a extracellular loop. Nevertheless, Cys152 plays a role in the structure and/or function of the loop because α coexpressed with mβ3a C152A was not resistant to IbTX (Fig. 5 M).
Cross-linking the mβ3a extracellular loop to α and IbTX resistance
Cys14 and Cys141 in the N-terminal extracellular segment of α (Fig. 1 A) normally form a disulfide (Liu et al., 2008a). When either Cys was mutated to Ala, Cys152 in mβ3a readily cross-linked to the free Cys, either Cys14 or Cys141 (Fig. 6 A). Even when both Cys14 and Cys141 are present (WT α), there was significant cross-linking to mβ3a Cys152, possibly by competition with the normal intrasubunit disulfide bond formation in α. Although the cross-links between Cys152 in mβ3a and Cys14 or Cys141 in α did not alter N-type inactivation (Fig. 6 B), the cross-links did affect the accessibility of IbTX to α. Channels containing mβ3a and either α C141A or α C14A were inhibited more quickly by IbTX than were channels containing mβ3a and pWT α (Fig. 6 C).
Figure 6.

Cross-linking of β3a Cys152 in the extracellular loop to α S1–S6. (A) β3a Cys152 cross-links to native Cys, Cys14, and Cys141 in α. The fraction of endogenous cross-linking between β3a Cys152 and α Cys14 or α Cys141 is shown at the bottom of each lane. Immunoblot with anti-BK α antibody. (B) Representative macroscopic currents conducted by WT α plus pWT mβ3a, pWT α plus pWT mβ3a, C14A α plus pWT mβ3a, and C141A α plus pWT mβ3a before (black trace) and after (red trace) 100 nM IbTBX. (C) Normalized current conducted by WT α, pWTα, C14A α, or C141Aα plus pWT mβ3a before and after 100 nM IbTX. Data points are means ± SEM; n > 3. pWT α without mβ3a is shown as a dashed blue line. (D) Extents of endogenous disulfide bond formation between Cys-substituted extracellular flanks of α S0–S6 and Cys152 of pWT β3a. The α residues substituted by Cys are shown on the left. The extents of disulfide bond formation are represented by bars in the horizontal direction. In the cases in which the mean extent of disulfide bond formation was zero, the value 0.5% was plotted to identify these pairs as tested. (E–G) Normalized current conducted by P137C α with or without pWT mβ3a, G260C α with or without pWT mβ3a, and K296C α with or without pWT mβ3a before and after 100 nM IbTX. pWTα plus pWT mβ3a is shown as a dashed line. Data are means ± SEM; n ≥ 3. (Insets) Representative macroscopic currents conducted by P137C α plus pWT mβ3a, G260C α plus pWT mβ3a, and K296C α plus pWT mβ3a before (black trace) and after (red trace) 100 nM IbTX.
Cys152 also formed cross-links with Cys substituted in the flanks of most of the TM helices of α, to the extent of 80–95% with the Cys in the first four positions flanking S0, more selectively but strongly with Cys in the flanks of S1, S2, S5, and S6, and weakly only with the Cys substituted in the S3–S4 loop (Fig. 6 D). Inactivation by mβ3a was unaffected by the disulfide cross-links between Cys152 and Cys substituted in S1 (P137C), S5 (G260C), and S6 (K296C) (Fig. 6, E–G, insets). These disulfide cross-links, however, variably affected the β3a-mediated resistance to IbTX (Fig. 6, E–G). Cross-linking of Cys152 to P137C in S1 caused a complete loss of resistance to IbTX (Fig. 6 E), similar to the cross-linking of Cys152 to α Cys141 (Fig. 6 C, C14A α + mβ3a). After cross-linking of Cys152 to α G260C in the flank of S5, protection against IbTX was decreased but not absent (Fig. 6 F). Cross-linking of mβ3a Cys 152 to α K296C in the flank of S6 increased protection against IbTX (Fig. 6 G). The steady-state block by IbTX of α K296C alone was less than that of WT α, but the rate of approach to this steady state was greater in the former case. Thus, Cys152 in the loop of β3a can reach α Lys296, which likely interacts with IbTX.
Locations of TM1 and TM2 of β3 relative to S0–S6 of α
With the background construct mβ3a C152A (pWT2 β3), we could determine the extent of endogenous cross-linking of Cys substituted in the flanks of mβ3a TM1 and TM2 to Cys substituted in the flanks of α S0–S6, with the latter mutations made in pWT α (containing C14A and C141A). Three examples of cross-linking, between the flanks of α S0 and β3a TM2, α S1 and β3a TM1, and α S2 and β3a TM1, are shown in Fig. 7 A. In the third panel, for example, N136C in the α-S1 flank cross-links endogenously to F76C in the mβ3a-TM1 flank to the extent of 64%. This cross-link is completely reduced by DTT and restored to the extent of 48% on the cell surface by the impermeant diamide derivative QPD.
Figure 7.

Position of extracellular flanks of mβ3a relative to the α S1–S6 TM helices. (A) Immunoblots illustrating endogenous cross-linking of Cys substituted in the extracellular flanks of α and C152A mβ3a. Expression of mutants, selection of channels transported to the cell surface, SDS gel electrophoresis, and blotting are described in Materials and methods. The blots were developed with an antibody against an epitope from the C terminus of α, which recognizes α (∼130 kD) and the cross-linked α–mβ3a complex (∼160 kD). Portions of each sample were reduced with DTT and oxidized for 20 min with 40 µM QPD. At the bottom of each lane is the fraction of the cross-linked α–β3 complex. (B and C) Extents of endogenous disulfide bond formation between Cys-substituted extracellular flanks of α S0–S6 with mβ3a TM1 (B) and mβ3a TM2 (C) flanks. The α residues substituted by Cys are color coded as shown. (D) Locations of the extracellular ends of mβ3a TM1 and TM2 relative to the extracellular ends of α S0–S6. The locations of the extracellular ends of mβ3a TM1 and TM2 are represented by labeled black and white circles. TM1 and TM2 from two mβ3a subunits are represented by white circles with black letters, and two mβ3a subunits are represented by black circles with white letters. For each pair of flanks, the mean of the top three extents of cross-linking (see Materials and methods) is coded by the color of the connecting line (see legend in panel). To indicate the distance of a cross-link from the ends of the TM helices, we added the number of residues out from the membrane of each of the two Cys. The mean of these distances for the top three extents of cross-linking is indicated by the thickness of the line (see legend in panel). The position of S0 relative to S1–S6 was determined as described in Liu et al. (2008a, 2010). P, pore.
Cys substitutions in the β3 TM1 flank formed disulfides most readily with Cys in the flanks of S1 and S2 (Fig. 7 B), whereas Cys in the flanks of TM2 formed disulfides most readily with Cys in the flanks of S0 (Fig. 7 C). Among the eight combinations tested of one Cys in the TM1 flank and one Cys in the S0 flank, one combination formed a disulfide to an extent ≥50% and two combinations formed disulfides to an extent ≥40%. We found similar levels of cross-linking of TM1 to S0 in β1 (Liu et al., 2008b), but not β2 (see below) or β4 (Wu et al., 2009). Among the eight combinations of a Cys in the TM1 flank and a Cys in the S1 flank, six combinations formed disulfides to an extent ≥50% and five combinations formed disulfides to an extent ≥60%. Slightly higher disulfide cross-linking was observed for S2-TM1 combinations: eight combinations formed disulfides to an extent ≥50%, and seven combinations formed disulfides to an extent ≥60%. Among the eight combinations of a Cys in the TM1 flank and a Cys in the S5 flank, significant cross-linking was observed for two combinations, both with G260C. Taking the mean of the top three extents of cross-linking for each pair of one flank in α and one flank in β as an indication of the proximity of the flanks, we found 46 ± 5% (mean ± SEM) for S0-TM1, 75 ± 4.2% for S1-TM1, 78 ± 1.3% for S2-TM1, and 47 ± 18.5% for S5-TM1. In contrast, the TM1 flank cross-linked much less to the flanks of S3–S4 and S6 (Fig. 7 B). Thus, the extracellular flank of TM1 is closest to the extracellular flank of S1 and S2, but it has substantial contact with the extracellular flank of S0, and to a lesser extent the extracellular flank of S5.
Among the eight combinations of one Cys in the TM2 flank and one Cys in the S0 flank, eight combinations formed disulfides to an extent ≥50% and three combinations formed disulfides to an extent ≥60%. Smaller extents of cross-linking were observed for the combinations with Cys in the S1 and the S2 flanks. In the S5 flank, G260C cross-linked to residues within the flank of TM2. The mean of the top three extents of cross-linking for S0-TM2 pairs was 69 ± 1.6%, for S1-TM2 pairs it was 21 ± 3.2%, for S2-TM2 pairs it was 35 ± 5.3%, and for S5-TM2 pairs it was 30 ± 17%. There was little cross-linking to the S3–S4 loop or to S6. Thus, the flank of TM2 is closest to the flank of S0 (Fig. 7 C), but it also has some contact with the flanks of S2 and S5. We represent the salient features of the cross-linking as lines linking the extracellular ends of the TM helices of α and mβ3a (Fig. 7 D). The circles labeled S1–S6 were superimposed on the extracellular ends of the helices determined in the x-ray crystallographic solution of the presumed open state of the potassium channel KV1.2 (Long et al., 2005a,b). The extracellular end of S0 is placed outside of the voltage-sensor domain, close to the short loop between S3 and S4 (Liu et al., 2010).
Locations of TM1 and TM2 of β2 relative to S0–S6 of α
A similar analysis of the proximities of the flanks of TM helices of α and β2 was performed (Fig. 8). The cross-linking of β2 is more sharply defined than that of β3a. β2 TM1 flank cross-links to α S1 and S2 and no other flanks of α TM helices (Fig. 8 B). Also, β2 TM2 cross-links to S0 and except for one residue in S5 to no other flanks in α (Fig. 8 C). Taking the mean of the top three extents of cross-linking for each pair of flanks as an indication of their proximity, we found 52 ± 3.5% for S1-TM1 and 71 ± 1.6% for S2-TM1. In contrast, the TM1 flank cross-linked much less to the flanks of S0, S3–S4, S5, and S6 (Fig. 8 B). Thus, the extracellular flank of TM1 is closest to the extracellular flank of S2, close to the extracellular flank of S1, but not close to the extracellular flanks of S0, S3–S4, S5, or S6.
Figure 8.

Position of extracellular flanks of β2 relative to the α S1–S6 TM helices. (A) Immunoblots illustrating endogenous cross-linking of Cys-substituted α and Cys-substituted β2. Portions of each sample were reduced with DTT and oxidized for 20 min with 40 µM QPD. At the bottom of each lane is the fraction of the cross-linked α–β2 complex. (B and C) Extents of endogenous disulfide bond formation between Cys-substituted extracellular flanks of α S0–S6 with β2 TM1 (B) and β2 TM2 (C) flanks. The α residues substituted by Cys are color coded as shown. The extent of disulfide bond formation is represented by bars in the horizontal direction. In the cases in which the mean extent of disulfide bond formation was zero, the value 0.5% was plotted to identify these pairs as tested. The residues closest to the membrane are underlined. (D) Locations of the extracellular ends of β2 TM1 and TM2 relative to the extracellular ends of α S0–S6. See Fig. 7 legend for details.
The mean of the top three extents of cross-linking for TM2 and S0 was 69 ± 0.6%. The mean of the top three extents of cross-linking for TM2 and S5 was 42 ± 11%, caused entirely by three cross-links to G260C in S5. There was little disulfide formation with the flanks of S1, S2, S3–S4 loop, or S6. Thus, the flank of TM2 is closest to the flank of S0, but it also has some contact with the top of S5 (Fig. 8 C). We represent the salient features of the cross-linking as lines linking the extracellular ends of the TM helices of α and β2 (Fig. 8 D).
N-type inactivation of α disulfide cross-linked to β2 and β3 TM1
Although the positions of the intracellular ends of β TM1 and TM2 relative to the intracellular ends of α S0–S6 are not known, the intracellular N termini of β2 and β3 are responsible for N-type inactivation of the BK channel complexes, which include them (Xia et al., 1999, 2003). These intracellular N-terminal segments contain ∼50 residues (Fig. 1 A). There is still a question of whether constraining the extracellular flanks of β2 and β3 TM1 by cross-links to S1 or S2 would affect the rates or extents of inactivation. Cross-linking of the pair S1 P137C and β2 TM1 Y73C (60%), the pair S2 F144C and β2 TM1 M74C (70%), and the pair S0 G18C and β2 TM2 N194C (70%) did not affect voltage-dependent inactivation (Fig. 9, A–E). Similarly, cross-linking of the pair S1 N136C and mβ3a TM1 F76C (80%), the pair S2 K146C and mβ3a TM1 F76C (80%), and the pair S0 R17C and mβ3a TM2 G195C (70%) did not affect voltage-dependent inactivation (Fig. 9, F–J). Either the cross-links constrain the extracellular ends of the β TM helices in close to native positions or the flexibility of the β2 and β3 TM helices or of the intracellular N-terminal segments is sufficient to compensate for any extracellular distortions.
Figure 9.

Inactivation of α–β2 and α–mβ3a cross-linked complexes. (A–D) Representative macroscopic currents conducted by pWTα plus pWTβ2 (A) and cross-linked complexes between α S1 and β2 TM1 (B), α S2 and β2 TM1 (C), and α S0 and β2 TM2 (D). Measurements were made with inside-out macropatches using 10 µM Ca2+ inside the pipette. Currents were elicited by stepping the membrane voltage from a holding potential of −80 mV to depolarized test potentials in +20-mV increments up to +140 mV. (E) Summary graph of τinactivation at +160 mV. Mean ± SEM; n > 4; P = not significant by one-way ANOVA. (F–I) Representative macroscopic currents conducted by pWTα plus pWT mβ3a (F) and cross-linked complexes between α S1 and mβ3a TM1 (G), α S2 and mβ3a TM1 (H), and α S0 and mβ3a TM2 (I). Measurements were obtained as described above. (J) Summary graph of τinactivation at +160 mV. Mean ± SEM; n > 4; P = not significant by one-way ANOVA.
DISCUSSION
Locations of the β TM helices relative to α S0–S6
For the extracellular ends of the TM helices of the β2 and β3a subunits, we infer from the extents of disulfide bond formation that TM1 is closer to α S1 and S2 than to S0, and that TM2 is closer to α S0 than to S1 and S2. These relative proximities are the same that we previously found for complexes of β1 and β4 with α (Liu et al., 2008b, 2010; Wu et al., 2009). There are some differences in the details, however. Referring in all cases to the extracellular flanks of the designated helix, in β1 and β3, TM1 cross-linked to S0 to the extent of 40–50%, whereas in β2 and β4, TM1 cross-linked to S0 only to the extent of 5–12%, close to background. Also in β1 and β3, TM2 cross-linked to S2 to the extent of 16–21%, whereas in β2 and β4, TM2 did not cross-link at all to S2. Thus, judging from the extents of cross-linking to the less close α-subunit helices, the extracellular flanks of the β1 and β3 TM helices appear more mobile than those of β2 and β4. Whether this difference reflects the mobilities of TM1 and TM2 themselves is unknown, as is the functional relevance of this mobility.
As discussed before, it is likely that in all β types, the loops cover part or all of the ChTX- and IbTX-binding site and perhaps even the pore itself. Nevertheless, Cys substituted within the first four to six residues in the flanks of TM1 and TM2 did not, for the most part, cross-link to Cys in the flanks of α S5 and S6, which surround the pore. An exception was α G260C four residues out from the membrane in the S5 flank. F76C three residues out from the membrane in the mβ3a TM1 flank cross-linked to α G260C to the extent of ∼80%. In contrast, Cys in the TM1 flanks of β1, β2, and β4 cross-linked weakly or not at all to α G260C. Cys in the TM2 flanks in β2, mβ3, and β4 (Wu et al., 2009) also cross-linked to G260C (Liu et al., 2008b). These results suggest flexibility in the S5 flank, which when matched with flexibility in the TM flanks, allows some cross-linking to S5. In contrast, Cys in the flanks of TM1 and TM2 of any of the four types of β did not cross-link to Cys in the flanks of S6. These results led to the placement of TM1 and TM2 in our model in the gap between adjacent voltage-sensing domains S0–S4 but not next to S5 and S6 (Figs. 7 D and 8 D).
Disulfide cross-linking pattern of conserved Cys within the extracellular loop of β subunits
None of the four Cys in the β1 extracellular loop are free: in unreduced β1, none of these Cys are labeled by N-biotin maleimide; after mutating one of the Cys to Ala, however, there was labeling (Hanner et al., 1998). The absence of disulfide cross-linking between pWT β1 and Cys substituted in the extracellular flanks of α (Liu et al., 2008b, 2010) also implies that the native Cys in the β1 loop are not free, i.e., engaged in disulfide bonds. Previous attempts to define the pattern of these disulfide bonds with combinations of double Cys mutants gave inconsistent results (Hanner et al., 1998). By a different approach, we determined the cross-linking pattern in epitope-tagged β1 subunits that contained the four native Cys and three Glu to Gln mutations. We showed that Cys53 (position 1) and Cys135 (position 8) form a disulfide (Fig. 3 E). Given that all Cys are disulfide bonded, Cys76 (position 5) and Cys103 (position 6) must also form a disulfide.
Both of the disulfides are required for stabilizing a conformation required for β1 interaction with 125I-ChTX (Hanner et al., 1998). The β1-induced shift in the G-V curve, however, survives disruption of either one of the two disulfides (Gruslova et al., 2012). In mβ3a, both of these disulfides are required for N-type inactivation and for conferring resistance to IbTX block (Fig. 5). The mβ3a-induced N-type inactivation is not dependent, however, on the disulfide between Cys105 (position 3) and Cys109 (position 4) (Fig. 5, G and H) or on the disulfide between Cys 2 and Cys 7.
The aligned sequences of the mβ3a subunit and of its human counterpart, hβ3a, are only 63% identical (Fig. 1 D) (Zeng et al., 2008). Cys152 (position 7) is unpaired only in rat and mouse, where a Phe is at position 2 (Zeng et al., 2008). Substituting a Cys for Phe101 in mβ3a, however, did not result in a fourth disulfide, as judged by the continued availability of free Cys for cross-linking to α. In the folded structure of mβ3a, these two Cys (positions 2 and 7) are not positioned to form a disulfide.
Rectification and protection against IbTX
Because mouse and human β3a equally protected against ChTX (Zeng et al., 2008), this function of the loop does not require a disulfide between the Cys 2 and Cys 7. Cys152 is required, however, for resistance to IbTX block because mutation of this residue to Ala prevented the mβ3 subunit from conferring resistance to IbTX block (Fig. 5). Mutation of Cys152 to Ala apparently alters the local conformation. Cys152 in mβ3a is aligned with Cys119 in hβ4, which is immediately adjacent in the sequence to Lys120, Arg121, and Lys125 (Fig. 1 D). These positively charged residues in β4 have been proposed to impede the approach of ChTX to the channel vestibule (Gan et al., 2008). Cys152 in mβ3a also aligns with Cys148 in hβ2, which is adjacent to several Lys shown to be required for β2-induced resistance to ChTX (Fig. 1 D) (Chen et al., 2008). Likewise, Cys105 and Cys109 (positions 3 and 4), or the disulfide that forms between these Cys, are required for protection by mβ3a against IbTX. In β1, the residue nearly aligned with mβ3a Cys109 is Lys69 (Fig. 1 D), the proximity of which to bound ChTX was demonstrated by cross-linking (Knaus et al., 1994a; Munujos et al., 1995).
The mβ3 subunit is less effective than hβ3 in causing outward rectification (Zeng et al., 2008). It was hypothesized that the slightly shorter loop of mβ3 compared with hβ3 might be responsible. Another possibility is that the less effective rectification might be caused by the absence of the fourth disulfide, given that reduction of the disulfides in hβ3 eliminated rectification (Zeng et al., 2003).
Protection against IbTX by the mβ3a loop cross-linked to α
In the Shaker K+ channel, Thr449 is essential for ChTX binding (Naranjo and Miller, 1996). Thr449 is in the linker between the ion selectivity filter and the top of S6 and aligns with Tyr294 in BK α. It was proposed that BK α Tyr294 and Phe266 contribute to the binding of ChTX (Gao and Garcia, 2003). Later work showed that the mutation of Phe266 to Leu or Ala did not affect ChTX inhibition of BK (Yao et al., 2006), but the mutation of Tyr294 to Val did markedly reduce protection against ChTX (Gan et al., 2008). We found that Cys substitution of the nearby Lys296 also reduced protection against IbTX, as did the mutation G260C (Fig. 6). In homology models of BK α, Gly260 is in the turret at the top of S5, which has been proposed to interact with ChTX and IbTX (Giangiacomo et al., 2008). Our result, however, that protection against IbTX block was decreased by cross-linking mβ3a Cys152 to G260C suggests that G260 is not a significant interaction site for IbTX; otherwise, cross-linking to that residue would block binding of IbTX. In addition, cross-linking Cys152 to either α Cys14 or α Cys141, both present and ordinarily cross-linked to each other in WT α (Liu et al., 2008a), diminished protection of mβ3a against IbTX. These cross-links may pull the loop away from the position in which it retards the binding of IbTX.
In contrast, protection against IbTX by mβ3a was increased by the cross-linking of Cys152 (position 7) to α K296C. This cross-link likely constrains the β loop in the vicinity of the IbTX-binding site on α. Overall, it appears that the β loop can assume various positions relative to the extracellular face of α. Only some of these positions are compatible with the protection against IbTX and ChTX by the β loop. In one of these positions, mβ3a Cys152 is close to α K296. In all of these positions of the loop constrained by cross-links, however, the intracellular N-type inactivation by the N terminus of β3a is normal.
What are the physiological implications of the endogenous cross-linking Cys152 to one of the two native extracellular Cys (14 and 141) in α? To the extent that this occurs in vivo, it could have an effect on rectification. Also, if mβ3a expression were limited, its cross-linking to α during assembly could stabilize the association.
Acknowledgments
We thank Christopher Lingle (Washington University) for providing the mβ3a cDNA.
This work was supported in part by National Institutes of Health (NIH) research grant awards P01 HL081172 and R01 HL68093 from the National Heart, Lung and Blood Institute and R01 NS054946 from the National Institute of Neurological Disorders and Stroke, and the Arlene and Arnold Goldstein Family Foundation. G. Liu is supported by an American Heart Association Scientist Developmental Award. R.S. Wu was supported by NIH T32 HL07854 and the Future Leaders in CV Medical Research award from Schering-Plough.
Sharona E. Gordon served as editor.
Footnotes
Abbreviations used in this paper:
- BK
- large-conductance voltage- and Ca2+-gated K+
- ChTX
- charybdotoxin
- Cys
- cysteine(s)
- DTT
- dithiothreitol
- IbTX
- iberiotoxin
- mβ3a
- mouse β3a
- pWT
- pseudo-WT
- QPD
- quaternary piperaziniumdiamide
- TM
- transmembrane
References
- Behrens R., Nolting A., Reimann F., Schwarz M., Waldschütz R., Pongs O. 2000. hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel beta subunit family. FEBS Lett. 474:99–106 10.1016/S0014-5793(00)01584-2 [DOI] [PubMed] [Google Scholar]
- Brenner R., Jegla T.J., Wickenden A., Liu Y., Aldrich R.W. 2000. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J. Biol. Chem. 275:6453–6461 10.1074/jbc.275.9.6453 [DOI] [PubMed] [Google Scholar]
- Butler A., Tsunoda S., McCobb D.P., Wei A., Salkoff L. 1993. mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science. 261:221–224 10.1126/science.7687074 [DOI] [PubMed] [Google Scholar]
- Chen M., Gan G., Wu Y., Wang L., Wu Y., Ding J. 2008. Lysine-rich extracellular rings formed by hbeta2 subunits confer the outward rectification of BK channels. PLoS ONE. 3:e2114 10.1371/journal.pone.0002114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Fernández J.M., Tomás M., Vázquez E., Orio P., Latorre R., Sentí M., Marrugat J., Valverde M.A. 2004. Gain-of-function mutation in the KCNMB1 potassium channel subunit is associated with low prevalence of diastolic hypertension. J. Clin. Invest. 113:1032–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan G., Yi H., Chen M., Sun L., Li W., Wu Y., Ding J. 2008. Structural basis for toxin resistance of beta4-associated calcium-activated potassium (BK) channels. J. Biol. Chem. 283:24177–24184 10.1074/jbc.M800179200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.D., Garcia M.L. 2003. Interaction of agitoxin2, charybdotoxin, and iberiotoxin with potassium channels: selectivity between voltage-gated and Maxi-K channels. Proteins. 52:146–154 10.1002/prot.10341 [DOI] [PubMed] [Google Scholar]
- Giangiacomo K.M., Becker J., Garsky C., Schmalhofer W., Garcia M.L., Mullmann T.J. 2008. Novel alpha-KTx sites in the BK channel and comparative sequence analysis reveal distinguishing features of the BK and KV channel outer pore. Cell Biochem. Biophys. 52:47–58 10.1007/s12013-008-9026-3 [DOI] [PubMed] [Google Scholar]
- Gruslova A., Semenov I., Wang B. 2012. An extracellular domain of the accessory β1 subunit is required for modulating BK channel voltage sensor and gate. J. Gen. Physiol. 139:57–67 10.1085/jgp.201110698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanner M., Vianna-Jorge R., Kamassah A., Schmalhofer W.A., Knaus H.G., Kaczorowski G.J., Garcia M.L. 1998. The beta subunit of the high conductance calcium-activated potassium channel. Identification of residues involved in charybdotoxin binding. J. Biol. Chem. 273:16289–16296 10.1074/jbc.273.26.16289 [DOI] [PubMed] [Google Scholar]
- Kaczorowski G.J., Knaus H.G., Leonard R.J., McManus O.B., Garcia M.L. 1996. High-conductance calcium-activated potassium channels; structure, pharmacology, and function. J. Bioenerg. Biomembr. 28:255–267 10.1007/BF02110699 [DOI] [PubMed] [Google Scholar]
- Knaus H.G., Eberhart A., Kaczorowski G.J., Garcia M.L. 1994a. Covalent attachment of charybdotoxin to the beta-subunit of the high conductance Ca(2+)-activated K+ channel. Identification of the site of incorporation and implications for channel topology. J. Biol. Chem. 269:23336–23341 [PubMed] [Google Scholar]
- Knaus H.G., Folander K., Garcia-Calvo M., Garcia M.L., Kaczorowski G.J., Smith M., Swanson R. 1994b. Primary sequence and immunological characterization of beta-subunit of high conductance Ca(2+)-activated K+ channel from smooth muscle. J. Biol. Chem. 269:17274–17278 [PubMed] [Google Scholar]
- Kosower E.M., Kosower N.S., Kenety-Londner H., Levy L. 1974. Glutathione. IX. New thiol-oxidizing agents: DIP, DIP+1, DIP+2. Biochem. Biophys. Res. Commun. 59:347–351 10.1016/S0006-291X(74)80213-5 [DOI] [PubMed] [Google Scholar]
- Lippiat J.D., Standen N.B., Harrow I.D., Phillips S.C., Davies N.W. 2003. Properties of BK(Ca) channels formed by bicistronic expression of hSloalpha and beta1-4 subunits in HEK293 cells. J. Membr. Biol. 192:141–148 10.1007/s00232-002-1070-0 [DOI] [PubMed] [Google Scholar]
- Liu G., Zakharov S.I., Yang L., Deng S.X., Landry D.W., Karlin A., Marx S.O. 2008a. Position and role of the BK channel α subunit S0 helix inferred from disulfide crosslinking. J. Gen. Physiol. 131:537–548 10.1085/jgp.200809968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Zakharov S.I., Yang L., Wu R.S., Deng S.X., Landry D.W., Karlin A., Marx S.O. 2008b. Locations of the beta1 transmembrane helices in the BK potassium channel. Proc. Natl. Acad. Sci. USA. 105:10727–10732 10.1073/pnas.0805212105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Niu X., Wu R.S., Chudasama N., Yao Y., Jin X., Weinberg R., Zakharov S.I., Motoike H., Marx S.O., Karlin A. 2010. Location of modulatory β subunits in BK potassium channels. J. Gen. Physiol. 135:449–459 10.1085/jgp.201010417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S.B., Campbell E.B., Mackinnon R. 2005a. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903 10.1126/science.1116269 [DOI] [PubMed] [Google Scholar]
- Long S.B., Campbell E.B., Mackinnon R. 2005b. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 309:903–908 10.1126/science.1116270 [DOI] [PubMed] [Google Scholar]
- Meera P., Wallner M., Song M., Toro L. 1997. Large conductance voltage- and calcium-dependent K+ channel, a distinct member of voltage-dependent ion channels with seven N-terminal transmembrane segments (S0-S6), an extracellular N terminus, and an intracellular (S9-S10) C terminus. Proc. Natl. Acad. Sci. USA. 94:14066–14071 10.1073/pnas.94.25.14066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meera P., Wallner M., Toro L. 2000. A neuronal beta subunit (KCNMB4) makes the large conductance, voltage- and Ca2+-activated K+ channel resistant to charybdotoxin and iberiotoxin. Proc. Natl. Acad. Sci. USA. 97:5562–5567 10.1073/pnas.100118597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munujos P., Knaus H.G., Kaczorowski G.J., Garcia M.L. 1995. Cross-linking of charybdotoxin to high-conductance calcium-activated potassium channels: identification of the covalently modified toxin residue. Biochemistry. 34:10771–10776 10.1021/bi00034a009 [DOI] [PubMed] [Google Scholar]
- Naranjo D., Miller C. 1996. A strongly interacting pair of residues on the contact surface of charybdotoxin and a Shaker K+ channel. Neuron. 16:123–130 10.1016/S0896-6273(00)80029-X [DOI] [PubMed] [Google Scholar]
- Schreiber M., Salkoff L. 1997. A novel calcium-sensing domain in the BK channel. Biophys. J. 73:1355–1363 10.1016/S0006-3495(97)78168-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold M.A., Wang B., Eng C., Kumar G., Beckman K.B., Sen S., Choudhry S., Meade K., Lenoir M., Watson H.G., et al. 2008. An African-specific functional polymorphism in KCNMB1 shows sex-specific association with asthma severity. Hum. Mol. Genet. 17:2681–2690 10.1093/hmg/ddn168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uebele V.N., Lagrutta A., Wade T., Figueroa D.J., Liu Y., McKenna E., Austin C.P., Bennett P.B., Swanson R. 2000. Cloning and functional expression of two families of beta-subunits of the large conductance calcium-activated K+ channel. J. Biol. Chem. 275:23211–23218 10.1074/jbc.M910187199 [DOI] [PubMed] [Google Scholar]
- Wallner M., Meera P., Toro L. 1996. Determinant for beta-subunit regulation in high-conductance voltage-activated and Ca(2+)-sensitive K+ channels: an additional transmembrane region at the N terminus. Proc. Natl. Acad. Sci. USA. 93:14922–14927 10.1073/pnas.93.25.14922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M., Meera P., Toro L. 1999. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane beta-subunit homolog. Proc. Natl. Acad. Sci. USA. 96:4137–4142 10.1073/pnas.96.7.4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Rothberg B.S., Brenner R. 2006. Mechanism of β4 subunit modulation of BK channels. J. Gen. Physiol. 127:449–465 10.1085/jgp.200509436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.S., Chudasama N., Zakharov S.I., Doshi D., Motoike H., Liu G., Yao Y., Niu X., Deng S.X., Landry D.W., et al. 2009. Location of the beta 4 transmembrane helices in the BK potassium channel. J. Neurosci. 29:8321–8328 10.1523/JNEUROSCI.6191-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X.M., Ding J.P., Lingle C.J. 1999. Molecular basis for the inactivation of Ca2+- and voltage-dependent BK channels in adrenal chromaffin cells and rat insulinoma tumor cells. J. Neurosci. 19:5255–5264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X.M., Ding J.P., Lingle C.J. 2003. Inactivation of BK channels by the NH2 terminus of the β2 auxiliary subunit: An essential role of a terminal peptide segment of three hydrophobic residues. J. Gen. Physiol. 121:125–148 10.1085/jgp.20028667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Li H., Gan G.L., Wu Y., Ding J.P. 2006. Residue Phe266 in S5-S6 loop is not critical for Charybdotoxin binding to Ca2+-activated K+ (mSlo1) channels. Acta Pharmacol. Sin. 27:945–949 10.1111/j.1745-7254.2006.00385.x [DOI] [PubMed] [Google Scholar]
- Zeng X.H., Xia X.M., Lingle C.J. 2003. Redox-sensitive extracellular gates formed by auxiliary beta subunits of calcium-activated potassium channels. Nat. Struct. Biol. 10:448–454 10.1038/nsb932 [DOI] [PubMed] [Google Scholar]
- Zeng X., Xia X.M., Lingle C.J. 2008. Species-specific differences among KCNMB3 BK β3 auxiliary subunits: Some β3 N-terminal variants may be primate-specific subunits. J. Gen. Physiol. 132:115–129 10.1085/jgp.200809969 [DOI] [PMC free article] [PubMed] [Google Scholar]