

Abstract
Reduced (FeII) Rhodopseudomonas palustris cytochrome c′ (Cyt c′) is more stable toward unfolding ([GuHCl]1/2 = 2.9(1) M) than the oxidized (FeIII) protein ([GuHCl]1/2 = 1.9(1) M). The difference in folding free energies (ΔΔGf° = 70 meV) is less than half of the difference in reduction potentials of the folded protein (100 mV vs. NHE) and a free heme in aqueous solution (≈−150 mV). The spectroscopic features of unfolded FeII–Cyt c′ indicate a low-spin heme that is axially coordinated to methionine sulfur (Met-15 or Met-25). Time-resolved absorption measurements after CO photodissociation from unfolded FeII(CO)–Cyt c′ confirm that methionine can bind to the ferroheme on the microsecond time scale [kobs = 5(2) × 104 s−1]. Protein folding was initiated by photoreduction (two-photon laser excitation of NADH) of unfolded FeIII–Cyt c′ ([GuHCl] = 2.02–2.54 M). Folding kinetics monitored by heme absorption span a wide time range and are highly heterogeneous; there are fast-folding (≈103 s−1), intermediate-folding (102–101 s−1), and slow-folding (10−1 s−1) populations, with the last two likely containing methionine-ligated (Met-15 or Met-25) ferrohemes. Kinetics after photoreduction of unfolded FeIII–Cyt c′ in the presence of CO are attributable to CO binding [1.4(6) × 103 s−1] and FeII(CO)–Cyt c′ folding [2.8(9) s−1] processes; stopped-flow triggered folding of FeIII–Cyt c′ (which does not contain a protein-derived sixth ligand) is adequately described by a single kinetics phase with an estimated folding time constant of ≈4 ms [ΔGf° = −33(3) kJ mol−1] at zero denaturant.
Energy-landscape descriptions of protein folding emphasize the roles of free energy and energy-surface ruggedness as key determinants of folding dynamics (1–3). The many correlations of folding times with free energy support this point of view, but it is difficult to reconcile the model with the seemingly simple kinetics that often are observed. Analyses of experimental data point to a relationship between the folding kinetics and the topology of the native protein structure (4–8). Proteins that, on average, have a large number of long-range contacts in the folded state tend to fold more slowly than those with a preponderance of short-range contacts. The questions remain: is it the depth and roughness of the funnel, the topology of the folded state, or some combination of the two that determines the folding mechanism?
Short-range contacts outnumber long-range interactions in helical bundles; if topology is the key, then these proteins should fold rapidly (4, 5). Highly helical ferrocytochrome b562 (FeII–Cyt b562) (9, 10), acyl-CoA binding protein (ACBP) (11–14), the E colicin binding immunity proteins Im7 and Im9 (15, 16), and the N-terminal domain of phage λ repressor (17) all fold on the millisecond time scale. These observations appear to support the notion that folded-state structural topology is of central importance in folding dynamics and that helical bundle structures are inherently fast folding.
We have shown previously that FeII–Cyt b562 folding can be triggered by electron transfer (ET), a method that takes advantage of the greater stabilities of reduced heme proteins in native states (9, 10, 18, 19). Although folded FeII–Cyt b562 was observed within milliseconds after reduction of the unfolded oxidized protein, no more than half of the reduced protein successfully developed native structure (10). Rapid heme dissociation from the polypeptide (kdiss ≈2–7 × 103 s−1) limited the yield of the folding reaction. The heme-loss step selects fast-folding conformations from the unfolded ensemble; if there are slow-folding components, they cannot be detected. Under these circumstances, the observed kinetics reflect heme-dissociation dynamics rather than folding.
Cytochrome c′ (Cyt c′) from the photosynthetic bacterium Rhodopseudomonas palustris is a monomeric, soluble, 125-residue, four-helix-bundle heme protein. Importantly, the porphyrin is bound to the polypeptide with two thioether links near the C terminus (Cys-113 and Cys-116) (20–22). Although sharing just 19% sequence identity and 40% similarity (23), Cyt c′ and Cyt b562 have quite similar folds (1.6-Å rms deviation in α-carbon position) (Fig. 1) (24, 25). Cyt b562 has a six-coordinate, lowspin heme with Met-7 and His-102 axial ligands [(H102N){PorN4FeIII}(M7S)+] and a reduction potential of 180 mV vs. NHE (24, 26). Cyt c′ has a high-spin, five-coordinate heme, axially ligated by His-117 [(H117N){PorN4FeIII}+] and a reduction potential of 100 mV (27–32). In Cyt c′, the side-chain of Leu-12 fills the space occupied by a sixth ligand in Cyt b562 (Fig. 1 Inset) (25); movement of this bulky group is necessary for ligand binding.
Figure 1.

Comparison of heme environments. In Cyt b562 (gray), the heme iron is axially ligated to His-102 and Met-7, whereas in Cyt c′ (black), the heme has only one axial ligand (His-117) with the side chain of Leu-12 at the other axial site. The nearest methionine residue (Met-15) in Cyt c′ is also shown. Backbone atoms of four α-helices of Cyt c′ (PDB 1A7V) are superimposed on the corresponding atoms in Cyt b562 (PDB 256B), with a calculated rms deviation of 1.6 Å.
The FeIII/II reduction potential is high enough to permit ET triggering of FeII–Cyt c′ folding in the 2.0–2.9 M guanidine hydrochloride (GuHCl) concentration range. FeII–Cyt c′ folding is highly heterogeneous, spanning a time range from milliseconds to several seconds. Importantly, the folding kinetics of FeIII–Cyt c′ and FeII(CO)–Cyt c′ are less complex, indicating that nonnative ligand traps may be responsible for the complexity of FeII–Cyt c′ folding.
Materials and Methods
GuHCl (Sigma, ultrapure grade), Tris(2,2′-bipyridine)ruthenium(II) chloride ([Ru(bpy)3]Cl2, Strem), and NADH (Sigma) were used as received.
Spectroscopic measurements were made as follows: absorption, Hewlett-Packard 8452 diode array spectrophotometer; CD, Aviv 62ADS spectropolarimeter; and luminescence, Hitachi F-4500 spectrofluorimeter (λex = 290 nm; λobsd = 300–500 nm). Mass spectral analyses were done at the Protein/Peptide Microanalytical Laboratory in the Beckman Institute at Caltech.
R. palustris strain 37 (ATCC 17007) Cyt c′ was isolated and purified by using minor modifications of published procedures (31, 33). Cyt c′ was chromatographed either on a CM Sepharose column equilibrated with 5 mM sodium phosphate and 10 mM NaCl (pH 7.0) in the presence of ferricyanide and eluted with the charging buffer, or on a column of hydroxyapatite (type I, Bio-Rad) equilibrated with 3 mM sodium phosphate and 15 mM NaCl (pH 7.0) in the presence of mercaptoethanol and eluted with a linear gradient from 10 mM to 100 mM sodium phosphate buffer (15 mM NaCl, pH 7.0). Proteins used in all experiments had purity indices (A280 nm/A398 nm, oxidized) between 0.22 and 0.25 (literature value, 0.21) (33). Although only a single fraction was isolated chromatographically, mass spectra revealed two components in the protein sample. The mass of one component matched that expected on the basis of the amino acid sequence (13,748 daltons); the other had a mass 17 atomic mass units lower than predicted. Protein concentrations were obtained from UV-visible absorption measurements (FeIII–Cyt c′, ɛ398 nm = 85 mM−1cm−1; FeII–Cyt c′, ɛ426 nm = 99 mM−1cm−1) (34). GuHCl concentrations were extracted from refractive index determinations (Abbe-3L refractometer, Milton Roy, Rochester, NY) (35).
Denaturation curves for FeII–Cyt c′ were determined in the presence of excess sodium dithionite. Transient absorption spectra and kinetics were measured by using an apparatus described previously (36). The excitation source was the third harmonic of a Q-switched Nd:YAG laser (355 nm) for electron photoinjection from NADH, and a Nd:YAG pumped optical parametric oscillator (480 nm) for injection from Ru(bpy)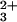 and for CO photolysis experiments. A 16-bit, 20 MS/s digital oscilloscope (CompuScope 1602, Gage Applied) was used to record the complete kinetics traces from 10−3 to 101 s. Samples for kinetics measurements were deoxygenated by repeated evacuation/argon-fill cycles on a Schlenk line. Typical samples contained Cyt c′ (100–200 μM), either Ru(bpy)
and for CO photolysis experiments. A 16-bit, 20 MS/s digital oscilloscope (CompuScope 1602, Gage Applied) was used to record the complete kinetics traces from 10−3 to 101 s. Samples for kinetics measurements were deoxygenated by repeated evacuation/argon-fill cycles on a Schlenk line. Typical samples contained Cyt c′ (100–200 μM), either Ru(bpy)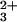 (1.0–1.5 equivalents) or NADH (2.0–2.5 equivalents), and GuHCl (1.8–3.5 M) in 100 mM sodium phosphate buffer (pH 7). The pH and index of refraction were measured after laser experiments. Reduced protein was stirred under 1 atm of CO for at least 30 min after deoxygenation to prepare FeII(CO)–Cyt c′.
(1.0–1.5 equivalents) or NADH (2.0–2.5 equivalents), and GuHCl (1.8–3.5 M) in 100 mM sodium phosphate buffer (pH 7). The pH and index of refraction were measured after laser experiments. Reduced protein was stirred under 1 atm of CO for at least 30 min after deoxygenation to prepare FeII(CO)–Cyt c′.
Measured kinetics traces were logarithmically compressed (100 points per time decade), normalized [I(t) − I(t∞)/I(t0) − I(t∞)], and fit by using a non-negative linear least-squares minimization algorithm to project the folding kinetics onto a basis of exponential decay functions with rate constants in the range 105 to 10−2 s−1 (10 rate constants per decade, spaced logarithmically).
The folding kinetics of FeIII–Cyt c′ were measured by using a BioLogic SFM-4S stopped-flow mixer, coupled via optical fiber to a monochromator fitted with a five-stage photomultiplier tube. Probe light was provided by a 200-W Hg/Xe arc lamp. The PMT output was recorded with the 16-bit digital oscilloscope. Kinetics traces were measured at 372 nm (formation of folded protein, Δɛ = 28 mM−1 cm−1) and 400 nm (disappearance of the unfolded protein, Δɛ = −44 mM−1 cm−1) for a time period up to 25 s, at a sampling rate of 2 or 5 kS/s. In all refolding experiments, unfolded FeIII–Cyt c′ (3.3 M GuHCl) was either diluted 6- or 12-fold with a combination of Hepes buffer ([Hepes] = 0.1 M, pH 7) and Hepes/GuHCl ([Hepes] = 0.1 M, [GuHCl] = 3.3 M, pH 7), giving final protein concentrations of 1.5 μM and varying GuHCl concentrations (0.6–1.80 M). Kinetics traces were fit to single exponential functions.
Results
Equilibrium Unfolding.
GuHCl titration curves generated from far-UV CD, Trp-72 fluorescence, and Soret absorbance spectra show that reduced Cyt c′ is more stable toward unfolding ([GuHCl]1/2 = 2.9(1) M) than oxidized Cyt c′ ([GuHCl]1/2 = 1.9(1) M) (Fig. 2 Inset). The stability of folded Cyt c′, judged by the folding free-energy change extrapolated to zero denaturant (ΔGf°), is measurably lower than that of Cyt b562 [Fe(III): ΔGf°(Cyt c′) = −33(3) kJ mol−1, ΔGf°(Cyt b562) = −42(3) kJ mol−1; Fe(II): ΔGf°(Cyt c′) = −40(4) kJ mol−1, ΔGf°(Cyt b562) ≈−78 kJ mol−1]. The difference in folding free energies between FeIII– and FeII–Cyt c′ (ΔΔGf° = 7 kJ mol−1 ≈70 meV) is less than half that predicted on the basis of the reduction potential of the folded protein (100 mV vs. NHE) and the potential expected for a heme in aqueous solution (−150 mV) (37). The titrations suggest that the potential of the unfolded protein is ≈0 V, possibly because a second axial ligand binds to the heme when the protein unfolds. Although the difference in folding free energies is smaller than anticipated, it is large enough to permit ET triggering of FeII–Cyt c′ folding between 2 and 2.9 M GuHCl.
Figure 2.

Absorption spectra of folded FeIII–Cyt c′ and FeII–Cyt c′ (bold lines), and unfolded FeIII–Cyt c′ and FeII–Cyt c′ (dashed lines). (Inset) GuHCl titration curves generated from CD, fluorescence, and Soret absorption data (χu is the unfolded-protein fraction).
Ligand Binding to Unfolded Cyt c′.
Addition of GuHCl to solutions of FeIII–Cyt c′ narrows the Soret absorption band (λmax = 400 nm, ɛ = 133 mM−1⋅cm−1) (Fig. 2). The absorption profile is analogous to that of the M80A mutant of cytochrome c (λmax = 400 nm, ɛ = 164 mM−1⋅cm−1, pH = 4.5) (38) and is consistent with the formation of a six-coordinate, high-spin heme (H117N) {PorN4FeIII}(OH2)+. The absorption spectrum in alkaline solution (pH >10, [GuHCl] ≥ 3.5 M) is typical of a low-spin FeIII heme (λmax = 406 nm, ɛ = 100 mM−1⋅cm−1) (38); in this case, it is possible that a lysine residue binds to form (H117N){PorN4FeIII}(KN)+. Unlike cytochrome c, Cyt c′ has no His residues (other than the native His-117 ligand) available to bind to the FeIII heme in the unfolded protein.
GuHCl denaturation induces a blue shift in the Soret absorption of FeII–Cyt c′ (folded: λmax = 426, 435 nm; unfolded: λmax = 420 nm), and two sharp Q-bands then appear (λmax = 524, 552 nm). These spectroscopic features are consistent with a six-coordinate, low-spin heme (Fig. 2). The absence of strongly shifted peaks in the 1H NMR spectrum of unfolded FeII–Cyt c′ also signals the presence of a diamagnetic heme. The absorption spectrum of the unfolded FeII protein is independent of pH in the range 4–10, although changes in Soret absorption at higher pH levels are consistent with lysine binding (λmax = 416, 520, 550 nm). Addition of N-acetyl-L-methionine to unfolded FeII–Cyt c′ causes little perturbation of the spectrum, suggesting methionine sulfur coordination [(H117N){PorN4FeII}(MS), Met-15 and/or Met-25] in unfolded FeII–Cyt c′. Methionine binding in unfolded FeII–Cyt c′ would raise the reduction potential and could explain the small difference between the folding free-energy changes in FeII– and FeIII–Cyt c′.
Misligated Met residues must dissociate from the heme before Cyt c′ can adopt its native folded structure. Indeed, the folding dynamics of FeII–Cyt c′ could reflect the kinetics of ligand substitutions. We have used two methods to examine the kinetics of MS binding to FeII in unfolded Cyt c′. Laser-excited Ru(bpy)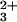 reduces unfolded FeIII–Cyt c′ ([GuHCl] = 4.4 M) in less than a microsecond. After the rapid [1.4(1) × 106 s−1] change in absorbance corresponding to heme reduction, there is no substantial variation in the spectrum of the reduced protein until charge recombination regenerates unfolded FeIII–Cyt c′. The transient difference spectrum recorded 20 μs after the laser flash reveals a mixture of high- and low-spin hemes, suggesting that a nonnative sixth ligand rapidly binds to the heme in the unfolded reduced protein. Ligand binding dynamics also were examined after photodissociation of CO from unfolded FeII(CO)-Cyt c′ (4.5 M GuHCl, pH 7). The transient absorption difference spectrum measured immediately after CO dissociation (t = 100 ns) exhibits a broad feature in the Q-band region (530–570 nm) corresponding to a five-coordinate heme (H117N) {PorN4FeII}. Subsequent absorption changes arise from the formation of a low-spin heme [kobs = 5(2) × 104 s−1] before CO rebinding. Both experiments indicate that MS can bind to the heme in unfolded FeII–Cyt c′ on the microsecond time scale (39–42).
reduces unfolded FeIII–Cyt c′ ([GuHCl] = 4.4 M) in less than a microsecond. After the rapid [1.4(1) × 106 s−1] change in absorbance corresponding to heme reduction, there is no substantial variation in the spectrum of the reduced protein until charge recombination regenerates unfolded FeIII–Cyt c′. The transient difference spectrum recorded 20 μs after the laser flash reveals a mixture of high- and low-spin hemes, suggesting that a nonnative sixth ligand rapidly binds to the heme in the unfolded reduced protein. Ligand binding dynamics also were examined after photodissociation of CO from unfolded FeII(CO)-Cyt c′ (4.5 M GuHCl, pH 7). The transient absorption difference spectrum measured immediately after CO dissociation (t = 100 ns) exhibits a broad feature in the Q-band region (530–570 nm) corresponding to a five-coordinate heme (H117N) {PorN4FeII}. Subsequent absorption changes arise from the formation of a low-spin heme [kobs = 5(2) × 104 s−1] before CO rebinding. Both experiments indicate that MS can bind to the heme in unfolded FeII–Cyt c′ on the microsecond time scale (39–42).
FeII-Cyt c′ Folding.
FeII–Cyt c′ folding was initiated by rapid electron injection (≈100 μs) into unfolded oxidized protein ([GuHCl] = 2.02–2.54 M) after two-photon laser excitation of NADH (43, 44). Under these conditions, heme reduction is slower than binding of the nonnative sixth ligand. The progress of the folding reaction was monitored by heme absorption in the Soret and Q-band regions from 10−4 to 1 s after excitation. The observed kinetics are highly heterogeneous. A small fraction (≈20%) of the population forms a high-spin heme species in about a millisecond. Complete formation of the fully folded ensemble requires several seconds (Fig. 3). Rate constants for FeII–Cyt c′ span a range from 103 to 10−1 s−1 revealing fast-folding (7.0 × 103 s−1, 8%; 5.7 × 103 s−1, 9%), intermediate-folding (9.0 × 102 to 1.5 × 101 s−1, 24%), and slow-folding (5.9 × 10−1 s−1, 16%; 4.8 × 10−1 s−1, 43%) components in the protein ensemble (Fig. 3 Inset). Although the relative populations and the number of different rate constants vary slightly for kinetics measured at different wavelengths, heterogeneous behavior is always observed. On very long time scales, oxidation of the reduced protein leads to some uncertainty in the extracted rate constants (although the samples were carefully deoxygenated, trace amounts of oxygen were always present). The transient difference spectrum recorded 100 μs after electron injection is characteristic of a mixture of five-coordinate, high-spin and six-coordinate, low-spin ferrohemes, suggesting that the slower folding populations are misligated. The difference spectra measured at 1 and 500 ms have been fit to a combination of the [(FeII-folded) − (FeIII-unfolded)] and [(FeII-unfolded) − (FeIII-unfolded)] molar difference spectra. The resulting folded and unfolded populations (1 ms, 25% folded, 75% unfolded; 500 ms, 52% folded, 48% unfolded) are consistent with the measured amplitude changes in the single wavelength kinetics.
Figure 3.

Normalized folding kinetics (observed at 440 nm, black line) fit to 80 rate constants spanning logarithmically 105 to 10−2 s−1 (gray line) by using a non-negative least-squares algorithm. (Upper) Residuals from the fit. (Inset) The projected population of rate constants [7.0 × 103 s−1 (8%); 5.7 × 103 (9%); 9.2 × 102 (1%); 2.2 × 102 (1%); 7.9 × 101 (11%); 1.9 × 101 (9%); 1.5 × 101 (2%); 5.9 × 10−1 (16%); 4.8 × 10−1 (43%)].
FeII-Cyt c′ Folding in the Presence of CO.
To determine whether misligated heme traps are responsible for the slow FeII–Cyt c′ folding, we examined ET-triggered folding of FeII–Cyt c′ in the presence of CO. In the unfolded protein (3.32 M GuHCl, pH 7), CO binding occurs with a millisecond time constant [1.0(4) × 103 s−1; 1 atm CO], whereas the folded protein binds CO much more slowly [1.4(4) s−1]. At lower denaturant concentration ([GuHCl] = 2.1 M, pH 7) in the presence of CO, reduction of unfolded FeIII–Cyt c′ is followed by two kinetics phases: a 1.4(6) × 103 s−1 step corresponding to CO binding, and 2.8(9) s−1 component due to FeII(CO)–Cyt c′ folding. The 1-ms transient difference spectrum can be modeled by two absorbing species: the major component is unfolded FeII(CO)–Cyt c′, and the minor fraction is five-coordinate, folded FeII–Cyt c′. Subsequent changes in absorbance are associated with the conversion of unfolded FeII(CO)– to folded FeII(CO)–Cyt c′ (Fig. 4).
Figure 4.

Schematic representation of heme coordination environment after electron-transfer triggered folding of FeII–Cyt c′ in the presence and absence of CO. The subscripts F and U refer to folded and unfolded polypeptides.
FeIII–Cyt c′ Folding.
Nonnative methionine-heme ligation is disfavored in unfolded FeIII–Cyt c′ (37), and the axial heme coordination sites are occupied by water and His-117. The stopped-flow triggered folding of FeIII–Cyt c′ is adequately described by a single kinetics phase with a rate constant that decreases exponentially with increasing denaturant concentration. The folding rate constant [1.0(2) s−1; [GuHCl] = 0.95 M; ΔGf = −16 kJ mol−1] is comparable to that of FeII(CO)–Cyt c′ and of the slower folding population of FeII–Cyt c′ ([GuHCl] = 2.16 M; ΔGf = −10 kJ mol−1). The extrapolated rate constant for folding FeIII–Cyt c′ in the absence of denaturant is 2.5 × 102 s−1 [ΔGf° = −33(3) kJ mol−1).
Discussion
It is reasonable to expect that topologically homologous proteins would fold at similar rates, yet cytochromes b562 and c′ display quite disparate folding kinetics. Apparently, there are factors beyond structural topology that must be considered to explain the folding kinetics of these four-helix bundles. Three key features of cytochromes b562 and c′ are likely to contribute to the differences in folding kinetics: covalent attachment of the porphyrin to the polypeptide, nonnative heme–ligand traps, and folding driving force.
The rapid dissociation of the heme from unfolded FeII–Cyt b562 limits the time scale for observation of folding kinetics. Because the porphyrin is covalently attached in Fe–Cyt c′, the folding kinetics cover a far wider time range and are considerably more complex. A fraction of the unfolded FeII–Cyt c′ ensemble refolds rapidly, but several seconds are required for the entire sample to fold. Proline isomerization is known to inhibit protein folding (45) but is unlikely to be responsible for the slow Fe–Cyt c′ kinetics. Although there are four proline residues in the Cyt c′ sequence, the observed folding rates depend on denaturant concentration and are substantially faster than typical proline isomerizations (45). It is interesting to note that the fast-folding fraction of FeII–Cyt c′ is roughly comparable to the yield of folded FeII–Cyt b562. It is possible, then, that if heme dissociation could be inhibited, FeII–Cyt b562 would display slower and more complex folding kinetics.
The refolding of FeII–Cyt c′ can be described qualitatively by a kinetic partitioning mechanism (46, 47). At the instant that folding is initiated, a fraction of the denatured proteins will have adopted conformations that can smoothly and rapidly fold to the native structure. The fast-folding population of FeII–Cyt c′ would correspond to this group. Folding of the remaining fraction is frustrated by transient trapping in local minima on the folding energy landscape. Escape from these misfolded structures is an activated process that slows formation of the native structure. The partition factor that determines the balance between fast- and slow-folding populations depends on the primary sequence as well as the refolding conditions (46–51). Simulated folding kinetics of model three-helix-bundle proteins (48, 49) point to a central role for the folding free-energy gap in determining the partitioning between fast- and slow-track folding. The heterogeneous folding kinetics of FeII–Cyt c′ are strikingly similar to the simulated folding kinetics of a small-gap three-helix bundle (49).
Nonnative methionine ligands in unfolded FeII–Cyt c′ contribute to the heterogeneity of the folding kinetics. In addition to the fast-folding population, there are intermediate (≈102 s−1) and slow-folding (<1 s−1) components. Iron–sulfur bond dissociation is not rate limiting because ferroheme–methionine ligand exchange is faster than either folding phase (40). In the presence of CO, the nonnative Met ligands are displaced from the ferroheme, and folding is dominated by a slow phase with a time constant of ≈350 ms. Similarly, FeIII–Cyt c′ folding is not complicated by heme–ligand binding, and a single 1-s phase predominates. These observations suggest that, in the absence of misligation, Cyt c′ requires 500 ms to 1 s to adopt a folded structure. The presence of an intermediate (≈102 s−1) folding phase in FeII–Cyt c′ implies that nonnative methionine ligation can, in some instances, facilitate refolding. The protein has two Met residues that can bind to the ferroheme when the protein unfolds. It is possible that the intermediate phase arises from protein populations with (H117N){PorN4FeII}(M15S) heme coordination, and the slow phase corresponds to populations with (H117N){PorN4FeII}(M25S) ligation (Fig. 4). Met-15 is in the distal heme pocket in the folded protein, so an intermediate with (H117N){PorN4FeII}(M15S) coordination might fold more readily than a (H117N){PorN4FeII}(M25S) form.
The Fe–Cyt c′ folding rates are compared in Fig. 5 to those of four structurally related proteins (11, 15, 19). The four-helix bundles Im7 and Im9 have 60% sequence identity and nearly identical three-dimensional structures, yet Im9 folds more than 10 times slower than Im7 (15). Curiously, Im9 displays the hallmarks of two-state folding, whereas intermediates are implicated in the faster Im7 folding process (15, 16). The folding of ACBP has been interpreted in terms of a two-state model (12, 13), although subsequent quenched-flow hydrogen exchange studies revealed that an intermediate is formed before the rate-limiting step (14). Among helical-bundle structures, there is a wide variation in measured folding rates as functions of −ΔGf (Fig. 5). It is clear that, even in the absence of misligation, Cyt c′ is the slowest folding protein in this structural class. The difference is even greater when account is taken of the fact that the Im7, Im9, and ACBP folding kinetics were measured at reduced temperatures. Cyt c′ is substantially larger than the other helical bundles (Im7, 85 residues; Im9, 85 residues; ACBP, 86 residues; and Cyt c′, 126 residues), and its size may be in part responsible for its slower folding. On a free-energy basis, the Fe–Cyt c′ folding kinetics more closely resemble those of Cyt c (52) than those of the nonheme helical-bundle proteins. Nonnative ligand binding clearly can perturb heme protein folding; it is likely that noncovalent, nonnative heme–polypeptide contacts represent additional sources of frustration.
Figure 5.

Measured folding rates of helical proteins as functions of folding driving force (ΔGf). Im7 (85 aa), pH 7, 10°C (■) (15); Im9 (85 aa) (PDB 1IMQ, upper right ribbon structure), pH 7, 10°C (□) (15); bovine ACBP (86 aa) (PDB 2ABD, upper left ribbon structure), pH 5.3, 5°C (▴) (12); FeII–Cyt c′ (126 aa), pH 7, 22°C, slowest phase (○); FeIII–Cyt c′ (PDB 1A7V, lower right ribbon structure), pH 7, 22°C (●); equine FeII–Cyt c (104 aa), pH 7, 22°C (◊) (52); yeast FeII–Cyt c (108 aa), pH 7, 22°C (▾) (52).
Acknowledgments
We thank Michael Cusanovich and Terry Meyer (University of Arizona) for assistance with protein isolation and purification, Brian Crane for help with protein structure modeling, and Akif Tezcan for discussions. J.C.L. thanks the Ralph M. Parsons Foundation for a graduate fellowship. This work was supported by National Science Foundation Grant MCB-9974477 and by the Arnold and Mabel Beckman Foundation.
Abbreviations
- Cyt b562
cytochrome b562
- ACBP
acyl-CoA binding protein
- Im
E colicin binding immunity protein
- Cyt c′
cytochrome c′
- ET
electron transfer
- GuHCl
guanidine hydrochloride
- bpy
2,2′-bipyridine
References
- 1.Bryngelson J D, Onuchic J N, Wolynes P G. Proteins Struct Funct Genet. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 2.Onuchic J N, Lutheyschulten Z, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 3.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 4.Plaxco K W, Simons K T, Baker D. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 5.Plaxco K W, Simons K T, Ruczinski I, David B. Biochemistry. 2000;39:11177–11183. doi: 10.1021/bi000200n. [DOI] [PubMed] [Google Scholar]
- 6.Dinner A R, Karplus M. Nat Struct Biol. 2001;8:21–22. doi: 10.1038/83003. [DOI] [PubMed] [Google Scholar]
- 7.Debe D A, Carlson M J, Goddard W A., III Proc Natl Acad Sci USA. 1999;96:2596–2601. doi: 10.1073/pnas.96.6.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muñoz V, Eaton W A. Proc Natl Acad Sci USA. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wittung-Stafshede P, Gray H B, Winkler J R. J Am Chem Soc. 1997;119:9562–9563. [Google Scholar]
- 10.Wittung-Stafshede P, Lee J C, Winkler J R, Gray H B. Proc Natl Acad Sci USA. 1999;96:6587–6590. doi: 10.1073/pnas.96.12.6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kragelund B B, Robinson C V, Knudsen J, Dobson C M, Poulsen F M. Biochemistry. 1995;34:7217–7224. doi: 10.1021/bi00021a037. [DOI] [PubMed] [Google Scholar]
- 12.Kragelund B B, Højrup P, Jensen M S, Schjerling C K, Juul E, Knudsen J, Poulsen F M. J Mol Biol. 1996;256:187–200. doi: 10.1006/jmbi.1996.0076. [DOI] [PubMed] [Google Scholar]
- 13.Kragelund B B, Osmark P, Neergaard T B, Schiødt J, Kristiansen K, Knudsen J, Poulsen F M. Nat Struct Biol. 1999;6:594–601. doi: 10.1038/9384. [DOI] [PubMed] [Google Scholar]
- 14.Teilum K, Kragelund B B, Knudsen J, Poulsen F. J Mol Biol. 2000;301:1307–1314. doi: 10.1006/jmbi.2000.4003. [DOI] [PubMed] [Google Scholar]
- 15.Ferguson N, Capaldi A P, James R, Kleanthous C, Radford S E. J Mol Biol. 1999;286:1597–1608. doi: 10.1006/jmbi.1998.2548. [DOI] [PubMed] [Google Scholar]
- 16.Capaldi A P, Shastry M C R, Kleanthous C, Roder H, Radford S E. Nat Struct Biol. 2001;8:68–72. doi: 10.1038/83074. [DOI] [PubMed] [Google Scholar]
- 17.Huang G S, Oas T G. Proc Natl Acad Sci USA. 1995;92:6878–6882. doi: 10.1073/pnas.92.15.6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pascher T, Chesick J P, Winkler J R, Gray H B. Science. 1996;271:1558–1560. doi: 10.1126/science.271.5255.1558. [DOI] [PubMed] [Google Scholar]
- 19.Telford J R, Wittung-Stafshede P, Gray H B, Winkler J R. Acc Chem Res. 1998;31:755–763. [Google Scholar]
- 20.Meyer T E, Kamen M D. Adv Protein Chem. 1982;35:105–212. doi: 10.1016/s0065-3233(08)60469-6. [DOI] [PubMed] [Google Scholar]
- 21.Moore G R, McClune G J, Clayden N J, Williams R J P, Alsaadi B M, Angstrom J, Ambler R P, van Beeumen J, Tempst P, Bartsch R G, et al. Eur J Biochem. 1982;123:73–80. doi: 10.1111/j.1432-1033.1982.tb06500.x. [DOI] [PubMed] [Google Scholar]
- 22.Moore G R, Pettigrew G W. Cytochromes c: Evolutionary, Structural, and Physicochemical Aspects. New York: Springer; 1990. [Google Scholar]
- 23.Ambler R P, Bartsch R G, Daniel M, Kamen M D, McLellan L, Meyer T E, Beeumen J V. Proc Natl Acad Sci USA. 1981;78:6854–6857. doi: 10.1073/pnas.78.11.6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamada K, Bethge P H, Mathews F S. J Mol Biol. 1995;247:947–962. doi: 10.1006/jmbi.1995.0192. [DOI] [PubMed] [Google Scholar]
- 25.Shibata N, Iba S, Misaki S, Meyer T E, Bartsch R G, Cusanovich M A, Morimoto Y, Higuchi Y, Yasuoka N. J Mol Biol. 1998;284:751–760. doi: 10.1006/jmbi.1998.2190. [DOI] [PubMed] [Google Scholar]
- 26.Moore G R, Williams R J P, Peterson J, Thomson A J, Mathews F S. Biochim Biophys Acta. 1985;829:83–96. doi: 10.1016/0167-4838(85)90071-8. [DOI] [PubMed] [Google Scholar]
- 27.Klerk H D, Bartsch R G, Kamen M D. Biochim Biophys Acta. 1965;97:275–280. doi: 10.1016/0304-4165(65)90092-9. [DOI] [PubMed] [Google Scholar]
- 28.Strekas T C, Spiro T G. Biochim Biophys Acta. 1974;351:237–245. doi: 10.1016/0005-2795(74)90186-x. [DOI] [PubMed] [Google Scholar]
- 29.Fujii S, Yoshimura T, Kamada H, Yamaguchi K, Suzuki S, Shidara S, Takakuwa S. BBA Protein Struct Mol Enzymol. 1995;1251:161–169. doi: 10.1016/0167-4838(95)00092-9. [DOI] [PubMed] [Google Scholar]
- 30.Clark K, Dugad L B, Bartsch R G, Cusanovich M A, Mar G N L. J Am Chem Soc. 1996;118:4654–4664. [Google Scholar]
- 31.Jackson J T, Mar G N L, Bartsch R G. J Biol Chem. 1983;258:1799–1805. [PubMed] [Google Scholar]
- 32.Barakat R, Strekas T C. Biochim Biophys Acta. 1982;679:393–399. [Google Scholar]
- 33.Bartsch R G. Methods Enzymol. 1971;23:344–363. [Google Scholar]
- 34.Bartsch R G. In: The Photosynthetic Bacteria. Clayton R K, Sistrom W R, editors. New York: Plenum; 1978. pp. 249–279. [Google Scholar]
- 35.Nozaki Y. Methods Enzymol. 1972;26:43–50. doi: 10.1016/s0076-6879(72)26005-0. [DOI] [PubMed] [Google Scholar]
- 36.Stowell M H B, Larsen R W, Winkler J R, Rees D C, Chan S I. J Phys Chem. 1993;97:3054–3057. [Google Scholar]
- 37.Tezcan F A, Winkler J R, Gray H B. J Am Chem Soc. 1998;120:13383–13388. [Google Scholar]
- 38.Bren K L, Gray H B. J Am Chem Soc. 1993;115:10382–10383. [Google Scholar]
- 39.Hagen S J, Hofrichter J, Szabo A, Eaton W A. Proc Natl Acad Sci USA. 1996;93:11615–11617. doi: 10.1073/pnas.93.21.11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagen S J, Hofrichter J, Eaton W A. J Phys Chem. 1997;101:2352–2365. [Google Scholar]
- 41.Lapidus L J, Eaton W A, Hofrichter J. Proc Natl Acad Sci USA. 2000;97:7220–7225. doi: 10.1073/pnas.97.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hagen S J, Carswell C W, Sjolander E M. J Mol Biol. 2001;305:1161–1171. doi: 10.1006/jmbi.2000.4366. [DOI] [PubMed] [Google Scholar]
- 43.Telford J R, Tezcan F A, Gray H B, Winkler J R. Biochemistry. 1999;38:1944–1949. doi: 10.1021/bi981933z. [DOI] [PubMed] [Google Scholar]
- 44.Orii Y. Biochemistry. 1993;32:11910–11911. doi: 10.1021/bi00095a021. [DOI] [PubMed] [Google Scholar]
- 45.Schmid F X, Mayr L M, Mücke M, Schönbrunner E R. Adv Protein Chem. 1993;44:25–66. doi: 10.1016/s0065-3233(08)60563-x. [DOI] [PubMed] [Google Scholar]
- 46.Thirumalai D, Klimov D K, Woodson S A. Theor Chem Acc. 1997;96:14–22. [Google Scholar]
- 47.Thirumalai D, Klimov D K. Curr Opin Struct Biol. 1999;9:197–207. doi: 10.1016/S0959-440X(99)80028-1. [DOI] [PubMed] [Google Scholar]
- 48.Zhou Y, Karplus M. Nature (London) 1999;401:400–403. doi: 10.1038/43937. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Y, Karplus M. J Mol Biol. 1999;293:917–951. doi: 10.1006/jmbi.1999.2936. [DOI] [PubMed] [Google Scholar]
- 50.Shea J-E, Onuchic J N, Brooks C L., III J Chem Phys. 2000;113:7663–7671. [Google Scholar]
- 51.Shea J-E, Onuchic J N, Brooks C L., III Proc Natl Acad Sci USA. 1999;96:12512–12517. doi: 10.1073/pnas.96.22.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mines G A, Pascher T, Lee S C, Winkler J R, Gray H B. Chem Biol. 1996;3:491–497. doi: 10.1016/s1074-5521(96)90097-6. [DOI] [PubMed] [Google Scholar]