

Abstract
Elucidating the mechanism of folding of polynucleotides depends on accurate estimates of free energy surfaces and a quantitative description of the kinetics of structure formation. Here, the kinetics of hairpin formation in single-stranded DNA are measured after a laser temperature jump. The kinetics are modeled as configurational diffusion on a free energy surface obtained from a statistical mechanical description of equilibrium melting profiles. The effective diffusion coefficient is found to be strongly temperature-dependent in the nucleation step as a result of formation of misfolded loops that do not lead to subsequent zipping. This simple system exhibits many of the features predicted from theoretical studies of protein folding, including a funnel-like energy surface with many folding pathways, trapping in misfolded conformations, and non-Arrhenius folding rates.
Hairpin loops are ubiquitous in single-stranded DNA and RNA. Knowing the time scales and mechanism of formation of these loops is an essential first step toward understanding the folding problem. Although the stability of hairpin loops and the kinetics of hairpin formation have been a subject of intense investigation for over 30 years (1–5), our understanding of the kinetics is limited. In particular, there is no simple physical model that describes in a consistent way both the thermodynamics and kinetics of hairpin formation. With the exception of some early work on the helix-to-coil transition, in which the kinetics were described in terms of a statistical mechanical kinetic “zipper” model (6, 7), the kinetics of hairpin-to-coil transition have been described more recently in terms of a two-state system with Arrhenius temperature dependence for the rates of hairpin formation and unwinding (5, 8). The equilibrium dynamics of hairpins, obtained from fluctuation correlation spectroscopy measurements of hairpins labeled with fluorescent donor and acceptor pairs (8, 9), have revealed a number of kinetic features that are not easily explained within the framework of a simple two-state analysis. First, the data of Libchaber and coworkers show that the rate coefficient corresponding to the closing of hairpins has a non-Arrhenius temperature dependence (8). Second, they report a puzzling result in which the apparent activation energy for forming hairpins with poly(dA) loops increases as the loop size increases. Third, Klenerman and coworkers report stretched exponential kinetics at temperatures well below the melting temperature (9). These observations suggest a failure of the simplest two-state analysis and require a modification of even the more rigorous kinetic “zipper” model in the form in which it was applied to helix–coil kinetics (6).
Here we present a model for the dynamics of hairpins that is consistent with many of the apparently anomalous kinetic observations. The dynamics are described as configurational diffusion along a free energy profile that we calculate from a statistical mechanical “zipper” model that describes the equilibrium melting profiles. The model we use to describe the dynamics is a continuous description of the discrete kinetic “zipper” model but with one important difference. We assume that the polynucleotide chain can be transiently trapped in “non-native” loops with mismatched stems, which do not lead to subsequent zipping of the stem. The assumption that non-native loops before the nucleation step can act as traps in the folding free energy surface is sufficient to explain the non-Arrhenius temperature dependence for the closing rates as well as the nonexponential nature of the kinetics at low temperatures.
The analysis presented in this paper is motivated by theoretical and computational studies of protein folding in which the search for the low energy structure has been described as configurational diffusion on an underlying free energy landscape (10–13). These studies have shown that the diffusion coefficient along an effective one-dimensional reaction coordinate depends on the local roughness of the energy surface, which arises from local minima corresponding to misfolded conformations. Here, we apply these theoretical ideas on a short DNA hairpin. The unwinding of the hairpin is initiated by a laser temperature jump (T-jump), and the kinetics are monitored by measuring the change in the fluorescence of 2-aminopurine (2AP), a fluorescent analog of the adenine base, which is substituted at various sites along the stem of the hairpin (Fig. 1). Our measurements and analysis show that folding of a hairpin occurs on an energy surface that has a “funnel-like” bias toward the native state, much like the energy surfaces predicted in the protein folding studies. The difference is that for proteins, the roughness of the energy surface increases as the protein approaches the native state, whereas for DNA hairpins, the energy surface is rough at the edges, in the initial stages before nucleation when the possibility of misfolds is the greatest, and essentially smooth and downhill when the chain folds into a correct nucleating loop. This simple system exhibits a number of features proposed in the context of folding kinetics of proteins: multiple pathways, misfolded traps early in the folding process, a transition “state” that corresponds to an ensemble of conformations, and non-Arrhenius folding rates.
Figure 1.

The hairpin sequence used in this study and its melting profiles. 2AP was substituted for A at the sites indicated on the hairpin. H1: substitution at site 1 only; H2: substitution at site 2 only, etc. H0 is the hairpin with no substitution; (black): melting profile of H0 from absorbance at 266 nm; (green): melting profile of H1 from absorbance at 266 nm; (red): melting profile of H1 from absorbance at 330 nm (where 2AP absorbs); (blue): melting profile of H1 from fluorescence of 2AP. The symbols are the data; the corresponding lines through the absorbance data are fits to a statistical mechanical “zipper” model; the line through the fluorescence data is a two-state van't Hoff fit. All melting profiles have been normalized by subtracting the Upper and Lower baselines determined from a fit to the raw data.
Materials and Methods
Materials.
DNA oligomer strands and the 2AP-substituted analogs were purchased from Oligos Etc. (Wilsonville, OR) and were HPLC purified. The buffer used in all experiments was 100 mM NaCL, 10 mM sodium phosphate, 0.1 mM EDTA, pH 7.5. The strand concentrations were ≈80 μM, sufficiently dilute to prevent bimolecular association.
Optical Melting Profiles.
Optical melting curves were obtained by measuring changes in absorbance at 266 and 330 nm (where 2AP absorbs). Absorbance curves were acquired by using a Hewlett–Packard 8452 diode array single-beam spectrophotometer. Melting profiles were also obtained by measuring the static fluorescence spectra of 2AP between 320 and 450 nm after excitation at 307 nm. The fluorescence spectra at each temperature were acquired by using a Fluoromax-2 spectrofluorometer (Jobin Yvon-Spex, Edison, NJ).
Laser T-Jump Spectrometer.
The laser T-jump apparatus consists of a Q-switched Nd/YAG laser (Continuum Surelite II, full width at half-maximum ≈6 ns, 600 mJ/pulse at 1.06 μm) that is used to pump a 2 m long Raman cell consisting of high-pressure methane gas, to yield 60–80 mJ/pulse at 1.54 μm. The infrared pulse is focused down to ≈1 mm full width at half-maximum on a quartz sample cell with 1-mm path length and is absorbed by the aqueous sample to yield a T-jump of ≈7–10°C. The probe source is a Xe/Hg 200-W lamp with a 307-nm interference filter to excite the 2AP fluorescence. The fluorescence emission intensities are detected by a photomultiplier tube (Hamamatsu R928, Middlesex, NJ) in a direction perpendicular to the incident light. The preamplifier is a 5-MHz current-to-voltage converter (Hamamatsu C1053–51). The voltages are digitized by a 500-MHz transient digitizer (Hewlett–Packard 54825A).
Analysis of Kinetics Measurements.
The observed kinetics at each temperature were fit to the functional form [I(0+, Tf) − I(∞, Tf)]exp(−kr(Tf)t) + I(∞, Tf), where I(∞,Tf) is the equilibrium intensity of 2AP fluorescence at the final temperature Tf, and I(0+,Tf) is the intensity immediately after the laser pulse. I(0+,Tf) differs from the prelaser baseline intensity I(0−, Ti) at the initial temperature Ti because of the intrinsic change in 2AP fluorescence with temperature (Fig. 2). Tf was determined from the ratio I(∞,Tf)/I(0−,Ti) calibrated on a fluorescence melting profile for each hairpin. The intensity I(0+,Tf) was determined by measuring the intrinsic temperature dependence of 2AP fluorescence incorporated in a three-nucleotide reference sample.
Figure 2.

Kinetics of unwinding/hairpin formation. The fluorescence of 2AP substituted at site 1 (hairpin H1) is monitored as a function of time following a laser temperature jump from 29 to 37°C. The characteristic relaxation rate kr obtained from a single exponential fit to the data is 1/(10.5 μs) at 37°C. The amplitude of the fluorescence extrapolated to zero time is smaller than the prelaser fluorescence as a result of a rapid decrease in the quantum yield of 2AP on change in temperature.
Two-State Arrhenius Description.
The equilibrium constant is defined as Keq = kc/ko, where kc is the rate coefficient for the step corresponding to the closing of hairpins, and ko is the rate coefficient for the opening (unwinding) step. Keq was determined from the equilibrium melting profiles at 266 nm. The measured relaxation rate kr = kc + ko = kc (1 + Keq)/Keq. In an earlier study of conformational relaxation in proteins, the prefactor in an Arrhenius description of the rates was found to depend on both the solvent friction (proportional to the solvent viscosity) and internal friction of the protein (14, 15). Here we ignore any contributions from internal friction and assume that the prefactor scales inversely with the solvent viscosity η
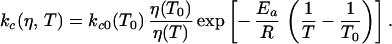 |
1 |
Configurational Diffusion Model.
The time-dependent evolution of the probability distribution on a free energy profile versus an effective reaction coordinate is calculated by using the method of Bicout and Szabo (16, 17). The characteristic relaxation rate is obtained from
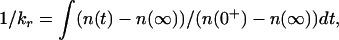 |
2 |
where n(t) is the center of mass of the probability distribution as a function of time. n(t) at t = 0+ is assumed a δ-function at θI = 0. The characteristic closing rate kc is calculated by using the double integral expression (18, 19):
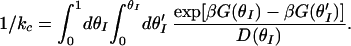 |
3 |
Here β = 1/(RT), θI is the fraction of intact base pairs and is defined as the effective reaction coordinate, G(θI) is the free energy along this reaction coordinate and is calculated from a statistical mechanical equilibrium “zipper” model, and D(θI) is the corresponding diffusion coefficient. The details of the “zipper” model and the calculation of G(θI) are available at www.uic.edu/∼ansari/zipper_model.pdf.
Results and Discussion
The thermodynamics and kinetics of a short DNA hairpin, formed from the self-complementary oligonucleotide sequence 5′-GGATAA(T4)TTATCC-3′, have been investigated in detail in this study. The hairpin has six base pairs in the stem and four thymine bases in the loop. 2AP, a fluorescent analog of the adenine base, was substituted at four different positions along the stem of the hairpin (Fig. 1). The fluorescence of 2AP decreases by about a factor of 2 when the base is hydrogen-bonded and stacked compared with the single-stranded state and provides both a local and a global probe for hairpin melting (20–22). Equilibrium melting profiles that monitor the average fraction of broken bonds (absorbance at 266 nm) as well as the fraction of broken bonds at the 2AP substituted site (absorbance at 330 nm) were monitored (Fig. 1). The melting profiles from these two sets of measurements overlap for each of the four 2AP-substituted analogs, indicating an all-or-none melting process. The melting profiles obtained from changes in the fluorescence of 2AP are, however, shifted to lower temperatures, suggesting a premelted state in which the bases are mobile while maintaining a time-averaged helical structure (21).
Kinetics of Hairpin Formation.
To study the kinetics of hairpin formation, we initiated the unwinding of an intact hairpin by using ≈6-ns laser T-jumps of ≈10°C. The characteristic time constant for attaining equilibrium after a T-jump perturbation is ≈10 μs at 37°C (Fig. 2). The measured relaxation rates show a weak temperature dependence for all three hairpin analogs whose kinetics were measured with 2AP substitution at sites 1, 2, and 3, respectively (Fig. 3). Assuming a two-state system with Arrhenius temperature dependence for the hairpin closing and opening rates yields a negative activation energy for the hairpin closing step (Ea ≈−11 kcal mol−1 averaged over the three hairpins). Negative activation energies have been observed for dimer formation in self-complementary oligonucleotides (6) and in β-hairpin and α-helical formation in polypeptides (23, 24). Negative activation energy implies that the transition “state” along the effective reaction coordinate has a lower enthalpy than the random coil state, and that the free energy barrier arises from a significant loss of entropy.
Figure 3.

Arrhenius plots of the relaxation rates (kr) and closing rates (kc). (a–c) The relaxation rates kr versus inverse temperature for hairpins with 2AP substitution at (a) site 1 (H1), (b) site 2 (H2), and (c) site 3 (H3), respectively. The circles are the measured relaxation rates. Red curves: fit to a two-state model with Arrhenius temperature dependence. The prefactors for the rate of hairpin formation (Eq. 1) are kc0 = 1.2 × 105 s−1, 0.4 × 105 s−1, 0.8 × 105 s−1 (at T0 = 25°C), and the apparent activation energies are Ea = −16.2 kcal mol−1, −7.3 kcal mol−1, −9.5 kcal mol−1 for the hairpins H1, H2, and H3, respectively. Blue curves: fit to the diffusion model. The fitting parameters are D0 = 2.1 × 105 s−1, 1.9 × 105 s−1, and 2.9 × 105 s−1 (at T0 = 25°) for H1, H2, and H3, respectively, and ΔE = 0.98 kcal mol−1. (d) Hairpin closing rates kc calculated from the diffusion model (Eq. 3) for H1 (continuous line), H2 (dashed line), and H3 (dash-dot-dot line).
Negative activation energies observed in our T-jump measurements as well as in earlier T-jump measurements on the helix–coil transition (6), at temperatures near the melting temperatures Tm, are in apparent contradiction with the positive activation energies of ≳5 kcal mol−1 reported by Libchaber and coworkers for similar hairpins (8). The activation energies in their measurements are obtained from Arrhenius fits to data primarily at temperatures lower than Tm (8). In the following discussion, we will show that this difference can be explained if we include kinetic trapping, from misfolding of loops, in the description of the dynamics.
Free Energy Profiles.
To calculate the free energy profiles, we first define an effective one-dimensional reaction coordinate, defined as the fraction of intact base pairs θI. A statistical mechanical equilibrium “zipper” model, in which all microstates of the hairpin with contiguous base pairs are included, is used to calculate the free energy corresponding to a given value of θI. The equilibrium model is based on the one-dimensional Ising model that allows for only two states for each base pair, broken or intact (25). Nearest-neighbor sequence dependence in the stacking interactions is included for each base pair, with parameters taken from the work of Benight and coworkers (4, 26). In our calculation, the statistical weight of the loop for each microstate in the ensemble depends on the size of the loop and is expressed in terms of a persistence length characterizing the flexibility of the single-stranded chain. In addition, stacking interactions among the bases in the loop and between the loop and the stem, and which themselves are found to exhibit a strong dependence on loop size, are also included in the loop free energy.§ This model describes very well the equilibrium melting profiles of hairpins with varying loop sizes and yields a value of ≈1.4 nm for the persistence length of single-stranded poly(dT) chains in 100 mM NaCl (S.V.K., Y.S., A. S. Benight & A.A., unpublished work; available at www.uic.edu/∼ansari/zipper_model.pdf).
Three important results from this analysis are summarized in Fig. 4. The first result is that the free energy barrier along this reaction coordinate corresponds to θI = 1/6 for the hairpin used in this study, which has six base pairs in the stem (Fig. 4a). For another hairpin with seven base pairs in the stem, the free energy barrier is located at θI = 1/7 (data not shown). Therefore, we postulate that the transition “state” in a two-state description of the kinetics consists of looped conformations, with one base pair closing the loop. The second result is that the height of the free energy barrier (≈2.8 kcal mol−1) is independent of the location of 2AP substitution (Fig. 4b), despite the fact that the melting temperature of the hairpin is lowered by 5–10°C as a result of 2AP substitution. This result is a strong indication that the transition “state” is an ensemble of looped conformations with varying loop sizes and not a specific sized loop, and that there are multiple paths to the “native” hairpin state. The third result is that we can estimate the enthalpy of the transition “state” ensemble, relative to the random coil, by extrapolating the height of the free energy barrier to T = 0K. This calculation yields a value of −11 ± 2.3 kcal mol−1 for each of the 2AP analogs, in remarkable agreement with the apparent activation energies observed in our kinetics measurements, thus lending further support to our identification of the transition “state.”
Figure 4.

The free energy landscape for hairpin formation. (a) The free energy profiles for the hairpin with no 2AP substitution are plotted versus the fraction of intact base pairs (θI), at three temperatures: 25°C (continuous line), 40°C (dashed line), and 55°C (dash-dot-dot line). The free energy profiles G(θI) are calculated from the statistical weights of the ensemble of microstates with a given value of θI, by using a statistical mechanical “zipper” model (details of the model and calculations are available on www.uic.edu/∼ansari/zipper_model. pdf). The discrete values of the free energies (at θI = 0, 1/6, 1/3, ½, 2/3, 5/6, and 1) are interpolated on a finer grid by using a cubic spline to get the semicontinuous profile. Diffusive dynamics on the free energy profile generated at each temperature yielded the characteristic rates for attaining equilibrium that are compared with the observed rates in Fig. 3 a–c. (b) The height of the free energy barrier at 25°C is plotted versus the location of the 2AP substitution.
Our conclusion regarding the nature of the transition state for hairpins differs from the conclusions reached by Crothers and coworkers in their analysis of the kinetics of dimer formation (6). Their results indicated that the stable nucleus for dimer formation consists of at least two to three base pairs before the downhill “zipping” of the rest of the chain. The results obtained from the analysis of dimer formation are not directly applicable to hairpins for the following very important reason. In dimer formation, the first base pair formed is not stabilized by stacking interactions and can easily dissociate. In hairpins, however, there are substantial stabilizing interactions between the bases in the loop and in the stem (27, 28), and it is very likely that the first base pair formed is already relatively stable. Short DNA fragments have been reported to form stable hairpin structures containing only two base pairs in the stem (29).
Configurational Diffusion.
If the reaction coordinate thus defined is appropriate and the free energy profiles are accurate, solving the diffusion equation along this coordinate should describe the observed kinetics. This approach was used to calculate the folding rates of model proteins represented on three-dimensional lattices (12, 13) and the folding rates of real proteins at ambient temperature (30). Predicting the temperature dependence of the measured rates requires knowledge of the free energy profile at each temperature and a model describing the temperature dependence and coordinate dependence of the effective diffusion coefficient. Here, we assume that in the nucleation step corresponding to the formation of the ensemble of looped conformations, the diffusion is impeded by the formation of “non-native” loops that have mismatched stems (Fig. 5). The energy of these misfolded loops is roughly the same as the energy of “native” loops; however, they serve as local traps that do not lead to subsequent zipping of the stem. These traps result in a “rough” energy landscape from θI = 0 to θI ≈ 1/6, with a corresponding decrease of the effective diffusion coefficient. When the correct loop is formed, the likelihood of misfolded structures in the zipping process, and hence the roughness, diminishes. We define two effective diffusion coefficients corresponding to the two steps, nucleation and zipping, as follows:
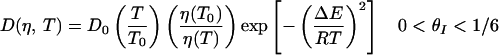 |
4a |
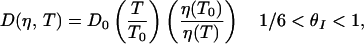 |
4b |
where D0 is a characteristic diffusion coefficient at T0 (=25°C), η(T) is the temperature-dependent viscosity of the solvent, and ΔE is the magnitude of the roughness in the nucleation step (12, 13, 19). The relaxation rates are readily calculated by solving the diffusion equation on the free energy profiles (16, 17).
Figure 5.

A schematic representation of the ensemble of microstates in the random coil, misfolded, transition “state,” and native-state conformations of the hairpin.
The fitting parameters in the diffusion model are D0 and ΔE. We assume ΔE is the same for all hairpins. The best-fit parameters yield D0 = 2.1 × 105 s−1, 1.9 × 105 s−1, 2.9 × 105 s−1 for the hairpins with 2AP substituted as sites 1, 2, and 3, respectively, and ΔE = 0.98 kcal mol−1. The value of ΔE is within a factor of 3 of the free energy cost of loop formation, consistent with the idea that the roughness arises from misfolded loops. The diffusion model describes remarkably well the temperature dependence of the measured relaxation rates with fewer parameters than for a two-state Arrhenius description (Fig. 3). The temperature dependence of the rates has two contributions in this model: one that comes from the temperature dependence of the free energy surface and another from the temperature dependence of the diffusion coefficient. The closing rates are found to exhibit a non-Arrhenius temperature dependence (Fig. 3d), consistent with the observations of Libchaber and coworkers (8). The non-Arrhenius temperature dependence is explained as follows: the closing rates are small at low temperatures as a result of getting stuck in the misfolded traps and are again small at high temperatures as a result of the uphill climb in free energy.
Non-Arrhenius temperature dependence for the folding step has been observed in lattice model simulations of protein folding (12, 13) and in experimental measurements of protein refolding (31, 32). This behavior is partially explained in terms of the unusual temperature dependence of the hydrophobic interaction, resulting in a large change in heat capacity on folding (31, 32). Scalley and Baker have deconvoluted the intrinsic temperature dependence of the folding reaction from the indirect effects of temperature on protein stability (33). Their results show that the effective diffusion coefficient in the preexponential of a simple transition-state theory expression is indeed strongly temperature dependent. Because of the narrow temperature range over which protein folding kinetics were measured, it is not possible from their analysis to distinguish between a simple Arrhenius temperature dependence for the diffusion coefficient or a super-Arrhenius dependence, as suggested in Eq. 4a. However, both theory (19) and numerical solutions (17) of diffusion in a one-dimensional potential show that relaxation dynamics in a rough potential are identical to dynamics in a smooth potential if the effective diffusion coefficient is rescaled as in Eq. 4a.
We can now reconcile the difference in the apparent activation energies between our measurements and those of Libchaber and coworkers. The roughness in the free energy surface and the probability of getting stuck in misfolded traps are expected to increase at temperatures well below Tm. Therefore, the apparent activation energies for the closing step obtained by Goddard et al. (8) are more likely dominated by the temperature dependence of the preexponential, which is proportional to the diffusion coefficient and measures the roughness of the free energy surface, and not by the enthalpy of the transition state. For the T-jump experiments presented here, the temperatures are close to Tm, and the roughness arising from misfolded loops is expected to be smaller. Therefore, apparent activation energies obtained from T-jump measurements reflect more accurately the enthalpy of the transition state.
Our model also provides a qualitative explanation for the intriguing result of Goddard et al. that the apparent activation energy for forming hairpins with poly(dA) loops increases as the length of the loop increases, a result that is counter to what one would expect from simple polymer theories describing loop closure for loops of a few persistence lengths. Poly(dA) chains have an increased propensity to stack, or “mis-stack” when the chain length increases. In our model, the oligonucleotides with long poly(dA) chains are expected to have a higher probability of misfolding before the correct nucleation, and the roughness in the free energy surface is expected to increase as the intervening chain length increases. Therefore, kinetic measurements below Tm would yield the result that the apparent activation energy increases with increasing loop length. Another set of equilibrium fluctuation measurements on hairpins with 30 poly(dA) bases in the loop reveal stretched exponential kinetics for hairpin formation (9). These experiments lend strong support to the hypothesis that at temperatures well below the melting temperatures, the dynamics of the hairpins are dominated by configurational diffusion among misfolded traps.
Finally, the configurational diffusion model described in this paper makes certain assumptions about the temperature and coordinate dependence of the diffusion coefficient. An important extension of this work would be to model the kinetics by solving the rate equations for a general discretized version that includes all possible microstates of the system. In previous applications of the kinetic “zipper” model to duplex formation, the possibility of mispairing, especially in the early stages of the nucleation, were not included explicitly (6). To apply the more general discrete model, including the possibility of trapping, would require enumerating all mispaired microstates in the ensemble.
Conclusions
We have shown that configurational diffusion on a free energy profile obtained from an equilibrium statistical mechanical model describes very well the kinetics of hairpin formation and explains a number of features that are not consistent with a simple two-state analysis with Arrhenius temperature dependencies. The main result from this work is that misfolded loops early in the nucleation step are responsible for decreasing the effective diffusion coefficient. This model naturally leads to non-Arrhenius temperature dependence for the hairpin formation step and also provides an explanation for the nonexponential nature of the kinetics at temperatures well below the melting temperatures for the hairpins. Our results and analysis highlight the need for models that elucidate the statistical nature of the folding process not only for proteins but also for polynucleotides. With few exceptions (34–36), the folding kinetics of RNA molecules have traditionally been described in terms of chemical kinetic schemes connecting sequential intermediates along some unique folding “pathway” (37–39). We have shown that even a simple system, a hairpin, folds via multiple paths and has the propensity to get stuck in misfolded conformations.
Acknowledgments
We thank Albert S. Benight and John F. Marko for illuminating discussions. We are also indebted to William A. Eaton, Timothy A. Keiderling, and Peter G. Wolynes for helpful comments on the manuscript. We are very grateful to Jeremy Fennema for his invaluable assistance in the design of the T-jump spectrometer, and to Peter M. Vallone for his help in sample preparation. This work was supported by the Donors of the Petroleum Research Fund administered by the American Chemical Society through grant ACS-PRF 32099-AC4, and by a Research Planning Grant and a CAREER award to A.A. from the National Science Foundation through Grants MCB-9707480 and MCB-9722295.
Abbreviations
- T-jump
temperature jump
- 2AP
2-aminopurine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Shen, Y., Kuznetsov, S. V. & Ansari, A. (2001) Biophys. J. 80, 481a.
References
- 1.Scheffler I E, Elson E L, Baldwin R L. J Mol Biol. 1968;36:291–304. doi: 10.1016/0022-2836(68)90156-3. [DOI] [PubMed] [Google Scholar]
- 2.Gralla J, Crothers D M. J Mol Biol. 1973;73:497–511. doi: 10.1016/0022-2836(73)90096-x. [DOI] [PubMed] [Google Scholar]
- 3.Uhlenbeck O C, Borer P N, Dengler B, Tinoco I., Jr J Mol Biol. 1973;73:483–496. doi: 10.1016/0022-2836(73)90095-8. [DOI] [PubMed] [Google Scholar]
- 4.Paner T M, Amaratunga M, Doktycz M J, Benight A S. Biopolymers. 1990;29:1715–1734. doi: 10.1002/bip.360291405. [DOI] [PubMed] [Google Scholar]
- 5.Bonnet G, Krichevsky O, Libchaber A. Proc Natl Acad Sci USA. 1998;95:8602–8606. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craig M E, Crothers D M, Doty P. J Mol Biol. 1971;62:383–401. doi: 10.1016/0022-2836(71)90434-7. [DOI] [PubMed] [Google Scholar]
- 7.Cohen R J, Crothers D M. J Mol Biol. 1971;61:525–542. doi: 10.1016/0022-2836(71)90063-5. [DOI] [PubMed] [Google Scholar]
- 8.Goddard N L, Bonnet G, Krichevsky O, Libchaber A. Phys Rev Lett. 2000;85:2400–2403. doi: 10.1103/PhysRevLett.85.2400. [DOI] [PubMed] [Google Scholar]
- 9.Wallace M I, Ying L, Balasubramanian S, Klenerman D. J Phys Chem B. 2000;104:11551–11555. [Google Scholar]
- 10.Bryngelson J D, Wolynes P G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leopold P E, Montal M, Onuchic J N. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryngelson J D, Onuchic J N, Socci N D, Wolynes P G. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 13.Socci N D, Onuchic J N, Wolynes P G. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- 14.Ansari A, Jones C M, Henry E R, Hofrichter J, Eaton W A. Science. 1992;256:1796–1798. doi: 10.1126/science.1615323. [DOI] [PubMed] [Google Scholar]
- 15.Ansari A. J Chem Phys. 1999;110:1774–1780. [Google Scholar]
- 16.Bicout D J, Szabo A. J Chem Phys. 1998;109:2325–2338. [Google Scholar]
- 17.Ansari A. J Chem Phys. 2000;112:2516–2522. [Google Scholar]
- 18.Szabo A, Schulten K, Schulten Z. J Chem Phys. 1980;72:4350–4357. [Google Scholar]
- 19.Zwanzig R. Proc Natl Acad Sci USA. 1988;85:2029–2030. doi: 10.1073/pnas.85.7.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nordlund T M, Andersson S, Nilsson L, Rigler R, Graslund A, McLaughlin L W. Biochemistry. 1989;28:9095–9103. doi: 10.1021/bi00449a021. [DOI] [PubMed] [Google Scholar]
- 21.Xu D, Evans K O, Nordlund T M. Biochemistry. 1994;33:9592–9599. doi: 10.1021/bi00198a027. [DOI] [PubMed] [Google Scholar]
- 22.Menger M, Eckstein F, Porschke D. Biochemistry. 2000;39:4500–4507. doi: 10.1021/bi992297n. [DOI] [PubMed] [Google Scholar]
- 23.Munoz V, Thompson P A, Hofrichter J, Eaton W A. Nature (London) 1997;390:196–199. doi: 10.1038/36626. [DOI] [PubMed] [Google Scholar]
- 24.Lednev I K, Karnoup A S, Sparrow M C, Asher S A. J Am Chem Soc. 1999;121:4076–4077. [Google Scholar]
- 25.Poland D, Scheraga H A. Theory of Helix-Coil Transitions in Biopolymers. New York: Academic; 1970. [Google Scholar]
- 26.Owczarzy R, Vallone P M, Goldstein R F, Benight A S. Biopolymers. 1999;52:29–56. doi: 10.1002/(SICI)1097-0282(1999)52:1<29::AID-BIP2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 27.Hilbers C W, Haasnoot C A, de Bruin S H, Joordens J J, van der Marel G A, van Boom J H. Biochimie. 1985;67:685–695. doi: 10.1016/s0300-9084(85)80156-5. [DOI] [PubMed] [Google Scholar]
- 28.Haasnoot C A, Hilbers C W, van der Marel G A, van Boom J H, Singh U C, Pattabiraman N, Kollman P A. J Biomol Struct Dyn. 1986;3:843–857. doi: 10.1080/07391102.1986.10508468. [DOI] [PubMed] [Google Scholar]
- 29.Hirao I, Kawai G, Yoshizawa S, Nishimura Y, Ishido Y, Watanabe K, Miura K. Nucleic Acids Res. 1994;22:576–582. doi: 10.1093/nar/22.4.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munoz V, Eaton W A. Proc Natl Acad Sci USA. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schindler T, Schmid F X. Biochemistry. 1996;35:16833–16842. doi: 10.1021/bi962090j. [DOI] [PubMed] [Google Scholar]
- 32.Tan Y J, Oliveberg M, Fersht A R. J Mol Biol. 1996;264:377–389. doi: 10.1006/jmbi.1996.0647. [DOI] [PubMed] [Google Scholar]
- 33.Scalley M L, Baker D. Proc Natl Acad Sci USA. 1997;94:10636–10640. doi: 10.1073/pnas.94.20.10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thirumalai D, Woodson S A. Acc Chem Res. 1996;29:433–439. [Google Scholar]
- 35.Pan J, Thirumalai D, Woodson S A. J Mol Biol. 1997;273:7–13. doi: 10.1006/jmbi.1997.1311. [DOI] [PubMed] [Google Scholar]
- 36.Isambert H, Siggia E D. Proc Natl Acad Sci USA. 2000;97:6515–6520. doi: 10.1073/pnas.110533697. . (First Published May 23, 2000, 10.1073/pnas.110533697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crothers D M, Cole P E, Hilbers C W, Shulman R G. J Mol Biol. 1974;87:63–88. doi: 10.1016/0022-2836(74)90560-9. [DOI] [PubMed] [Google Scholar]
- 38.Zarrinkar P P, Williamson J R. Science. 1994;265:918–924. doi: 10.1126/science.8052848. [DOI] [PubMed] [Google Scholar]
- 39.Fang X, Pan T, Sosnick T R. Biochemistry. 1999;38:16840–16846. doi: 10.1021/bi991700n. [DOI] [PubMed] [Google Scholar]