

Abstract
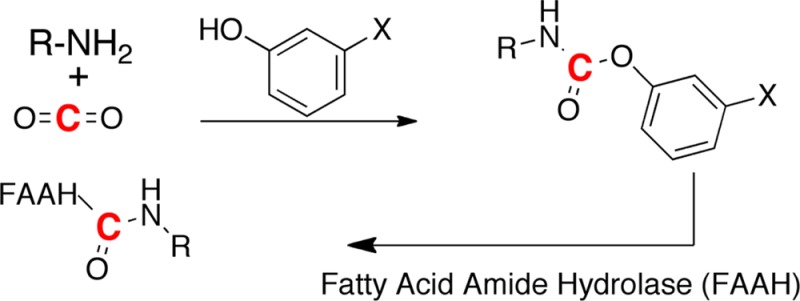
Fatty acid amide hydrolase (FAAH) plays a key role in regulating the tone of the endocannabinoid system. Radiotracers are required to image and quantify FAAH activity in vivo. We have synthesized a series of potent FAAH inhibitors encompassing two classes of N-alkyl-O-arylcarbamates and radiolabeled eight of them with carbon-11. The [11C-carbonyl]-radiotracers were evaluated in vitro and ex vivo in rats as potential FAAH imaging agents for positron emission tomography (PET). Both sets of [11C]O-arylcarbamates showed good to excellent brain penetration and an appropriate regional distribution. Pretreatments with a FAAH inhibitor demonstrated that 80–95% of brain uptake of radioactivity constituted binding of the radiotracers to FAAH. Brain extraction measurements showed that binding to FAAH was irreversible and kinetically different for the two classes of carbamates. These promising results are discussed in terms of the requirements of a suitable radiotracer for the in vivo imaging of FAAH using PET.
Introduction
The lipid character of endocannabinoids, such as anandamide and 2-arachidonoyl glycerol, dictates that these signaling molecules for the cannabinoid CB1 and CB2 receptors cannot be stored in vesicles, unlike polar G-protein coupled receptor neurotransmitters such as dopamine, acetylcholine, or serotonin. Rather, they are synthesized on demand and require efficient and rapid degradation to control their endogenous levels.1 In the central nervous system, levels of anandamide are primarily controlled by the degradative enzyme fatty acid amide hydrolase (FAAH), which converts anandamide into arachidonic acid and ethanolamine.2 Levels of the other major signaling endocannabinoid, 2-arachidonoyl glycerol, are similarly controlled by monoacylglycerol lipase (MAGL), although other hydrolytic enzymes can contribute to its metabolism.3 While very few selective inhibitors of MAGL have been reported,4 selective and potent inhibitors of FAAH are plentiful and have been useful tools in determining the role of FAAH in pain, anxiety, addiction, and psychiatric disorders.5
Measurement of FAAH activity in the living human brain, like other targets which exist in nanomolar concentrations, requires an imaging technique such as positron emission tomography (PET) or single photon emission computed tomography (SPECT), whereby a small amount of a positron-emitting or single photon emitting radiotracer is administered to a subject and the rate and extent of localization (binding) of the radiotracer to the target is measured using specialized cameras sensitive to the emitted radiation.6 The growing appreciation of the importance of FAAH in a variety of disorders has, in turn, led to an increased interest in developing suitable radiotracers for PET and SPECT imaging of this target.
An optimal radiotracer for human brain imaging studies requires balancing several factors including lipophilicity, affinity, specificity, metabolism, and pharmacokinetics.7 The last is especially important for the short-lived radionuclides typically used in PET scan imaging (carbon-11, t1/2 20.4 min; fluorine-18, t1/2 109.8 min), as the kinetics of the radiotracer (brain penetration rate, binding to target, washout rate, etc.) have to match the half-life in order for meaningful information to be extracted from the scan. While initial attempts were unsuccessful,8 we recently reported the first carbon-11 radiotracer that successfully tagged FAAH in vivo, namely [11C-carbonyl]N-cyclohexyl-5-hydroxy[1,1′-biphenyl]-3-ylcarbamic acid ester ([11C]CURB, [11C]1, Table 1).9 This carbamate radiotracer showed good brain penetration in rats which could be blocked by pretreatment with the prototypical FAAH inhibitor 2 (URB597, Table 1),5e and we are currently evaluating [11C]1 in human PET imaging studies.10 Very recently, a fluorine-18 FAAH inhibitor which shows some promise has also been synthesized.11
Table 1. Properties of Synthesized FAAH Inhibitors.
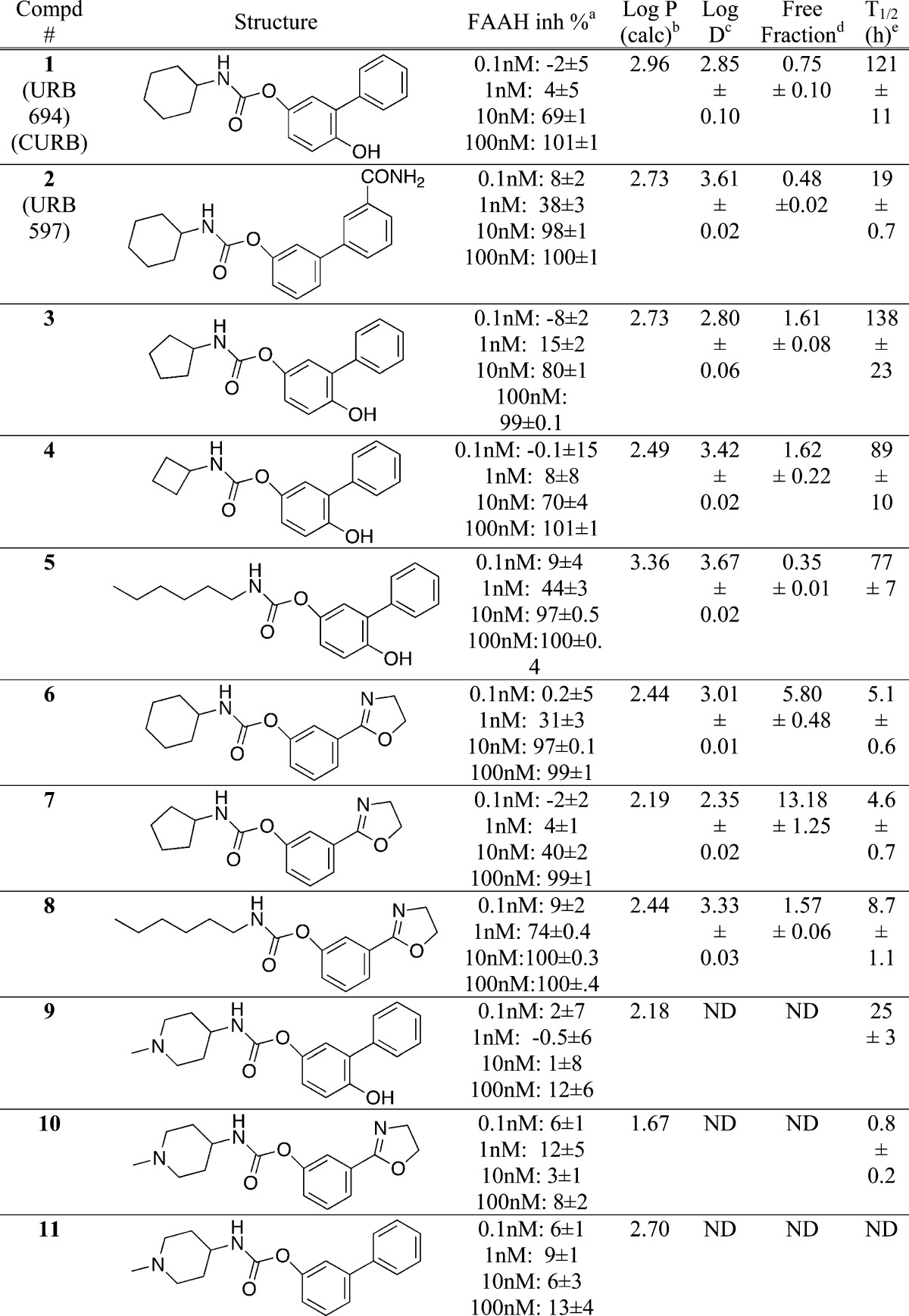
Inhibition of FAAH in rat brain homogenates using 0.5 μM [3H]anandamide as substrate. Values are % inhibition ± SEM, n = 3 at the inhibitor concentrations shown and with a preincubation time of 60 min.
Lipophilicity calculated using the Moriguchi method.14
Measured at pH 7.4.
Plasma free fraction measured using human plasma.
Rate of hydrolysis in pH 7.4 phosphate buffer at 37 °C.
To explore the relationships between structure, affinity, and in vivo binding of putative FAAH radiotracers, we have prepared a small library of O-arylcarbamates, radiolabeled them with carbon-11, and studied their binding characteristics to FAAH in rats. Our findings expand the library of available radiotracers for in vivo FAAH imaging as well as providing insight into the kinetics of FAAH inhibition in vivo.
Results
We synthesized the putative and the known FAAH inhibitors 2–11 (Table 1), as well as previously reported 1,9 by the straightforward methods outlined in Scheme 1. Where available, commercial isocyanates were reacted directly with the appropriate phenol (12, 13, or 14) to yield the desired O-arylcarbamates.
Scheme 1. Synthesis of O-Arylcarbamates Studied as FAAH Inhibitors.
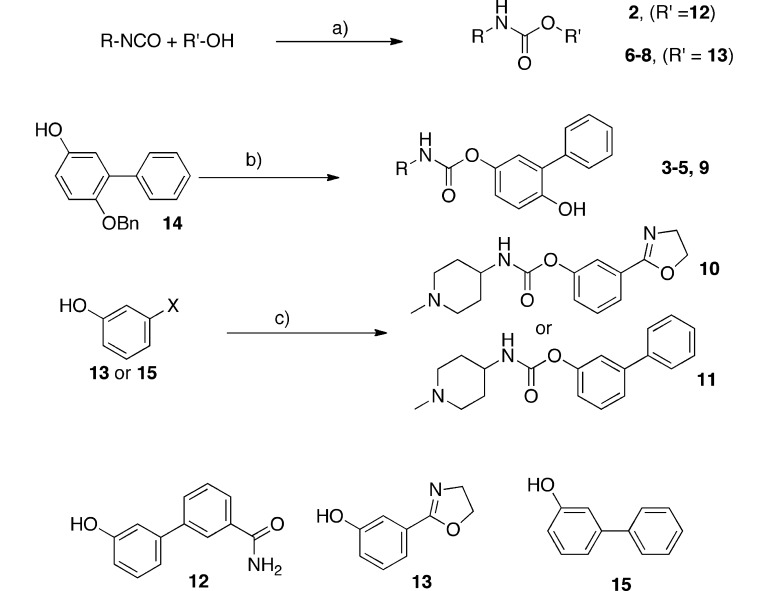
Reagents and conditions: (a) TEA, CH3CN, 90 min; (b) (i) COCl2, toluene, 0 °C to ambient, 30 min, (ii) R-NH2, TEA, CH3CN, 30 min, (iii) 10%Pd/C, ammonium formate, MeOH, reflux, 15 min; (c) (i) 4-nitrophenylchloroformate, DIPEA, DCM, 30 min, (ii) R-NH2, 1 h.
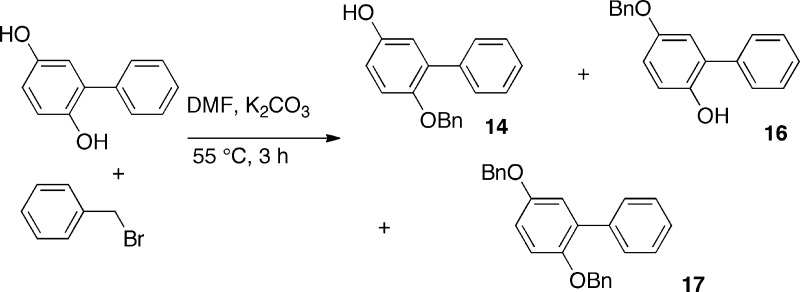
For the synthesis of biphenylcarbamates (3–5, 9), 2-phenyl-1,4-dihydroquinone, protected in the 1-position by a benzyl group (14), was reacted with phosgene to generate the chloroformate which was combined with the appropriate amine, yielding the O-arylcarbamates after deprotection. Alternatively, the 4-nitrophenylcarbonates of the required phenol were employed to generate carbamates 10 and 11. The synthesis of the monobenzyl ether of 2-phenyl-1,4-dihydroquinone (14) (Scheme 2) was tedious and inefficient, but its use, in place of the unprotected dihydroquinone, avoided the formation of hard-to-separate carbamate regioisomers. Moreover, the desired carbamate regioisomer was always the minor product when unprotected 2-phenyl-1,4-dihydroquinone was reacted with an isocyanate.
Scheme 2. Synthesis of Benzyl Protected Dihydroquinone, 14.
We screened the synthesized O-arylcarbamates for their ability to inhibit FAAH activity using rat brain homogenates and [3H]anandamide as substrate. All the simple N-alkyl and N-cycloalkylcarbamates (1–8) were potent inhibitors of FAAH with effective inhibitory concentrations in low nanomolar region (Table 1), regardless of whether the O-aryl group was of the biphenyl or dihydrooxazole type. The two most potent compounds were 5 and 8, both bearing linear N-hexyl substitutents, suggesting that the N-alkyl cabamates are somewhat more efficient inhibitors of FAAH than their cycloalkyl congeners, and also more lipophilic. We have not indicated IC50 values in view of the limited number of data points per compound and because for presumed covalent inhibitors the IC50 value is dependent upon the preincubation time used. A more detailed investigation with eight concentrations of 8 ranging from 0.1 to 30 nM returned pI50 values of 9.26 ± 0.02 and 8.93 ± 0.02 following preincubation times of 90 and 45 min, respectively, at 37 °C, corresponding to IC50 values of 0.55 and 1.2 nM, respectively. For comparative purposes, the pI50 values for 1, determined concomitantly and using six concentrations in the range 1–50 nM, were 8.31 ± 0.02 and 8.02 ± 0.03 following preincubation times of 90 and 45 min, respectively, at 37 °C, corresponding to IC50 values of 4.9 and 9.4 nM, respectively. Both compounds were approximately 4-fold less potent in the absence of a preincubation phase than after a 45 min preincubation phase (see Figure S1 in Supporting Information).
While it is known that both long-chain N-alkyl and N-alkylaryl groups are well tolerated, and bulky alkyl groups are not,12 there is a paucity of structure–activity information on the FAAH inhibiting activity of heteroatom containing N-alkyl carbamates. In this regard, we found that all three N-piperidinyl O-arylcarbamates (9–11) were impotent as FAAH inhibitors despite differing from known potent FAAH inhibitors only by the inclusion of a tertiary nitrogen in the cyclohexylring (Table 1). This was disappointing, as the piperidinyl nitrogen would have provided a convenient handle with which to attach simple and readily available synthons such as [11C]iodomethane or [18F]fluoroalkyl halides.13 Interestingly, a 6-morpholinohexyl group has also been reported as unfavorable for FAAH inhibition compared with a N-phenylhexyl group.12b
We radiolabeled the potent FAAH inhibitors 2–8 with carbon-11 in the carbonyl position via synthesized [11C]isocyanates by a [11C]CO2 fixation method developed in-house.9,15 Cyclotron-produced [11C]CO2 was reacted with the appropriate amine in the presence of BEMP, an efficient CO2 fixation agent,15 and the subsequent carbamate salts dehydrated to the [11C]isocyanates using POCl3.16 Subsequent reaction of the intermediate [11C]isocyanates with the appropriate phenol and purification by HPLC generated the target radiotracers in ample quantities for animal studies (Scheme 3). It must be noted that the position of the radiolabel in these FAAH inhibitors, at the carbonyl carbon, is crucial for their ability to image FAAH in vivo as the mechanism of inhibition involves covalent attachment of the serine-241 residue to the carbamoyl carbon with expulsion of the O-aryl residue, resulting in a covalent labeling of the enzyme with carbon-11.17
Scheme 3. Radiosynthesis of [11C-Carbonyl]O-arylcarbamates and Their Subsequent Tagging of FAAH.
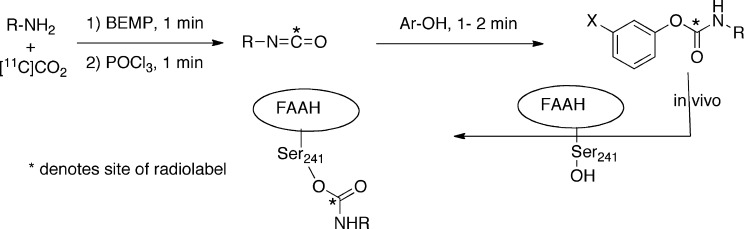
We explored the potential of the carbon-11 radiotracers as FAAH imaging agents in conscious rats. Animals were administered the radiotracer via the tail vein and the uptake of radioactivity in various brain regions measured by ex vivo dissection at an early (2 min) and a late (40 min) time point postinjection. Specificity of regional brain uptake was determined by pretreatment of animals with compound 2 (Table 1), the prototypical FAAH inhibitor in this class.5g The brain regional distribution for [11C]3 and [11C]8 typical examples are shown in Figure 1. Regional distribution for all the other radiotracers are provided in the Supporting Information.
Figure 1.
Regional uptake of radioactivity in rat brain of two representative [11C]-radiotracers at 2 and 40 min post iv injection. (A, left): [11C]7; (B, right): [11C]3. The blocked groups were pretreated with compound 2 (2 mg/kg, ip). Each value represents the mean (n = 5) ± SD.
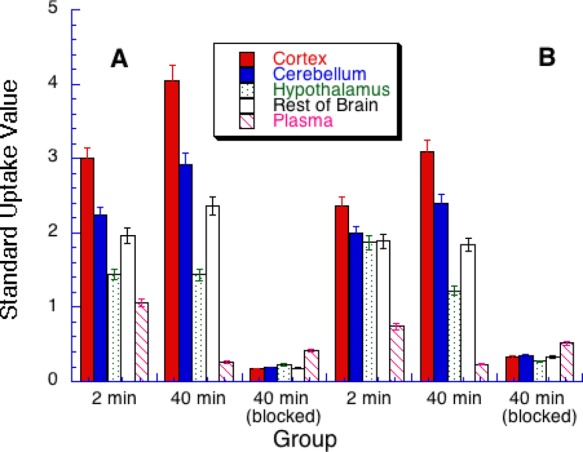
All the radiotracers rapidly penetrated the brain with moderate ([11C]2) to excellent ([11C]6) brain uptake. In general, the dihydrooxazoles (6–8) had higher brain uptake than the biphenyl carbamates (1–5) (Table 2). In every case, uptake of radioactivity was higher in the cortex, a region of higher FAAH density than in the FAAH-poor hypothalamus (Table 2).18 Moreover, levels of radioactivity were substantially (80–95% in cortex at 40 min postinjection) reduced by pretreatment with 2 (termed “blocked” in Table 2 and Figures 1 and 2) in all cases (Table 2 and Figure 1), showing that radioactivity uptake in the brain was mediated by FAAH.
Table 2. Regional Brain Uptake (In Standard Uptake Values) of [11C-Carbonyl]aryl Carbamates at 2 and 40 min Post iv Injection in Rat.
| compd no | cortex (2 min) | hypothalamus (2 min) | cortex (40 min) | hypothalamus (40 min) | cortex (40 min, blocked)a |
|---|---|---|---|---|---|
| [11C]1 | 2.40 ± 0.18 | 1.61 ± 0.23 | 2.49 ± 0.25 | 1.14 ± 0.13 | 0.36 ± 0.04b |
| [11C]2 | 0.79 ± 0.04 | 0.47 ± 0.03 | 0.94 ± 0.11 | 0.51 ± 0.10 | 0.17 ± 0.04b |
| [11C]3 | 2.36 ± 0.13 | 1.87 ± 0.08 | 3.09 ± 0.24 | 1.22 ± 0.15 | 0.33 ± 0.05b |
| [11C]4 | 2.68 ± 0.26 | 1.81 ± 0.29 | 3.37 ± 0.07 | 1.48 ± 0.08 | 0.23 ± 0.05b |
| [11C]5 | 1.63 ± 0.09 | 1.21 ± 0.07 | 2.49 ± 0.15 | 1.25 ± 0.04 | 0.27 ± 0.03b |
| [11C]6 | 4.52 ± 0.32 | 2.10 ± 0.17 | 6.61 ± 0.67 | 2.19 ± 0.10 | 0.74 ± 0.41b |
| [11C]7 | 3.00 ± 0.12 | 1.44 ± 0.07 | 4.05 ± 0.38 | 1.43 ± 0.08 | 0.17 ± 0.02b |
| [11C]8 | 3.58 ± 0.04 | 2.19 ± 0.14 | 5.30 ± 0.20 | 2.88 ± 0.13 | 0.20 ± 0.03b |
Pretreated with compound 2 (2 mg/kg, ip).
Significantly different from controls (p < 0.01). Each value represents the mean (n = 5) ± SD.
Figure 2.
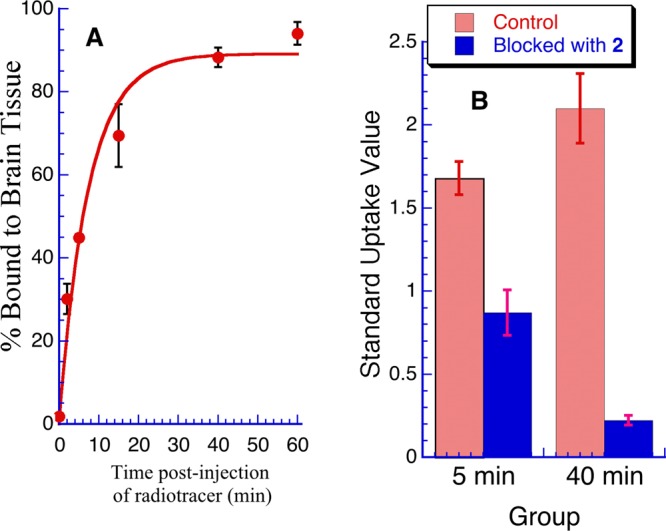
Amounts of radioactivity irreversibly bound to rat brain parenchyma postintravenous injection of [11C]3 (n = 3–4/group). (A) % bound at various time points. (B) Amount bound at 5 and 40 min postinjection and after pretreatment with compound 2 (2 mg/kg, ip).
We examined four of the radiotracers ([11C]1, [11C]3, [11C]6, and [11C]7) to determine the rate of binding to FAAH in vivo in rat brain and to explore further the irreversible nature of the binding.19 At various times, post radiotracer injection whole brains were excised, homogenized, and the rat parenchyma exhaustively extracted with aqueous acetonitrile. The amounts of radioactivity extracted (the unbound soluble fraction) and the amount bound to the tissue (covalently bound to protein fraction) were then counted. Figure 2A shows the rate and extent of irreversible binding of [11C]3. Pretreatment of rats with 2 reduced the amount of radioactivity bound to tissue by >90% (Figure 2B), demonstrating that the irreversible binding was FAAH mediated. The two biphenyl carbamates, [11C]3 and [11C]1, irreversibly bound to rat brain parenchyma, with half-lives of 5.5 and 6.6 min, respectively, and the % bound was about 90% at 60 min postinjection in both cases (Figure 2A and Figure S9 in Supporting Information). In contrast, the dihydrooxazole radiotracers [11C]6 and [11C]7 bound much more rapidly, with half-lives of 0.4 and 0.65 min, respectively, and the % bound at 60 min postinjection was >95% (Figures S7 and S8 in Supporting InformationI).
Discussion and Conclusions
We have synthesized and compared eight radiotracers having high affinity in vitro for FAAH comprising two classes of O-arylcarbamates, including the prototypical FAAH inhibitor 2 for reference purposes. Preparing [11C-carbonyl]-carbamates and ureas has, until recently, been limited to the very few facilities which had access to [11C]phosgene.20 The method employed herein, directly fixing cyclotron-produced [11C]CO2, makes these classes of radiotracers far more accessible to researchers. Thus far we have applied this method to three radiotracers for human imaging studies. The fact that all eight of the O-aryl carbamates described here were prepared in sufficient quantities and qualities for animal studies, without any individual tailoring of reaction parameters, demonstrates the broad applicability of the method.
Rat brain biodistribution studies following tail vein injection of radiotracers showed that both classes of O-aryl carbamates demonstrate favorable properties as potential imaging agents for FAAH. We observed good brain uptake, regional heterogeneity reflecting FAAH CNS distribution, and challenge studies with 2 indicated high specificity of binding to FAAH. Both classes also bound irreversibly to rat tissue in a time-dependent manner as demonstrated by ex vivo extraction measurements (Figure 2A). Pretreatment with the prototypical FAAH inhibitor, 2, which abolished >90% of irreversibly bound radioactivity clearly showed that this binding was mediated by FAAH (Figure 2B). However, we found tangible differences between the biphenyl and the dihydrooxazole carbamate radiotracers. The latter demonstrate somewhat higher brain uptake and lower nonspecific binding. In addition, the rate of irreversible binding of the dihydrooxazoles to FAAH is considerably faster than to the biphenyls. Both of these differences may reflect the lower lipophilicities of the dihydrooxazoles which may also be instrumental in their lower plasma protein binding (Table 1). The dihydrooxazoles are also inherently more labile than the biphenyl carbamates, as shown by their more rapid hydrolysis in aqueous solution (Table 1).
In addition to possessing good target delivery, high specificity, and low nonspecific binding, a useful imaging radiotracer must also be sensitive to changes in the target density or activity. If the radiotracer has too high an affinity to the target, then it can be rendered inefficient, i.e., radiotracer uptake in target regions is more determined by blood flow (delivery) than by the amount or activity of the target (which is the parameter we want to measure in vivo).21 In this context, it is possible that the dihydrooxazole radiotracers may not be ideal to image FAAH by PET given their very rapid brain uptake and irreversible binding to the FAAH enzyme in rat brain. Two points attenuate this view, however. All three [11C]dihydrooxazoles were capable of differentiating between a brain region of high FAAH activity (cortex) and a region of low FAAH activity (hypothalamus). Indeed, the cortex to hypothalamus ratios of uptake (range 2–3) were not significantly different between the two classes of compounds (Table 2). In addition, the amount of FAAH in human brain is reported to be only one-third of that in rat brain, suggesting that radiotracers with higher affinity and/or more rapid kinetics may be more suitable for human than rat.22 Nevertheless, until human or perhaps nonhuman primate imaging studies are performed with the [11C]dihydrooxazoles using full kinetic modeling, their rapid binding characteristics will remain a cause for concern despite their otherwise favorable characteristics such as ease of radiolabeling, high specific binding, and specificity for FAAH. Kinetic analysis of [11C]CURB binding to FAAH in human studies, presently underway, should also shed light on which properties should be pursued.
In summary, we have synthesized a series of [11C-carbonyl]-radiolabeled O-aryl carbamates and evaluated them as radiotracers for imaging FAAH with PET. All eight radiotracers have prima facie characteristics that should be favorable for in vivo PET imaging such as good brain penetration, high target specificity, and appropriate regional distribution. The range of other properties displayed by the radiotracer series such as fast or slow rates of uptake and varying levels of nonspecific binding provides us with a palette from which to choose a suitable candidate radiotracer for human imaging of FAAH with PET.
Experimental Section
A Scanditronix MC 17 cyclotron was used for radionuclide production. [11C]CO2, produced by the 14N(p,α)11C nuclear reaction, was concentrated from the gas target in a stainless steel coil cooled to −178 °C. Upon warming, the [11C]CO2 in a stream of N2 gas was passed through a NOx trapping column and a drying column of P2O5 prior to use.23 Purifications and analyses of radioactive mixtures were performed by high performance liquid chromatography (HPLC) with an in-line UV (254 nm) detector in series with a NaI crystal radioactivity detector (purifications) or a Bioscan Flowcount coincidence radioactivity detector (analyses). Isolated radiochemical yields were determined with a dose calibrator (Capintec CRC-712M). Automated radiosyntheses were controlled by Labview software. Unless stated otherwise, all radioactivity measurements were normalized for radioactive decay. POCl3 was distilled under reduced pressure prior to use. Cyclohexylamine, cyclopentylamine, cylclobutylamine, and hexylamine were distilled from NaOH under reduced pressure. Proton and carbon-13 NMR spectra were recorded at 25 °C on a Varian Mercury 400 MHz spectrometer. Electrospray ionization mass spectrometry was conducted with MDS Sciex QStar mass spectrometer to obtain the HRMS. All tested compounds had a purity of >95% as determined by reverse-phase HPLC. All animal experiments were carried out under humane conditions, with approval from the Animal Care Committee at the Centre for Addiction and Mental Health and in accordance with the guidelines set forth by the Canadian Council on Animal Care. Rats (male, Sprague–Dawley, 300–350 g) were kept on a reversed 12 h light/12 h dark cycle and allowed food and water ad libitum.
General Procedure for Synthesis of Carbamates 2, 6–8
A solution of the alkyl isocyanate (12 mmol) in acetonitrile (5 mL) was added to a stirred solution of the appropriate phenol 12(24) or 13(12c) (10 mmol) and triethylamine (0.5 g, 5 mmol) in acetonitrile (30 mL) at ambient temperature. Stirring was continued for 90 min, and then the reaction mixture quenched with 70 mL of water. The precipitate was collected by vacuum filtration, washed well with water, and recrystallized from 80% aqueous ethanol in 51–65% yields.
3′-Carbamoyl-[1,1′-biphenyl]-3-yl Cyclohexylcarbamate (2)
Obtained as a white solid in 51% yield; mp 186–190 °C (lit.24 178 °C). 1H NMR (CD3OD): δ ppm 1.16–1.45 (m, 5H), 1.59–1.71 (m, 1H), 1.75–1.85 (m, 2H), 1.90–2.02 (m, 2H), 3.39–3.50 (m, 1H), 7.12 (d, J = 7.02 Hz, 1H), 7.39–7.43 (m, 1H), 7.46 (t, J = 7.80 Hz, 1H), 7.51–7.58 (m, 2H), 7.78–7.89 (m, 2H), 8.14 (s, 1H). 13C NMR (DMSO-d6): δ ppm 26.32, 26.77, 34.17, 51, 121.69, 122.29, 125.04, 127.40, 128.05, 130.35, 131.0, 131.56, 135.78, 142.04, 143.07, 153.41, 156.52, 172.30. HRMS (ESI+) m/z calcd for [m + H]+ C20H23N2O3, 339.1705; found, 339.1703.
3-(4,5-Dihydrooxazol-2-yl)phenyl Cyclohexylcarbamate (6)
Obtained as a white solid in 60% yield; mp 151–155 °C (lit.12c 151–152 °C). 1H NMR was in agreement with the literature.12c
3-(4,5-Dihydrooxazol-2-yl)phenyl Cyclopentylcarbamate (7)
Obtained as a white solid in 65% yield; mp 168–171 °C (lit.12c 166–168 °C). 1H NMR was in agreement with the literature.12c
3-(4,5-Dihydrooxazol-2-yl)phenyl Hexylcarbamate (8)
Obtained as a white solid in 64% yield; mp 94–97 °C. 1H NMR (DMSO-d6): δ ppm 0.83–0.93 (m, 3 H), 1.21–1.35 (m, 6 H), 1.40–1.53 (m, 2 H), 3.0–3.10 (m, 2 H), 3.96 (t, J = 9.36 Hz, 2 H), 4.41 (t, J = 9.56 Hz, 2 H), 7.24–7.30 (m, 1 H), 7.47 (t, J = 7.80 Hz, 1 H), 7.52–7.56 (m, 1 H), 7.66–7.72 (m, 1 H), 7.82 (t, J = 5.66 Hz, 1 H). 13C NMR (DMSO-d6): δ ppm 14.35, 22.49, 26.34, 29.55, 31.38, 40.92, 54.89, 67.97, 121.26, 124.56, 125.07, 129.06, 130.12, 151.52, 154.45, 162.75. HRMS (ESI+) m/z calcd for [m + H]+ C16H23N2O3, 291.17087; found, 291.17128.
General Procedure for Synthesis of Carbamates 3–5, 9
A mixture of 14 (1.0 g, 3.62 mmol) in toluene (30 mL) and 5N aq NaOH (30 mL) was stirred at 0 °C and 10 mL of 20% phosgene in toluene added (CAUTION! Work in well ventilated fume hood with protective clothing and gloves). The ice-bath was removed and stirring continued at ambient temperature for 30 min. The aqueous layer was discarded and the toluene layer washed with 1N aq and HCl, dried (MgSO4), and about 70% of the toluene removed on the rotary evaporator at 40 °C. The remainder (about 10 mL) containing the phenylchloroformate of 14 was stored at −15 °C until required. A solution of the appropriate amine (0.4 mmol) in acetonitrile (1 mL) was added to 1 mL of the phenylchloroformate solution followed by triethylamine (TEA) (150 μL). After 30 min of stirring, 10 mL of 10% aq HCl was added. The mixture was extracted twice with EtOAc, the combined organics evaporated, and the residue taken up in MeOH (5 mL) and treated with 10%Pd/C and 1g of ammonium formate. This mixture was heated to reflux for 15 min, cooled, and filtered through a small pad of diatomaceous earth. Dilution of the filtrate with water (100 mL) precipitated crude product, which was collected by vacuum filtration, dried in vacuo, and recrystallized from ethylacetate/hexane.
6-Hydroxy-[1,1′-biphenyl]-3-yl Cyclopentylcarbamate (3)
Obtained as a white solid in 67% yield (from cyclopentylamine); mp 82–86 °C. 1H NMR (CDCl3): δ ppm 1.42–1.54 (m, 2H), 1.56–1.77 (m, 4H), 1.96–2.07 (m, 2H), 4.00–4.11 (m, 1H), 4.95 (d, J = 6.63 Hz, 1H), 6.87–6.92 (m, 1H), 6.96–7.00 (m, 1H), 7.00–7.02 (m, 1H), 7.38 (dq, J = 8.50, 4.31 Hz, 1H), 7.43–7.49 (m, 4H). 13C NMR (CDCl3): δ ppm 23.77, 33.41, 58.81, 116.61, 122.30, 123.33, 128.18, 128.79, 129.27, 129.37, 136.81, 144.63, 150.09. 163.44. HRMS (ESI+) m/z calcd for [m + H]+ C18H20NO3;, 298.14432; found, 298.14427.
6-Hydroxy-[1,1′-biphenyl]-3-yl Cyclobutylcarbamate (4)
Obtained as a white solid in 42% yield (from cylobutylamine): mp 140–142 °C. 1H NMR (CD3OD): δ ppm 1.62–1.78 (m, 2H), 1.94–2.09 (m, 2H), 2.22–2.35 (m, 2H), 4.04–4.18 (m, 1H), 6.85–6.87 (m, 2H), 6.95 (d, J = 3.12 Hz, 1H), 7.25–7.31 (m, 1H), 7.37 (t, J = 7.41 Hz, 2H), 7.50–7.57 (m, 2H). 13C NMR (DMSO-d6): δ ppm 15.78, 31.68, 47.73, 117.40, 122.58, 124.72, 128.10, 129.19, 130.46, 130.58, 139.68, 145.47, 152.92, 156.85. HRMS (ESI+) m/z calcd for [m + H]+ C17H18NO3, 284.1273; found, 284.1281.
6-Hydroxy-[1,1′-biphenyl]-3-yl Hexylcarbamate (5)
Obtained as an off-white solid in 55% yield (from hexylamine): mp 144–148 °C. 1H NMR (CDCl3): δ ppm 0.85–0.95 (m, 3 H), 1.28–1.41 (m, 6 H), 1.50–1.62 (m, 2 H), 3.16–3.38 (m, 2 H), 5.00 (br s, 1 H), 5.33 (br s, 1 H), 6.89–6.94 (m, 1 H), 6.97–7.04 (m, 2 H), 7.35–7.52 (m, 5 H). 13C NMR (CDCl3): δ ppm 13.99, 22.53, 26.40, 29.78, 31.43, 41.31, 116.37, 122.05, 123.07, 127.96, 128.55, 129.02, 129.16,136.53, 144.43, 149.85, 155.05. HRMS (ESI+) m/z calcd for [m + H]+ C19H24NO3, 314.17562; found, 314.17561.
6-Hydroxy-[1,1′-biphenyl]-3-yl (1-Methylpiperidin-4-yl)carbamate (9)
Obtained as a light-brown powder in 34% yield: mp 174–175 °C (dec.). 1H NMR (CDCl3): δ ppm 1.49–1.64 (m, 2H), 1.97–2.04 (m, 2H), 2.08–2.16 (m, 2H), 2.28 (s, 3H), 2.79 (d, J = 11.74 Hz, 2H), 3.50–3.66 (m, 1H), 4.92 (d, J = 7.82 Hz, 1H), 6.89–6.95 (m, 1H), 6.97–7.04 (m, 2H), 7.36–7.51 (m, 5H). 13C NMR (CDCl3): δ ppm 14.24, 32.35, 46.14, 54.32, 116.53, 122.05, 123.15, 128.00, 129.10, 129.18, 131.05, 136.67, 144.39, 155.10, 158.94. HRMS (ESI+) m/z calcd for [m + H]+ C19H23N2O3, 327.17087; found, 327.17219.
3-(4,5-Dihydrooxazol-2-yl)phenyl (1-Methylpiperidin-4-yl)carbamate (10)
To a stirred solution of 13 (500 mg, 3.1 mmol) and DIPEA (535 μL, 3.1 mmol) in 5:1 v/v CH3CN:DMSO (12 mL) at ambient temperature under N2 was added 4-nitrophenylchloroformate (617 mg, 3.1 mmol) as a solid. After 30 min, 4-amino-N-methylpiperidine (385 μL, 3.1 mmol) was added dropwise. After 1 h, the reaction was concentrated in vacuo and the residue was partitioned between saturated aqueous sodium bicarbonate (15 mL) and chloroform (15 mL). The aqueous layer was extracted twice with chloroform (15 mL), and the combined organic layers were dried (MgSO4) and filtered and the solvent was removed. The yellow residue was recrystallized from ethyl acetate:hexane to give a white solid (360 mg, 39%): mp 129–130 °C. 1H NMR (CDCl3): δ ppm 1.58 (m, 2H), 2.42 (m, 4H), 2.29 (s, 3H), 2.79 (d, J = 11.6 Hz, 2H), 3.58 (m, 1H), 4.06 (t, J = 9.5 Hz, 2H), 4.43 (t, J = 9.6 Hz, 2H), 4.96 (d, J = 7.3 Hz, 1H), 7.24 (m, 1H), 7.39 (t, J = 8.0 Hz, 1H), 7.70 (t, J = 1.8 Hz, 1H), 7.78 (d, J = 7.8 Hz, 1H). 13C NMR (CDCl3): δ ppm 32.4, 46.2, 48.0, 59.3, 60.0, 67.7, 121.5, 124.6, 125.0, 129.1, 129.3, 150.9, 153.4, 163.9. HRMS (ESI+) m/z calcd for [m + H]+ C16H22N3O3, 304.16612; found, 304.16655.
[1,1′-Biphenyl]-3-yl (1-Methylpiperidin-4-yl)carbamate (11)
To a stirred solution of 15 (500 mg, 2.9 mmol) and DIPEA (511 μL, 2.9 mmol) in DCM (12 mL) under N2 at ambient temperature was added 4-nitrophenylchloroformate (592 mg, 2.9 mmol) as a solid. After 30 min, 4-amino-N-methylpiperidine (366 μL, 2.9 mmol) was added dropwise. After 1 h, the reaction was concentrated in vacuo and the residue was partitioned between saturated aqueous sodium bicarbonate (15 mL) and chloroform (15 mL). The aqueous layer was extracted twice with chloroform (15 mL), the combined organic layers were dried (MgSO4) and filtered, and the solvent was removed. The yellow residue was recrystallized from EtOAc:hexane to give a white solid (298 mg, 33%): mp 132–134 °C. 1H NMR (CDCl3): δ ppm 1.60 (m, 2H), 2.07 (m, 4H), 2.30 (s, 3H), 2.81 (d, J = 11.0 Hz, 2H), 3.61 (m, 1H), 4.97 (d, J = 6.6 Hz, 1H), 7.11 (m, 1H), 7.37 (m, 5H), 7.56 (m, 2H). 13C NMR (CDCl3): 32.0, 45.8, 47.8, 54.2, 120.4, 124.1, 127.0, 127.2, 127.6, 128.8, 129.6, 140.3, 142.7, 151.3, 153.8. HRMS (ESI+) m/z calcd for [m + H]+ C19H23N2O2, 311.175695; found, 311.17565.
6-(Benzyloxy)-[1,1′-biphenyl]-3-ol (14)
To a stirred mixture of 1,1′-[biphenyl]-2,5-diol (10g, 0.054 mol) and K2CO3 (22.3g, 0.16 mol) in DMF (150 mL) at 50–60 °C was added benzyl bromide (6.42 mL, 0.053 mol) dropwise over 1 h. After 2 h, solids were filtered off, the DMF removed on a rotary evaporator, and the residue purified by flash chromatography (DCM:hexane (2/1)) to yield 17 (RF 0.88, 5.5 g, 28%), 16 (RF 0.39, 2.23 g, 15%), and the desired 14 (RF 0.15, 2.07 g, 14%).%): mp 123–126 °C. 1H NMR (CDCl3): δ ppm 4.87 (s, 1H), 4.94 (s, 2H), 6.73 (dd, J = 8.79, 3.09 Hz, 1H), 6.84 (d, J = 3.09 Hz, 1H), 6.91 (d, J = 8.79 Hz, 1H), 7.21–7.46 (m, 7H), 7.53–7.61 (m, 2H). 1H NMR (DMSO-d6) d ppm 4.97 (s, 2H) 6.70–6.76 (m, 2H) 7.01 (d, J = 8.52 Hz, 1H) 7.24–7.35 (m, 5H) 7.36–7.43 (m, 2H) 7.48–7.54 (m, 2H) 9.11 (s, 1H). 13C NMR (CDCl3): δ ppm 71.29, 115.39, 116.09, 117.78, 127.55, 127.88, 128.18, 128.61, 128.93, 129.86, 132.10, 138.18, 138.95, 148.70, 152.32. HRMS (ESI+) m/z calcd for [m + H]+ C19H17O2, 277.12285; found, 277.12226.
Radiosynthesis of [11C-Carbonyl]aryl Carbamates
Ten min prior to cyclotron end-of-bombardment (EOB), a conical vial (1 mL) sealed with a Teflon-lined silicone septum was swept with N2 (10 mL/min for 5 min) and then charged with 100 μL of a solution of the appropriate amine (1 mg/mL) and 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP, 5 μL) in CH3CN. Cyclotron-produced [11C]CO2 was passed through the amine solution, via a 2″ × 21G needle, in a stream of N2 (10 mL/min) until trapped radioactivity peaked as measured by a proximal small radiation detector. One min later, a freshly prepared (same day) solution of POCl3 (0.2% v/v, 100 μL) in CH3CN was added, followed 30 s later by a solution of the appropriate phenol (1,1′-[biphenyl]-2,5-diol, 12, 13, or 15) (2 mg) in either CH3CN (100 μL, for 12 and 15) or DMSO/CH3CN (for 13, 100 μL. 1/1 (v/v)). One min (for 12 or 15) or two min (for 13) later, the reaction mixture was quenched with HPLC eluent/water (1/1 (v/v), 750 μL) and injected onto an HPLC column for purification. The product was collected and evaporated to dryness at 70 °C under vacuum in a rotary evaporator and reconstituted in saline (10 mL) containing 0.25% Tween 80 and 5% ethanol. Sterile filtration into a sterile and pyrogen-free vial provided the final formulated radiotracers, sterile and pyrogen-free, suitable for animal and PET imaging studies. Product identities, radiochemical purities, and specific activities were established using analytical HPLC and by coinjections with standards under a variety of conditions (columns, eluents, pH). From 850 mCi (31 GBq) of [11C]CO2 49–105 mCi (1.8–3.9 GBq) of formulated radiotracer were obtained in 25–30 min after radionuclide production with specific activities of 2300–6000 mCi/μmol (85.1–222 GBq/μmol) at end-of-synthesis. Radiochemical purities were >98%. It should be noted that when 1,1′-[biphenyl]-2,5-diol was used as the phenol (for the radiosynthesis of [11C]3, [11C]4, [11C]5, and [11C]9), the major product was the undesired regioisomer (see Figure S10 in Supporting Information), but more than sufficient quantities of the desired regioisomer were isolated for animal studies.
Biodistribution Studies of Radiotracers in Rat Brain
Groups of conscious rats in a restraining box (n = 5/time point) were injected with 30–60 MBq of high specific activity [11C]-radiotracer (70–170 GBq/μmol; 0.3–0.8 nmol) in 0.3 mL of buffered saline containing 0.5% Tween 80 via the tail vein, vasodilated in a warm water bath. Rats were sacrificed by decapitation at various time points postinjection, blood collected from the trunk in a heparinized tube, and the whole brain surgically removed from the skull and stored on ice. Brain regions were excised, blotted, and weighed. Radioactivities in tissues were assayed in an automated gamma counter, back-corrected to time of injection, using diluted aliquots of the initial injected dose as standards. For challenge studies, rats were pretreated with compound 2 (2 mg/kg) at 2 mL/kg, in saline containing Tween 80, ip, or vehicle alone, 30 min prior to radiotracer injection. Values are reported as standard uptake values (SUVs, mean ± standard deviation) defined as % injected dose/g of tissue divided by rat weight in kg. Treated groups were compared to those of the vehicle-treated group by Student’s t test. Statistical comparisons were considered significant when p < 0.05.
Metabolite Studies
Groups of rats (n = 3–4) were injected with [11C]-radiotracer as described above. Blood was collected from the trunk in a heparinized tube, and the whole brain surgically removed from the skull and stored on ice. Brain tissue was exhaustively extracted of all nonbound radioactivity using a previously described method.10 Blood was centrifuged to separate the plasma, which was treated with 10% by volume of acetic acid then used directly for HPLC analysis using minor modifications of the method described by Hilton.10,25 The rate of irreversible binding of [11C]-radiotracer to FAAH was calculated using a one-phase exponential association equation (Y = Ymax(1 – exp(−KX)) with GraphPad Prism software.
Log D Measurements
The partition coefficients of [11C]-radiotracers, between 1-octanol and 0.02 M phosphate buffer at pH 7.4, were determined by a previously described method and replicated six times.7a
Plasma Protein Binding
Binding of [11C]-radiotracers to human plasma proteins was performed using an ultrafiltration technique described previously.26 Briefly, human blood samples (n = 6) were centrifuged and the plasma spiked with [11C]-radiotracer. After incubation at ambient temperature for 10 min, the plasma was then filtered by centrifugation through an ultrafiltration unit (AmiconCentrifree; Millipore) and the ultrafiltrate and plasma counted for radioactivity and weighed. Binding to the filtration apparatus was monitored and corrected for by substituting phosphate buffer pH 7.4 for plasma in the procedure.
Kinetics of Hydrolysis of Carbamates
Solutions of the carbamates 1–10 (ca. 5 μM) in 0.05 N phosphate buffer, pH 7.4, containing 1% DMSO, were prepared. These were incubated at 37 °C. The rate of hydrolysis was monitored by HPLC (Phenomenex Kinetix C18, 3 μ) at 254 nm. All reactions followed first-order kinetics out to >4 half-lives.
In Vitro FAAH Inhibition Enzyme Assays
The ability of the compounds to inhibit the FAAH-catalyzed hydrolysis of [3H]anandamide was determined in rat brain homogenates using the method of Boldrup et al.27 Briefly, test compounds were preincubated with 0.5 μg protein for 60 min (unless otherwise stated) prior to addition of substrate (0.5 μM [3H]anandamide, labeled in the ethanolamine side chain part of the molecule, American Radiolabeled Chemicals, Inc., St. Louis, MO, USA) and incubation for a further 10 min at 37 °C (assay volume 200 μL). Following the incubation phase, reactions were stopped by addition of 400 μL of active charcoal mixture (80 μL of charcoal + 320 μL of 0.5 M HCl), and after vortexing and centrifugation, aliquots of the aqueous phase containing the [3H]ethanolamine produced by substrate hydrolysis were collected and counted for tritium content by liquid scintillation spectrometry with quench correction.
Acknowledgments
We thank Armando Garcia, Winston Stableford, Min Wong, Alvina Ng, and Laura Nguyen for their assistance with radiochemistry and animal dissection experiments, and Eva Hallin for her assistance with the in vitro FAAH assays. This work was supported by National Institutes of Health (NIH) grant no. 1 R21 MH 094424 01 and an infrastructure grant from the Canadian Foundation for Innovation. The in vitro FAAH assays were supported by the Swedish Research Council grant no. 12158, medicine (to C.J.F.).
Glossary
Abbreviations Used
- BEMP
2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine
- DIPEA
diisopropylethylamine
- MAGL
monoacylglyceroyl lipase
- pI50
–log of half-maximum inhibitory concentration
- SD
standard deviation
- SUV
standard uptake value
- TEA
triethylamine
Supporting Information Available
1H and 13C NMR spectra of all new compounds reported, representative radio-HPLC chromatograms of reactions mixtures, and brain biodistribution plots. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Author Present Address
∥ Division of Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital, Boston Massachusetts 02114, United States.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Piomelli D. The molecular logic of endocannabinoid signalling. Nature Rev. Neurosci. 2003, 4, 873–884. [DOI] [PubMed] [Google Scholar]; b Ahn K.; McKinney M.; Cravatt B. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 2008, 108, 1687–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cravatt B. F.; Giang D. K.; Mayfield S. P.; Boger D. L.; Lerner R. A.; Gilula N. B. Molecular characterization of an enzyme that degrades neuromodulatory fatty acid amides. Nature 1996, 384, 83–87. [DOI] [PubMed] [Google Scholar]; b Giang D. K.; Cravatt B. F. Molecular characterization of human and mouse fatty acid amide hydrolases. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 2238–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Blankman J.; Simon G.; Cravatt B. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dinh T. P.; Carpenter D.; Leslie F. M.; Freund T. F.; Katona I.; Sensi S. L.; Kathuria S.; Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 10819–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fowler C. J. Monoacylglycerol lipase—a target for drug development?. Br. J. Pharmacol. 2012, 166, 1568–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chang J. W.; Niphakis M. J.; Lum K. M.; Iii A. B. C.; Wang C.; Matthews M. L.; Niessen S.; Buczynski M. W.; Parsons L. H.; Cravatt B. F. Highly Selective Inhibitors of Monoacylglycerol Lipase Bearing a Reactive Group that Is Bioisosteric with Endocannabinoid Substrates. Chem. Biol. 2012, 19, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Long J.; Li W.; Booker L.; Burston J.; Kinsey S.; Schlosburg J.; Pavón F.; Serrano A.; Selley D.; Parsons L. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nature Chem. Biol. 2008, 5, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a McKinney M.; Cravatt B. Structure and function of fatty acid amide hydrolase. Annu. Rev. Biochem. 2005, 74, 411–432. [DOI] [PubMed] [Google Scholar]; b Sipe J. C.; Waalen J.; Gerber A.; Beutler E. Overweight and obesity associated with a missense polymorphism in fatty acid amide hydrolase (FAAH). Int. J. Obes. 2005, 29, 755–759. [DOI] [PubMed] [Google Scholar]; c Fowler C. J. The cannabinoid system and its pharmacological manipulation—a review, with emphasis upon the uptake and hydrolysis of anandamide. Fundam. Clin. Pharmacol. 2006, 20, 549–562. [DOI] [PubMed] [Google Scholar]; d Jayamanne A.; Greenwood R.; Mitchell V. A.; Aslan S.; Piomelli D.; Vaughan C. W. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br. J. Pharmacol. 2006, 147, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Pillarisetti S.; Alexander C. W.; Khanna I. Pain and beyond: fatty acid amides and fatty acid amide hydrolase inhibitors in cardiovascular and metabolic diseases. Drug Discovery Today 2009, 14, 1098–1111. [DOI] [PubMed] [Google Scholar]; f Schlosburg J. E.; Kinsey S. G.; Lichtman A. H. Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J. 2009, 11, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Piomelli D.; Tarzia G.; Duranti A.; Tontini A.; Mor M.; Compton T. R.; Dasse O.; Monaghan E. P.; Parrott J. A.; Putman D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006, 12, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Tarzia G.; Duranti A.; Gatti G.; Piersanti G.; Tontini A.; Rivara S.; Lodola A.; Plazzi P. V.; Mor M.; Kathuria S.; Piomelli D. Synthesis and structure–activity relationships of FAAH inhibitors: cyclohexylcarbamic acid biphenyl esters with chemical modulation at the proximal phenyl ring. ChemMedChem 2006, 1, 130–139. [DOI] [PubMed] [Google Scholar]; i Seierstad M.; Breitenbucher J. G. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J. Med. Chem. 2008, 51, 7327–7343. [DOI] [PubMed] [Google Scholar]
- a Dannals R. F.; Ravert H. T.; Wilson A. A.. Chemistry of Tracers for Positron Emission Tomography. In Nuclear Imaging in Drug Discovery, Development, and Approval; Burns H. D., Gibson R. E., Dannals R. F., Siegl P., Eds.; Birkhäuser: Boston, 1993; [Google Scholar]; b Fowler J. S.; Wolf A. P. Working against Time: Rapid Radiotracer Synthesis and Imaging the Human Brain. Acc. Chem. Res. 1997, 30, 181–188. [Google Scholar]; c Miller P. W.; Long N. J.; Vilar R.; Gee A. D. Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew. Chem., Int. Ed. Engl. 2008, 47, 8998–9033. [DOI] [PubMed] [Google Scholar]; d Wahl R. L.Principles and Practice of PET and PET/CT, 2nd ed.; Lippincott Williams&Wilkins: Piladelphia, PA, 2009. [Google Scholar]
- a Wilson A. A.; Jin L.; Garcia A.; DaSilva J. N.; Houle S. An Admonition When Measuring the Lipophilicity of Radiotracers using Counting Techniques. Appl. Radiat. Isot. 2001, 54, 203–208. [DOI] [PubMed] [Google Scholar]; b Waterhouse R. N. Determination of lipophilicity and its use as a predictor of blood–brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2003, 5, 376–389. [DOI] [PubMed] [Google Scholar]; c Eckelman W. The Use of in Vitro Models to Predict the Distribution of Receptor Binding Radiotracers in vivo. Int. J. Radiat. Appl. Instrum., Part B 1989, 16, 233–245. [DOI] [PubMed] [Google Scholar]; d Laruelle M.; Slifstein M.; Huang Y. Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol. Imaging Biol. 2003, 5, 363–375. [DOI] [PubMed] [Google Scholar]; e Pike V. W. PET radiotracers: crossing the blood–brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyffels L.; Muccioli G. G.; De Bruyne S.; Moerman L.; Sambre J.; Lambert D. M.; De Vos F. Synthesis, in vitro and in vivo evaluation, and radiolabeling of aryl anandamide analogues as candidate radioligands for in vivo imaging of fatty acid amide hydrolase in the brain. J. Med. Chem. 2009, 52, 4613–4622. [DOI] [PubMed] [Google Scholar]
- Wilson A. A.; Garcia A.; Houle S.; Sadovski O.; Vasdev N. Synthesis and application of isocyanates radiolabeled with carbon-11. Chem.—Eur. J. 2011, 17, 259–264. [DOI] [PubMed] [Google Scholar]
- Wilson A. A.; Garcia A.; Parkes J.; Houle S.; Tong J.; Vasdev N. [11C]CURB: Evaluation of a novel radiotracer for imaging fatty acid amide hydrolase by positron emission tomography. Nucl. Med. Biol. 2011, 38, 247–253. [DOI] [PubMed] [Google Scholar]
- Skaddan M. B.; Zhang L.; Johnson D. S.; Zhu A.; Zasadny K. R.; Coelho R. V.; Kuszpit K.; Currier G.; Fan K. H.; Beck E. M.; Chen L.; Drozda S. E.; Balan G.; Niphakis M.; Cravatt B. F.; Ahn K.; Bocan T.; Villalobos A. The synthesis and in vivo evaluation of [18F]PF-9811: a novel PET ligand for imaging brain fatty acid amide hydrolase (FAAH). Nucl. Med. Biol. 2012, 39, 1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tarzia G.; Duranti A.; Tontini A.; Piersanti G.; Mor M.; Rivara S.; Plazzi P. V.; Park C.; Kathuria S.; Piomelli D. Design, synthesis, and structure–activity relationships of alkylcarbamic acid aryl esters, a new class of fatty acid amide hydrolase inhibitors. J. Med. Chem. 2003, 46, 2352–2360. [DOI] [PubMed] [Google Scholar]; b Mor M.; Lodola A.; Rivara S.; Vacondio F.; Duranti A.; Tontini A.; Sanchini S.; Piersanti G.; Clapper J. R.; King A. R.; Tarzia G.; Piomelli D. Synthesis and quantitative structure–activity relationship of fatty acid amide hydrolase inhibitors: modulation at the N-portion of biphenyl-3-yl alkylcarbamates. J. Med. Chem. 2008, 51, 3487–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Myllymaki M. J.; Kasnanen H.; Kataja A. O.; Lahtela-Kakkonen M.; Saario S. M.; Poso A.; Koskinen A. M. Chiral 3-(4,5-dihydrooxazol-2-yl)phenyl alkylcarbamates as novel FAAH inhibitors: insight into FAAH enantioselectivity by molecular docking and interaction fields. Eur. J. Med. Chem. 2009, 44, 4179–4191. [DOI] [PubMed] [Google Scholar]
- a Crouzel C.; Längström B.; Pike V. W.; Coenen H. H. Recommendations for a practical production of [11C]methyl iodide. Int. J. Appl. Instrum., Part A 1987, 38, 601–603. [Google Scholar]; b Chi D.; Kilbourn M.; Katzenellenbogen J.; Welch M. A Rapid and Efficient Method for the Fluoroalkylation of Amines and Amides. Development of a Method Suitable for Incorporation of the Short-Lived Positron Emitting Radionuclide Fluorine-18. J. Org. Chem. 1987, 52, 658–664. [Google Scholar]
- Moriguchi I. Development of quantitative structure–activity relationships and computer-aided drug design. Yakugaku Zasshi 1994, 114, 135–146. [DOI] [PubMed] [Google Scholar]
- Wilson A. A.; Garcia A.; Houle S.; Vasdev N. Direct fixation of [11C]-CO2 by amines: formation of [11C-carbonyl]-methylcarbamates. Org. Biomol. Chem. 2010, 8, 428–432. [DOI] [PubMed] [Google Scholar]
- McGhee W. D.; Pan Y.; Talley J. J. Conversion of amines to carbamoyl chlorides using carbon dioxide as a phosgene replacement. Tetrahedron Lett. 1994, 35, 839–842. [Google Scholar]
- Alexander J. P.; Cravatt B. F. Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem. Biol. 2005, 12, 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Egertova M.; Giang D. K.; Cravatt B. F.; Elphick M. R. A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc. Biol. Sci. 1998, 265, 2081–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Thomas E.; Cravatt B.; Danielson P.; Gilula N.; Sutcliffe J. Fatty acid amide hydrolase, the degradative enzyme for anandamide and oleamide, has selective distribution in neurons within the rat central nervous system. J. Neurosci. Res. 1997, 50, 1047–1052. [DOI] [PubMed] [Google Scholar]; c Ueda N.; Puffenbarger R. A.; Yamamoto S.; Deutsch D. G. The fatty acid amide hydrolase (FAAH). Chem. Phys. Lipids 2000, 108, 107–121. [DOI] [PubMed] [Google Scholar]
- Fowler J. S.; Wolf A. P.; MacGregor R. R.; Dewey S. L.; Logan J.; Schlyer D. J.; Langstrom B. Mechanistic positron emission tomography studies: demonstration of a deuterium isotope effect in the monoamine oxidase-catalyzed binding of [11C]l-deprenyl in living baboon brain. J. Neurochem. 1988, 51, 1524–1534. [DOI] [PubMed] [Google Scholar]
- Brown G.; Henderson D.; Steel C.; Luthra S.; Price P.; Brady F. Two routes to [11C-carbonyl] organo-isocyanates utilizing [11C] phosgene ([11C] organo-isocyanates from [11C] phosgene). Nucl. Med. Biol. 2001, 28, 991–998. [DOI] [PubMed] [Google Scholar]
- a Kilbourn M. R.; Snyder S. E.; Sherman P. S.; Kuhl D. E. In vivo studies of acetylcholinesterase activity using a labeled substrate, N-[11C]methylpiperdin-4-yl propionate ([11C]PMP). Synapse 1996, 22, 123–131. [DOI] [PubMed] [Google Scholar]; b Koeppe R. A.; Frey K. A.; Snyder S. E.; Meyer P.; Kilbourn M. R.; Kuhl D. E. Kinetic modeling of N-[11C]methylpiperidin-4-yl propionate: alternatives for analysis of an irreversible positron emission tomography trace for measurement of acetylcholinesterase activity in human brain. J. Cereb. Blood Flow Metab. 1999, 19, 1150–1163. [DOI] [PubMed] [Google Scholar]
- Li W. P.; Sanabria-Bohorquez S.; Aniket J.; Cook J.; Holahan M.; Posavec D.; Purcell M.; DeVita R.; Chobanian H.; Liu P.; Chioda M.; Nargund R.; Lin L. N.; Zeng Z. Z.; Miller P.; Chen T. B.; O’Malley S.; Riffel K.; Williams M.; Bormans G.; Van Laere K.; De Groot T.; Evens N.; Serdons K.; Depre M.; de Hoon J.; Sullivan K.; Hajdu R.; Shiao L. L.; Alexander J.; Blanchard R.; DeLepeleire I.; Declercq R.; Hargreaves R.; Hamill T. The Discovery and Characterization of [C-11]MK-3168, A Novel PET Tracer for Imaging Fatty Acid Amide Hydrolase (FAAH). J. Labelled Compd. Radiopharm. 2011, 54, S38–S38. [Google Scholar]
- Tewson T. J.; Banks W.; Franceschini M.; Hoffpauir J. A Trap for the Removal of Nitrogen Oxides from Carbon-11 Carbon Dioxide. Int. J. Radiat. Appl. Instrum., Part A 1989, 40, 765–768. [Google Scholar]
- Mor M.; Rivara S.; Lodola A.; Plazzi P.; Tarzia G.; Duranti A.; Tontini A.; Piersanti G.; Kathuria S.; Piomellis D. Cyclohexylcarbamic Acid 3-or 4-Substituted Biphenyl-3-yl Esters as Fatty Acid Amide Hydrolase Inhibitors: Synthesis, Quantitative Structure– Activity Relationships, and Molecular Modeling Studies. J. Med. Chem. 2004, 47, 4998–5008. [DOI] [PubMed] [Google Scholar]
- Hilton J.; Yokoi F.; Dannals R. F.; Ravert H. T.; Szabo Z.; Wong D. F. Column-switching HPLC for the analysis of plasma in PET imaging studies. Nucl. Med. Biol. 2000, 27, 627–630. [DOI] [PubMed] [Google Scholar]
- Price J. C.; Mayberg H. S.; Dannals R. F.; Wilson A. A.; Ravert H. T.; Sadzot B.; Rattner Z.; Kimball A.; Feldman M. A.; Frost J. J. Measurement of Benzodiazepine Receptor Number and Affinity in Humans Using Tracer Kinetic Modeling, Positron Emission Tomography, and [C-11]Flumazenil. J Cereb. Blood Flow. Metab. 1993, 13, 656–667. [DOI] [PubMed] [Google Scholar]
- Boldrup L.; Wilson S. J.; Barbier A. J.; Fowler C. J. A simple stopped assay for fatty acid amide hydrolase avoiding the use of a chloroform extraction phase. J. Biochem. Biophys. Methods 2004, 60, 171–177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.