

Abstract
Viruses are strictly dependent on cells to propagate and many incorporate host proteins in their viral particles, but the significance of this incorporation is poorly understood. Recently, we performed the first comprehensive characterization of the mature herpes simplex virus type 1 (HSV-1) in which up to 49 distinct cellular proteins were identified by mass spectrometry. In the present study, we sought to identify if these cellular factors are relevant for the HSV-1 life cycle. To this end, we performed a small interfering RNA functional screen and found that 15 of these host proteins altered HSV-1 proliferation in cell culture, without any significant effect on cell viability. Moreover, the siRNA used had no negative consequences for Adenovirus type 5 propagation (with one exception) indicating that the modulation was specific for HSV-1 and not merely due to unhealthy cells. The positive host proteins include several Rab GTPases and other intracellular transport components as well as proteins involved in signal transduction, gene regulation and immunity. Remarkably, in most cases when virions were depleted for one of the above proteins, they replicated more poorly in subsequent infections in wild type cells. This highlights for the first time that both the cellular and virion-associated pools of many of these proteins actively contribute to viral propagation. Altogether, these findings underscore the power and biological relevance of combining proteomics and RNA interference to identify novel host-pathogen interactions.
Introduction
Herpes simplex virus type 1 (HSV-1) virions are composed of a DNA core within an icosahedral capsid surrounded by a heterogeneous and poorly characterized layer of proteins called tegument, which is itself wrapped in an envelope. Many of the tegument components are critical at an early stage of the infection. For example, the binding of incoming viral capsids to microtubules and their transport to the nucleus are dependent on components of the tegument, including the viral proteins UL36 and UL37 [1], [2], [3], [4]. Furthermore, the incoming virion host shut off protein (Vhs; UL41) quickly down regulates the expression of several host proteins following viral entry [5], [6] while VP16, also a tegument protein, regulates the impending cascade of viral gene expression [7]. Interestingly, two other transactivators, namely ICP0 and ICP4, have also been reported in the viral tegument and may play an early role upon entry of the incoming virus [8]. In principle, the incorporation of these molecules should be beneficial to the virus to facilitate the next round of infection.
The importance and complexity of the HSV-1 tegument is illustrated by a recent mass spectrometry study of highly purified extracellular virions, which revealed they contain 23 potential viral teguments and up to 49 distinct cellular proteins [9]. This analysis showed that roughly half of the host proteins found in HSV-1 virions are proteins that had not yet been reported in any herpesviruses. In contrast, the presence of members of the annexin and heat shock protein families as well as cyclophilin A, DDX3X and components of the cytoskeleton have been documented in other Herpesviridae [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], suggesting a common function for these proteins. Moreover, host proteins have been documented in numerous other viral particles, including human immunodeficiency virions (HIV [21], [22], [23]), influenza [24], vesicular stomatitis virus [25] and vaccinia [26]. However their biological relevance is, at best, unclear.
The incorporation of host proteins within mature viral particles presumably benefits the virus and may for example jump-start its replication cycle. In contrast, viruses also avoid the incorporation of proteins that can inhibit their replication. One such case is the HIV protein Vif that binds to the host APOBEC3G protein to prevent its inclusion into nascent virions and prevent deamination of the viral genome by this RNA editing protein [21]. It is thus likely that the incorporation of host proteins in viruses is not random but rather a regulated process. Unfortunately, few of these studies have examined the relevance of this phenomenon in the course of an infection [23], [26], [27], [28], [29].
In the present study, we aimed to identify amidst the 49 host proteins found in HSV-1 mature particles those that influence its replication and proliferation. To this end, we designed and validated a functional screening assay using small interfering RNAs (siRNA). We now report that 15 of these proteins have a significant impact on HSV-1 propagation in cell culture, with limited siRNA-associated toxicity or effect on the propagation of another double-stranded DNA virus, the human Adenovirus type 5. Positive hits include proteins involved in vesicular transport, gene regulation, signaling and immunity. Furthermore, we show functional evidence that the incorporation of most of these proteins within mature virions is biologically relevant for HSV-1 infectivity.
Materials and Methods
Cells and viruses
143B tk− (ATCC CRL8303) and Vero (ATCC CCL81) cells were grown in Dulbecco's modified Eagle's medium (Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS, HyClone) and 2 mM L-glutamine (Invitrogen) in 5% CO2. 143B cells were also supplemented with 15 µg/ml 5-bromo-2 deoxyuridine (BrdU; Sigma) except prior to transfection and infection. The previously described HSV-1 K26GFP mutant (strain KOS) has been provided by Prashant Desai [30]. All viruses were propagated on BHK cells and titrated on Vero cells as previously described. Finally, the pIX-ΔE3 EGFP human Adenovirus type 5 [31] was propagated on 293 cells.
Antibodies
Primary antibodies were provided and diluted as follows: the anti-DDX3 R648 (1∶1000) was a kind gift from Dr A. Patel [32], while anti-VP16 (1∶1000) and the Remus V anti-HSV-1 antibodies were kindly provided by Dr H. Browne and Dr B. Sodeik respectively. Other antibodies were purchased from commercial sources, including anti-eIF4H (1∶1000, Cell signaling), anti-VP5 (1∶2000; Cedarlane), anti-ICP0 (1∶1000; Abcam), anti-ICP4 (1∶800; Abcam) and anti γ-tubulin (1∶1000; Sigma-Aldrich). Goat anti-mouse and anti-rabbit secondary antibodies were purchased from Jackson ImmunoReasearch.
Inhibition of HSV-1 life cycle by drugs
143B cells were seeded overnight in a 24-well plate at a 2.5×105 cells/well density. Cells were then mock treated or infected at a multiplicity of infection (MOI) of 5 with HSV-1 K26GFP. After adsorption, cells were washed twice in PBS and fed with complete DMEM containing MG132 (25 µM, Calbiochem), phosphonoacetic acid (PAA; 400 µg/ml; Sigma-Aldrich) or Brefeldin A (BFA; 5 µg/ml; Sigma-Aldrich). In the case of cells treated with MG132, the drug was first added 15 minutes prior to infection and during adsorption in order to prevent the transport of capsids to the nucleus. The infection proceeded for 24 hours at which time supernatants were collected and viral output measured as below (see HSV siRNA screen).
HSV siRNA screen
siGENOME SMARTpool and select ON-TARGETplus siRNAs targeting the human proteins previously identified in mature extracellular virions [9] were purchased from Dharmacon (Table 1; Thermo Fisher Scientific). The siRNA targeting the HSV-1 protein VP16 was used as a positive control [33] and a scrambled sequence of the VP16 siRNA was used as a negative control. Twenty-four hours prior to transfection, 143B cells were seeded in 24-well plates at a concentration of 5×104 cells/well. siRNA transfections were then performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions, using 25 or 100 nM/well of siRNA diluted in Opti-MEM or only Opti-MEM as control. The siRNA treated cells were incubated for 5 hours before the medium was replaced with complete DMEM and incubated for 43 more hours for a total of 48 hours of transfection. At that point, cells were mock treated or infected with HSV-1 K26GFP at a MOI of 5. After a one hour adsorption period, cells were washed twice in phosphate-buffered saline (PBS), complete DMEM was added to the wells and the cells were incubated for a further 24 hours. Cells and supernatants were then collected. The viruses in supernatants were inactivated using formaldehyde (Sigma), concentrated 2 hours at 18000× g and the resulting viral pellets resuspended in 100 µl of PBS pH 8. They were then transferred into a 96-well black bottom μClear plate (Greiner Bio-One) and their fluorescence was measured using a Gemini EM spectrofluorometer and SoftMax Pro 4 (Molecular Devices). Alternatively, the infectious virions were titrated on Vero cells as described before [34]. Each experiment was performed in triplicates and repeated three times.
Table 1. Proteins tested in the siRNA screen.
| Protein Name | Gene ID | Symbol | Catalog Number |
| 14-3-3 protein epsilon | 7531 | YWHAE | M-017302-02 |
| 14-3-3 protein gamma | 7532 | YWHAG | M-008844-00 |
| 14-3-3 protein zeta/delta | 7534 | YWHAZ | M-003332-01 |
| Alpha actin | 58 | ACTA1 | M-011194-00 |
| Beta actin | 60 | ACTB | M-003451-03 |
| Gamma actin | 71 | ACTG1 | M-005265-01 |
| Adaptor protein 1 - beta 1 subunit | 162 | AP1 | M-011200-00 |
| Adaptor protein 2 - beta 1 subunit | 163 | AP2 | M-003627-02 |
| Adaptor protein 3 - delta 1 subunit | 8943 | AP3 | M-016014-02 |
| Adaptor protein 4 - epsilon 1 subunit | 23431 | AP4 | M-021474-01 |
| Arf1 | 375 | ARF1 | M-011580-01 |
| Arf3 | 377 | ARF3 | M-011581-00 |
| Arf4 | 378 | ARF4 | M-011582-01 |
| Arf5 | 381 | ARF5 | M-011584-01 |
| ATP dependant RNA helicase DDX3X | 1654 | DDX3X | M-006874-01 |
| Casein kinase 2 | 1457 | CSNK2A1 | M-003475-03 |
| Cofilin 1 | 1072 | CFL1 | M-012707-00 |
| Cyclophilin A | 5478 | PPIA | M-004979-01 |
| Cystein-glycin rich protein 1 | 1465 | CSRP1 | M-011839-02 |
| Eukaryotic translation initiation factor 4H | 7458 | EIF4H | M-013054-00 |
| Golgi-associated, gamma adaptin ear containing, ARF binding protein 1 | 26088 | GGA1 | M-013694-01 |
| Golgi-associated, gamma adaptin ear containing, ARF binding protein 2 | 23062 | GGA2 | M-012908-01 |
| Golgi-associated, gamma adaptin ear containing, ARF binding protein 3 | 23163 | GGA3 | M-012881-00 |
| Glyceraldehyde-3-phosphate dehydrogenase | 2597 | GAPDH | M-004253-02 |
| Growth factor receptor bound protein 2 | 2885 | GRB2 | M-019220-00 |
| Heat Shock 70 protein 8a | 3312 | HSPA8 | M-017609-01 |
| Keratin 1 | 3848 | KRT1 | M-012865-02 |
| Keratin 10 | 3858 | KRT10 | M-023057-00 |
| Macrophage Migration Inhibitory Factor | 4282 | MIF | M-011335-01 |
| CD59b | 966 | CD59 | M-004537-01 |
| NM23Ac | 4830 | NME1 | M-006821-01 |
| NM23Bc | 4831 | NME2 | M-005102-02 |
| Peroxiredoxin-1 | 5052 | PRDX1 | M-010338-00 |
| Peroxiredoxin-2 | 7001 | PRDX2 | M-008178-03 |
| Profilin-1 | 5216 | PFN1 | M-012003-01 |
| Programmed cell death protein 6 | 10016 | PDCD6 | M-004440-03 |
| Rab2A | 5862 | RAB2A | M-010533-01 |
| Rab2B | 84932 | RAB2B | M-010370-00 |
| Rab4B | 53916 | RAB4B | M-008780-03 |
| Rab5A | 5868 | RAB5A | M-004009-00 |
| Rab5B | 5869 | RAB5B | M-004010-01 |
| Rab5C | 5878 | RAB5C | M-004011-01 |
| Rab6A | 5870 | RAB6A | M-008975-01 |
| Rab6B | 51560 | RAB6B | M-008548-02 |
| Rab6C | 84084 | RAB6C | M-009031-02 |
| Rab7A | 7879 | RAB7 | M-010388-00 |
| Rab10 | 10890 | RAB10 | M-010823-01 |
| Rab11A | 8766 | RAB11A | M-004726-02 |
| Rab11B | 9230 | RAB11B | M-004727-02 |
| Rab15 | 376267 | RAB15 | M-031564-00 |
| Rab33B | 83452 | RAB33B | M-008909-02 |
| Rab35 | 11021 | RAB35 | M-009781-00 |
| Rab-like protein 3 | 285282 | RABL3 | M-018128-01 |
| S100 calcium protein binding A11 | 6282 | S100A11 | M-012138-00 |
| SEC14-like 4 | 284904 | SEC14L4 | M-019246-00 |
| Tetraspanin 13 | 27075 | TSPAN13 | M-012516-01 |
| Transferrin Receptor (CD71) | 7037 | TFRC | M-003941-02 |
| Transgelin 2 | 8407 | TAGLN2 | M-011468-02 |
| Translocase of inner mitochondrial membrane 50 homolog | 92609 | TIMM50 | M-023692-01 |
| Triosephosphatase isomerase | 7167 | TPI1 | M-009776-02 |
| Ubiquitin C | 7316 | UBC | M-019408-01 |
| Ubiquitin-conjugating enzyme E2L 3 | 7332 | UBE2L3 | M-010384-01 |
HSP70 (Loret, 2008).
Membrane attack complex inhibition factor (Loret, 2008).
Nucleoside diphosphate kinase A/B (Loret, 2008).
Adenovirus siRNA screen
143B cells transfected for 48 hours with Lipofectamine alone or 25 nM siRNA as described above were mock treated or infected with the Adenovirus pIX-ΔE3 EGFP at an equal MOI for 48 hours. Supernatant were collected and viral DNA was purified using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich) according to the manufacturer's instructions. qPCR was performed with a Rotor-Gene 6000 (Corbett) using the FastStart SYBR Green Master (Roche Diagnostics). For the amplification, the primers pairs specific for EGFP were used: forward, 5′ ACG TAA ACG GCC ACA AGT TC 3′; reverse, 5′ AAG TCG TGC TGC TTC ATG TG 3′. The data are expressed in relative genome copy compared to the Lipofectamine-treated control and were pooled from three independent experiments. For these experiments, siRNA targeting VP16 served as negative control whereas a siRNA targeting the Adenoviral hexon protein [35] was used as positive control.
Viability assays
143B cells were mock treated with Lipofectamine alone or transfected with siRNA for 44 hours. Cells were washed twice and the medium replaced. alamarBlue (AbD Serotec) was subsequently added to the cells or to DMEM medium as a control according to the manufacturer's instruction. The cells were further incubated in accordance with the manufacturers' instructions before viability was measured by spectrofluorometry.
Two-steps infection assays
143B cells were transfected for 48 hours as in the siRNA screen or with Lipofectamine only. Cells were then infected for a further 24 hours with HSV-1 K26GFP at a MOI of 5. Supernatants, containing viruses depleted of individual host proteins, were collected and titrated on Vero cells. Fresh 143B cells were either mock treated or transfected with the siRNA for 48 hours. They were then infected with the depleted virions at an MOI of 0.1 for 48 hours. The supernatants form this second round of transfection/infection were collected and titrated once again on Vero cells.
Western blotting
43B cells were transfected for 48 hours with Lipofectamine alone or siRNAs then were mock treated or infected with K26GFP at a MOI of 5 for 24 hours. After a centrifugation of 5 min at 1000×g, the cells were collected, washed in PBS and lysed by 3 cycles of rapid freeze-thaw. Infected cell's supernatants were concentrated for 2 hours at 18 000× g and resuspended in 10 µl of PBS. Unless specified otherwise, equal amount of proteins were analyzed by SDS-PAGE [9] [12]. Antibodies against the viral proteins VP5 and VP16 or the human DDX3X, ARF1, eIF4H and γ-tubulin proteins were used to blot the respective proteins. When indicated, protein expression levels were quantified using ImageJ (version 1.45b).
Statistical analysis
Fluorescence and virus titers were normalized to the values obtained for the controls as indicated in the figure legends. Bilateral Student T tests were performed using GraphPad Prism 5 (GraphPad Software).
Results
Validation of the siRNA-based assay
Our strategy to first examine the physiological relevance of the host proteins identified in mature HSV-1 virions involved a novel viral propagation assay based on RNA interference and HSV-1 K26GFP, a wild-type virus in which the Aequorea victoria green fluorescent protein (GFP) is fused to the capsid protein VP26 [30] (Figure 1A). This approach enabled us to easily and rapidly measure viral output and to quantitatively screen many targets without resorting to the classical but time-consuming and cumbersome plaque assays. We selected a human cell line for this screen because it is the HSV-1 natural reservoir, it is compatible with our previous proteomics report [9] and a human siRNA library is commercially available. We opted for the human osteosarcoma-derived 143B cell line since it is more resistant to the cytopathic effects of the virus and produces significantly greater quantities of extracellular viruses upon infection than the HeLa cells originally used in our proteomic study ([34], [36] and data not shown). In addition, 143B cells have a greater than 80% siRNA transfection rate (data not shown). Cell plating density, infection conditions, harvesting time, assay buffers, plate format and parameters of the plate reader software were all extensively optimized (data not shown) to ensure that quantification of the virus from the supernatant was accurate, linear and sufficiently sensitive to detect extracellular virions (Figure 1B).
Figure 1. Screening method.
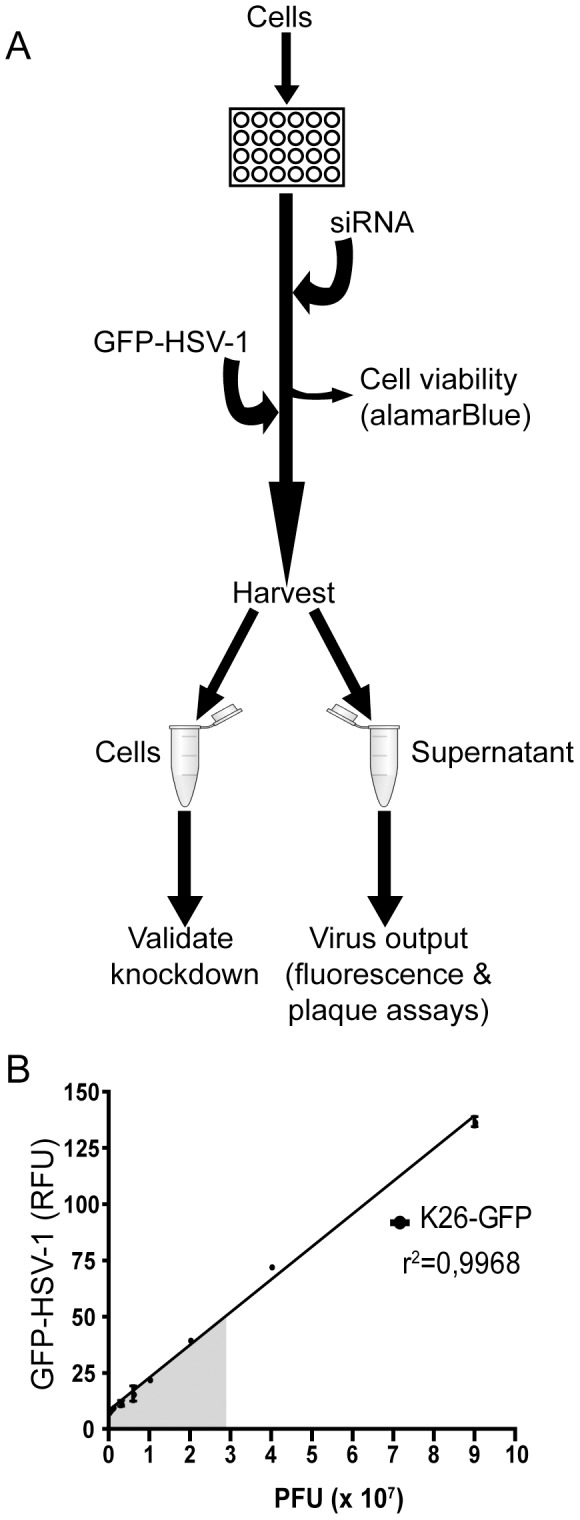
A) 143B cells were seeded in 24-well plates 24 hours prior to transfection. Cells were then transfected with siRNA and incubated 48 hours before being infected with HSV-1 K26GFP for an additional 24 hours. Supernatants were collected and their fluorescence was measured using a Gemini EM spectrofluorometer. As a cytotoxicity control, cell viability was assessed in parallel using alamarBlue 2 days post-transfection. In addition, the cells were lysed and used for Westerns to validate the siRNA knockdowns. B) The fluorescence of pre-titered infectious HSV-1 K26GFP particles serially diluted in PBS was quantified using a spectrofluorometer. The fluorescence obtained was linear with the titers through the entire selection (r2 = 0.9968), thus demonstrating the advanced sensitivity of the device. The gray area denotes the typical of values obtained in the RNA interference screen below. RFU: relative fluorescence units.
We next sought to validate that the assay could indeed detect the impact of known inhibitors of the HSV-1 life cycle. We therefore pretreated cells with MG132, an inhibitor of the proteasome that perturbs the post-entry delivery of HSV-1 to the nucleus [37], phosphonoacetic acid (PAA) which prevents viral replication [38] and brefeldin A (BFA) which arrests viral egress of newly synthesized viral particles [39], [40]. As expected, HSV-1 output was drastically lower in drug-treated cells than in untreated ones (Figure 2A). As a second control, cells were transfected with siRNA targeting the HSV-1 protein VP16 (UL48), since its inhibition by siRNA is known to efficiently reduce VP16 expression and viral production [33]. Cells were thus transfected for 48 hours prior to infection with the single most effective siRNA targeting VP16 [33] or with Lipofectamine only. A scrambled sequence of the VP16 siRNA was used as negative control (scVP16) Since the scVP16 siRNA does not have any homology to any human or viral sequence as determined by blast (data not shown), it also served as a non-targeting control. The knockdown of VP16 expression was assessed by Western blotting and quantified with ImageJ, which revealed the high efficacy of the siRNA employed (up to 88% inhibition; Figure 2B). Though the inhibition of VP16 was not absolute, the extracellular viral yields were significantly lower upon treatment with siRNA against VP16 than for cells treated with the transfection agent only or the scrambled control (Figure 2C). Taken together these results demonstrate the sensitivity and specificity of the assay.
Figure 2. Validation of the assay.

143B cells were seeded in 24-well plates and infected with HSV-1 K26GFP and A) treated with drugs targeting HSV-1 post-entry (MG132), replication (PAA) and egress (BFA). B–C) As above except that cells were instead transfected for 48 hours prior to infection with Lipofectamine only, a scrambled version of the VP16 siRNA or siRNA targeting VP16. For panel A and C, the fluorescent viruses in the supernatants were concentrated by centrifugation at 24 hpi and quantified by spectrofluorometry. For panel B, 25 µg of proteins from cell lysates were tested by Western blotting for VP16 knockdown. γ-tubulin was used as loading control. The error bars show the standard errors of the mean (SEM) of three independent experiments. Bilateral Student's T-tests were performed to detect significant hits compared to the siRNA free control (***: p<0.0001).
Assessment of host proteins critical for the viral infection
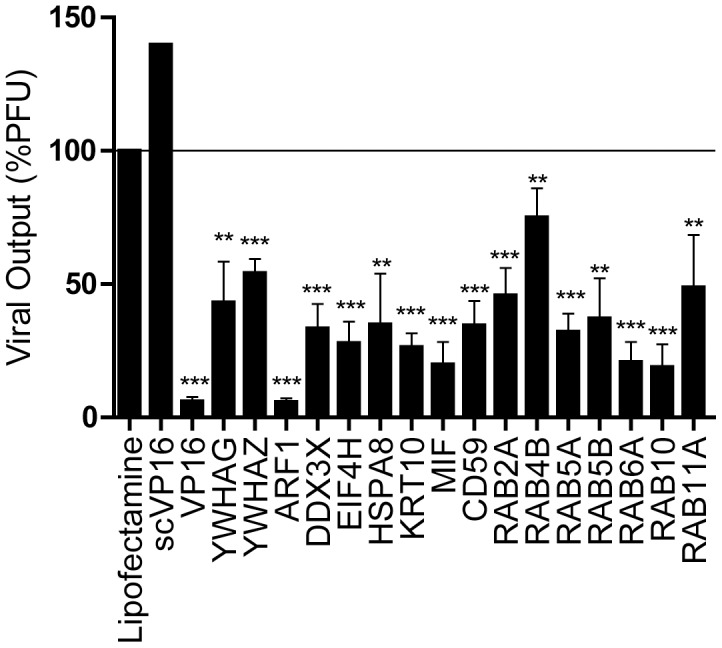
To evaluate the functional relevance of the cellular proteins identified in mature extracellular virions, we examined them using the above siRNA screen. Since some of the peptides that were identified in the course of the proteomic study are common to different protein isoforms, we screened them together but also individually, thus lengthening our original list of proteins to 55 (Table 1). Furthermore, we added two additional specificity controls that were not in our initial proteomics study. They are the adaptor proteins (AP1 to AP4) and GGA proteins (1 to 3), all molecules regulating protein transport and relevant in the view of the many transport molecules reported here. The siRNA targeting VP16 and its scrambled version (scVP16) were once again used as controls. For each of the human targets identified in our proteomic study [9], we used a pool of four different siRNA duplexes to ensure the most effective knockdowns. These siRNA pools are commercially available and predefined by the manufacturer according to their proprietary algorithm (see Table 1 for catalogue numbers). Cells were transfected in triplicates with these siRNA for 48 hours, to allow endogenous levels of the host proteins to significantly decrease, and subsequently infected with GFP tagged HSV-1. Twenty-four hours later, viral yields were measured. Though both the intracellular and extracellular viral pools could be quantified, we opted to only probe the latter since free VP26-GFP is inevitably present in infected cells and would contribute to a higher background. Each experiment was normalized to the mean fluorescence obtained in supernatants from cells transfected with the transfection agent only and results from three independent screens were compiled. This method allowed us to identify several cellular proteins whose absence statistically altered viral production (p<0.05 or better; Figure 3). To evaluate the toxicity of the siRNA reagents and rule out nonspecific effects, the viability of the cells was monitored in parallel using the redox indicator alamarBlue, which quantitatively monitors cell metabolism and, consequently, overall cell viability (Figure 3, dots). Among the proteins that affected viral output, only the inhibition of ubiquitin C resulted in high cytotoxicity under our experimental conditions and was not pursued further. It is relevant to note that targeting some of the proteins with siRNA resulted in lower cellular viability without any impact on viral production (e.g. KRT1 and RAB5C), further highlighting the genuine implication of the other cellular proteins on the HSV-1 viral life cycle.
Figure 3. siRNA screen against the host proteins identified in mature extracellular HSV-1 virions.

143B cells were transfected for 48 hours with Lipofectamine alone or with siRNA duplexes targeting the indicated cellular proteins (gene names are indicated on the left). Forty-eight hours later, cellular viability was assessed by alamarBlue or cells were infected in parallel with K26GFP at a MOI of 5 for a further 24 hours. The fluorescence in the supernatant was quantified using a spectrofluorometer. The data was normalized to the mean value obtained with Lipofectamine only samples. Bilateral Student's T-tests were performed to detect significant hits compared to the Lipofectamine only control (*: p<0.05, **: p<0.001, ***: p<0.0001). The solid line represents the relative fluorescence intensity of the Lipofectamine treated samples while the dotted lines indicate ±25% viability. The error bars show the standard errors of the means (SEM) of three independent experiments, each done in triplicates.
Given that the GFP-based assay used in this study detects total viral particles rather than infectious virions, single positive isoforms were confirmed by conventional plaque assays to measure viral titers following siRNA treatment. 143B cells were transfected with siRNA, infected as before and the infectious particles in the supernatant were titrated on Vero cells. As expected, infected cells treated with the scrambled control produced similar levels of extracellular infectious particles as the Lipofectamine-treated sample. Moreover, all the siRNA tested lead to a statistically significant reduction in viral yields and were thus fully coherent with the GFP detection assay (Figure 4). Since Rab4B inhibition reduced viral yields by less than 25%, it was therefore excluded from further analysis. Taken together, these data validated the GFP-based readouts and show that the individual depletion of host proteins incorporated in mature HSV-1 virions impacts both total and infectious particle yields.
Figure 4. Impact of host protein depletion on infectious particles.
143B cells were transfected with siRNA and infected with K26GFP as before for the samples that were positive hits in Figure 3. Supernatants were individually collected and their viral content titrated on Vero cells. The titers were normalized to the mean value obtained with samples treated with Lipofectamine only. The error bars show the standard errors of the means (SEM) of three independent experiments, each done in duplicates. Bilateral Student's T-tests were performed to detect significant hits compared to the Lipofectamine only control (**: p<0.001, ***: p<0.0001). PFU: Plaque forming units.
Thus far, our data support the potential involvement of several host proteins in the HSV-1 life cycle. To evaluate if these cellular proteins specifically affect HSV-1 propagation rather than non-specifically perturb cell metabolism, 143B cells were transfected with siRNAs targeting the positive hits and infected with human Adenovirus type 5, another double-stranded DNA virus that replicates in the nucleus. In this experiment, the siRNA targeting the HSV-1 protein VP16 served as negative control whereas a siRNA targeting the hexon protein [35] was used as positive control. As expected, siRNA targeting the adenoviral hexon reduced viral yields (68% inhibition) while siRNA targeting VP16 did not (Figure 5). Interestingly, only Arf1, Rab6A and Rab10 statistically influenced Adenovirus proliferation. This strongly suggests that the positive hits found in our screen specifically modulated HSV-1 proliferation and supports the conclusion that the effects observed were not due to some general cellular defects.
Figure 5. Impact of the HSV-1 virion-incorporated cellular proteins on the life cycle of Adenoviruses.
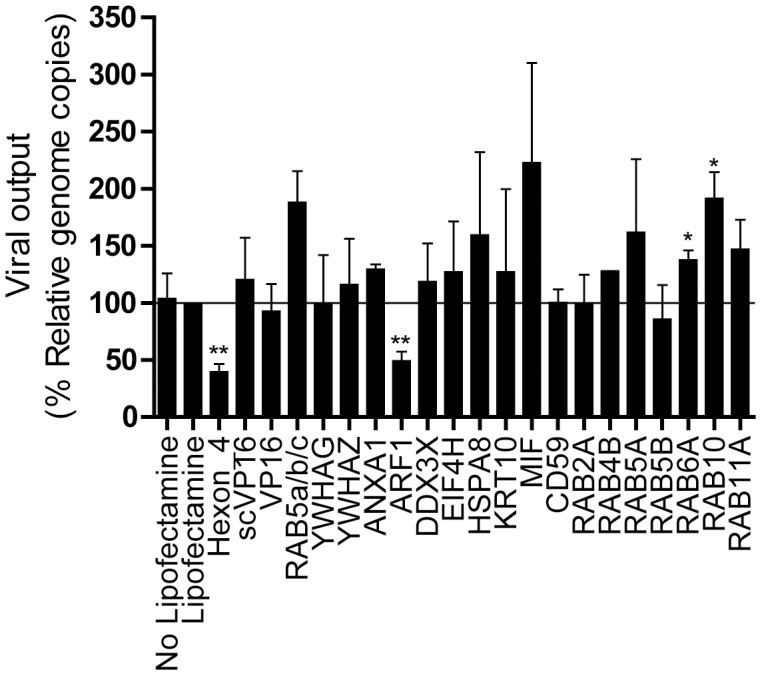
143B cells were transfected with siRNA and infected with HAd pIX ΔE3 GFP as in materials and methods for the samples that were positive hits in Figure 3. Supernatants were individually collected and their viral content titrated by qPCR. The titers were normalized to the mean value obtained with samples treated with Lipofectamine only. The error bars show SEM of three independent experiments. Bilateral Student's T-tests were performed to detect significant hits compared to the Lipofectamine only control (*: p<0.05, **: p<0.001).
Importance of the proteins present in the mature virions
Several viruses have been reported to incorporate cellular proteins but the significance of this incorporation is often unclear. There are various reasons why cellular proteins could be incorporated in virions. Some of them may simply be incorporated in virions because they are abundant and/or happen to be at the right place and time but are not valuable to the virus. A second possibility is they may be included as a consequence of their function during viral assembly, intracellular transport or egress. In such cases, the proteins would not necessarily be important to initiate the next round of infection. Finally, some proteins may specifically be required to promote the infection of neighboring cells. In this exciting scenario, the depletion of the cellular proteins from the viral particles should stop or delay the next replication cycle, much the same way VP16 is incorporated into mature virions to act early during the infection as transactivator of immediate early genes [7]. We thus wished to directly address the functional significance of the virion-associated host proteins.
Since the virion-incorporated host proteins are present in both mature virions and the cell, the respective contribution of both of these pools to the viral life cycle was assessed. To this end, a two-step approach was employed. In the first step, viruses individually depleted for each of the positive targets identified above were produced on siRNA-treated cells. As controls, we also produced virions on Lipofectamine-treated cells or cells treated with VP16 or scVP16 siRNAs. The efficacy of the depletion was then examined by Western blotting for four of the proteins. As shown in figure 6, the siRNA strongly reduced the amount of the proteins in extracellular virions . It was thus possible to produce stocks of virions depleted on a given host protein. These viruses were then titrated and used to infect a new monolayer of cells (second step; Figure 7A). To reduce the risk of complementation between depleted virions and wild type virions produced by cells that may not have taken up the siRNA, a low MOI of 0.1 was employed. To sort out the importance of the viral and cellular pools of the proteins, all four combinations of wild type/depleted virions and wild type/depleted cells were probed. As expected, the infection of VP16 siRNA-treated cells with wild type virus harvested from Lipofectamine-treated cells strongly reduced viral yields, in accordance with the idea that newly expressed VP16 does regulate the viral life cycle (Figure 7B). Though not as extensive, the infection of Lipofectamine-treated cells with VP16-depleted virions also significantly reduced viral production, consistent with past reports that the virion associated pool of VP16 is actively involved in the early stages of the infection process [7]. Moreover, the depletion of both newly expressed and virion-associated VP16 (i.e. infection of VP16 siRNA-treated cells with VP16-depleted virions) further reduced viral output compared to the virion-only depleted sample, hence reiterating the importance of both newly expressed and virion-associated VP16 pools during the HSV-1 infection. Interestingly, the individual depletion from the mature virions of 13 of the 15 target tested statistically reduced viral production in Lipofectamine-treated cells, 8 of which did so by at least 50% (DDX3X, HSPA8, KRT10, MIF, RAB5A, RAB6A, RAB10 and YWHAZ (Figure 7B). One notable case was Arf1, for which the cellular pool clearly participated in greater proportion to viral propagation, despite the significant contribution of the viral pool of this protein. Given the above findings occurred in the presence of a normal complement of host proteins, this reemphasized the specific involvement of these proteins in HSV-1 replication (Figures 3 and 4) rather than an indirect metabolic defect. Altogether, this directly demonstrates for the first time that both the cellular and the virion-incorporated pools of these proteins do indeed participate in the HSV-1 replication cycle.
Figure 6. Virions harvested from siRNA treated cells are efficiently depleted of the siRNA-targeted protein.
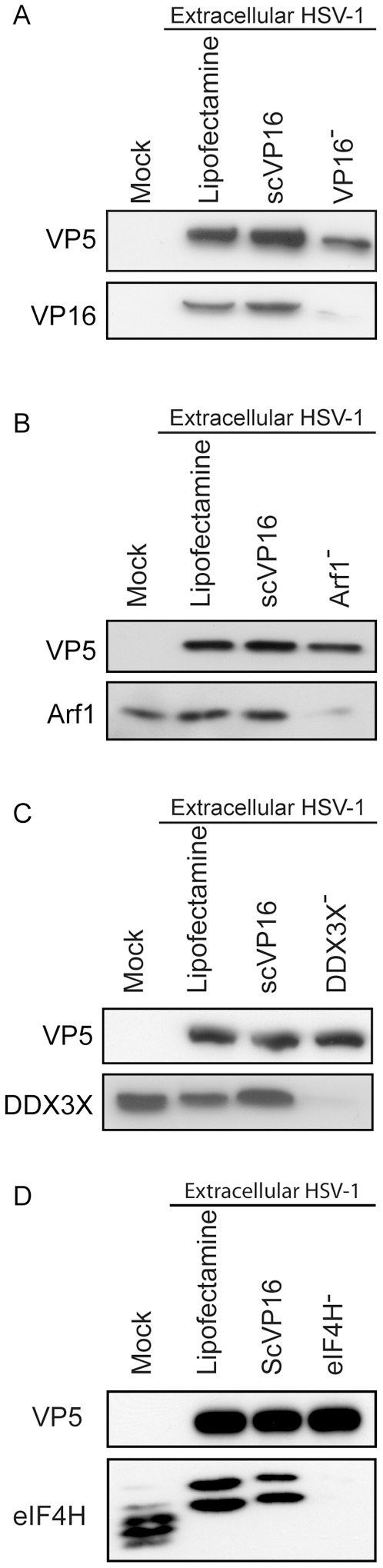
143B cells were transfected with Lipofectamine only or siRNAs against the control VP16 or the positive hits Arf1, DDX3X and eIF4H. They were then infected at a MOI of 5 with HSV-1 K26GFP and the supernatants collected at 24 hours. Concentrated supernatants corresponding to equal amounts of VP5 were analyzed and 20 µg of Lipofectamine-treated cell lysates were used as control. The reduced levels of A) VP16, B) ARF1, C) DDX3X and D) eIF4H in newly produced virions in the supernatants were confirmed by Western blotting.
Figure 7. Importance of the virion-incorporated pools of the proteins.

A) As described in materials and methods, a two step approach was used. In the first step, virions were harvested from 143B cells individually treated with siRNA against 15 cellular proteins (DDX3X, MIF, RAB5A, HSPA8, KRT10, RAB6A, RAB10, YWHAZ, RAB5B, RAB2A, CD59, YWHAG, ARF1, eIF4H and RAB11A) and infected with wild type HSV-1 K26GFP at a MOI of 5 for 24 hpi. As control, viruses produced on Lipofectamined-treated cells were also produced. The extracellular virions were collected, titrated and used to infect a fresh layer of 143B cells transfected or not with siRNA at an MOI of 0.1 for 48 hours (second Step). The presence of the cellular proteins in the virus or in the cells is indicated with +/− under each condition of infection. B) The supernatants form this second infections were collected and titrated on Vero cells. Viral titers were normalized to the mean value of Lipofectamine-treated cells infected with wild type virus (+/+). The presence or absence of the cellular proteins in the cell, in the virus or in both is presented with + and − under each graphic. To statistically evaluate if the viral pool of the host proteins modulate the propagation of HSV-1, the depleted virions (3rd bar) were compared to the Lipofectamine only control (1st bar) using bilateral Student's T-tests (*: p<0.05, **: p<0.001, ***: p<0.0001).
Discussion
Viruses are obligatory intracellular parasites that depend on many cellular functions to complete their life cycle. These interactions are complex and often poorly understood. While the contribution of host proteins to HSV-1 has been partially elucidated, genomic screens with other viruses suggest this may only be the tip of the iceberg [41], [42], [43], [44], [45]. Recently, we reported the detection of up to 49 distinct cellular proteins within mature extracellular HSV-1 virions [9], adding an additional layer of complexity to this scenario. Since some of these proteins may play pivotal roles during the viral life cycle, they constitute prime targets to identify and characterize novel host-pathogen interactions in the context of an HSV-1 infection.
The siRNA assay reported in this study is sensitive, rapid, linear and correlates with classical plaque assays (Figures 1 to 4). The data obtained by both fluorometry and plaque assays suggest that at least 15 of the cellular proteins tested are involved in the HSV-1 replication cycle in cell culture (Figures 3 and 4). Moreover, the incorporation into mature extracellular virions of most of these proteins seems necessary to ensure an optimal new round of infection (Figure 7). It should be noted that overexpression of several of these proteins did not influence viral yields, presumably because they are not rate limiting in the cell (data not shown). These results are unlikely due to side effects of the depletion of the host proteins within the cells or to off-targeting effects of the siRNA. Indeed, first of all, only limited cytotoxicity was noted under our experimental conditions, which is consistent with other genomic screens [44], [46]. Secondly, even when siRNA were somewhat toxic (e.g. KRT1 and Rab5C), this did not cause a reduction of viral yields. Only the inhibition of ubiquitin C, which did perturb HSV-1 output, correlated with high siRNA-associated cytotoxicity and was therefore not considered further, though it may still be relevant for the HSV-1 replication cycle (Figure 3). Third, we could single out specific protein isoforms that affect viral proliferation, while other isoforms of the same proteins had no impact. For instance, inhibition of Arf1 significantly reduced viral production while the inhibition of Arf3, 4 or 5 failed to do so, hinting at the specificity of the assay and the lack of pleiotropic effect of the siRNAs (Figure 3). Forth, the same siRNA, with the sole exception of Arf1, Rab6A and Rab10, did not significantly impact the replication of Adenovirus type 5 (Figure 5). In that case, it remains to be seen if these proteins are essential to viral growth under our experimental conditions or if both HSV-1 and Ad5 propagation depends on these host proteins. Finally, depleting these host proteins from the virions, but not the cell, still had an impact on viral yields (Figure 7). We therefore conclude that these molecules are most likely involved in the viral life cycle and that our observations cannot be credited to some pleiotropic effect.
It is important to note that the actual number of proteins regulating the HSV life cycle in the original RNA interference screen is likely higher than reported in the present study. Missed targets could include cellular proteins with long half-lives unperturbed by the siRNA, proteins with more subtle impacts on the virus as well as proteins that may be complemented by their virion-associated counterpart or by other cellular proteins. Consistent with this latter scenario, it is worth noting that depletion of either the cellular and virion pools of the proteins often affected viral propagation. Furthermore, the screen only considers host proteins involved in HSV-1 replication in tissue culture and may exclude some virulence factors and immune or latency-related proteins. Finally, the screen initially measured GFP output and quantified total viral particle release rather than infectious virions as determined by plaque assays. It is thus possible that some siRNA reduced the production of infectious viruses without affecting the total amount of viral particles, as noted when VP16 is inactivated [47]. Despite these limitations, the positive rate from this study is very high at 31% (i.e. 15/49). For comparison, genome-wide siRNA screens for HIV, influenza and West Nile virus have positive rates below 1.5% [41], [42], [43], [44], [45], [48]. The data thus constitute a strong validation of the relevance of our strategy to unmask novel cellular players by combining proteomics [9] and a targeted siRNA screen (this study). Furthermore, this study and those of others [5], [33], [49], [50], [51], [52] highlight the potency of siRNA as a useful tool to study HSV-1 host-pathogen interactions, despite of the ability of the virus to downregulate RNA-induced gene silencing [53].
The current GFP-based assay is less sensitive than traditional plaque assays but is a good complementary approach to screen large numbers of targets. While it is clear that the reductions are not as spectacular as when some viral genes are deleted, important differences have to be considered when analyzing the impact of host proteins instead of viral proteins. For instance, host proteins are often complemented by another gene copy, another isoform, a parallel pathway or other means, which implies that smaller effects upon depletion should not be surprising. This is even true for some HSV-1 genes, for which multiple deletions are required to see a significant phenotype. Moreover, cellular proteins finely exert their actions with impacts typically in the lower fold range (2–4 folds) rather than logs (10–100 folds). In agreement with this view, a study by Hill and colleagues [54] showed that HSV-1 itself modulates the expression of 3,123 host proteins by only 1.4 to 14.2 folds, changes that are presumably beneficial for the virus. Our results are also in the same range (i.e. less than a log difference) observed by others in recent studies analyzing the impact of cellular proteins on HSV or HIV proliferation [55], [56], [57], [58], [59], [60], [61]. Multi-log inhibitions where therefore not expected in the present study but it does naturally make it more difficult to ascribe a functional role to present host proteins. However, the significant, albeit moderate, impact of depleting the host proteins in the virions themselves, while leaving the cellular pools intact, strongly supports their contribution to the HSV-1 replication cycle. The present data suggest that many of the tested proteins are indeed subtle modulators of viral production.
The positive hits found in this study identified several potential players in the HSV-1 life cycle. Many of these proteins are involved in pathways that are most likely used in the infection process (Figure 8). Among them, several Rab GTPases and other intracellular transport components as well as proteins involved in signal transduction were identified, some of which have already been proven necessary for viral production. For instance, it has recently been shown that Rab6A is necessary for the cellular trafficking of HCMV viral protein pp150 and efficient virus assembly [57], while Rab11 modulates the intracellular transport of influenza genome to the plasma membrane [62]. Similarly, Rab1 and Rab43 have been implicated in HSV-1 secondary envelopment [55]. It is thus highly possible that some of the proteins identified in the present study are involved in the numerous transport events occurring in the course of HSV-1 infection. The present screen is therefore very useful to pursue novel host-pathogen interactions in the context of HSV-1 infections. Moreover, 6 of the proteins; 14-3-3 ζ/δ (YWHAZ), Arf1 (ARF1), DDX3X, eIF4H, keratin 10 (KRT10) and CD59 have previously been found in HCMV, KSHV and/or PRV viral particles in addition to HSV-1 [10], [12], [13], [14], [17], suggesting that they might have a common roles throughout the herpesvirus family.
Figure 8. Cellular functions of virion-incorporated host proteins.
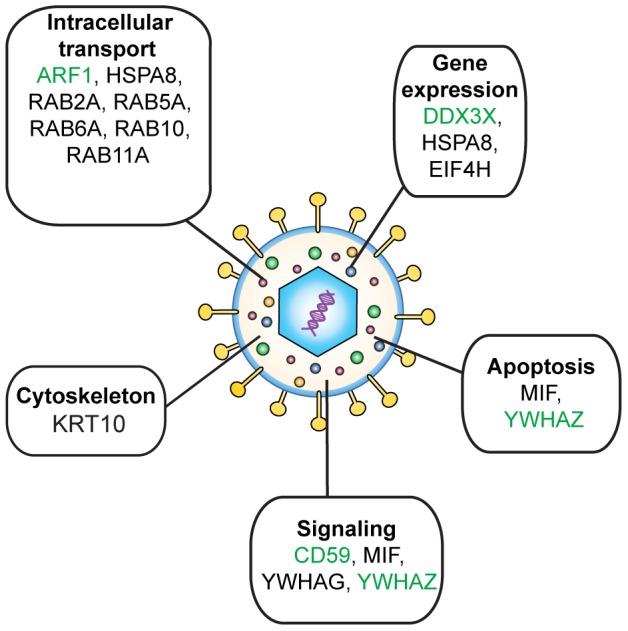
Several of the positive hits of our functional screen are implicated in pathways that are most likely relevant for the virus. While all of them were found in HSV-1 mature viral particles, many of these proteins (in green) have also been found incorporated into other herpesviruses, suggesting a common role for these proteins throughout the family [12], [13], [14], [17].
Many studies have reported the incorporation of host proteins within mature virions but very few have addressed the significance of the incorporated fraction of cellular proteins in viral infectivity. To verify the importance of the pool of host proteins present in the extracellular virion, we depleted these proteins from virions by RNA interference and used them to re-infect untreated cells. This method enabled us to identify for the first time 8 proteins whose absence from the virions reduced viral production by at least 50%, with evidence of contributions by another 5 host proteins (Figure 7). This highlights the very close relationship between the virus and its host. In contrast, the depletion of some host proteins (i.e. Rab11A and YWHAG) in the virions caused no significant impact on viral production. It may be relevant to highlight that the present study was performed in 143B cells and it is likely that the pool of host proteins incorporated in mature virions or their roles varies among cell types. Though difficult to perform, a comprehensive study of host proteins in virions produced on neuronal cells would be particularly interesting.
The present study highlights the very close relationship between HSV-1 and its host. However, the full extent of host-pathogen interactions between HSV-1 and the cell remains elusive at the moment. Our approach constitutes an innovative and powerful strategy to identify potential novel cellular proteins that modulate the HSV-1 life cycle. The characterization of their functions at the molecular level is therefore of the upmost interest. Since all of these proteins have previously been detected in mature extracellular HSV-1 virions [9], they constitute a new class of proteins to study as potential therapeutic targets.
Acknowledgments
We are indebted to Drs Prashant Desai, Arwind Patel, Takeshi Sekiguchi, Helena Browne and Beate Sodeik for antibodies, viruses and cell lines. We would also like to thank members of the laboratory for their critical reading of the manuscript and suggestions.
Funding Statement
This work was funded by a grant from the Canadian Institutes of Health Research (MOP 82921). YY was supported by a Canada Frederick Banting and Charles Best studentship from the CIHR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Döhner K, Wolfstein A, Prank U, Echeverri C, Dujardin D, et al. (2002) Function of dynein and dynactin in herpes simplex virus capsid transport. Mol Biol Cell 13: 2795–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sodeik B, Ebersold MW, Helenius A (1997) Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol 136: 1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luxton GW, Haverlock S, Coller KE, Antinone SE, Pincetic A, et al. (2005) Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc Natl Acad Sci U S A 102: 5832–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, et al. (2010) Plus- and Minus-End Directed Microtubule Motors Bind Simultaneously to Herpes Simplex Virus Capsids Using Different Inner Tegument Structures. PLoS Pathog 6: e1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sarma N, Agarwal D, Shiflett LA, Read GS (2008) Small interfering RNAs that deplete the cellular translation factor eIF4H impede mRNA degradation by the virion host shutoff protein of herpes simplex virus. J Virol 82: 6600–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lam Q, Smibert CA, Koop KE, Lavery C, Capone JP, et al. (1996) Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J 15: 2575–2581. [PMC free article] [PubMed] [Google Scholar]
- 7. Weinheimer SP, Boyd BA, Durham SK, Resnick JL, O'Boyle DR 2nd (1992) Deletion of the VP16 open reading frame of herpes simplex virus type 1. J Virol 66: 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loret S, Lippé R (2012) Biochemical Analysis of ICP0, ICP4, UL7 and UL23 Incorporated into Extracellular Herpes Simplex Type 1 Virions. J Gen Virol 93: 624–634. [DOI] [PubMed] [Google Scholar]
- 9. Loret S, Guay G, Lippé R (2008) Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82: 8605–8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lippé R (2012) Deciphering novel host–herpesvirus interactions by virion proteomics. Front Microbio 3: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bechtel JT, Winant RC, Ganem D (2005) Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol 79: 4952–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, et al. (2004) Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78: 10960–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spear G, Lurain N, Parker C, Ghassemi M, Payne G, et al. (1995) Host cell-derived complement control proteins CD55 and CD59 are incorporated into the virions of two unrelated enveloped viruses. Human T cell leukemia/lymphoma virus type I (HTLV-I) and human cytomegalovirus (HCMV). The Journal of Immunology 155: 4376–4381. [PubMed] [Google Scholar]
- 14. Kramer T, Greco TM, Enquist LW, Cristea IM (2011) Proteomic Characterization of Pseudorabies Virus Extracellular Virions. J Virol 85: 6427–6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Del Rio T, Ch'ng TH, Flood EA, Gross SP, Enquist LW (2005) Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J Virol 79: 3903–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baldick CJ Jr, Shenk T (1996) Proteins associated with purified human cytomegalovirus particles. J Virol 70: 6097–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu FX, Chong JM, Wu L, Yuan Y (2005) Virion proteins of Kaposi's sarcoma-associated herpesvirus. J Virol 79: 800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, et al. (2004) Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A 101: 16286–16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dry I, Haig DM, Inglis NF, Imrie L, Stewart JP, et al. (2008) Proteomic analysis of pathogenic and attenuated alcelaphine herpesvirus 1. J Virol 82: 5390–5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kattenhorn LM, Mills R, Wagner M, Lomsadze A, Makeev V, et al. (2004) Identification of proteins associated with murine cytomegalovirus virions. J Virol 78: 11187–11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cantin R, Methot S, Tremblay MJ (2005) Plunder and Stowaways: Incorporation of Cellular Proteins by Enveloped Viruses. J Virol 79: 6577–6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ott DE (2008) Cellular proteins detected in HIV-1. Reviews in Medical Virology 18: 159–175. [DOI] [PubMed] [Google Scholar]
- 23. Gurer C, Cimarelli A, Luban J (2002) Specific Incorporation of Heat Shock Protein 70 Family Members into Primate Lentiviral Virions. J Virol 76: 4666–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shaw ML, Stone KL, Colangelo CM, Gulcicek EE, Palese P (2008) Cellular Proteins in Influenza Virus Particles. PLoS Pathog 4: e1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moerdyk-Schauwecker M, Hwang SI, Grdzelishvili VZ (2009) Analysis of virion associated host proteins in vesicular stomatitis virus using a proteomics approach. Virol J 6: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krauss O, Hollinshead R, Hollinshead M, Smith GL (2002) An investigation of incorporation of cellular antigens into vaccinia virus particles. J Gen Virol 83: 2347–2359. [DOI] [PubMed] [Google Scholar]
- 27. Lodish HF, Porter M (1980) Specific incorporation of host cell surface proteins into budding vesicular stomatitis virus particles. Cell 19: 161–169. [DOI] [PubMed] [Google Scholar]
- 28. Zhang C, Xue C, Li Y, Kong Q, Ren X, et al. (2010) Profiling of cellular proteins in porcine reproductive and respiratory syndrome virus virions by proteomics analysis. Virology Journal 7: 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sokolskaja E, Sayah DM, Luban J (2004) Target Cell Cyclophilin A Modulates Human Immunodeficiency Virus Type 1 Infectivity. J Virol 78: 12800–12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Desai P, Person S (1998) Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J Virol 72: 7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Le LP, Everts M, Dmitriev IP, Davydova JG, Yamamoto M, et al. (2004) Fluorescently labeled adenovirus with pIX-EGFP for vector detection. Mol Imaging 3: 105–116. [DOI] [PubMed] [Google Scholar]
- 32. Angus AG, Dalrymple D, Boulant S, McGivern DR, Clayton RF, et al. (2010) Requirement of cellular DDX3 for hepatitis C virus replication is unrelated to its interaction with the viral core protein. J Gen Virol 91: 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang YQ, Lai W, Li H, Li G (2008) Inhibition of herpes simplex virus type 1 by small interfering RNA. Clin Exp Dermatol 33: 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turcotte S, Letellier J, Lippé R (2005) Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J Virol 79: 8847–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eckstein A, Grossl T, Geisler A, Wang X, Pinkert S, et al. (2010) Inhibition of adenovirus infections by siRNA-mediated silencing of early and late adenoviral gene functions. Antiviral Res 88: 86–94. [DOI] [PubMed] [Google Scholar]
- 36. Campadelli G, Brandimarti R, Di Lazzaro C, Ward PL, Roizman B, et al. (1993) Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the Golgi apparatus after infection with herpes simplex virus 1. Proc Natl Acad Sci U S A 90: 2798–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delboy MG, Roller DG, Nicola AV (2008) Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J Virol 82: 3381–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hay J, Brown SM, Jamieson AT, Rixon FJ, Moss H, et al. (1977) The effect of phosphonoacetic acid on herpes viruses. J Antimicrob Chemother 3 Suppl A: 63–70. [DOI] [PubMed] [Google Scholar]
- 39. Dasgupta A, Wilson DW (2001) Evaluation of the primary effect of brefeldin A treatment upon herpes simplex virus assembly. J Gen Virol 82: 1561–1567. [DOI] [PubMed] [Google Scholar]
- 40. Cheung P, Banfield BW, Tufaro F (1991) Brefeldin A arrests the maturation and egress of herpes simplex virus particles during infection. J Virol 65: 1893–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, et al. (2010) Human host factors required for influenza virus replication. Nature 463: 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, et al. (2008) Identification of host proteins required for HIV infection through a functional genomic screen. Science 319: 921–926. [DOI] [PubMed] [Google Scholar]
- 43. Karlas A, Machuy N, Shin Y, Pleissner K-P, Artarini A, et al. (2010) Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463: 818–822. [DOI] [PubMed] [Google Scholar]
- 44. König R, Zhou Y, Elleder D, Diamond TL, Bonamy GMC, et al. (2008) Global Analysis of Host-Pathogen Interactions that Regulate Early-Stage HIV-1 Replication. Cell 135: 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, et al. (2008) RNA interference screen for human genes associated with West Nile virus infection. Nature 455: 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tai AW, Benita Y, Peng LF, Kim S-S, Sakamoto N, et al. (2009) A Functional Genomic Screen Identifies Cellular Cofactors of Hepatitis C Virus Replication. Cell host & microbe 5: 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ace CI, McKee TA, Ryan JM, Cameron JM, Preston CM (1989) Construction and characterization of a herpes simplex virus type 1 mutant unable to transinduce immediate-early gene expression. J Virol 63: 2260–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Clemente R, Sisman E, Aza-Blanc P, de la Torre JC (2010) Identification of host factors involved in borna disease virus cell entry through a small interfering RNA functional genetic screen. J Virol 84: 3562–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bhuyan PK, Kariko K, Capodici J, Lubinski J, Hook LM, et al. (2004) Short interfering RNA-mediated inhibition of herpes simplex virus type 1 gene expression and function during infection of human keratinocytes. J Virol 78: 10276–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhe R, Mei-Ying Z, Kitazato K, Kobayash N, Qin-Chang Z, et al. (2008) Effect of siRNA on HSV-1 plaque formation and relative expression levels of UL39 mRNA. Arch Virol 153: 1401–1406. [DOI] [PubMed] [Google Scholar]
- 51. Durand LO, Roizman B (2008) Role of cdk9 in the optimization of expression of the genes regulated by ICP22 of herpes simplex virus 1. J Virol 82: 10591–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. English L, Chemali M, Duron J, Rondeau C, Laplante A, et al. (2009) Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol 10: 480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu Z, Zhu Y, Bisaro DM, Parris DS (2009) Herpes simplex virus type 1 suppresses RNA-induced gene silencing in mammalian cells. J Virol 83: 6652–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Clement C, Popp MP, Bloom DC, Schultz G, Liu L, et al. (2008) Microarray analysis of host gene expression for comparison between naive and HSV-1 latent rabbit trigeminal ganglia. Molecular Vision 14: 1209–1221. [PMC free article] [PubMed] [Google Scholar]
- 55. Zenner HL, Yoshimura S, Barr FA, Crump CM (2011) Analysis of Rab GTPase-activating proteins indicates that Rab1a/b and Rab43 are important for herpes simplex virus 1 secondary envelopment. J Virol 85: 8012–8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maric M, Shao J, Ryan RJ, Wong CS, Gonzalez-Alegre P, et al. (2011) A functional role for TorsinA in herpes simplex virus 1 nuclear egress. J Virol 85: 9667–9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Indran SV, Britt WJ (2011) A Role for the Small GTPase Rab6 in Assembly of Human Cytomegalovirus. J Virol 85: 5213–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Karasneh GA, Ali M, Shukla D (2011) An important role for syndecan-1 in herpes simplex virus type-1 induced cell-to-cell fusion and virus spread. PLoS ONE 6: e25252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou Q, Zhu M, Zhang H, Yi T, Klena JD, et al. (2012) Disruption of the p53-p21 pathway inhibits efficiency of the lytic-replication cycle of herpes simplex virus type 2 (HSV-2). Virus Res [DOI] [PubMed] [Google Scholar]
- 60. Dziuba N, Ferguson MR, O'Brien WA, Sanchez A, Prussia AJ, et al. (2012) Identification of cellular proteins required for replication of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses 28: 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Naji S, Ambrus G, Cimermancic P, Reyes JR, Johnson JR, et al. (2012) Host cell interactome of HIV-1 Rev includes RNA helicases involved in multiple facets of virus production. Mol Cell Proteomics 11: M111 015313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eisfeld AJ, Kawakami E, Watanabe T, Neumann G, Kawaoka Y (2011) RAB11A Is Essential for Transport of the Influenza Virus Genome to the Plasma Membrane. J Virol 85: 6117–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]