

Abstract
The cag pathogenicity island is a well-characterized virulence determinant. It is composed of 32 genes that encode a type IV bacterial secretion system and is linked with a more severe clinical outcome. The following chapters will explore the manipulation of bacterial factors in order to understand their role in gastric mucosal disease.
Keywords: Helicobacter pylori, cag pathogenicity island
1. The Helicobacter pylori cag Pathogenicity Island
H. pylori strains exhibit a high degree of genetic heterogeneity due to genomic rearrangements, point mutations, gene insertions, and/or deletions (1–4). Genetically unique variants of a single strain are present within the stomachs of each human host, and the genetic composition of these populations can evolve over time (5). The identification of bacterial factors clearly associated with disease outcomes has been hindered because of this high level of genetic diversity; however, specific loci have been identified that augment the risk for carcinogenesis. The cag pathogenicity island (cag PAI) is a 40 kb DNA insertion element, containing approximately 32 genes that encode a bacterial type IV secretion system (4, 6–8). The cag PAI is a well-characterized H. pylori virulence determinant that is present in approximately 60–70% of Western H. pylori strains and virtually 100% of East-Asian H. pylori strains (4, 6–8). Although all H. pylori strains induce gastritis, strains that harbor the cag PAI (cag+) augment the risk for severe gastritis, atrophy, dysplasia, and gastric adenocarcinoma compared to strains that lack the cag island (cag−) (9–20). H. pylori cag− strains are found predominantly in the mucus gel layer while cag+ strains are found adjacent to or adherent to gastric epithelial cells, indicating that cag genotype influences the topography of colonization within the stomach (21).
The cag type IV secretion system allows for the delivery of bacterial effector molecules into host gastric epithelial cells. One of these proteins, CagE, is a structural component of the functional type IV secretion system and inactivation of this gene product abrogates delivery of H. pylori proteins into host cells. Another component of the secretion system, CagL, functions as a specialized bacterial adhesin that binds to and activates α5β1 integrin receptors, triggering the delivery of bacterial effector molecules into the cytoplasm of host cells (22). CagL bridges the type IV secretion system to α5β1 integrins on target cells and activates host focal adhesion kinase (FAK) and Src (Fig. 1). In addition to α5β1 integrin, CagL can also bind integrin and fibronectin, although the downstream consequences of binding to these receptors remains undefined. Recently, additional Cag proteins (CagA, CagI, CagY) have been shown to bind β1 integrin and induce conformational changes of integrin heterodimers, permitting translocation of bacterial effectors (23).
Fig. 1.
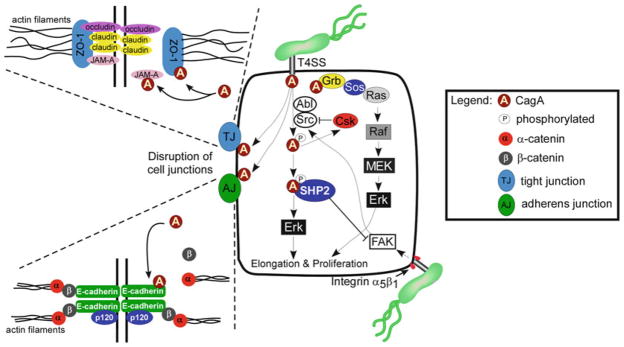
Molecular signaling alterations induced by intracellular delivery of CagA. Translocation of CagA by the H. pylori cag type IV secretion system leads to activation of host signaling pathways that promote epithelial responses with carcinogenic potential. CagA is phosphorylated by Src and Abl kinases, which is followed by a decrease in levels of phosphorylated-CagA via the inhibitory kinase c-src kinase (Csk). Phosphorylated CagA activates SHP2 and Erk leading to morphological changes, such as cellular elongation. Additionally, the interaction between phosphorylated-CagA and SHP2 results in inactivation of focal adhesion kinase (FAK), which can activate Src. Unphosphorylated CagA also leads to changes in epithelial cell motility and proliferation through binding Grb/Sos/Ras and activation of the Raf/MEK/Erk pathway. Unmodified CagA can also associate with the tight junction proteins ZO-1 and JAM-A as well as the adherens junction protein E-cadherin, leading to dysregulated junctional complexes.
2. CagA
The terminal gene product of the cag island, CagA, is translocated into host cells by the cag type IV secretion system following bacterial attachment (24). Transgenic mice that overexpress CagA develop gastric epithelial cell hyperproliferation and gastric adenocarcinoma (25), implicating this molecule as a bacterial oncoprotein. CagA is a 120–140 kD protein that contains tyrosine phosphorylation motifs (glutamate-proline-isoleucine-tyrosine-alanine, EPIYA) within the carboxyl-terminal variable region of the protein (26). To date, four distinct EPIYA motifs have been identified within the carboxyl-terminal polymorphic region of CagA. These motifs are designated EPIYA -A, -B, -C, or -D and are distinguished by the amino acid sequences flanking the EPIYA motif (27–29). Most variants of CagA throughout the world contain EPIYA-A and -B motifs, while EPIYA-C motifs are predominantly found in strains from Western countries (Europe, North America, and Australia). The number of EPIYA-C sites can vary; however, most CagA proteins contain a single EPIYA-C site (A-B-C-type). The EPIYA-A and -B motifs are phosphorylated to a lesser extent than EPIYA-C motifs and in Western strains an increased number of CagA EPIYA-C motifs correlates with increased gastric cancer risk (30, 31). EPIYA-D motifs are found almost exclusively in East-Asian H. pylori strains (Japan, Korea, and China) and are phosphorylated to a greater extent than all other EPIYA motifs (27). H. pylori strains containing EPIYA-D motifs induce significantly higher levels of IL-8 release from gastric epithelial cells compared to strains harboring Western A-B-C-type CagA (27, 32). Thus, the majority of cag+ Western strains express A-B-C-type CagA, and the number of EPIYA-C regions may vary between 1 and 3 repeated copies among different strains, while East-Asian strains typically express A-B-D-type CagA (27).
3. CagA Phosphorylation-Dependent Perturbation of Host Cell Signaling
Following its injection into epithelial cells, CagA undergoes tyrosine phosphorylation at EPIYA motifs by members of the Abl and Src family of kinases (24, 33–37) (Fig. 1). Intracellular, phosphorylated-CagA, in turn, activates a eukaryotic phosphatase (SHP-2) and extracellular signal-regulated kinase 1 and 2 (Erk1/2), leading to cell scattering, robust actin reorganization, and other morphologic changes reminiscent of unrestrained stimulation by growth factors (24, 26, 33–41). Specifically, CagA transfection studies have demonstrated that phosphorylated-CagA-SHP-2 interactions contribute to cytoskeletal rearrangements and cell elongation by activation of the Erk-signaling pathway (38). East-Asian A-B-D-type CagA exhibits a higher binding affinity for SHP-2 than Western A-B-C-type CagA and, therefore, induces a more robust morphologic response by gastric epithelial cells (39).
H. pylori CagA proteins tightly and specifically regulate the activity of Src and Abl family kinases in a time-dependent manner. Src is activated during the initial stages of infection and is then rapidly inactivated, while Abl is continuously activated by H. pylori with enhanced activities at later time points, supporting a model of successive phosphorylation of CagA by Src and Abl family kinases (42). Phosphorylated-CagA can also inhibit Src via recruitment of C-terminal Src kinase (Csk), a negative-regulator of Src that acts rapidly to initiate a negative feedback loop to downregulate Src signaling (41, 43) (Fig. 1). As Src is the primary kinase activated by CagA, inhibition of Src by phosphorylated-CagA generates a negative feedback loop that carefully regulates the amount of intracellular, phosphorylated-CagA. The catalytic activity of Src is inhibited by phosphorylated-CagA, leading to tyrosine dephosphorylation of the actin-binding proteins cortactin, ezrin, and vinculin, which ultimately results in cellular rearrangements and elongation (43–45).
In AGS human gastric epithelial cells, translocation and subsequent phosphorylation of CagA results in cell elongation and scattering, known as the “hummingbird” phenotype (35, 46). In this cell line, interactions between phosphorylated-CagA and SHP-2 increase the duration of Erk activation in a Ras- and PI3K-independent manner, resulting in cell elongation (38). The interaction between phosphorylated-CagA and SHP-2 also results in dephosphorylation and inactivation of FAK, which again leads to morphologic aberrations (47) (Fig. 1).
4. CagA Phosphorylation-Independent Perturbation of Host Cell Signaling
Non-phosphorylated CagA also exerts numerous effects within gastric epithelial cells that contribute to pathogenesis. CagA translocation, but not phosphorylation, leads to disruption of apical–junctional complexes (Fig. 1). Non-phosphorylated CagA associates with the epithelial tight-junction scaffolding protein zona occludens 1 (ZO-1) and the transmembrane protein junctional adhesion molecule A (JAM-A), leading to nascent but incomplete assembly of tight junctions (TJ) at ectopic sites of bacterial attachment (48). In addition, non-phosphorylated CagA disrupts adherens junctions (AJ) leading to aberrant activation of β-catenin and an overall loss of barrier function and cellular polarity (48–53), alterations that play an important role in carcinogenesis. Non-phosphorylated CagA interacts with the cell adhesion protein E-cadherin, the hepatocyte growth factor receptor c-Met, the phospholipase PLC-γ, and the adaptor protein Grb2 (51, 52, 54, 55), which leads to proinflammatory and mitogenic responses, disruption of cellular junctions, and loss of cellular polarity. Recently, non-phosphorylated CagA was shown to directly bind PAR1b/MARK2, a central regulator of cell polarity, and inhibit its kinase activity. This interaction induced the dysregulation of mitotic spindle formation, promoting a loss of cellular polarity (52, 56, 57). These events were dependent on conserved 16 amino acid repeat motifs embedded within the 3 terminus of CagA and which are known as CagA multimerization (CM) sites (58), the conserved repeat responsible for phosphorylation-independent activity (CRPIA) (59), or the PAR1b/MARK2 kinase inhibitor (MKI) (60) motifs. These motifs, which vary in number among H. pylori strains, bind PAR1b/MARK2 and mediate homodimerization of CagA, conferring heightened SHP-2 binding and activation. A recent co-crystallography analysis of the CagA-PAR1b/MARK2 complex demonstrated that the PAR1b/MARK2-binding site resides in the initial 14 amino acids of the CagA CM motif, and binding leads to inhibition of PAR1b/MARK2 kinase activity (60).
5. Peptidoglycan
Another consequence of H. pylori cag pathogenicity island-mediated host cell contact is the production of pro-inflammatory cytokines. In certain H. pylori strains, CagA can induce IL-8 expression via NF-κB activation (61–63); however, the ability of CagA to induce IL-8 expression is not universal across all cag PAI-bearing strains (64–67). In addition to CagA, H. pylori peptidoglycan (PGN) is delivered into host cells via the cag type IV secretion system and outer membrane vesicles (OMV) (68), where they are sensed by the nucleotide-binding oligomerization domain 1 (NOD1), an intracytoplasmic pathogen pattern-recognition molecule (69, 70) (Fig. 2). NOD1 activation by H. pylori peptidoglycan stimulates the production of proinflammatory cytokines MIP-2, β-defensin, and IL-8 through induction of host cells signaling molecules, nuclear factor κB (NF-κB), p38, and Erk (70, 71). Furthermore, NOD1 activation by H. pylori peptidoglycan also regulates the production of type I interferon (IFN), which likely affects Th1 cell differentiation (72). NOD1-deficient mice develop an attenuated mucosal cytokine response following infection with H. pylori cag+ strains (73), implicating peptidoglycan-NOD1 signaling as an important mediator of H. pylori pathogenesis.
Fig. 2.
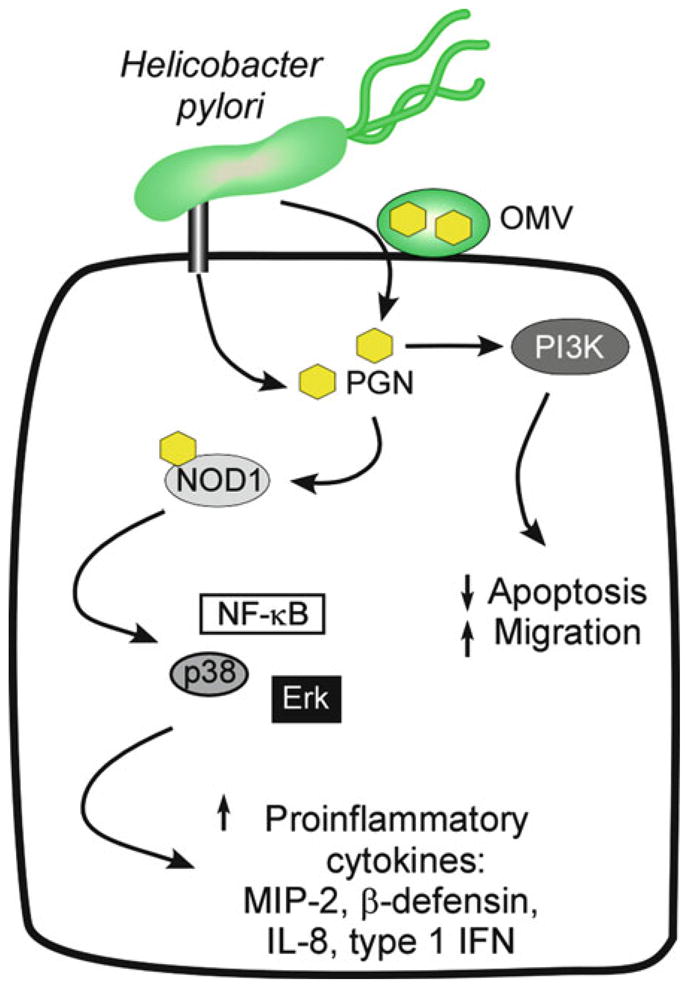
Molecular signaling alterations induced by intracellular delivery of peptidoglycan. In addition to CagA, the H. pylori cag type IV secretion system can deliver peptidoglycan (PGN) into host cells. Another mechanism of PGN delivery is via outer membrane vesicles (OMV). Delivery of PGN results in activation of the intracellular receptor nucleotide oligomerization domain 1 (NOD1) and triggers multiple signaling pathways that culminate in NF-κB activation and subsequent production of inflammatory and immune effectors, such as IL-8 and Type 1 IFN. Further, PGN can also activate PI3K, leading to decreased levels of apoptosis and increased cell migration.
Delivery of peptidoglycan components into host cells induces additional epithelial responses with carcinogenic potential, such as activation of PI3K and cell migration (Fig. 2). The H. pylori gene slt encodes a soluble lytic transglycosylase that is required for peptidoglycan turnover and release (70), thereby regulating the amount of peptidoglycan translocated into host cells. Inactivation of slt has now been shown to inhibit H. pylori-induced PI3K signaling and cell migration (74). The protein encoded by the H. pylori gene HP0310 deacetylates N-acetylglucosamine peptidoglycan residues and is required for peptidoglycan synthesis (75). Loss of HP0310, leading to decreased peptidoglycan production, reciprocally augments delivery of the other major cag secretion system substrate, CagA, into host cells, suggesting that functional interactions occur between H. pylori translocated effector molecules (76). Further, H. pylori peptidoglycan deacetylation by HP0310 is an important mechanism for mitigating host immune detection, which facilitates bacterial persistence and colonization (77). In total, these findings indicate that contact between cag+ strains and host cells activates multiple signaling pathways that regulate oncogenic cellular responses, which may heighten the risk for transformation, particularly over prolonged periods of H. pylori infection.
References
- 1.Alm RA, Bina J, Andrews BM, Doig P, Hancock RE, Trust TJ. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect Immun. 2000;68:4155–4168. doi: 10.1128/iai.68.7.4155-4168.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Go MF, Kapur V, Graham DY, Musser JM. Population genetic analysis of Helicobacter pylori by multilocus enzyme electrophoresis: extensive allelic diversity and recombinational population structure. J Bacteriol. 1996;178:3934–3938. doi: 10.1128/jb.178.13.3934-3938.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salama N, Guillemin K, McDaniel TK, Sherlock G, Tompkins L, Falkow S. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc Natl Acad Sci USA. 2000;97:14668–14673. doi: 10.1073/pnas.97.26.14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 5.Israel DA, Salama N, Krishna U, Rieger UM, Atherton JC, Falkow S, et al. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc Natl Acad Sci USA. 2001;98:14625–14630. doi: 10.1073/pnas.251551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akopyants NS, Clifton SW, Kersulyte D, Crabtree JE, Youree BE, Reece CA, et al. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol Microbiol. 1998;28:37–53. doi: 10.1046/j.1365-2958.1998.00770.x. [DOI] [PubMed] [Google Scholar]
- 7.Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 8.Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, et al. Cag, a pathogenicity island of Helicobacter pylori, encodes type I specific and disease-associated virulence factors. Proc Natl Acad Sci USA. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–2115. [PubMed] [Google Scholar]
- 10.Cover TL, Dooley CP, Blaser MJ. Characterization of and human serologic response to proteins in Helicobacter pylori broth culture supernatants with vacuolizing cytotoxin activity. Infect Immun. 1990;58:603–610. doi: 10.1128/iai.58.3.603-610.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crabtree JE, Taylor JD, Wyatt JI, Heatley RV, Shallcross TM, Tompkins DS, et al. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration, and gastric pathology. Lancet. 1991;338:332–335. doi: 10.1016/0140-6736(91)90477-7. [DOI] [PubMed] [Google Scholar]
- 12.Crabtree JE, Wyatt JI, Sobala GM, Miller G, Tompkins DS, Primrose JN, et al. Systemic and mucosal humoral responses to Helicobacter pylori in gastric cancer. Gut. 1993;34:1339–1343. doi: 10.1136/gut.34.10.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuipers EJ, Perez-Perez GI, Meuwissen SG, Blaser MJ. Helicobacter pylori and atrophic gastritis: importance of the cagA status. J Natl Cancer Inst. 1995;87:1777–1780. doi: 10.1093/jnci/87.23.1777. [DOI] [PubMed] [Google Scholar]
- 14.Parsonnet J, Replogle M, Yang S, Hiatt R. Seroprevalence of CagA-positive strains among Helicobacter pylori-infected, healthy young adults. J Infect Dis. 1997;175:1240–1242. doi: 10.1086/593680. [DOI] [PubMed] [Google Scholar]
- 15.Peek RM, Jr, Miller GG, Tham KT, Perez-Perez GI, Zhao X, Atherton JC, et al. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab Invest. 1995;73:760–770. [PubMed] [Google Scholar]
- 16.Queiroz DM, Mendes EN, Rocha GA, Oliveira AM, Oliveira CA, Magalhaes PP, et al. CagA-positive Helicobacter pylori and risk for developing gastric carcinoma in Brazil. Int J Cancer. 1998;78:135–139. doi: 10.1002/(sici)1097-0215(19981005)78:2<135::aid-ijc1>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 17.Rudi J, Kolb C, Maiwald M, Zuna I, von Herbay A, Galle PR, et al. Serum antibodies against Helicobacter pylori proteins VacA and CagA are associated with increased risk for gastric adenocarcinoma. Dig Dis Sci. 1997;42:1652–1659. doi: 10.1023/a:1018849112533. [DOI] [PubMed] [Google Scholar]
- 18.Shimoyama T, Fukuda S, Tanaka M, Mikami T, Munakata A, Crabtree JE. CagA seropositivity associated with development of gastric cancer in a Japanese population. J Clin Pathol. 1998;51:225–228. doi: 10.1136/jcp.51.3.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torres J, Perez-Perez GI, Leal-Herrera Y, Munoz O. Infection with CagA+ Helicobacter pylori strains as a possible predictor of risk in the development of gastric adenocarcinoma in Mexico. Int J Cancer. 1998;78:298–300. doi: 10.1002/(SICI)1097-0215(19981029)78:3<298::AID-IJC6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 20.Vorobjova T, Nilsson I, Kull K, Maaroos HI, Covacci A, Wadstrom T, et al. CagA protein seropositivity in a random sample of adult population and gastric cancer patients in Estonia. Eur J Gastroenterol Hepatol. 1998;10:41–46. doi: 10.1097/00042737-199801000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Camorlinga-Ponce M, Romo C, Gonzalez-Valencia G, Munoz O, Torres J. Topographical localisation of cagA positive and cagA negative Helicobacter pylori strains in the gastric mucosa; an in situ hybridisation study. J Clin Pathol. 2004;57:822–828. doi: 10.1136/jcp.2004.017087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449:862–866. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- 23.Jimenez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, et al. Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathogens. 2009;5:e1000684. doi: 10.1371/journal.ppat.1000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 25.Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA. 2008;105:1003–1008. doi: 10.1073/pnas.0711183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ, Covacci A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Microbiol. 2002;43:971–980. doi: 10.1046/j.1365-2958.2002.02781.x. [DOI] [PubMed] [Google Scholar]
- 27.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–694. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 28.Higashi H, Yokoyama K, Fujii Y, Ren S, Yuasa H, Saadat I, et al. EPIYA motif is a membrane-targeting signal of Helicobacter pylori virulence factor CagA in mammalian cells. J Biol Chem. 2005;280:23130–23137. doi: 10.1074/jbc.M503583200. [DOI] [PubMed] [Google Scholar]
- 29.Naito M, Yamazaki T, Tsutsumi R, Higashi H, Onoe K, Yamazaki S, et al. Influence of EPIYA-repeat polymorphism on the phosphorylation-dependent biological activity of Helicobacter pylori CagA. Gastroenterology. 2006;130:1181–1190. doi: 10.1053/j.gastro.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 30.Basso D, Zambon CF, Letley DP, Stranges A, Marchet A, Rhead JL, et al. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology. 2008;135:91–99. doi: 10.1053/j.gastro.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 31.Schneider N, Krishna U, Romero-Gallo J, Israel DA, Piazuelo MB, Camargo MC, et al. Role of Helicobacter pylori CagA molecular variations in induction of host phenotypes with carcinogenic potential. J Infect Dis. 2009;199:1218–1221. doi: 10.1086/597416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Argent RH, Hale JL, El-Omar EM, Atherton JC. Differences in Helicobacter pylori CagA tyrosine phosphorylation motif patterns between western and East Asian strains, and influences on interleukin-8 secretion. J Med Microbiol. 2008;57:1062–1067. doi: 10.1099/jmm.0.2008/001818-0. [DOI] [PubMed] [Google Scholar]
- 33.Asahi M, Azuma T, Ito S, Ito Y, Suto H, Nagai Y, et al. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J Exp Med. 2000;191:593–602. doi: 10.1084/jem.191.4.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Backert S, Ziska E, Brinkmann V, Zimny-Arndt U, Fauconnier A, Jungblut PR, et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2000;2:155–164. doi: 10.1046/j.1462-5822.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- 35.Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci USA. 1999;96:14559–14564. doi: 10.1073/pnas.96.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Selbach M, Moese S, Hauck CR, Meyer TF, Backert S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem. 2002;277:6775–6778. doi: 10.1074/jbc.C100754200. [DOI] [PubMed] [Google Scholar]
- 37.Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci USA. 2000;97:1263–1268. doi: 10.1073/pnas.97.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem. 2004;279:17205–17216. doi: 10.1074/jbc.M309964200. [DOI] [PubMed] [Google Scholar]
- 39.Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, et al. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci USA. 2002;99:14428–14433. doi: 10.1073/pnas.222375399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–686. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- 41.Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M. Attenuation of Helicobacter pylori CagA-SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J Biol Chem. 2002;21:21. doi: 10.1074/jbc.M208155200. [DOI] [PubMed] [Google Scholar]
- 42.Tammer I, Brandt S, Hartig R, Konig W, Backert S. Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology. 2007;132:1309–1319. doi: 10.1053/j.gastro.2007.01.050. [DOI] [PubMed] [Google Scholar]
- 43.Selbach M, Moese S, Hurwitz R, Hauck CR, Meyer TF, Backert S. The Helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c-Src inactivation. EMBO J. 2003;22:515–528. doi: 10.1093/emboj/cdg050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moese S, Selbach M, Brinkmann V, Karlas A, Haimovich B, Backert S, et al. The Helicobacter pylori CagA protein disrupts matrix adhesion of gastric epithelial cells by dephosphorylation of vinculin. Cell Microbiol. 2007;9:1148–1161. doi: 10.1111/j.1462-5822.2006.00856.x. [DOI] [PubMed] [Google Scholar]
- 45.Selbach M, Moese S, Backert S, Jungblut PR, Meyer TF. The Helicobacter pylori CagA protein induces tyrosine dephosphorylation of ezrin. Proteomics. 2004;4:2961–2968. doi: 10.1002/pmic.200400915. [DOI] [PubMed] [Google Scholar]
- 46.Moese S, Selbach M, Kwok T, Brinkmann V, Konig W, Meyer TF, et al. Helicobacter pylori induces AGS cell motility and elongation via independent signaling pathways. Infect Immun. 2004;72:3646–3649. doi: 10.1128/IAI.72.6.3646-3649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsutsumi R, Takahashi A, Azuma T, Higashi H, Hatakeyama M. Focal adhesion kinase is a substrate and downstream effector of SHP-2 complexed with Helicobacter pylori CagA. Mol Cell Biol. 2006;26:261–276. doi: 10.1128/MCB.26.1.261-276.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bagnoli F, Buti L, Tompkins L, Covacci A, Amieva MR. Helicobacter pylori CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proc Natl Acad Sci USA. 2005;102:16339–16344. doi: 10.1073/pnas.0502598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. 2005;102:10646–10651. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26:4617–4626. doi: 10.1038/sj.onc.1210251. [DOI] [PubMed] [Google Scholar]
- 52.Saadat I, Higashi H, Obuse C, Umeda M, Murata-Kamiya N, Saito Y, et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447:330–333. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki M, Mimuro H, Suzuki T, Park M, Yamamoto T, Sasakawa C. Interaction of CagA with Crk plays an important role in Helicobacter pylori-induced loss of gastric epithelial cell adhesion. J Exp Med. 2005;202:1235–1247. doi: 10.1084/jem.20051027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Churin Y, Al-Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann M. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the mitogenic response. J Cell Biol. 2003;161:249–255. doi: 10.1083/jcb.200208039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol Cell. 2002;10:745–755. doi: 10.1016/s1097-2765(02)00681-0. [DOI] [PubMed] [Google Scholar]
- 56.Lu H, Murata-Kamiya N, Saito Y, Hatakeyama M. Role of partitioning-defective 1/microtubule affinity-regulating kinases in the morphogenetic activity of Helicobacter pylori CagA. J Biol Chem. 2009;284:23024–23036. doi: 10.1074/jbc.M109.001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Umeda M, Murata-Kamiya N, Saito Y, Ohba Y, Takahashi M, Hatakeyama M. Helicobacter pylori CagA causes mitotic impairment and induces chromosomal instability. J Biol Chem. 2009;284:22166–22172. doi: 10.1074/jbc.M109.035766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurashima Y, Murata-Kamiya N, Kikuchi K, Higashi H, Azuma T, Kondo S, et al. Deregulation of beta-catenin signal by Helicobacter pylori CagA requires the CagA-multimerization sequence. Int J Cancer. 2008;122:823–831. doi: 10.1002/ijc.23190. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, Morikawa H, et al. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe. 2009;5:23–34. doi: 10.1016/j.chom.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 60.Nesic D, Miller MC, Quinkert ZT, Stein M, Chait BT, Stebbins CE. Helicobacter pylori CagA inhibits PAR1-MARK family kinases by mimicking host substrates. Nat Struct Mol Biol. 2010;17:130–132. doi: 10.1038/nsmb.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA. 2005;102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim SY, Lee YC, Kim HK, Blaser MJ. Helicobacter pylori CagA transfection of gastric epithelial cells induces interleukin-8. Cell Microbiol. 2006;8:97–106. doi: 10.1111/j.1462-5822.2005.00603.x. [DOI] [PubMed] [Google Scholar]
- 63.Lamb A, Yang XD, Tsang YH, Li JD, Higashi H, Hatakeyama M, et al. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009;10:1242–1249. doi: 10.1038/embor.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keates S, Keates AC, Warny M, Peek RM, Murray PG, Kelly CP. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag+ and cag− Helicobacter pylori. J Immunol. 1999;163:5552–5559. [PubMed] [Google Scholar]
- 65.Keates S, Sougioultzis S, Keates AC, Zhao D, Peek RM, Shaw LM, et al. Cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Biol Chem. 2001;276:48127–48134. doi: 10.1074/jbc.M107630200. [DOI] [PubMed] [Google Scholar]
- 66.Meyer-Ter-Vehn T, Covacci A, Kist M, Pahl HL. Helicobacter pylori activates mitogen-activated protein kinase cascades and induces expression of the proto-oncogenes c-fos and c-jun. J Biol Chem. 2000;275:16064–16072. doi: 10.1074/jbc.M000959200. [DOI] [PubMed] [Google Scholar]
- 67.Naumann M, Wessler S, Bartsch C, Wieland B, Covacci A, Haas R, et al. Activation of activator protein 1 and stress response kinases in epithelial cells colonized by Helicobacter pylori encoding the cag pathogenicity island. J Biol Chem. 1999;274:31655–31662. doi: 10.1074/jbc.274.44.31655. [DOI] [PubMed] [Google Scholar]
- 68.Kaparakis M, Turnbull L, Carneiro L, Firth S, Coleman HA, Parkington HC, et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol. 2010;12:372–385. doi: 10.1111/j.1462-5822.2009.01404.x. [DOI] [PubMed] [Google Scholar]
- 69.Boughan PK, Argent RH, Body-Malapel M, Park JH, Ewings KE, Bowie AG, et al. Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection. J Biol Chem. 2006;281:11637–11648. doi: 10.1074/jbc.M510275200. [DOI] [PubMed] [Google Scholar]
- 70.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 71.Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol. 2009;183:8099–8109. doi: 10.4049/jimmunol.0900664. [DOI] [PubMed] [Google Scholar]
- 72.Watanabe T, Asano N, Fichtner-Feigl S, Gorelick PL, Tsuji Y, Matsumoto Y, et al. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest. 2010;120:1645–1662. doi: 10.1172/JCI39481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol. 2006;6:9–20. doi: 10.1038/nri1747. [DOI] [PubMed] [Google Scholar]
- 74.Nagy TA, Frey MR, Yan F, Israel DA, Polk DB, Peek RM. Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J Infect Dis. 2009;199:641–651. doi: 10.1086/596660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang G, Olczak A, Forsberg LS, Maier RJ. Oxidative stress-induced peptidoglycan deacetylase in Helicobacter pylori. J Biol Chem. 2009;284:6790–6800. doi: 10.1074/jbc.M808071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Franco AT, Friedman DB, Nagy TA, Romero-Gallo J, Krishna U, Kendall A, et al. Delineation of a carcinogenic Helicobacter pylori proteome. Mol Cell Proteomics. 2009;8:1947–1958. doi: 10.1074/mcp.M900139-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang G, Maier SE, Lo LF, Maier G, Dosi S, Maier RJ. Peptidoglycan deacetylation in Helicobacter pylori contributes to bacterial survival by mitigating host immune responses. Infect Immun. 2010;78:4660–4666. doi: 10.1128/IAI.00307-10. [DOI] [PMC free article] [PubMed] [Google Scholar]