

Abstract
Telomeres protect the natural ends of chromosomes from being repaired as deleterious DNA breaks. In fission yeast, absence of Taz1 (homologue of human TRF1 and TRF2) renders telomeres vulnerable to DNA repair. During the G1 phase, when non-homologous end joining (NHEJ) is upregulated, taz1Δ cells undergo telomere fusions with consequent loss of viability. Here, we show that disruption of the fission yeast MRN (Rad23MRE11-Rad50-Nbs1) complex prevents NHEJ at telomeres and, as a result, rescues taz1Δ lethality in G1. Neither Tel1ATM activation nor 5′-end resection was required for telomere fusion. Nuclease activity of Rad32MRE11 was also dispensable for NHEJ. Mutants unable to coordinate metal ions required for nuclease activity were proficient in NHEJ repair. In contrast, Rad32MRE11 mutations that affect binding and/or positioning of DNA ends leaving the nuclease function largely unaffected also impaired NHEJ at telomeres and restored the viability of taz1Δ in G1. Consistently, MRN structural integrity but not nuclease function is also required for NHEJ of independent DNA ends in a novel split-molecule plasmid assay. Thus, MRN acts to tether unlinked DNA ends, allowing for efficient NHEJ.
Keywords: DNA repair, fission yeast, MRN, NHEJ, telomeres
Introduction
Eukaryotic chromosome ends are composed of specialized structures known as telomeres. When telomeres are lost, chromosome ends are processed inappropriately and are often fused by cellular DNA repair machineries. Chromosome fusions can lead to an uneven distribution of the genetic content and chromosomal aberrations that compromise genomic integrity and may trigger tumorigenesis. The two main pathways of DNA double strand break (DSB) repair, homologous recombination (HR) and non-homologous end joining (NHEJ), are tightly regulated throughout the cell cycle, with NHEJ being predominant in G1 and HR being dominant in S/G2. Thus, the challenges posed to unprotected telomeres vary throughout the cell cycle, and their outcomes carry important consequences for the cell (Jain and Cooper, 2010).
Taz1, the orthologue of shelterin proteins TRF1 and TRF2 (Li et al, 2000), plays a critical role in chromosome end protection in fission yeast. In the absence of Taz1, telomeres are recognized as DSBs, triggering a DNA repair response (Ferreira and Cooper, 2001; Carneiro et al, 2010). During vegetative growth, taz1Δ telomeres undergo constant HR processing, leading to the exchange of subtelomere sequences (Rog et al, 2009). Conversely, NHEJ repair is upregulated in the G1 phase (Ferreira and Cooper, 2004). Because fission yeast exerts size control in G2, its cell cycle exhibits a characteristically short G1 phase. Consequently, wild-type or taz1Δ cells do not upregulate NHEJ and do not undergo telomere fusion during vegetative growth (Ferreira and Cooper, 2004). However, prolonged G1 periods result in taz1Δ lethality generated by telomere fusion, via the NHEJ pathway (Ferreira and Cooper, 2001). Thus, the taz1Δ telomere provides an excellent reporter for DSB repair and a unique internal substrate for the NHEJ pathway.
MRN (Mre11(Rad32)/Rad50/Nbs1) is a heterotrimeric complex with well-characterized functions in DNA repair, checkpoint signalling, DNA replication, telomere length maintenance and meiosis (Williams et al, 2010). The importance of the MRN complex in genome stability is evident in humans who carry hypomorphic mutations on the gene responsible for Nijmegen breakage syndrome (NBS1), a rare autosomal disease characterized by immunodeficiency, microcephaly and a propensity to cancer. In addition, MRE11 hypomorphic mutations result in ataxia-telangiectasia-like disorder (ATLD), with symptoms that resemble ATM deficiency, such as ataxia and neurodegeneration.
MRN is at the hub of DNA damage responses. As a DSB sensor, MRN binds and activates Tel1ATM kinase signalling via its adaptor subunit Nbs1 (Falck et al, 2005; You et al, 2005). Rad50 forms a dimer composed of an ATPase head and a hinge region, separated by a long coiled-coil domain involved in bridging DNA ends. The Mre11 subunit is central for DNA binding and end processing. Mre11 has both ssDNA endonuclease and 3′-5′ exonuclease activities (Paull and Gellert, 1998). In all organisms tested, MRN and its nuclease partner CtIP/Sae2/Ctp1 have been found to be associated with HR repair. Ctp1CtIP promotes DNA end processing and initiates 5′-end resection at DSBs (Limbo et al, 2007; Huertas and Jackson, 2009; Langerak et al, 2011). Subsequent exonucleolytic activities (provided by Exo1 or Dna2) generate a 3′-ssDNA end required for homology searches (Mimitou and Symington, 2011). In budding yeast and in mammalian cells, MRN was also shown to be required for NHEJ repair (Boulton and Jackson, 1998; Wang et al, 2003). Mre11 forms a dimer that is responsible for tethering DNA ends together, helping to coordinate subsequent reactions (Williams et al, 2008). Thus, MRN may be involved in synapsis and cleaning DNA ends for end-joining reactions. In contrast, studies in fission yeast using plasmid-based NHEJ assays have concluded that MRN is dispensable for NHEJ (Manolis et al, 2001; Porter-Goff and Rhind, 2009).
In a number of model systems, NHEJ occurring at dysfunctional telomeres requires the function of MRN. Budding yeast defective for the telomere-binding protein Rap1 undergo NHEJ, which is dependent on MRX (Pardo and Marcand, 2005). In mammalian cells, the disruption of the shelterin component TRF2 results in ATM activation, the phosphorylation of Chk2 and H2AX, the formation of 53BP1-associated telomere-induced DNA damage foci (TIF) and NHEJ-mediated telomere fusions (Karlseder et al, 1999; Celli and de Lange, 2005). MRN is required to activate ATM kinase in the presence of dysfunctional telomeres, and this function does not depend on Mre11 nuclease activity (Deng et al, 2009; Dimitrova and de Lange, 2009). During G1, when the large majority of telomere fusions occur (Konishi and de Lange, 2008), MRN and consequent ATM activation are required for NHEJ at dysfunctional telomeres (Konishi and de Lange, 2008; Dimitrova and de Lange, 2009). However, in a parallel study, the ability of MRN to promote NHEJ also appeared to depend on Mre11 nuclease function, possibly to remove 3′-overhangs prior to end joining, but not on the activation of ATM or 53BP1 (Deng et al, 2009). After the S phase, Mre11 and its nuclease domain may have an opposite role by preventing NHEJ from occurring at newly generated telomeres (Deng et al, 2009; Dimitrova and de Lange, 2009). MRN-dependent 5′ resection of blunt-ended telomeres produces a telomeric 3′-overhang, recruits ssDNA telomere proteins and, thus, prevents NHEJ.
We investigated the role of the MRN complex in the repair of taz1Δ dysfunctional telomeres. Consistent with previous studies in fission yeast, we found that MRN was dispensable for NHEJ assays based on plasmid-end religation. However, MRN was required for NHEJ repair at unprotected telomeres. In contrast to mammalian cell studies, we excluded the requirement of MRN in Tel1ATM (or Rad3ATR) activation and its role in DNA resection. However, even though Mre11 nuclease function was per se dispensable, rad32MRE11 nuclease mutants that also affect DNA end coordination drastically reduced NHEJ at dysfunctional telomeres and suppressed the lethality of taz1Δ in G1-arrested cells. Using a novel plasmid repair assay, we show that, similar to MRN-deleted mutants, both rad32MRE11 dimerization and phosphoesterase motif II and III mutants are impaired in the ability to join two distinct DNA fragments in vivo by NHEJ. Collectively, our results point towards a chromosomal tethering function for MRN during the NHEJ repair of dysfunctional telomeres.
Results
Fission yeast MRN is required for NHEJ at dysfunctional telomeres
HR and NHEJ are reciprocally regulated throughout the cell cycle, with increased levels of HR in the S/G2 phases of the cell cycle and high levels of NHEJ in the G1 phase (Ferreira and Cooper, 2004). Because fission yeast lacks a prolonged G1 phase, HR is the pathway of choice during normal growth. The two repair pathways compete for DNA ends not only at DSBs but also at telomeres (Frank-Vaillant and Marcand, 2002). HR repair involves the generation of ssDNA by 5′- to 3′-end resection. Resected DNA ends are refractory to Ku70/80 heterodimer binding and thus block NHEJ. HR prevents taz1Δ telomere fusion by NHEJ, and rad22RAD52 gene deletion leads to an accumulation of taz1Δ telomere fusions during S/G2 (Ferreira and Cooper, 2001).
taz1Δ telomeres possess long 3′-overhangs as a result of extensive resection by MRN and the flap endonuclease Dna2 (Tomita et al, 2004). Because the disruption of telomeric 5′-end resection in mrnΔ taz1Δ double mutants could provide a template for NHEJ (and inhibit HR), we initially hypothesized that the disruption of MRN would lead to increased levels of telomere fusions in G1 and the appearance of fusions in S/G2 cells as a result of a reduction in HR repair. The S. pombe genome is packaged into three chromosomes, and telomere fusions can be detected by pulsed-field gel electrophoresis (PFGE; Figure 1A). We deleted each one of the three subunits of the MRN complex in a taz1Δ background, and to our surprise, the disruption of MRN not only failed to increase telomere fusions in S/G2 phases but also abolished fusions in G1-arrested cells (Figure 1B). Contrary to our prediction, we found that MRN was required for NHEJ at dysfunctional telomeres in fission yeast, similarly to its mammalian and S. cerevisiae counterparts.
Figure 1.
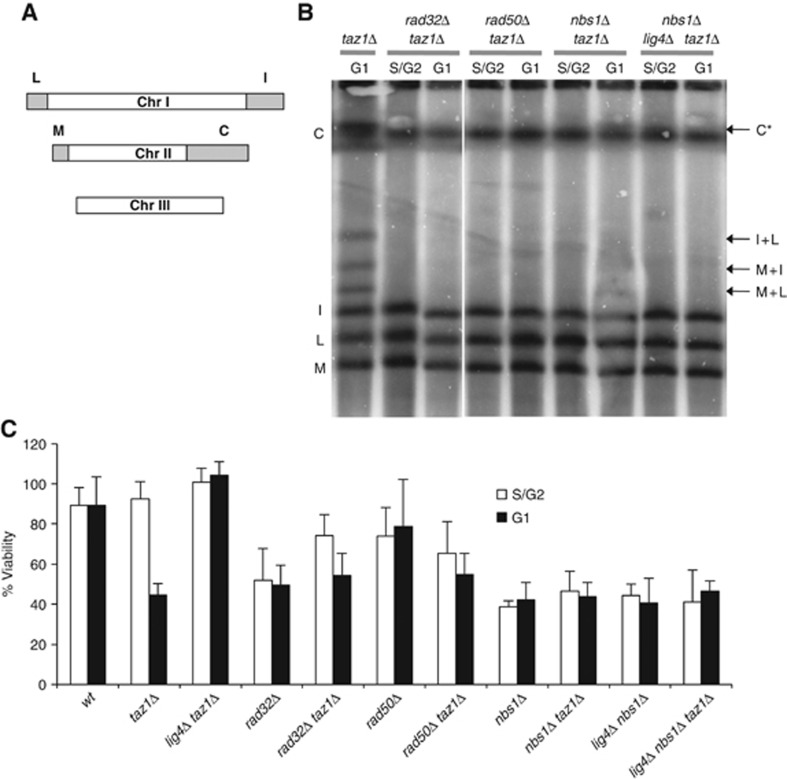
MRN is required for NHEJ repair of unprotected taz1Δ telomeres. (A) Scheme of telomeric NotI restriction fragments. Chromosomes I and II each release two telomeric restriction fragments (C, I, L and M). Chromosome III lacks NotI restriction sites. (B) Disruption of the MRN complex prevents taz1Δ NHEJ-mediated telomeric fusions. NotI digests of genomic DNA of the indicated strains were separated by PFGE and chromosomal end-to-end fusions were detected by Southern blot using a telomere probe (arrows indicate the positions of the resolved telomere fusions). C* indicates a C restriction fragment migrating alongside the unresolved C+I, L and M fusion bands. Please note that lanes come from the same Southern blot and that only irrelevant lanes have been removed in the interest of clarity. Full-sized source images of the original scans can be found as an online supplement to this paper. (C) Nitrogen starvation-induced lethality of taz1Δ cells is rescued by deletion of mrn+. Logarithmically growing and nitrogen-starved cells were plated on rich medium, and their ability to form colonies was scored after a 5-day incubation at 32°C. A minimum of three independent experiments was performed. Error bars represent 2 × s.e.m.
The lethality of taz1Δ G1-arrested cells is suppressed by the deletion of genes involved in NHEJ repair (Ferreira and Cooper, 2001), such as pku70+ and lig4+. Because MRN deletion prevents taz1Δ telomere fusion, it might also prevent the lethality resulting from a G1 arrest. Consistent with an absence of telomere fusions, the deletion of MRN genes, though causing reduced viability, suppressed the lethality conferred by taz1+ deletion in cells that have undergone a G1 arrest (Figure 1C). Thus, the loss of viability caused by the fusion of dysfunctional telomeres similarly depends on MRN as it does on NHEJ components.
MRN-dependent 5′–3′ resection is not involved in telomere fusions
To better understand the role of MRN at unprotected taz1Δ telomeres, we took a genetic approach to abolish each of its known functions, aiming to identify the one required for NHEJ at chromosome ends. Ctp1CtIP was recently found to act cooperatively with MRN to catalyse the 5′–3′ single-strand resection required for efficient HR repair in fission yeast and human cells (Limbo et al, 2007; Huertas et al, 2008; Langerak et al, 2011). Ctp1 contributes to the cell-cycle regulation of HR and displays epistasis with MRN in HR repair and DNA damage sensitivities. Although Ctp1 is not expressed in G1 phase of the cell cycle (Limbo et al, 2007), it could be required to process telomeres during S/G2 prior to G1 phase. Therefore, we evaluated the effect of ctp1+ deletion on NHEJ at dysfunctional telomeres. In contrast to MRN, Ctp1 was dispensable for taz1Δ telomere fusions in G1-arrested cells (Figure 2A). Consistent with the accumulation of telomere fusion bands in ctp1Δ taz1Δ double mutants, ctp1+ deletion was unable to rescue the lethality caused by NHEJ in taz1Δ G1-arrested cells (Figure 2A). Thus, in contrast to many other functions of this complex, NHEJ repair at unprotected telomeres requires MRN, but not Ctp1, activity.
Figure 2.
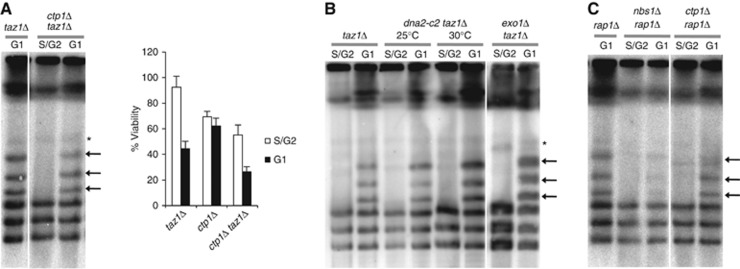
Telomere 5′- to 3′-end resection is not required for telomere fusions. (A) Deletion of ctp1+does not prevent taz1Δ NHEJ-mediated telomere fusions in G1-arrested cells and does not suppress the lethality of taz1Δ cells after G1 arrest. NotI-digested genomic DNA was analysed by PFGE followed by Southern blot analysis using a telomere probe. Non-specific, non-telomere bands on the PFGE are indicated by a star. Viability assays were performed as in Figure 1C. (B) Impairment of Exo1 or Dna2 nuclease activities do not affect taz1Δ telomere fusions. The dna2-c2 taz1Δ was cultured either at the permissive temperature of 25°C or at the semipermissive temperature of 30°C either in vegetative growth or following nitrogen arrest. Please note that lanes in (A) and (B) come from the same Southern blots and that only irrelevant lanes have been removed in the interest of clarity. Full-sized source images of the original scans can be found as an online supplement to this paper. (C) PFGE analysis revealed that NHEJ-mediated fusions of rap1Δ telomeres in G1 require MRN but not Ctp1.
In addition to Ctp1, MRN recruits other nucleases that further promote the 5′–3′ resection required for HR repair. We analysed the effect of the inactivation of Exo1 and Dna2 nucleases in telomere fusions. Exo1 is required to process DSBs generated by DNA damage but not telomere ends (Tomita et al, 2003). Deletion of exo1+ did not affect NHEJ at taz1Δ telomeres in G1 (Figure 2B). Inactivating Dna2 by growing a dna2-c2 temperature-sensitive mutant at the semipermissive temperature of 30°C greatly reduces telomeric 3′-overhangs at both wild-type and taz1Δ telomeres (Tomita et al, 2004). Because the MRN complex, like Dna2, is required for the generation of 3′-overhangs in taz1Δ cells, one could speculate that telomeric overhangs are required for telomere fusions. However, like Ctp1 and Exo1, inactivating Dna2 did not prevent telomere fusions in taz1Δ cells arrested in G1 (Figure 2B). These results indicate that 5′–3′ resection is not involved in processing dysfunctional telomeres for NHEJ repair.
Rap1, a telomeric protein recruited by Taz1, shares some of its functions in telomere protection. Similarly to Taz1, Rap1 protects telomeres from DNA repair and regulates telomere size and 3′-overhang formation (Miller et al, 2005). However, Taz1, but not Rap1, is required for efficient telomere replication, and its inactivation leads to replication fork stalling at telomere sequences. MRN is involved in the DNA repair of collapsed replication forks that arise during DNA replication (Naito et al, 1998; Costanzo et al, 2001; Trenz et al, 2006). Thus, the function of MRN at taz1Δ telomeres could consist of processing stalled replication forks prior to end-joining reactions. If this event were the case, then rap1Δ telomere fusions would not require the function of MRN because fork stalling does not occur at these dysfunctional telomeres. To test this hypothesis, we asked whether rap1Δ nbs1Δ cells would accumulate telomere fusions upon G1 arrest. Similarly to in mrnΔ taz1Δ cells, we observed a drastic reduction in the amount of rap1Δ chromosome-end fusions upon deletion of nbs1+, and these fusions did not require ctp1 activity (Figure 2C). Thus, MRN is required for the repair of dysfunctional telomeres by NHEJ, regardless of replication fork stalling.
To rule out fusions between short telomeres that would be difficult to detect using telomere probes, we re-probed the Southern blots using LMIC subtelomere probes, which maintain their intensity levels irrespective of telomere size. PFGE analysis using either telomere or subtelomere probes yielded similar results, revealing that the lack of fusion bands was not due to very short telomeres in strains carrying dysfunctional telomeres (Supplementary Figure 1).
Tel1ATM and Rad3ATR are dispensable for NHEJ at dysfunctional telomeres
MRN has a dual role in activating DNA damage responses. It both recruits the Tel1ATM checkpoint sensor kinase to DSBs and initiates 5′–3′ resection. This function, in turn, leads to the accumulation of ssDNA and ultimately to the activation of the Rad3ATR kinase. The C-terminus of Nbs1 interacts with Tel1ATM, and this interaction is required for the activation of Tel1ATM and its localization to sites of DNA damage (You et al, 2005). In mammalian cells, the loss of TRF2 leads to telomere deprotection and chromosomal fusions. These fusions are mediated via NHEJ and are dependent on ATM activity (Denchi and de Lange, 2007). Therefore, the lack of recruitment of Tel1ATM to taz1Δ telomeres in mrnΔ cells could explain the effect of deleting MRN on taz1Δ telomere fusions. To test this hypothesis, we used two alleles of nbs1+, nbs1-9 and nbs1-10, that carry mutations in the C-terminus that block the interaction with Tel1ATM (You et al, 2005). We found that disrupting the interaction between MRN and Tel1ATM in both nbs1-9 taz1Δ and nbs1-10 taz1Δ mutants had no impact on NHEJ at dysfunctional telomeres (Figure 3A).
Figure 3.
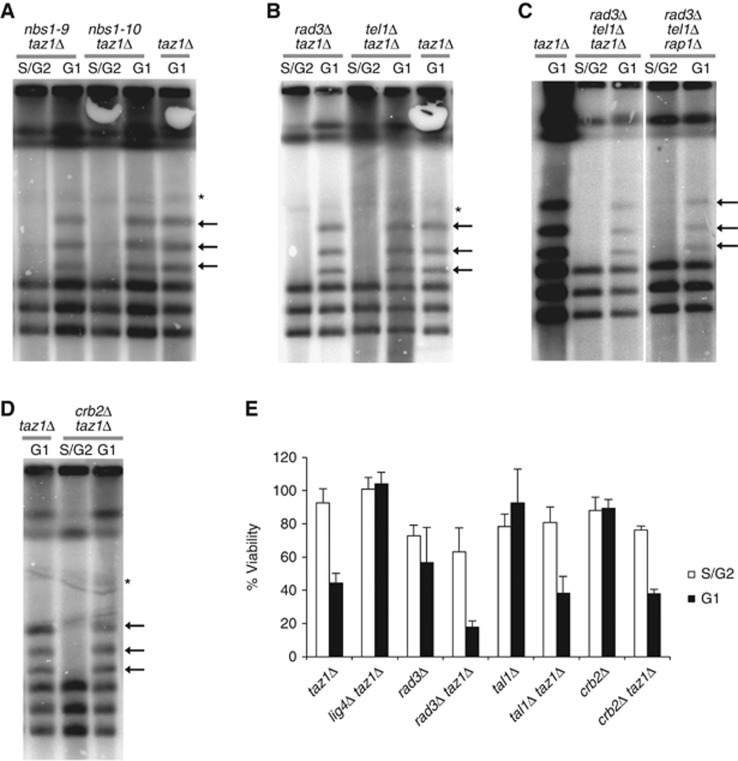
Tel1ATM recruitment by MRN and checkpoint activation are dispensable for chromosome end fusions. (A) Mutations in nbs1+ that impair the Nbs1 interaction with Tel1ATM do not prevent taz1Δ NHEJ-mediated telomere fusions in G1-arrested cells. Strains of the indicated genotypes were analysed by PFGE in G1-arrested or dividing cells, and fusions were detected using a telomere probe. Non-specific, non-telomere bands are indicated by an asterisk. (B) rad3ATR and tel1ATM deletions do not impair taz1Δ telomere repair in G1-arrested cells. (C) tel1ATM and rad3ATR are not redundant in preventing chromosomal end fusions at taz1Δ or rap1Δ unprotected telomeres. Please note that lanes come from the same Southern blot and that only irrelevant lanes have been removed in the interest of clarity. Full-sized source images of the original scans can be found as an online supplement to this paper. (D) Deletion of crb2+does not prevent taz1Δ NHEJ-mediated telomere fusions in G1-arrested cells. (E) The viability of taz1Δ after a G1 arrest is not suppressed by rad3ATR, tel1ATM or crb253BP1 deletions. Viability assays were performed as in Figure 1C.
To further investigate the role of both checkpoint kinases, we investigated whether they themselves were required for chromosome end fusions. Because neither Tel1ATM nor Rad3ATR is essential in fission yeast, we subjected tel1Δ taz1Δ and rad3Δ taz1Δ double mutants to G1 arrest by nitrogen starvation. PFGE analysis revealed that, in either case, telomere fusions were abundantly present (Figure 3B). Thus, in contrast to mammalian cells, the deletion of Tel1ATM (or even Rad3ATR) cannot prevent NHEJ at dysfunctional fission yeast telomeres. Because Tel1ATM and Rad3ATR are partially redundant in checkpoint signalling and telomere maintenance, we constructed a triple mutant with dysfunctional telomeres to completely rule out possible redundancies. tel1Δ rad3Δ mutants are unable to activate telomerase and, consequently, lose telomeres over several passages, similar to trt1Δ cells (Naito et al, 1998). However, tel1Δ rad3Δ taz1Δ mutants lose telomeres rapidly because replication fork stalling at chromosome ends requires constant telomerase activity (Miller et al, 2006). In contrast, tel1Δ rad3Δ rap1Δ mutants lose telomeres at a slower rate. In order to investigate the complete absence of both Tel1ATM and Rad3ATR on NHEJ repair, we generated triple mutants by knock-out of rad3+ in tel1Δ taz1Δ and in tel1Δ rap1Δ strains. The two strains created were immediately grown in order to prevent the complete loss of telomeres. Southern blotting using telomere probes revealed that, although less intense, both tel1Δ rad3Δ taz1Δ and tel1Δ rad3Δ rap1Δ mutants exhibited telomere fusion bands while arrested in G1 phase (Figure 3C). The lower intensity of the telomere signal in the fusion bands is due to shorter telomeres in these strains, as observed by Southern blot analysis and subtelomere (LMIC) probing (Supplementary Figure 2a and b). Comparison of the signal obtained from telomere and subtelomere probes revealed that tel1Δ rad3Δ taz1Δ telomeres already exhibited chromosome-end fusions in cycling S/G2 cells. However, these fusions occurred between chromosomes that had completely lost telomere repeats. Thus, simultaneous deletion of both Tel1ATM and Rad3ATR does not block NHEJ at taz1Δ or rap1Δ dysfunctional telomeres.
Another component of the DNA damage checkpoint, 53BP1, is required for efficient NHEJ-mediated fusions of TRF2−/− telomeres in mouse cells (Denchi and de Lange, 2007). We tested whether the fission yeast 53BP1 structurally related, crb2+, is also required for taz1Δ fusions. Similarly to tel1Δ taz1Δ and rad3Δ taz1Δ, mutants carrying crb2Δ taz1Δ mutations accumulated telomere fusions when arrested in G1 to the same extent as taz1Δ single mutants (Figure 3D). Consistent with the detection of telomere fusions in checkpoint signalling mutants, deletion of these genes did not suppress the loss of viability exhibited by G1-arrested taz1Δ cells (Figure 3E). Thus, genetic requirements for NHEJ repair of unprotected telomeres appear to differ between fission yeast and mammalian cells with respect to the checkpoint pathways.
The nuclease activity of Rad32MRE11 is dispensable for telomere fusions
The requirement for Mre11 nuclease activity in NHEJ repair has been ambiguous in the organisms investigated. A study using MRE11 nuclease-deficient mouse embryo fibroblasts (Mre11H129N/Δ) showed that dysfunctional telomeres failed to undergo NHEJ-mediated fusions, even though the cells were able to activate ATM and recruit 53BP1 (Deng et al, 2009). However, a previous study in S. cerevisiae, the homologous mutation (mre11-H125N) was shown to be dispensable for NHEJ in plasmid ligation assays (Moreau et al, 1999). To ascertain whether the nuclease activity of Rad32MRE11 is required for NHEJ repair of dysfunctional telomeres in fission yeast, we used strains carrying a rad32-D65N mutation that precludes active-site Mn2+ binding (Hartsuiker et al, 2009) and two mutations, rad32-H68S and rad32-H134S that disrupt phosphoesterase motifs II and III, respectively (Williams et al, 2008). None of these single amino-acid substitutions behave as null mutants as observed by their reduced DNA damage sensitivities when compared to a deletion mutant (Supplementary Figure 4).
Both taz1Δ and taz1Δ rad32 nuclease double mutant strains were arrested in the G1 phase using nitrogen starvation. PFGE and Southern blot analysis revealed abundant telomere end-to-end fusions in G1-arrested cells in rad32-D65N taz1Δ (Figure 4A). Accordingly, the rad32-D65N mutation failed to suppress the taz1Δ lethality incurred during the G1 arrest (Figure 4C). In contrast, both rad32-H68S taz1Δ and rad32-H134S taz1Δ presented a reduced number of telomere fusions and, thus, alleviated the G1-specific lethality of taz1Δ (Figure 4A and C). These results argue that the nuclease function of fission yeast Rad32MRE11 is not required, as revealed by the ion coordination rad32-D65N mutant. However, other aspects intimately related to Rad32MRE11 nuclease function, such as DNA end coordination, may be required for efficient NHEJ at telomeres. This possibility may help conciliate the conflicting results obtained in budding yeast and mammalian cells harbouring the MRE11 phosphoesterase motif III mutations.
Figure 4.
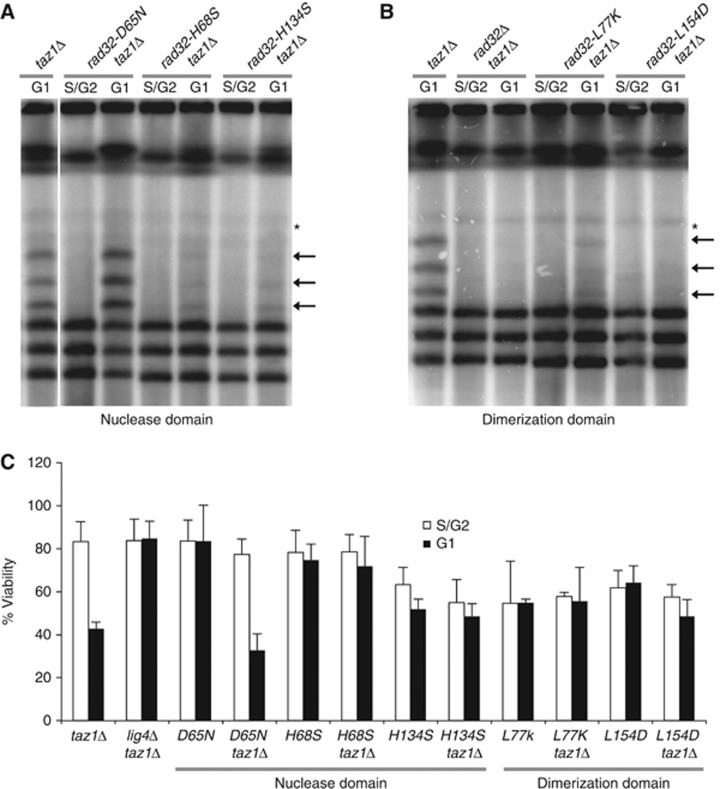
Rad32 dimerization, but not nuclease activity, is required for NHEJ-dependent telomere fusions. (A) The rad32-D65N nuclease-dead mutant exhibits abundant taz1Δ telomere fusions in G1-arrested cells. In contrast, rad32-H68S and rad32-H134S mutants that disrupt phosphoesterase motif II and III greatly reduce the amount of telomere fusions. PFGE was performed as in Figure 1A. Please note that lanes come from the same Southern blots and that only irrelevant lanes have been removed in the interest of clarity. Full-sized source images of the original scans can be found as an online supplement to this paper. (B) The rad32-L77K and rad32-L154D alleles, which impair Rad32 self-dimerization, significantly reduce taz1Δ telomere fusions in G1-arrested cells. Non-specific bands are indicated by an asterisk. (C) The viability of taz1Δ upon a G1 arrest is not suppressed by the rad32-D65N allele but is rescued by the rad32-H68S, rad32-H134S, rad32-L77K and rad32-L154D alleles. Viability assays were performed as in Figure 1C.
Rad32MRE11 complex architecture is required for efficient NHEJ
Our previous result suggested that coordination of DNA ends could be required for ensuing subsequent NHEJ reactions. Rad32MRE11 functions as a dimer that can bind both sides of a DSB and stabilize them in close proximity. The rad32-L77K and rad32-L154D alleles prevent the Rad32MRE11 subunit from self-interacting while preserving both endo- and exonuclease activity (Williams et al, 2008). To understand whether the tethering function of MRN is required for telomere repair, we analysed the effect of impaired Rad32MRN DNA binding ability on taz1Δ telomere fusions. In contrast with all the other mutants tested, these mutations drastically reduced the accumulation of taz1Δ telomere fusions in G1 (Figure 4B). Consistent with the lack of telomere fusions, the rad32-L77K and rad32-L154D alleles suppressed the taz1Δ loss of viability in G1 (Figure 4C). This result suggests that the function of MRN in NHEJ repair of unprotected telomeres is to facilitate the synapsis of DNA ends for end-joining reactions.
MRN is required for NHEJ repair in a novel plasmid-based tethering assay
Our results showing that MRN is required for NHEJ at dysfunctional telomeres contrast to those results obtained using established plasmid-based assays, where MRN has been shown to be dispensable for NHEJ repair (Boulton and Jackson, 1996; Manolis et al, 2001; Porter-Goff and Rhind, 2009). To ascertain whether MRN is specifically required for NHEJ at telomeres, we repeated the previously published experiments in both S/G2 and G1-arrested cells. We confirmed that, while the in vivo ligation of cut plasmids requires canonical NHEJ machinery (i.e., pku70+, pku80+ and lig4+), it does not rely on MRN because rad32Δ, rad50Δ and nbs1Δ cells are proficient in plasmid NHEJ repair in the G1 phase (Figure 5A). However, ctp1+ is not required for NHEJ repair, in agreement with a previous report (Limbo et al, 2007) and all rad32MRE11 nuclease and homodimerization mutants were similarly dispensable for the classical plasmid NHEJ repair throughout the cell cycle (Figure 5A and C). Altogether, these data suggest that the requirement for MRN in NHEJ is telomere specific. However, the inherent close proximity of the DNA ends in the plasmid assay may obviate the need for a mechanism to bring free ends together.
Figure 5.
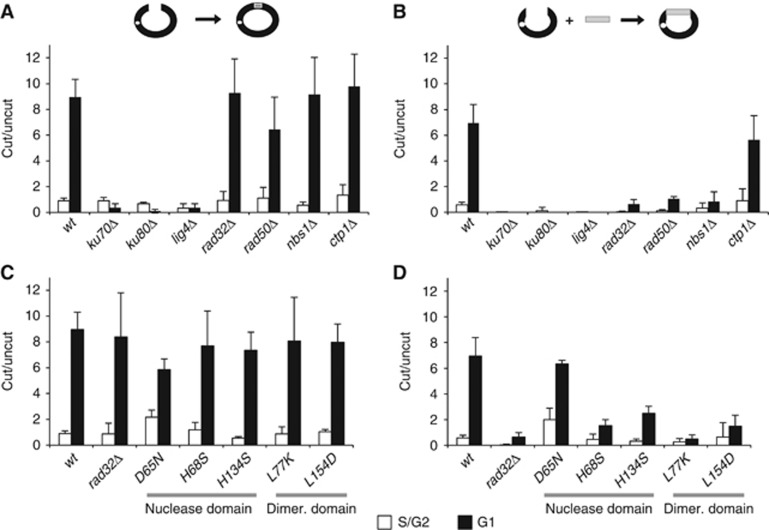
MRN is required for NHEJ repair of free DNA fragments in vivo. (A) MRN and ctp1 null mutants are proficient in single molecule plasmid NHEJ repair. NHEJ plasmid repair assays were performed in strains from the indicated genotypes. Results are plotted as the ratio of the number of transformants obtained when transforming with linearized plasmid over those transformed with uncut plasmid. (B) MRN is required tether independent DNA fragments for NHEJ repair in vivo. Strains from the indicated genotypes were co-transformed with KpnI digested plasmid carrying resistance to G418 and an equimolar amount of a DNA fragment encoding for LEU2. After co-transformation, results were scored as the number of colonies in double selection media. The values plotted represent the ratio of colonies grown in double selection media over the ones obtained in parallel transformations using the uncut pKan1 plasmid. (C) The nuclease and tethering activities of Rad32 are dispensable for in vivo single plasmid NHEJ repair. Single plasmid repair assays were performed in strains from the indicated genotypes as in Figure 5A. (D) The dimerization domain is required for tethering unlinked DNA fragments for NHEJ repair. Nuclease domain of Rad32 is differentially required, whereas rad32-D65N nuclease dead is proficient in NHEJ, both rad32-H68S and rad32-H134S mutants are unable to join free DNA ends. Split molecule plasmid repair assays were performed in strains from the indicated genotypes as in Figure 5B. Results for both the single and split molecule plasmid assays are presented as the average of at least three experiments for each strain. Error bars represent 2 × s.e.m.
In light of our results highlighting the role of MRN in bridging chromosome ends to allow for NHEJ repair, we developed a new split molecule plasmid assay in which we could directly evaluate the tethering of free DNA ends. This assay differs from previous NHEJ plasmid assays because we co-transform both the linear plasmid and a second DNA fragment encoding a distinct marker and lacking an origin of replication. The ability of two DNA ends to undergo an end-joining reaction is scored by the number of colonies grown in double selection media (measured as a ratio with the uncut vector). PCR analysis confirmed that transformants that were able to grow in double selection media indeed joined the two DNA fragments in vivo (Supplementary Figure 3). As expected, our split molecule plasmid assay relied on NHEJ repair genes (pku70+, pku80+ and lig4+), and, despite a somewhat lower efficiency in end joining (Figure 5B and D). Similarly, NHEJ levels were upregulated in G1-blocked cells. In contrast to the single plasmid repair assay, the deletion of rad32+, rad50+ or nsb1+, but not ctp1+, significantly impaired the ability of cells to join and repair the two free DNA fragments in vivo (Figure 5B).
We next analysed the nuclease function of Rad32MRE11. We observed that, similarly to NHEJ repair at unprotected telomeres, rad32-D65N nuclease mutant is dispensable for both single molecule and split molecule NHEJ assays (Figure 5D). However, both rad32-H68S and rad32-H134S mutants showed reduced levels in the split molecule NHEJ assay (Figure 5D). These results suggest that, even though nuclease mutants may disrupt complex architecture to different extents, nuclease activity per se is not required for assembling independent DNA ends for the ensuing NHEJ reaction. Consistent with a function of Rad32MRE11 in tethering free DNA ends, both the rad32-L77K and rad32-L154D homodimerization mutants that are nonetheless proficient in nuclease activity greatly reduced the end-joining reaction (Figure 5D). The inability of MRN to join independent DNA molecules was not simply due to its requirement for processing incompatible ends in our split molecule assay. All mutants tested were able to perform NHEJ repair in single plasmids bearing non-cohesive ends (Supplementary Figure 5). Thus, loss of MRN, or mutations that impair its ability to tether DNA ends, limits NHEJ repair of independent DNA ends. Nevertheless, MRN is dispensable for NHEJ repair of plasmid ends, which are inherently proximal. These results indicate that rather than being telomere specific, the role of fission yeast MRN in NHEJ encompasses a function in synapsis of free DNA ends to secure them for the subsequent end-joining reaction.
Discussion
We have revealed an unanticipated requirement for the MRN complex in NHEJ repair in fission yeast. Even though MRN function during HR has been well characterized, conventional plasmid re-ligation assays have shown that this central complex for DSB repair is dispensable for NHEJ in S. pombe (Manolis et al, 2001; Porter-Goff and Rhind, 2009). Our observation that MRN function is essential for NHEJ at dysfunctional telomeres is at odds with the current literature. We analysed an extensive list of MRN-dependent functions and concluded that the requirement for this complex in NHEJ is not through its role in 5′–3′ DNA resection, end processing of ‘dirty’ DNA ends via the nuclease activity of Rad32MRE11, or Tel1ATM activation or initiation of DNA damage checkpoints.
The recent crystal structure of the Mre11-Rad50 core complex in Pyrococcus furiosus has provided vital information about its function (Williams et al, 2008, 2011). Along with the other studies, the structure revealed a major role of the MRN complex in bridging DNA ends, involving a concerted action of two components: one action coordinated by the Mre11 nuclease domain responsible for the synapsis of the two ends of a DSB and a second action mediated by the long-range tethering of two DNA molecules by Rad50. Structural studies were accompanied by a fruitful functional analysis of a series of newly generated mutants in fission yeast (Williams et al, 2008, 2011). These studies led to the identification of mutations on the surface of Rad32MRE11 that prevented homodimerization and stable DNA interaction leaving both exo- and endonuclease function largely unaffected (Williams et al, 2008). Our results suggest that whole complex architecture and stable DNA interaction but not nuclease activity are key elements for NHEJ repair. Even though Rad32MRE11 homodimerization may be helped by Rad50 head interactions (Williams et al, 2011), the function of the whole complex remains impaired in the mutants, as indicated by their DNA damage sensitivities (Supplementary Figure 4; Williams et al, 2008).
Analysis of Rad32MRE11 nuclease requirements in telomere fusions and plasmid-based NHEJ assays revealed a complex pattern. A strong nuclease dead mutant rad32-D65N (responsible for Mn2+ coordination) was clearly proficient in NHEJ repair at telomeres and in split-molecule plasmid assays, supportive of a nuclease-independent tethering role of the MRN complex for NHEJ. However, nuclease mutants that may additionally affect the general architecture of the complex such as rad32-H134S (phosphoesterase motif III required for phosphate rotation) and a second phosphoesterase mutant in motif II, rad32-H68S (predicted to be exonuclease, but not endonuclease, deficient) were required for NHEJ repair of free DNA ends. Thus, even though the actual nuclease deficiencies are yet to be confirmed in vitro in fission yeast, our results suggest that DNA end bridging performed by the nuclease domain of MRN, rather than the nuclease activity per se, is the important feature for the ensuing NHEJ repair reaction. Further studies will allow us to clarify the role of this multifaceted complex in NHEJ repair.
As expected from previous results obtained with mrn deletions (Manolis et al, 2001; Porter-Goff and Rhind, 2009), none of the Rad32MRE11 mutants exhibit lower levels of plasmid end rejoining in the classical NHEJ assay. The plasmids used in these assays are typically shorter than 10 kb (Boulton and Jackson, 1998; Manolis et al, 2001; Porter-Goff and Rhind, 2009), so the DNA ends available for NHEJ are always in close proximity. However, using our redesigned NHEJ assay, we measured a significant decrease in the ability of these mutants to join DNA ends that are not closely linked together. Likewise, dimerization mutants abrogate NHEJ at dysfunctional telomeres and rescue the lethality inflicted by chromosome end fusions. Thus, we hypothesize that this function is critical for NHEJ repair in fission yeast when DNA ends are not in close proximity. Even though yeast telomeres tend to cluster at the nuclear periphery, we anticipate mechanisms involved in search and increased mobility to facilitate in the repair of dysfunctional chromosome ends. Such a role has been proposed for ATM and 53BP1 in NHEJ repair reactions involving distant sites, including the joining of dysfunctional telomeres (Dimitrova and de Lange, 2009) and DSBs generated in class-switch recombination—a function that also requires MRN (Dinkelmann et al, 2009).
While telomere fusions in mammalian cells may be detected throughout the cell cycle, NHEJ of uncapped telomeres takes place primarily during G1 (Konishi and de Lange, 2008). MRN is essential for NHEJ at TRF2 dysfunctional telomeres, and it requires both ATM and 53BP1 checkpoint proteins (Denchi and de Lange, 2007). Checkpoint responses comprise several steps that include chromatin remodelling at the upstream point for signal propagation and further alterations required for subsequent DNA repair. MRN may initiate several events, some of which may be observable only if the previous ones have been satisfied. Because of reduced levels of NHEJ throughout almost the entirety of the cell cycle, dysfunctional telomeres subsist in fission yeast (Ferreira et al, 2004; Carneiro et al, 2010). In the absence of Taz1, chromatin at telomeres and neighbouring sequences is severely affected, allowing for de-repression of transcriptional silencing. Thus, open chromatin is a constant feature at taz1Δ telomeres, and this may obviate the need for upstream MRN-dependent activities identified in mammalian cells, such as ATM activation. Consequently, in our studies, MRN would not be required for early checkpoint-dependent chromatin remodelling, but only for the later event of tethering chromosome ends. This function may likewise be required in higher eukaryotes. However, this function may not be revealed unless initial steps are bypassed and its specific requirement is tested.
Although our work discards a role for both Tel1ATM and Rad3ATR (and even Crb253BP1) in NHEJ of taz1Δ telomeres, it does emphasize the function of MRN in bridging unlinked DNA ends in end-joining reactions. Our work also highlights the need for better assays in measuring NHEJ repair at different substrates and different parts of the cell cycle. During every cell cycle, chromosome ends unfold to allow for the passage of the replication fork, and innumerable DSBs are formed by genotoxic stress. All these events require specific responses and, surprisingly, are mediated by very few proteins, including MRN. The regulation of NHEJ repair at DNA ends, including at the natural ends of chromosomes, is a decisive step in cell metabolism, and its outcome may be the difference between cancer-prone genome instability or error-free DNA repair.
Materials and methods
Yeast strains
Strains used in this study are described in Table I.
Table 1. Strains used in this work.
| Strain | Genotype | Source |
|---|---|---|
| CTN23-22 | h− ade6-M216 ura4-D18 leu1-32 tel1::LEU2 | F Ishikawa |
| TN1138 | h− ade6-M216 leu1-32 ura4-D18 his3-D1 rad32::kanMX6 | T Nakamura |
| TN1155 | h− ade6-M216 leu1-32 ura4-D18 his3-D1 rad32::kanMX6 taz1::ura4+ | T Nakamura |
| KT120 | h− ade6-M210 leu1-32 ura4D-18 rad50::LEU2 | M Ueno |
| KT121 | h+ ade6-M216 leu1-32 ura4D-18 rad50::LEU2 taz1::ura4+ | M Ueno |
| 805 | h− smt0 ura4-D18 rad32-D65N | E Hartsuiker |
| OL4121 | h− leu1-32 ura4-D18 ctp1::kanMX6 | P Russell |
| JW4167 | h+ leu1-32 ura4-D18 mre11-L77K-13myc::kanMX6 | P Russell |
| JW4168 | h+ leu1-32 ura4-D18 mre11-L154D-13myc::kanMX6 | P Russell |
| JW4170 | h− leu1-32 ura4-D18 mre11-H68S-13myc::kanMX6 | P Russell |
| JW4171 | h− leu1-32 ura4-D18 mre11-H134S-13myc::kanMX6 | P Russell |
| MGF10 | h− ade6-M210 his3-D1 leu1-32 ura4-D18 | JP Cooper |
| MGF21 | h+ taz1::kanMX6 | JP Cooper |
| MGF44 | h− rap1::kanMX6 | JP Cooper |
| MGF255 | h− ade6-M216 leu1-32 ura4-D18 tel1::LEU2 taz1::kanMX6 | JP Cooper |
| MGF271 | h− ura4-D18 dna2-C2 taz1::ura4+ | M Ueno |
| MGF294 | h− ura4-D18 exo1::ura4 taz1::kanMX6 | This study |
| MGF864 | h− nbs1::natMX6 taz1::hphMX6 | This study |
| MGF1023 | h− nbs1::natMX6 taz1::hphMX6 lig4::kanMX6 | This study |
| MGF472 | h− ade6-M210 his3-D1 leu1-32 ura4-D18 taz1::kan | This study |
| MGF631 | h− leu1-32 ura4-D18 ctp1::kanMX6 taz1::ura4+ | This study |
| MGF724 | h− rap1::kanMX6 nbs1::natMX6 | This study |
| MGF865 | h+ ctp1::kanMX6 rap1::hphMX6 | This study |
| MGF1022 | h− taz1::kanMX6 lig4::hphMX6 | This study |
| MGF400 | h+ ade6-M216 his3-D1 leu1-32 ura4+-D18 nbs1::ura4+ | This study |
| MGF866 | h− taz1::kanMX6 rad3::natMX6 | This study |
| MGF516 | h− ade6-M210 his3-D1 leu1-32 ura4-D18 crb2::hphMX6 | This study |
| MGF517 | h+ taz1::kan crb2::hphMX6 | This study |
| MGF727 | h+ ade6-M210 leu1-32 ura4-D18 his3-D1 rad3::natMX6 | This study |
| MGF1548 | h− ade6-M216 leu1-32 ura4-D18 tel1::LEU2 taz1::kanMX6 rad3::hphMX6 | This study |
| MGF1594 | h+ ade6-M216 leu1-32 tel1::LEU2 his3-D1 ura4-D18 rap1::kanMX6 rad3::hphMX6 | This study |
| MGF470 | h− smt0 ura4-D18 rad32-D65N taz1::kanMX6 | This study |
| MGF717 | h+ leu1-32 ura4-D18 mre11-L77K-13myc::kanMX6 taz1::ura4+ | This study |
| MGF719 | h+ leu1-32 ura4-D18 mre11-L154D-13myc::kanMX6 taz1::ura4+ | This study |
| MGF720 | h− leu1-32 ura4-D18 mre11-H68S-13myc::kanMX6 taz1::ura4+ | This study |
| MGF721 | h− leu1-32 ura4-D18 mre11-H134S-13myc::kanMX6 taz1::ura4+ | This study |
| MGF1016 | h− ade6-M210 his3-D1 leu1-32 ura4-D18 lig4::hphMX6 | This study |
| MGF1260 | h− ade6-M216 leu1-32 ura4-D18 his3-D1 rad32::hphMX6 | This study |
| MGF1405 | h− ade6-M210 his3-D1 leu1-32 ura4-D18 rad50::hphMX6 | This study |
| MGF1895 | h− ade6-M210 his3-D1 leu1-32 ura4-D18 ctp1::hphMX6 | This study |
| MGF1230 | h+ leu1-32 ura4-D18 mre11-L77K-13myc::hphMX6 | This study |
| MGF1232 | h+ leu1-32 ura4-D18 mre11- L154D-13myc::hphMX6 | This study |
| MGF1233 | h− leu1-32 ura4-D18 mre11- H68S-13myc::hphMX6 | This study |
| MGF1234 | h− leu1-32 ura4-D18 mre11- H134S-13myc::hphMX6 | This study |
| MGF722 | h− leu1-32 ura4-D18 nbs1-9-TAP::kanMX6 taz1::ura4+ | This study |
| MGF723 | h− leu1-32 ura4-D18 nbs1-10-TAP::kanMX6 taz1::ura4+ | This study |
| MGF495 | h+leu1-32 ura4-D18 dna2-C2 exo1::ura4+ taz1::kanMX6 | This study |
| MGF1191 | h+ade6-M210 his3-D1 leu1-32 ura4-D18 lig4::natMX6 nbs1::ura4+ | This study |
| MGF1842 | h− ade6-M210 leu1-32 ura4-D18 ku70::hphMX6 | This study |
| MGF2025 | h+ade6-M216 leu1-32 ura4-D18 ku80::hphMX6 | This study |
Pulsed-field gel electrophoresis
Agarose chromosome plugs were prepared as previously described (Ferreira and Cooper, 2004). NotI-digested plugs were loaded onto 1% agarose gels and separated on a BioRad CHEF DR-III system in 0.5 × TBE at 14°C using the following program: 24 h run time, 60–120 s switch time, 120° angle, at 6 V/cm. After electrophoresis, DNA was visualized by ethidium bromide staining, and gels were processed for Southern blotting, which was performed using telomere probes (Rog et al, 2009) or LMIC probes (Miller et al, 2006).
Nitrogen starvation and cell viability assays
Nitrogen starvation and viability assays were essentially performed as previously described (Ferreira and Cooper, 2001). Cells were grown to logarithmic phase in EMM and then washed with three volumes of EMM-N. Overnight EMM-N cultures were subsequently used for chromosomal sample preparation for PFGE, plasmid transformations, or evaluation of the effect of G1 arrest in cell viability. Viability assays were performed by plating 300 cells/plate, each in triplicate. Colonies were counted 5 days after plating, and cell viability was scored as the ratio of colonies formed to cells plated. The average and 2 × s.e.m. of at least three replicates of each experiment is presented.
Plasmid assays
Plasmid NHEJ repair assays were performed exactly as previously described (Ferreira and Cooper, 2004). Plasmid tethering assays were performed by transforming exponentially growing and nitrogen-starved yeast cells auxotrophic for leu1+. One microgram of undigested pKan1 plasmid was transformed in order to normalize for different transformation efficiencies. In parallel, the same yeast cells were co-transformed with 1 μg of KpnI-linearized pKan1 plasmid and an equimolar amount of a HindIII fragment of the LEU2 gene of S. cerevisiae. The LEU2 gene and its regulatory sequences were obtained by HindIII digestion of pREP41 plasmid and agarose gel purification. Transformed cells were recovered in non-selective rich media for 1 h at 32°C and plated in triplicate in double selection media. Colonies were scored after a 6-day incubation at 32°C. The ability to repair two independent DNA fragments in vivo was calculated as the ratio of colonies that were formed after co-transformation with Kpn1-linearized pKan1 plasmid and HindIII LEU2 fragment over uncut pKan1 plasmid transformation colony number. The average and 2 × s.e.m. of at least three replicates of each transformation is presented. To measure NHEJ between incompatible ends, pKan plasmid was double digested with KpnI and BglII and agarose gel purified. One microgram of uncut plasmid and 1 μg of KpnI/BglII-linearized pKan1 were transformed in parallel.
PCR analysis of in vivo plasmid ligation reactions
Colony PCR was performed in yeast cells derived from co-transformation of LEU2 fragment and digested pKan1 plasmid. Primer KanF (5′-TTCGCCTCGACATCATCTGC-3′) was used in combination with either primer Leu2F (5′-GTTAAAAAGGTTTTGGATGC-3′) or Leu2R (5′-AAAACGACGATCTTCTTAGG-3′) in order to amplify the product of the two possible directions of in vivo ligation.
DNA damage sensitivities
Ten-fold serial dilutions (5 μl each) of log-phase cells were spotted in YES-rich media plates containing the indicated doses of HU or camptothecin or treated with UV immediately after cell spotting. Plates were incubated for 4 days at 32°C.
Supplementary Material
Acknowledgments
We are indebted to Julie Cooper for her valuable contributions at the beginning of this project. We are grateful to Julie Cooper and Lars Jansen for critically reading the manuscript. We thank E Hartsuiker, P Russell, F Ishikawa, T Nakamura, M Ueno and JP Cooper for strains and reagents. CCR is supported by Fundação para a Ciência e a Tecnologia (FCT) postdoctoral fellowship. This work was supported by the FCT (PTDC/SAU-OBD/66438/2006 and PTDC/BIA-BCM/099367/2008) and Prémio Simbiontes (Associação Viver a Ciência). MGF is an HHMI International Early Career Scientist.
Author Contributions: CCR designed and executed the experiments and helped in writing the paper. SB performed the plasmid NHEJ assays. MGF conceived the study, helped with plasmid NHEJ assays and wrote the paper.
Footnotes
The authors declare that they have no conflict of interest
References
- Boulton SJ, Jackson SP (1996) Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res 24: 4639–4648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP (1998) Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J 17: 1819–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro T, Khair L, Reis CC, Borges V, Moser BA, Nakamura TM, Ferreira MG (2010) Telomeres avoid end detection by severing the checkpoint signal transduction pathway. Nature 467: 228–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli GB, de Lange T (2005) DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol 7: 712–718 [DOI] [PubMed] [Google Scholar]
- Costanzo V, Robertson K, Bibikova M, Kim E, Grieco D, Gottesman M, Carroll D, Gautier J (2001) Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol Cell 8: 137–147 [DOI] [PubMed] [Google Scholar]
- Denchi EL, de Lange T (2007) Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448: 1068–1071 [DOI] [PubMed] [Google Scholar]
- Deng Y, Guo X, Ferguson DO, Chang S (2009) Multiple roles for MRE11 at uncapped telomeres. Nature 460: 914–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, de Lange T (2009) Cell cycle-dependent role of MRN at dysfunctional telomeres: ATM signaling-dependent induction of nonhomologous end joining (NHEJ) in G1 and resection-mediated inhibition of NHEJ in G2. Mol Cell Biol 29: 5552–5563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO (2009) Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol 16: 808–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605–611 [DOI] [PubMed] [Google Scholar]
- Ferreira MG, Cooper JP (2001) The fission yeast Taz1 protein protects chromosomes from Ku-dependent end-to-end fusions. Mol Cell 7: 55–63 [DOI] [PubMed] [Google Scholar]
- Ferreira MG, Cooper JP (2004) Two modes of DNA double-strand break repair are reciprocally regulated through the fission yeast cell cycle. Genes Dev 18: 2249–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MG, Miller KM, Cooper JP (2004) Indecent exposure: when telomeres become uncapped. Mol Cell 13: 7–18 [DOI] [PubMed] [Google Scholar]
- Frank-Vaillant M, Marcand S (2002) Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell 10: 1189–1199 [DOI] [PubMed] [Google Scholar]
- Hartsuiker E, Mizuno K, Molnar M, Kohli J, Ohta K, Carr AM (2009) Ctp1CtIP and Rad32Mre11 nuclease activity are required for Rec12Spo11 removal, but Rec12Spo11 removal is dispensable for other MRN-dependent meiotic functions. Mol Cell Biol 29: 1671–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP (2008) CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455: 689–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Jackson SP (2009) Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 284: 9558–9565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain D, Cooper JP (2010) Telomeric strategies: means to an end. Annu Rev Genet 44: 243–269 [DOI] [PubMed] [Google Scholar]
- Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T (1999) p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 283: 1321–1325 [DOI] [PubMed] [Google Scholar]
- Konishi A, de Lange T (2008) Cell cycle control of telomere protection and NHEJ revealed by a ts mutation in the DNA-binding domain of TRF2. Genes Dev 22: 1221–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langerak P, Mejia-Ramirez E, Limbo O, Russell P (2011) Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet 7: e1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Oestreich S, de Lange T (2000) Identification of human Rap1: implications for telomere evolution. Cell 101: 471–483 [DOI] [PubMed] [Google Scholar]
- Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, Russell P (2007) Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell 28: 134–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolis KG, Nimmo ER, Hartsuiker E, Carr AM, Jeggo PA, Allshire RC (2001) Novel functional requirements for non-homologous DNA end joining in Schizosaccharomyces pombe. EMBO J 20: 210–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KM, Ferreira MG, Cooper JP (2005) Taz1, Rap1 and Rif1 act both interdependently and independently to maintain telomeres. EMBO J 24: 3128–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KM, Rog O, Cooper JP (2006) Semi-conservative DNA replication through telomeres requires Taz1. Nature 440: 824–828 [DOI] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS (2011) DNA end resection—unraveling the tail. DNA Repair (Amst) 10: 344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S, Ferguson JR, Symington LS (1999) The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol 19: 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito T, Matsuura A, Ishikawa F (1998) Circular chromosome formation in a fission yeast mutant defective in two ATM homologues. Nat Genet 20: 203–206 [DOI] [PubMed] [Google Scholar]
- Pardo B, Marcand S (2005) Rap1 prevents telomere fusions by nonhomologous end joining. EMBO J 24: 3117–3127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Gellert M (1998) The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell 1: 969–979 [DOI] [PubMed] [Google Scholar]
- Porter-Goff ME, Rhind N (2009) The role of MRN in the S-phase DNA damage checkpoint is independent of its Ctp1-dependent roles in double-strand break repair and checkpoint signaling. Mol Biol Cell 20: 2096–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rog O, Miller KM, Ferreira MG, Cooper JP (2009) Sumoylation of RecQ helicase controls the fate of dysfunctional telomeres. Mol Cell 33: 559–569 [DOI] [PubMed] [Google Scholar]
- Tomita K, Kibe T, Kang HY, Seo YS, Uritani M, Ushimaru T, Ueno M (2004) Fission yeast Dna2 is required for generation of the telomeric single-strand overhang. Mol Cell Biol 24: 9557–9567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Matsuura A, Caspari T, Carr AM, Akamatsu Y, Iwasaki H, Mizuno K, Ohta K, Uritani M, Ushimaru T, Yoshinaga K, Ueno M (2003) Competition between the Rad50 complex and the Ku heterodimer reveals a role for Exo1 in processing double-strand breaks but not telomeres. Mol Cell Biol 23: 5186–5197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenz K, Smith E, Smith S, Costanzo V (2006) ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J 25: 1764–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Perrault AR, Takeda Y, Qin W, Iliakis G (2003) Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res 31: 5377–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Lees-Miller SP, Tainer JA (2010) Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 9: 1299–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, Limbo O, Guenther G, SilDas S, Hammel M, Russell P, Tainer JA (2011) ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat Struct Mol Biol 18: 423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA (2008) Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135: 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Chahwan C, Bailis J, Hunter T, Russell P (2005) ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol 25: 5363–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.