

Abstract
Polyethylenimine (PEI) is one of the most broadly used polycations for gene delivery due to its high transfection efficiency and commercial availability but materials are cytotoxic and often polydisperse. The goal of current work is to develop an alternative family of polycations based on controlled living radical polymerization (CLRP) and to optimize the polymer structure for efficient gene delivery. In this study, well-defined poly(glycidyl methacrylate)(P(GMA)) homopolymers were synthesized using reversible addition fragmentation chain transfer (RAFT) polymerization followed by decoration using three different types of oligoamines, i.e., tetraethylenepentamine (TEPA), pentaethylenehexamine (PEHA), and tris(2-aminoethyl)amine (TREN), respectively, to generate various P(GMA-oligoamine) homopolycations. The effect of P(GMA) backbone length and structure of oligoamine on gene transfer efficiency was then determined. The optimal polymer, P(GMA-TEPA)50, provided comparable transfection efficiency but lower cytotoxicity than PEI. P(GMA-TEPA)50 was then used as the cationic block in di-block copolymers containing hydrophilic N-(2-hydroxypropyl) methacrylamide (HPMA) and oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA). Polyplexes of block copolymers were stable against aggregation in physiological salt condition and in Opti-MEM due to the shielding effect of P(HPMA) and P(OEGMA). However, the presence of the HPMA/OEGMA block significantly decreased the transfection efficacy of P(GMA-TEPA)50homopolycation. To compensate for reduced cell uptake caused by the hydrophilic shell of polyplex, the integrin-binding peptide, RGD, was conjugated to the hydrophilic chain end of P(OEGMA)15-b-P(GMA-TEPA)50 copolymer by Michael-type addition reaction. At low polymer to DNA ratios, the RGD-functionalized polymer showed increased gene delivery efficiency to HeLa cells compared to analogous polymers lacking RGD.
Keywords: Brush-like polycation, non-viral gene vector, targeted gene delivery
INTRODUCTION
Gene therapy is a promising approach for the treatment of various human diseases,1–3 but clinical translation of gene transfer technologies has been hindered by the lack of safe, efficient, and cost-effective gene delivery vectors.4 Non-viral vectors, especially polycations-based materials, have attracted increasing attention due to their affordable production at large scale and possibility of tailor-made structures and functionalities.5–8 Branched polyethylenimine (bPEI, 25kDa), with its excellent transfection efficiency and commercial availability, is one of the most extensively used polycations, but the drawback for its therapeutic use is its inherent cytotoxicity.9,10 Although low-molecular weight PEI exhibits significantly reduced cytotoxicity than its high-molecular weight counterpart, it also suffers from decreased transfection efficiency.11,12 Because of this, the molecular weight and molecular weight distribution (MWD, also termed as polydispersity index (PDI)) of the polymer also affects the cytotoxicity and transfection efficacy significantly.13–15 Hence, there is considerable scope for the development of novel polycations with narrow PDI, good transfection activity and low cytotoxicity.
The rapid advances in controlled living radical polymerization (CLRP)16, such as atom transfer radical polymerization (ATRP)17, and reversible addition fragmentation chain transfer (RAFT)18 polymerization, have enabled the synthesis of well-defined and relatively uniform cationic polymers, such as poly(2-(dimethylamino) ethyl methacrylate) (P(DMAEMA))19, for gene delivery applications20,21. Poly(glycidyl methacrylate) (PGMA) is another poly(methacrylate) that provides access to various functional groups (e.g. amine and carboxylic acids) through its pendant reactive epoxy group. Thus PGMA has been used as a parent polymer for the generation of diverse functional daughter polymers. For example, Li et al. prepared different oligoamine-decorated poly(ethylene glycol)-b-poly(GMA) (PEG-b-P(GMA)) block copolymers by ATRP and post-polymerization decoration using ethyldiamine (EDA), diethylenetriamine (DETA), triethylenetetramine (TETA), and low-molecular weight PEI (Mw~400), and further evaluated the transfection efficacy of these materials.22 Recently, Xu et al. reported the synthesis of ethanolamine (EA)-functionalized comb-shaped PGMA polymers that are effective gene carriers by a combination of ATRP and ring-opening reactions.23
RAFT is an alternative polymerization approach to ATRP that utilizes a wide range of potential monomers21,24,25, including hydrophilic monomers, N-(2-hydroxypropyl) methacrylamide(HPMA)26, oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA)27 and 2-(methacryloxy)ethyl phosphorylcholine28 (a zwitterionic example), which can be polymerized to provide well-hydrated polymers that are attractive alternative to poly(ethylene glycol) (PEG). Recently, for example, our group prepared various functional peptide-grafted copolymers as gene vectors using RAFT copolymerization of HPMA and peptide-based macromonomer.29,30 These brush-shaped polymers based on oligolysine were significantly more efficient than linear poly(L)lysine polymers. However, a drawback of this approach is the relatively costly synthesis of oligolysine monomers.
The goal of this current work is to develop an alternative family of polycations with narrow PDI by RAFT polymerization, and to further optimize the polymer structure for gene delivery. To this end, P(GMA) homopolymers with pendent oligoamine side chains were synthesized by RAFT polymerization followed by post-polymerization decoration. The structure of the homopolycation that provides efficient gene transfer with relatively low cytotoxicity was first determined by testing P(GMA) polymers functionalized with three different oligoamines and polymerized to two different lengths. The selected polycation was then synthesized as a diblock copolymer with a hydrophilic HPMA or OEGMA block to impart salt stability to formulated complexes. In addition, to partially compensate for reduced cell uptake caused by the hydrophilic shell of polyplex, an RGD (integrin-binding) peptide was introduced to the P(OEGMA) block by Michael-type addition reaction between the terminal free thiol group of the P(OEGMA) chain and maleimide-modified RGD, and evaluated for targeted gene delivery.
EXPERIMENTAL SECTION
Materials
Glycidylmethacrylate(GMA, 97%, Aldrich) was purified by vacuum distillation before use. N-(2-hydroxypropyl) methacrylamide (HPMA) was purchased from Polysciences (Warrington, PA) and used as received. Oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA, Mn= 300 g/mol and pendent EO units DP~4.5) from Aldrich was purified by passing through a column filled with basic alumina to remove the inhibitor. RAFT CTA 4-cyanopentanoic acid dithiobenzoate (CPADB), N,N′-Azobisisobutyronitrile (AIBN), tetraethylenepentamine (TEPA), pentaethylenehexamine (PEHA), tris(2-aminoethyl)amine (TREN), N,N′-dimethylacetamide (DMAc, HPLC, 99.9%), poly(ethylenimine) (PEI, 25kDa, branched) and all other chemicals were purchased from Sigma-Aldrich and used as received. Endotoxin-free plasmid pCMV-Luc (Photinuspyralis luciferase under control of the cytomegalovirus (CMV) enhancer/promoter) was produced with the Qiagen Plasmid Giga kit (Qiagen, Hilden, Germany) according to the manufacturer’s recommendations.
Preparation of P(GMA) homopolymer
P(GMA) homopolymers with degree of polymerization (DP) of 50 and 100 were synthesized by RAFT polymerizations in DMAc using AIBN as the primary radical source and CPADB as the CTA. Solutions of (a) GMA(0.45 g, 3.2mmol), CPADB (17.8 mg, 6.27 × 10−5mol), AIBN (2.05 mg, 1.25 × 10−5mol) in DMAc (4 mL), and (b) GMA (0.23 g, 1.6 mmol), CPADB (4.47 mg, 1.6× 10−5mol), AIBN (0.87 mg, 5.3× 10−6mol) in DMAc (2 mL) were prepared in a 5 mL round-bottom flask equipped with a magnetic stirring bar to obtain [Monomer (M)]/[CTA] ratios of 50 or 100, respectively. The flask was sealed with a rubber septum, purged by nitrogen for 10 min, and then immersed in a preheated oil bath at 70 °C to start the polymerization. After reaction for 24 h, the flask was taken away from the oil bath and the reaction mixture was cooled to room temperature; the reaction mixture was poured into ice-cold diethyl ether to precipitate the product. The product was separated by centrifugation and further purified twice by redissolving/reprecipitating with DMAc/diethyl ether, and finally dried in a vacuum oven for 24 h, yielding a pink powdery polymer.
Preparation of P(HPMA)-b-P(GMA) and P(OEGMA)-b-P(GMA) block copolymer
Block copolymers of P(HPMA)/P(OEGMA) and P(GMA) were synthesized by RAFT polymerizations of HPMA/OEGMA in DMAc using AIBN as the primary radical source and P(GMA) as the macro-CTA. Solutions of (a) HPMA (0.616 g, 4.3mmol), PGMA50 macro-CTA (102 mg, 1.437× 10−5mol), AIBN (0.7854 mg, 4.79× 10−6mol) in DMAc (4.3mL), and(b)OEGMA(1.357 g, 4.5mmol), PGMA50 macro-CTA (160.5 mg, 2.26× 10−5mol), AIBN (1.236 mg, 7.53× 10−6mol) in DMAc (4.5 mL) were prepared to obtain [M]/[CTA] ratios of 300 (for HPMA) and 200 (for OEGMA), respectively. The solution was split in equal volumes into several glass vials. After putting in a stirbar and sealing with a rubber septum, the solution was purged with nitrogen for 5 min and then immersed in a preheated oil bath at 70 °C. The polymerization was quenched in liquid nitrogen at predetermined time intervals. After thawing, the polymer solution was precipitated in diethyl ether. The product was separated by centrifugation and further purified by redissolving/reprecipitating with DMAc/diethyl ether (for HPMA) and THF/diethyl ether (for OEGMA) twice. After drying in a vacuum oven for 24 h, pink powdery block copolymers were obtained. The conversion of polymerization and composition of block copolymers were determined by 1H NMR as listed in Table S1 & 2 (See supporting information (SI)).
Decoration of P(GMA) homopolymer and block copolymer by oligoamines
P(GMA) homopolymers with DP of 50 and 100 were decorated by either TEPA, PEHA, or TREN oligoamines. Block copolymers of P(HPMA)/P(OEGMA)-b-P(GMA)50 were decorated by the optimized oligoamine TEPA in a 30-fold molar excess. In a typical procedure, TEPA (2.0 mL, 10.5mmol)was dissolved in 10 mL of dry DMAc in a 25 mL round-bottom flask equipped with a magnetic stirring bar. PGMA50(50 mg, 7.04× 10−6mol) in 2 mL of DMAc was added dropwise into the TEPA solution. After reaction at 60 °C for 7 h, the mixture was poured into diethyl ether to precipitate the product. The product was separated by centrifugation and further purified by dialysis against 4 L of distilled water, which was replaced three times per day over the course of 3 days. Finally, the oligoamine-decorated polymers were collected by freeze-drying for further characterization.
Conjugation of RGD targeting peptide
RGD peptide was synthesized by solid phase peptide synthesis as described previously.31 Prior to the conjugation of RGD peptide, the presence of free thiol in the terminal of P(OEGMA)15-b-P(GMA-TEPA)50 polymer chain was confirmed by Ellman’s study. It was found that ~90 % of polymer chains are free thiol-terminated. P(OEGMA)15-b-P(GMA-TEPA)50 block copolymer (10 mg) and maleimide-modified RGD (10 mg) were dissolved in 2 mL of PBS (pH 6.1), and mixture was stirred at room temperature overnight. The product was purified by dialysis to remove excess RGD and any impurities, and further collected by freeze-drying. The amount of RGD peptide incorporated into the polymer was determined by comparing the UV absorbance of polymer with and without RGD at 280 nm followed by quantification using the following standard curve of RGD in Ellman working buffer,
where ARGD-polymer and Apolymer are the UV absorbances of RGD-conjugated polymer and RGD-free polymer in Ellman working buffer (1 mg/mL) at 280 nm, respectively and C is the concentration of maleimide-RGD (μg/mL).
Characterization of polymer
1H NMR spectra were recorded on a Bruker AV 500 nuclear magnetic resonance (NMR) instrument using d6-DMSO, CDCl3and D2O as a solvent. The molecular weight and molecular weight distribution of the polymers were determined by size exclusion chromatography (SEC). To prepare materials for analysis, the purified polymer was dissolved at 10 mg/mL in running buffer (0.15 M sodium acetate buffered to pH 4.4 with acetic acid) for analysis by SEC.29,30 Samples were then applied to an OHpak SB-804 HQ column (Shodex) in line with a miniDAWN TREOS light scattering detector (Wyatt) and a OptiLab rEX refractive index detector (Wyatt). Absolute molecular weight averages (Mw and Mn), and dn/dc were calculated using ASTRA software (Wyatt).
Preparation and characterization of DNA polyplexes
The pCMV-Luc2 plasmid was diluted in double-distilled H2O to a concentration of 0.1 mg/mL and mixed with an equal volume of polymer (also diluted in double-distilled H2O) at different N/P ratios, as previously described.32 After mixing, the polyplexes were allowed to incubate for 10 min at room temperature. Each polyplex solution (containing 2 μg DNA) was mixed with 40 μl of PBS (where PBS was used to dilute polyplex solutions to a final ionic strength of 150 mM), and then used to determine the particle size of the polyplexes by dynamic light scattering(DLS) performed on a Brookhaven Instruments Corp ZetaPALS instrument at a wavelength of 659.0 nm and a detection angle of 90°. The measurements were performed in triplicate. The morphology of the polyplexes was observed by JEOL 1230 TEM at an acceleration voltage of 100 kV.
Gel retardation assay
The DNA binding ability of the polymers was investigated by agarose gel retardation assay. The polymer/DNA complexes prepared at varying N/P (polymer protonatable nitrogens to DNA phosphates) ratios from 0.5/1 to5.5/1 were electrophoresed through a 1% (w/v) agarose gel containing ethidium bromide at 100 V in TAE buffer solution (40 mMTris-HCl,1 v/v % acetic acid, and 1 mM EDTA).
Acid–base titration
The buffering capacity of various P(GMA-oligoamine) homopolycations was determined by acid–base titration over a pH range from 2.0 to 11.0. Briefly, polymer was dissolved in 0.15M NaCl aqueous solution (0.2 mg/mL). The solution was brought to a starting pH of 10.0 with 0.1 M NaOH and then was titrated with 0.1 M HCl using a pH meter. As a reference,25kDa PEI was also titrated following the same method. The buffering capacity was determined as μmol of H+ per mg of polymer required to decrease the pH of 0.2 mg/mL polymer solution from 7.4 to 5.0.
Cytotoxicity study
The cytotoxicity of various P(GMA-oligoamine) homopolycations was evaluated in vitro using the MTS assay. HeLa cells were plated overnight in96-well plates at a density of 3000 cells per well per 0.1 mL. Polymers were prepared in serial dilutions in water and then diluted 10-fold in Opti-MEM medium (Invitrogen). The cells were rinsed once with PBS and incubated with 40 μl of the polymer solution for 4 h at 37 °C, 5%CO2. Cells were rinsed with PBS and the medium was replaced with100 μl complete growth medium. At 48 h, 20 μl of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium(MTS) (Promega) were added to each well. Cells were then incubated at 37 °C, 5% CO2 for 4 h. The absorbance of each well was measured at 490 nm using a plate reader. IC50 values were measured on a Tecan Safire2 plate reader (Männerdorf, Switzerland) with excitation at 491 nm and emission at 509 nm. The fluorescence signal for each N/P ratio was normalized to the N/P ratio=0 (DNA only) signal.
In vitro transfection study
Transfection studies in cultured cells were conducted as previously described.29,30 HeLa cells were seeded at a density of 15,000 cells/well in MEM medium supplemented with 10% FBS and 1% antibiotic/antimicrobial in a 24-well plate. Cells were allowed to attach for 24 h at 37 °C, 5% CO2. Polyplexes were formed at different N/P ratios using 1 μg of pCMV-Luc2 plasmid DNA in 20 μl total volume. Each sample was diluted to 200 μl with OptiMEM medium. The cells were washed once with PBS and the transfection solutions were added. The cells were incubated at 37 °C, 5% CO2 for 4 h. The cells were then washed twice with PBS and the polyplex solution was replaced with complete cell culture media. After an additional 44 h at 37 °C, 5% CO2, luciferase expression was quantified with a luciferase assay kit (Promega Corp.) according to the manufacturer’s instructions, except that a freeze-thaw cycle at −80 °C was included after the addition of the lysis buffer to ensure complete cell lysis. Luminescence intensity was measured on the plate reader with integration for 1 s. The total protein content in each well was measured by a BCA Protein Assay Kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s instructions so that the luciferase activity was normalized to the total protein content in each well after subtracting the background signal from cell lysate. Each sample was tested with a sample size (n)=3 or 6.
RESULTS AND DISCUSSION
Polymer synthesis
Both RAFT and ATRP techniques have offered good control over polymerization of GMA monomers22,23,33; however, RAFT polymerization was selected for these studies because it does not involve the use of any copper catalyst that might cause toxicity in vivo if not properly removed.16,34–36
Well-defined P(GMA) homopolymers were therefore synthesized by RAFT polymerization using CPADB as a CTA in DMAc (Scheme 1). Figure S1 shows the 1H NMR spectrum of purified PGMA50. By comparing the integral ratio of two protons (signal 6) in GMA units and the RAFT agent end group protons (ωend chemical shifts at 7.5–8.0 ppm ascribed to the phenyl protons of the RAFT agent Z-fragment), the DP was determined to be 50. PGMA with DP of 100 was also synthesized by varying the feed ratio of monomer and CTA. P(GMA) homopolymers (PGMA50and PGMA100) with pendant oligoamine side chains were prepared by the nucleophilic reaction of oligoamines with the epoxy group of GMA in DMAc at 60 °C for 7 h in the presence of a molar excess (30 equiv. relative to GMA unit) of oligoamine. The excess oligoamine was removed by extensive dialysis. It should be noted that the terminal dithioester RAFT group was reduced to free thiol by aminolysis during the oligoamine decoration. The free thiols generated in the terminal of polymer chain could be further functionalized by various methods. Herein, a targeting ligand was incorporated by Michael-type addition between the free thiol and maleimide-modified targeting peptide.
Scheme 1.
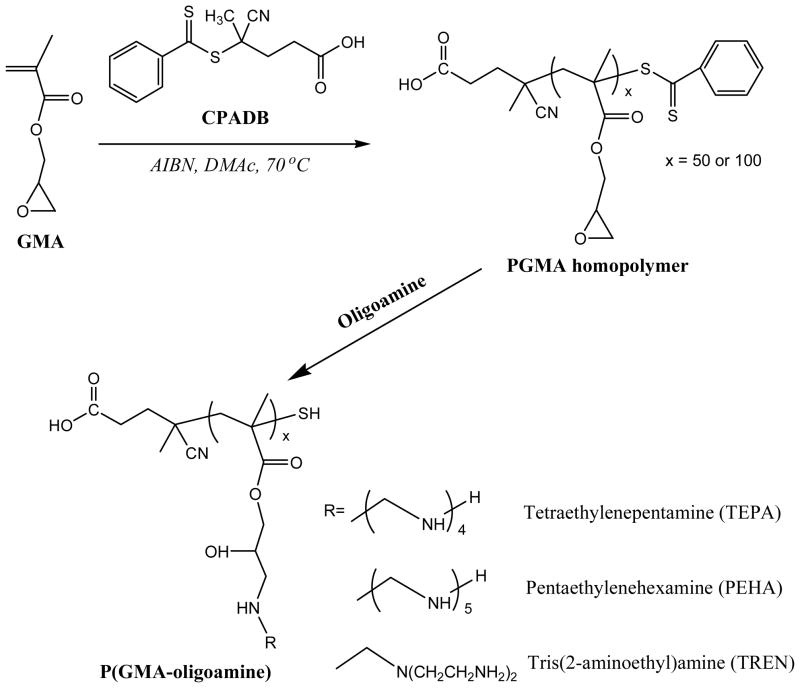
Synthesis of various P(GMA-oligoamine)s.
Three different types of oligoamines-TEPA, PEHA, and TREN-were used to generate six homopolycation P(GMA-oligoamine)s with different PGMA backbones and pendant oligoamine side chains. These oligoamines were selected for their similarities to ethylenimine and potential to buffer at endosomal pH when polymerized. TEPA and PEHA contain primary and secondary amine, while TREN contains primary and tertiary amines. Complete reaction of epoxy groups with oligoamine was confirmed by 1H NMR (Figure S1). The resonances assignable to the epoxy group (3.26, 2.87, and 2.66 ppm) completely disappear, whereas a new peak attributable to the ethylene protons connecting to amines (2.5–3.0 ppm) is clearly recorded. In addition, two characteristic peaks of ethylene adjacent to the epoxy group at 4.32 and 3.83 ppm are replaced by a new signal centered at 4.0 ppm due to the disappearances of the space isomerization of the epoxy group. The results confirm the successful synthesis of P(GMA-TEPA)50 cationic block copolymer with amino derivative side chains. SEC-MALLS were used to characterize the molecular weight and molecular weight distribution of these homopolycations. All the polymers exhibit uni-modal and narrowly-distributed molecular weight (Table 1 & Figure 1c), demonstrating well-controlled RAFT and post-polymerization decoration process.
Table 1.
Characteristics of various P(GMA-oligoamine)50/100.
| Mn (kDa)a | PDIa | dn/dca | Buffering capacity (μmol of H+ per mg of polymer)b | |
|---|---|---|---|---|
| P(GMA-TEPA)50 | 22.5 | 1.11 | 0.216 | 2.93 |
| P(GMA-PEHA)50 | 23.8 | 1.09 | 0.226 | 3.28 |
| P(GMA-TREN)50 | 23.3 | 1.35 | 0.213 | 3.26 |
| P(GMA-TEPA)100 | 41.9 | 1.23 | 0.214 | 3.39 |
| P(GMA-PEHA)100 | 47.6 | 1.20 | 0.218 | 3.54 |
| P(GMA-TREN)100 | 37.8 | 1.20 | 0.210 | 3.20 |
Determined by SEC-MALLS;
Determined by titration curves presented in Figure S2.
Figure 1.
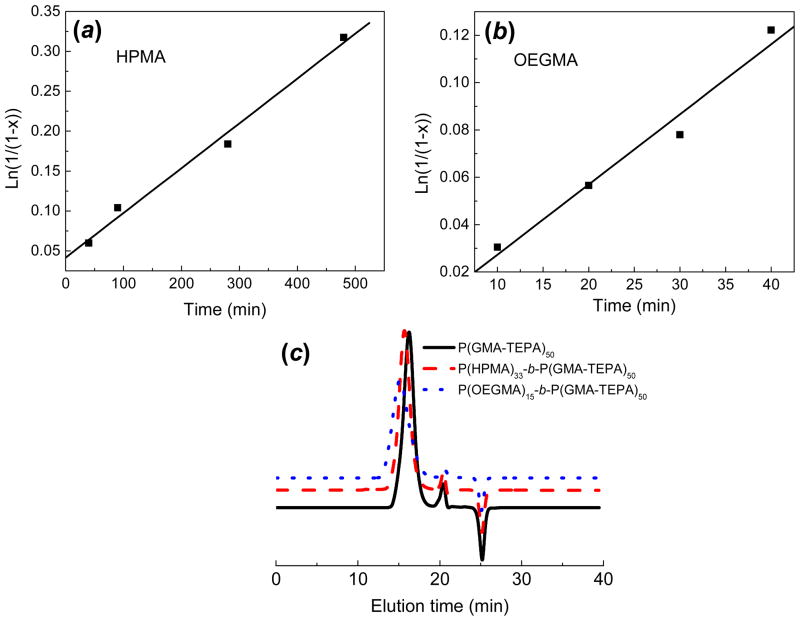
Pseudofirst-order kinetic plots of the polymerization of(a) HPMA ([M]:[macro-CTA]:[AIBN] = 300:1:0.33, [M] = 1.0 M, T = 70 °C, DMAc) and (b) OEGMA ([M]:[macro-CTA]:[AIBN] = 200:1:0.33, [M] = 1.0 M, T = 70 °C, DMAc) using PGMA50 as a macro-CTA. (c) SEC traces of P(GMA-TEPA)50 (Mn = 22.5kDa, PDI = 1.11, dn/dc =0.216), P(HPMA)33-b-P(GMA-TEPA)50 (Mn = 28.0kDa, PDI = 1.07, dn/dc =0.194), and P(OEGMA)15-b-P(GMA-TEPA)50 copolymer (Mn = 30.1 kDa, PDI = 1.28, dn/dc = 0.201).
To further incorporate HPMA and OEGMA segments, P(GMA)50 was then used as a macro-CTA to carry out RAFT polymerization of HPMA and OEGMA300 monomers (Scheme 2) by using a molar feed monomer: macro-RAFT agent: initiator ratio of 300:1:0.33 and 200:1:0.33 for HPMA and OEGMA, respectively. Table S1 & 2 summarize the properties of the block copolymers obtained. Both monomers were polymerized with pseudo first-order kinetics (Figure 1a & b) and linear growth of polymer chain, demonstrating excellent synthesis control via RAFT polymerization. Representative 1H NMR spectra are shown for two block copolymers: 1H NMR spectra of P(HPMA)33-b-P(GMA)50 (No. 2, Table S1), and P(OEGMA)15-b-P(GMA)50(No. 2, Table S2). Compared with the spectrum of P(GMA), the appearance of new characteristic signals of P(HPMA) block (the peaks at 2.89, 3.65, and 4.70 ppm are assigned to the HPMA repeating units) and P(OEGMA) block (the peaks at 4.15, 3.60, and 3.40 are ascribed to the OEGMA repeating units) confirms the successful synthesis of P(HPMA)33-b-P(GMA)50 and P(OEGMA)15-b-P(GMA)50 block copolymer.
Scheme 2.
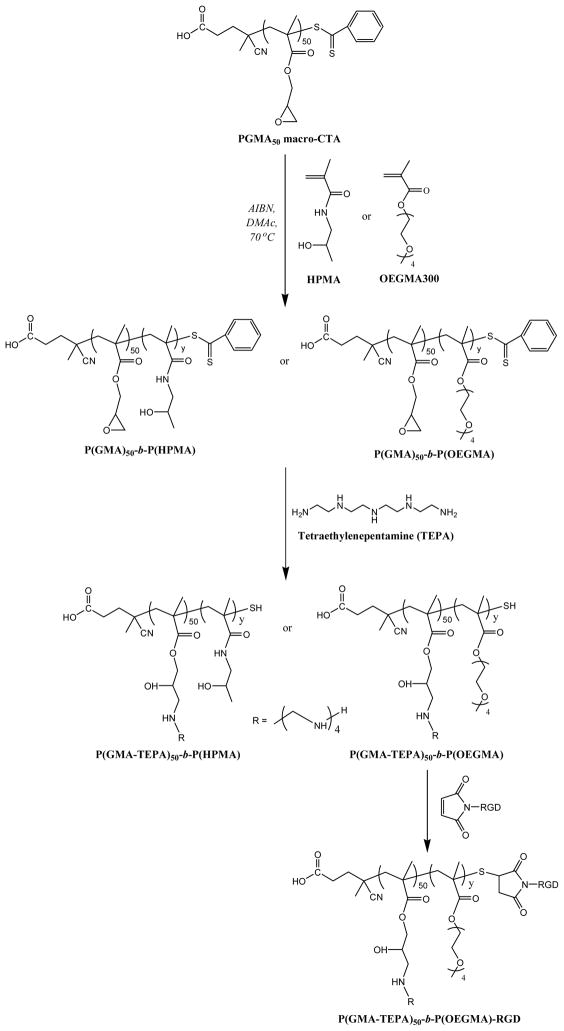
Synthesis of P(HPMA)-b-P(GMA-TEPA)50 and P(OEGMA)-b-P(GMA-TEPA)50, and subsequent conjugation of maleimide-RGD.
The GMA units in the block copolymers were decorated by oligoamine TEPA to generate cationic block copolymers with different chain lengths of hydrophilic blocks. Similar to the P(GMA-TEPA)50 homopolycation, the complete decoration of reactive epoxy groups was also confirmed by1H NMR (Figure S1). Compared to the chromatogram of P(GMA-TEPA)50, a shift of SEC traces towards shorter elution time was recorded for both P(GMA-TEPA)50-b-P(HPMA)33 and P(GMA-TEPA)50-b-P(OEGMA)15 (Figure 1c). Combined with the unimodal characteristics of all the chromatograms with narrow PDI (< 1.3), the results strongly indicate an efficient chain extension by a well-controlled RAFT process using PGMA50 as a macro-CTA.
Optimization of P(GMA-oligoamine)homopolycations
Efficient escape from endosomes is a critical attribute of successful polymeric gene vectors.37 It is hypothesized that the high transfection activity of PEI is associated with its intrinsic proton sponge effect, which facilitates endosomal escape of its DNA complexes.10,38,39 In this study, acid-base titration was performed to assess the proton buffering capacity of various P(GMA-oligoamine) homopolycations. The buffering capacity determined from the acid–base titration curves (Figure S2) was expressed as the μmol of H+ per mg of polymer required to decrease the pH of a 0.2 mg/mL polymer solution from 7.4 to 5.0. The results (Table 1) indicate that the P(GMA-oligoamine)s possess similar buffering capacity, approaching that of bPEI (3.85). Next, agarose gel electrophoresis was performed to investigate the binding capability of various P(GMA-oligoamine)s with pDNA. As shown in Figure S3, all the polymers exhibit good DNA binding property. The complete retardation of DNA was observed at nitrogen/phosphate (N/P) ratios of 2–4, which is typical of most polycations.19,23,40 For the same type of oligoamine, homopolycations with longer PGMA backbone (DP 100) display slightly stronger DNA binding ability, fully complexing plasmid at lower N/P ratios. The observation that higher molecular weight polycations more effectively bind DNA has been reported for other systems.22,23,30,41 For the same type of PGMA backbone, oligoamine TREN decorated-homopolycations show the best DNA binding ability with complete inhibition of DNA migration at N/P ratios of 2 (P(GMA-TREN)100) and 2.5 (P(GMA-TREN)50).
The in vitro gene transfection efficiency and cytotoxicity of various P(GMA-oligoamine)/pDNA complexes was assessed using luciferase as a gene reporter in HeLa cell line. First, the P(GMA-oligoamine)50/100polymers were complexed with plasmid DNA at N/P ratios of 3, 5, 7, 10, 20 and tested for gene delivery efficiencies (data not shown). Transfection efficiency was optimal at an N/P ratio of 10 for all the homopolycations. Lower N/P ratios (3,5,7) led to decreased transfection efficacy, and higher N/P ratio (20) resulted in significant cytotoxicity. No reporter gene expression was observed by delivery of plasmid alone. A comparison of the transfection efficacy (Figure 2A) and cytotoxicity (Figure 2B) of all the P(GMA-oligoamine)s at their optimal N/P ratio of 10 show that the transfection activity is dependent on the molecular weight of homopolycation, and for the same type of oligoamine, homopolycations with shorter PGMA backbone (DP 50) display higher transfection efficacy. P(GMA-TEPA)50 and P(GMA-PEHA)50 show the highest transfection efficacy, comparable to that of bPEI at its optimal N/P ratio of 5 but with less associated cytotoxicity. It is interesting that these polymers are less efficient at DNA condensation compared to P(GMA-TREN)50 (Figure S3). These results indicate that besides buffering capacity, balanced DNA condensation ability may be a factor for cationic polymers to achieve efficient gene transfer.42 The best polymeric vectors, on one hand, should effectively condense DNA, protecting DNA from degradation and facilitating cellular uptake, while also be readily displaced in the intracellular environment so that DNA transcription can occur.43
Figure 2.
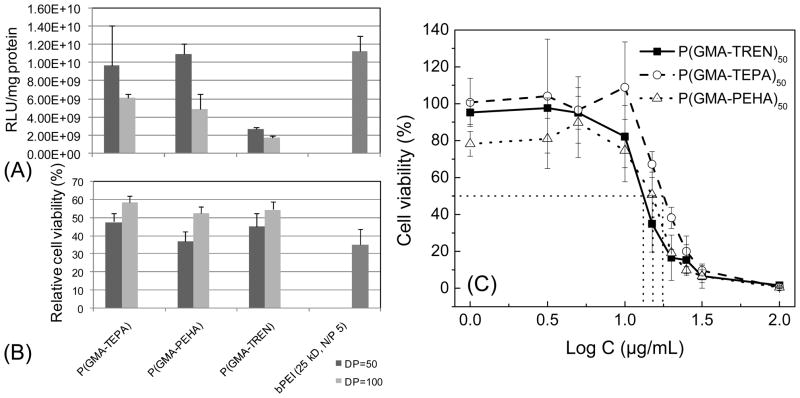
Transfection efficiency (A) and relative cell compatibility (B) of polyplexes formed by various P(GMA-oligoamine)50/100 in HeLacells at an N/P ratio of 10. Data are shown as mean ± SD (n = 3);(C)Cytotoxicity of various P(GMA-oligoamine)50 at different concentrations in HeLa cells determined by MTS study. Cell viabilities are shown as mean ± SD (n = 4).
Given that P(GMA-oligoamine)s with DP of 50 show better transfection efficiency than the counterparts with DP of 100, a tetrazolium-based metabolic assay, MTS assay, was used to determine the cytotoxicity of P(GMA-TEPA)50, P(GMA-PEHA)50, and P(GMA-TREN)50 to HeLa cells. A range of polymer concentrations was evaluated in order to determine the IC50 value (concentration of polymer for 50%cell survival) for each polymer. The results (Figure 2C) show that P(GMA-TEPA)50(17.78μg/mL) exhibits the lowest cytotoxicity, compared to P(GMA-PEHA)50(15.14 μg/mL) and P(GMA-TREN)50 (13.18 μg/mL).
Taking together the transfection efficacy and cytotoxicity results, we identified P(GMA-TEPA)50, which offers comparable transfection efficacy to PEI and displaying the lowest cytotoxicity among the tested polymers, to be the optimal cationic block. Note that we also assessed the transfection efficacy of block copolymer formulations with two different oligoamine grafts, P(GMA-TEPA)50-b-P(HPMA)33and P(GMA-PEHA)50-b-P(HPMA)33. The results (FigureS4) reveal that P(GMA-TEPA)50-b-P(HPMA)33 displays similar transfection efficacy but lower cytotoxicity compared to P(GMA-PEHA)50-b-P(HPMA)33, which agrees well with the transfection results of homopolycations, and further confirms that the optimized cationic segment remains optimal in the di-block formulations.
Figure 4.

TEM images of polyplexes formed by (a) P(GMA-TEPA)50 and (b) P(HPMA)33-b-P(GMA-TEPA)50 at an N/P ratio of 5 (Scale bar: 100 nm).
Incorporation of hydrophilic HPMA/OEGMA shell for stabilization of polyplexes
Polyplexes formed by the P(GMA-TEPA)50 homopolymer aggregate rapidly in salt medium (PBS, 150 mM, pH 7.4) due to the lack of a shielding corona, leading to the formation of large structures with diameter>1000 nm within 1 h.44 Salt stability is necessary for intravenous application of polyplexes, as colloidal aggregation causes toxicity after systemic administration.6,45 To address the aggregation issue, diblock copolymers of P(GMA-TEPA)50 with either P(HPMA) (Table S1) or P(OEGMA) (Table S2) of various chain lengths were synthesized.
The average hydrodynamic diameters of polyplexes in 150 mM PBS were determined by dynamic light scattering (DLS). Compared to the polyplexes of P(GMA-TEPA)50, all the diblock copolymers were able to condense DNA into smaller polyplexes with lower PDIs in the salt medium (Figure 3& S5), demonstrating that both P(HPMA) and P(OEGMA) shielding blocks can significantly enhance the salt stability of polyplex formed by the homopolycation. Notably, the polyplex sizes decrease with increasing chain length of HPMA/OEGMA block, indicating that polyplexes with longer hydrophilic chain were less prone to salt-induced aggregation. The increase of N/P ratios within the polyplex formulations also led to better salt stability, as evidenced by the decrease of polyplex sizes. Block copolymers of P(HPMA)33-b-P(GMA-TEPA)50and P(OEGMA)15-b-P(GMA-TEPA)50 were able to form DNA polyplexes that were stable at physiological salt concentrations with diameter < 200 nm at all the tested N/P ratios. Similar results were obtained using Opti-MEM transfection media (results not shown). Thus, the 4.7 kDa P(HPMA) block and 4.5 kDa P(OEGMA) block possess similar capability in stabilizing polyplex, and provide sufficient extracellular colloidal stability for polyplexes of P(GMA-TEPA)50 homopolycation.
Figure 3.
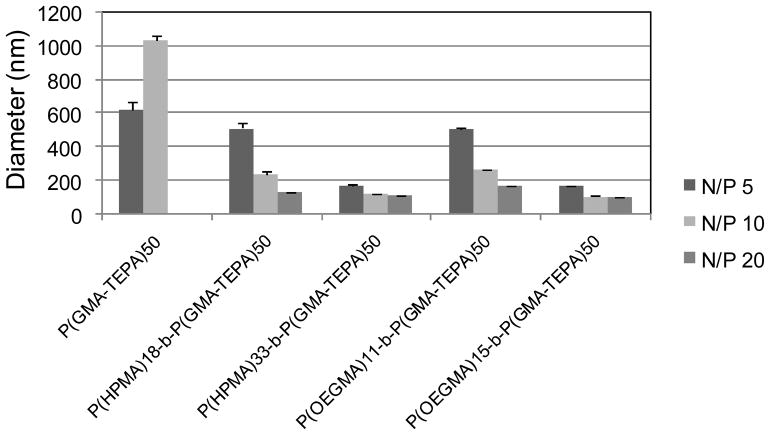
Average hydrodynamic diameter of various polyplexes formed at different N/P ratios in 150 mM PBS.
The morphologies of P(GMA-TEPA)50 and P(HPMA)33-b-P(GMA-TEPA)50 polyplexes prepared in distilled water were visualized by TEM (Figure 4) at an N/P of 5. Both materials condensed pDNA into compact particles with diameter<50 nm. Polyplexes formed using block copolymers were more compact than those formed by the P(GMA-TEPA)50 homopolymer. Similar results were observed for the polyplexes of P(OEGMA)15-b-P(GMA-TEPA)50 as well.44
DNA binding ability of block polymers was also investigated by agarose gel retardation assay. Compared to the P(GMA-TEPA)50homopolymer, block copolymers of P(HPMA)33-b-P(GMA-TEPA)50 and P(OEGMA)15-b-P(GMA-TEPA)50display decreased DNA binding capability (Figure S3), demonstrating the incorporation hydrophilic block disfavors DNA condensation. The in vitro transfection efficacy of block copolymers was further investigated to evaluate the effect of the hydrophilic block on the transfection activity of P(GMA-TEPA)50. First, the effect of hydrophilic block length on transfection efficacy (Figure 5(A)) and cell viability (Figure 5(B)) was testing using the P(HPMA)18/33-b-P(GMA-TEPA)50 polymers. Second, the effect of hydrophilic block composition on transfection efficacy (Figure (5C)) and cell viability (Figure 5(D)) was tested by comparing P(HPMA)33-b-P(GMA-TEPA)50with P(OEGMA)15-b-P(GMA-TEPA)50. The hydrophilic block lengths of these two polymers are 4.7 kDa and 4.5 kDa, respectively. The results reveal that: (1) Incorporation of a hydrophilic block significantly decreases the transfection efficacy of P(GMA-TEPA)50 homopolymer, and such loss of efficacy cannot be recovered by high polymer to DNA charge ratios, and (2) All the block copolymers studied exhibit similar transfection efficacy at the N/P ratios above 10; surprisingly, no significant difference in either transfection efficacy or cell viability was observed between block copolymers containing the linear P(HPMA) stabilizer or the brush-structured P(OEGMA) stabilizer. The decreased transfection efficacy of block copolymer is expected since polyplexes shielded against protein adsorption and aggregation have altered internalization and intracellular trafficking by cells, compromising transfection efficiencies,46 and this trade-off between particle stabilization and transfection efficiency resulting from the introduction of hydrophilic OEGMA blocks has been reported previously for several other polycations used for gene delivery.47,48
Figure 5.
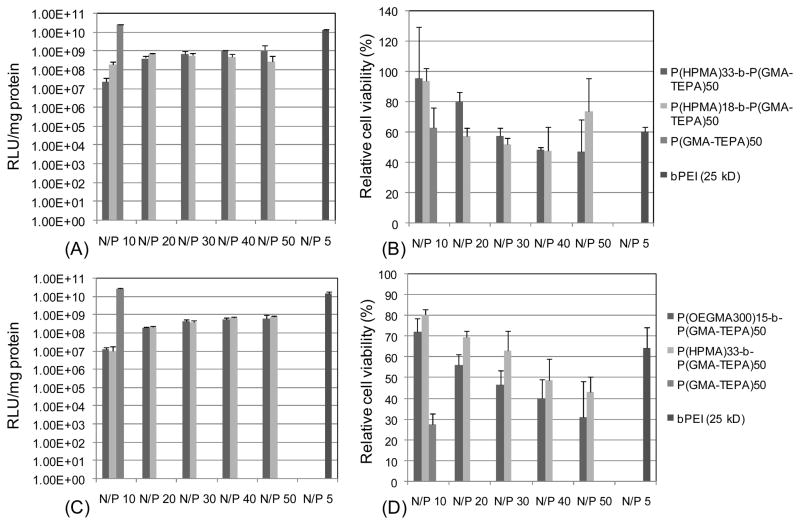
Transfection efficiency (A) and relative cell viability (B) of polyplexes formed by P(HPMA)18/33-b-P(GMA-TEPA)50; Transfection efficiency (C) and relative cell viability (D) of polyplexes formed by P(HPMA)33-b-P(GMA-TEPA)50, and P(OEGMA)15-b-P(GMA-TEPA)50. Data are shown as mean ± SD (n = 3).
To address the decreased polyplex uptake, we further incorporated a RGD targeting peptide to the surface of polyplexes. The RGD peptide motif is recognized by integrins αvβ3, αvβ5, which play a critical role in the adhesion and migration of tumorcells.49 Therefore short RGD-containing peptide has been extensively used to facilitate the attachment to cancer cell lines in the field of targeted drug50,51 or gene52,53 delivery. An N-terminus maleimide-functionalized RGD was conjugated by Michael-type addition to the terminal free thiols of P(OEGMA)15-b-P(GMA-TEPA)50 (Scheme 2). The conjugation efficacy of RGD peptide was determined to be ~50%, that’s to say, there were on average ~0.50 mol RGD per mol of polymer chain. The targeted transfection efficacy of RGD-conjugated polyplexes was further assessed in HeLa cells. Notably, the RGD-conjugated polyplexes mediates 3.4-fold higher transfection activity (p< 0.05) than the corresponding polymer lacking RGD at an N/P of 5 (Figure 6). However, at high charge ratios (N/P ≥8), in vitro transfection efficiency becomes RGD-independent, i.e., the transfection activity of RGD-conjugated polymer is similar to that of RGD-free polymer (data not shown). The ligand-independent transfection at high charge ratios may be attributed to (a) transfection dominated by charge-mediated transfection via proteoglycan interaction54 or (b) at higher N/P, the increased amount of free RGD-conjugated polymer55 effectively compete with polyplex for receptor binding. Despite the increase in transfection efficacy afforded by the targeting ligand, transfection efficacy remains ~2 orders of magnitude lower than the homopolycation. Thus, the shielding polymer block, in addition to reducing cell uptake, likely impairs intracellular trafficking or plasmid release. Reversibly-shielded polyplexes have been shown to offer both in vivo friendly, stable formulation, while maintaining good transfection efficiencies.44,56–58
Figure 6.
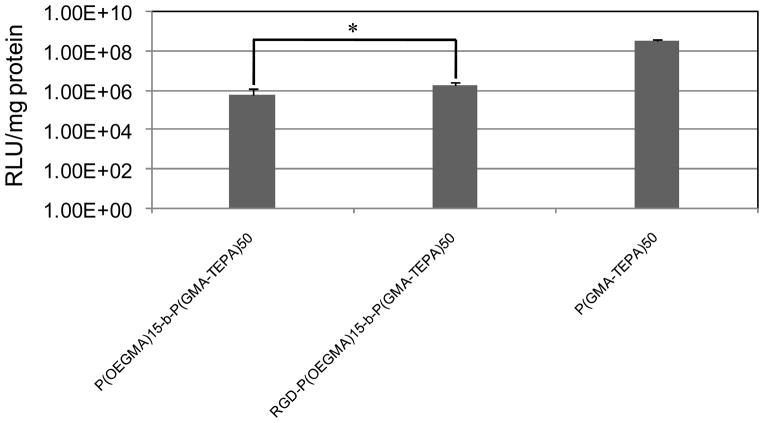
Transfection efficiency of polyplexes formed by P(OEGMA)15-b-P(GMA-TEPA)50, RGD-P(OEGMA)15-b-P(GMA-TEPA)50, and P(GMA-TEPA)50 in HeLa cells at an N/P ratio of 5. Data are shown as mean ± SD (n = 6; student’s t test, *p< 0.05).
CONCLUSION
In summary, a series of well-defined homopolycation P(GMA-oligoamine)s with pendant oligoamine side chains were synthesized by RAFT polymerization and post-polymerization decoration. We tested various types of oligoamine and lengths of polymer backbone for optimal cell transfection efficiency, and identified P(GMA-TEPA)50, with in vitro transfection efficacy comparable to bPEI and the lowest cytotoxicity, to be the most effective cationic carrier. Hydrophilic HPMA or OEGMA blocks were then incorporated by consecutive RAFT using PGMA50 as macro-CTA to improve the poor colloidal stability of P(GMA-TEPA)50/DNA polyplexes in salt medium. The shielding block with proper chain length significantly enhanced the salt stability of polyplexes, but dramatically decreased the transfection efficacy of homopolycation. To further compensate for the reduced cell uptake caused by the hydrophilic shielding layer, a RGD targeting peptide was conjugated to the hydrophilic terminus of the block copolymer. The RGD-conjugated polyplexes mediate higher transfection activity than the corresponding polymer lacking RGD at an N/P of 5. The RAFT-synthesized polycations developed herein are viable alternative to bPEI.
Supplementary Material
Acknowledgments
This work was funded by NIH 1R01NS064404 and NSF DMR 1206426. Technical help from David S.H. Chu and Dr. Joan G. Schellinger was greatly appreciated.
Footnotes
Supporting Information Available: Summary of polymer properties, 1H NMR spectra of polymers, buffering capacity of various P(GMA-oligoamine)50/100, DNA binding ability of polymers, transfection efficacy and cell viability of P(HPMA)33-b-P(GMA-TEPA/PEHA)50, and Average size distributions (PDIs) of various polyplexes formed at different N/P ratios in 150 mM PBS. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Niidome T, Huang L. Gene Ther. 2002;9:1647–1652. doi: 10.1038/sj.gt.3301923. [DOI] [PubMed] [Google Scholar]
- 2.Zuber G, Dauty E, Nothisen M, Belguise P, Behr JP. Adv Drug Delivery Rev. 2001;52:245–253. doi: 10.1016/s0169-409x(01)00213-7. [DOI] [PubMed] [Google Scholar]
- 3.Anderson WF. Nature. 1998;392:25–30. doi: 10.1038/32058. [DOI] [PubMed] [Google Scholar]
- 4.Kaiser J. Science. 2007;317:580–580. doi: 10.1126/science.317.5838.580. [DOI] [PubMed] [Google Scholar]
- 5.Pack DW, Hoffman AS, Pun S, Stayton PS. Nat Rev Drug Discovery. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 6.Jeong JH, Kim SW, Park TG. Prog Polym Sci. 2007;32:1239–1274. [Google Scholar]
- 7.Mintzer MA, Simanek EE. Chem Rev. 2009;109:259–302. doi: 10.1021/cr800409e. [DOI] [PubMed] [Google Scholar]
- 8.Mastrobattista E, van der Aa M, Hennink WE, Crommelin DJA. Nat Rev Drug Discovery. 2006;5:115–121. doi: 10.1038/nrd1960. [DOI] [PubMed] [Google Scholar]
- 9.Bonetta L. Nat Methods. 2005;2:875–883. [Google Scholar]
- 10.Boussif O, Lezoualch F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. Proc Natl Acad Sci U S A. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunath K, von Harpe A, Fischer D, Peterson H, Bickel U, Voigt K, Kissel T. J Controlled Release. 2003;89:113–125. doi: 10.1016/s0168-3659(03)00076-2. [DOI] [PubMed] [Google Scholar]
- 12.Godbey WT, Wu KK, Mikos AG. J Biomed Mater Res. 1999;45:268–275. doi: 10.1002/(sici)1097-4636(19990605)45:3<268::aid-jbm15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 13.Georgiou TK, Vamvakaki M, Patrickios CS, Yamasaki EN, Phylactou LA. Biomacromolecules. 2004;5:2221–2229. doi: 10.1021/bm049755e. [DOI] [PubMed] [Google Scholar]
- 14.Fischer D, Bieber T, Li YX, Elsasser HP, Kissel T. Pharm Res. 1999;16:1273–1279. doi: 10.1023/a:1014861900478. [DOI] [PubMed] [Google Scholar]
- 15.Nemoto Y, Borovkov A, Zhou YM, Takewa Y, Tatsumi E, Nakayama Y. Bioconjugate Chem. 2009;20:2293–2299. doi: 10.1021/bc900283h. [DOI] [PubMed] [Google Scholar]
- 16.Braunecker WA, Matyjaszewski K. Prog Polym Sci. 2007;32:93–146. [Google Scholar]
- 17.Matyjaszeski K, Xia J. Chem Rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 18.Boyer C, Bulmus V, Davis TP, Ladmiral V, Liu JQ, Perrier S. Chem Rev. 109:5402–5436. doi: 10.1021/cr9001403. 009. [DOI] [PubMed] [Google Scholar]
- 19.Zhu CH, Zheng M, Meng FH, Mickler FM, Ruthardt N, Zhu XL, Zhong ZY. Biomacromolecules. 2012;13:769–778. doi: 10.1021/bm201693j. [DOI] [PubMed] [Google Scholar]
- 20.Chu DSH, Schellinger JG, Shi J, Convertine AJ, Stayton PS, Pun SH. Acc Chem Res. 2012;45:1089–1099. doi: 10.1021/ar200242z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed M, Narain R. Prog Polym Sci. [DOI] [Google Scholar]
- 22.Ma M, Li F, Chen FJ, Cheng SX, Zhuo RX. Macromol Biosci. 2010;10:183–191. doi: 10.1002/mabi.200900183. [DOI] [PubMed] [Google Scholar]
- 23.Yang XC, Chai MY, Zhu Y, Yang WT, Xu FJ. Bioconjugate Chem. 2012;23:618–626. doi: 10.1021/bc200658r. [DOI] [PubMed] [Google Scholar]
- 24.Barner-Kowollik C, Perrier S. J Polym Sci Part A: Polym Chem. 2008;46:5715–5723. [Google Scholar]
- 25.Moad G, Rizzardo E, Thang SH. Aust J Chem. 2006;59:669–692. [Google Scholar]
- 26.Talelli M, Rijcken CJF, van Nostrum CF, Storm G, Hennink WE. Adv Drug Delivery Rev. 2010;62:231–239. doi: 10.1016/j.addr.2009.11.029. [DOI] [PubMed] [Google Scholar]
- 27.Dey S, Kellam B, Alexander MR, Alexander C, Rose FRAJ. J Mater Chem. 2011;21:6883–6890. [Google Scholar]
- 28.Hemp ST, Smith AE, Bryson JM, Allen MH, Long TE. Biomacromolecules. 2012;13:2439–2445. doi: 10.1021/bm300689f. [DOI] [PubMed] [Google Scholar]
- 29.Johnson RN, Burke RS, Convertine A, Hoffman AS, Stayton PS, Pun SH. Biomacromolecules. 2010;11:3007–3013. doi: 10.1021/bm100806h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson RN, Chu DSH, Shi J, Schellinger JG, Carlson PM, Pun SH. J Controlled Release. 2011;155:303–311. doi: 10.1016/j.jconrel.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quan CY, Chang C, Wei H, Chen CS, Xu XD, Cheng SX, Zhang XZ, Zhuo RX. Nanotechnology. 2009;20:335101. doi: 10.1088/0957-4484/20/33/335101. [DOI] [PubMed] [Google Scholar]
- 32.Shi J, Johnson RN, Schellinger JG, Carlson PM, Pun SH. Int J Pharm. 2012;427:113–122. doi: 10.1016/j.ijpharm.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gudipati CS, Tan MBH, Hussain H, Liu Y, He CB, Davis TP. Macromol Rapid Commun. 2008;29:1902–1907. doi: 10.1002/marc.200800799. [DOI] [PubMed] [Google Scholar]
- 34.Jia ZF, Wong LJ, Davis TP, Bulmus V. Biomacromolecules. 2008;9:3106–3113. doi: 10.1021/bm800657e. [DOI] [PubMed] [Google Scholar]
- 35.Geng J, Mantovani G, Tao L, Nicolas J, Chen GJ, Wallis R, Mitchell DA, Johnson BRG, Evans SD, Haddleton DM. J Am Chem Soc. 2007;129:15156–15163. doi: 10.1021/ja072999x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mantovani G, Lecolley F, Tao L, Haddleton DM, Clerx J, Cornelissen JJLM, Velonia K. J Am Chem Soc. 2005;127:2966–2973. doi: 10.1021/ja0430999. [DOI] [PubMed] [Google Scholar]
- 37.Suk JS, Suh J, Lai SK, Hanes J. Exp Biol Med. 2007;232:461–469. [PubMed] [Google Scholar]
- 38.Behr JP. Chimia. 1997;51:34–36. [Google Scholar]
- 39.Thomas M, Klibanov AM. Proc Natl Acad Sci USA. 2002;99:14640–14645. doi: 10.1073/pnas.192581499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng Q, Zhong ZL, Zhuo RX. Bioconjugate Chem. 2008;19:499–506. doi: 10.1021/bc7003236. [DOI] [PubMed] [Google Scholar]
- 41.Dai ZJ, Wu C. Macromolecules. 2012;45:4346–4353. [Google Scholar]
- 42.Grigsby CL, Leong KW. J R Soc, Interface. 2010;7:S67–S82. doi: 10.1098/rsif.2009.0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaffer DV, Fidelman NA, Dan N, Lauffenburger DA. Biotechnol Bioeng. 2000;67:598–606. doi: 10.1002/(sici)1097-0290(20000305)67:5<598::aid-bit10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 44.Wei H, Schellinger JG, Chu DSH, Pun SH. J Am Chem Soc. 2012;134:16554–16557. doi: 10.1021/ja3085803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaffert D, Wagner E. Gene Ther. 2008;15:1131–1138. doi: 10.1038/gt.2008.105. [DOI] [PubMed] [Google Scholar]
- 46.Mishra S, Webster P, Davis ME. Eur J Cell Biol. 2004;83:97–111. doi: 10.1078/0171-9335-00363. [DOI] [PubMed] [Google Scholar]
- 47.Georgiou TK, Vamvakaki M, Phylactou LA, Patrickios CS. Biomacromolecules. 2005;6:2990–2997. doi: 10.1021/bm050307w. [DOI] [PubMed] [Google Scholar]
- 48.Georgiou TK, Phylactou LA, Patrickios CS. Biomacromolecules. 2006;7:3505–3512. doi: 10.1021/bm060657y. [DOI] [PubMed] [Google Scholar]
- 49.Mukhopadhyay S, Barnés CM, Haskel A, Short SM, Barnes KR, Lippard SJ. Bioconjugate Chem. 2008;19:39–49. doi: 10.1021/bc070031k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duneau AB, Saulnier P, Hindre FO, Clavreul A, Leroux JC, Benoit JP. Biomaterials. 2007;28:4978–4990. doi: 10.1016/j.biomaterials.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 51.Tan YC, Hettiarachchi K, Siu M, Pan YR, Lee AP. J Am Chem Soc. 2006;128:5656–5658. doi: 10.1021/ja056641h. [DOI] [PubMed] [Google Scholar]
- 52.Moore NM, Barbour TR, Sakiyama-Elbert SE. Mol Pharm. 2008;5:140–150. doi: 10.1021/mp700072n. [DOI] [PubMed] [Google Scholar]
- 53.Chen XY, Plasencia C, Hou YP, Neamati N. J Med Chem. 2005;48:1098–1106. doi: 10.1021/jm049165z. [DOI] [PubMed] [Google Scholar]
- 54.Mislick KA, Baldeschwieler JD. Proc Natl Acad Sci USA. 1996;93:12349–12354. doi: 10.1073/pnas.93.22.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boeckle S, von Gersdorff K, van der Piepen S, Culmsee C, Wagner E, Ogris M. J Gene Med. 2004;6:1102–1111. doi: 10.1002/jgm.598. [DOI] [PubMed] [Google Scholar]
- 56.Takae S, Miyata K, Oba M, Ishii T, Nishiyama N, Itaka K, Yamasaki Y, Koyama H, Kataoka K. J Am Chem Soc. 2008;130:6001–6009. doi: 10.1021/ja800336v. [DOI] [PubMed] [Google Scholar]
- 57.Walker GF, Fella C, Pelisek J, Fahrmeir J, Boeckle S, Ogris M, Wagner E. Molecular Ther. 2005;11:418–425. doi: 10.1016/j.ymthe.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 58.Yang XZ, Du JZ, Dou S, Mao CQ, Long HY, Wang J. ACS Nano. 2012;6:771–781. doi: 10.1021/nn204240b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.