

Background: High dietary fat is linked to cardiac oxidative stress, however, little is known about the endogenous antioxidant response.
Results: High fat feeding and fasting rapidly up-regulate catalase.
Conclusion: Up-regulation of catalase is designed to protect mitochondria from oxidative damage while not perturbing H2O2-mediated signaling.
Significance: Coupling fatty acid oxidation to H2O2 production creates a mechanism for sensing and communicating diet composition.
Keywords: Antioxidants, Heart, Metabolism, Mitochondria, Obesity, Peroxisomes, Catalase, High Fat Diet, Quantitative Proteomics, Selected Reaction Monitoring
Abstract
Obesity is a predictor of diabetes and cardiovascular disease. One consequence of obesity is dyslipidemia characterized by high blood triglycerides. It has been proposed that oxidative stress, driven by utilization of lipids for energy, contributes to these diseases. The effects of oxidative stress are mitigated by an endogenous antioxidant enzyme network, but little is known about its response to high fat utilization. Our experiments used a multiplexed quantitative proteomics method to measure antioxidant enzyme expression in heart tissue in a mouse model of diet-induced obesity. This experiment showed a rapid and specific up-regulation of catalase protein, with subsequent assays showing increases in activity and mRNA. Catalase, traditionally considered a peroxisomal protein, was found to be present in cardiac mitochondria and significantly increased in content and activity during high fat feeding. These data, coupled with the fact that fatty acid oxidation enhances mitochondrial H2O2 production, suggest that a localized catalase increase is needed to consume excessive mitochondrial H2O2 produced by increased fat metabolism. To determine whether the catalase-specific response is a common feature of physiological conditions that increase blood triglycerides and fatty acid oxidation, we measured changes in antioxidant expression in fasted versus fed mice. Indeed, a similar specific catalase increase was observed in mice fasted for 24 h. Our findings suggest a fundamental metabolic process in which catalase expression is regulated to prevent damage while preserving an H2O2-mediated sensing of diet composition that appropriately adjusts insulin sensitivity in the short term as needed to prioritize lipid metabolism for complete utilization.
Introduction
Long term consumption of a diet high in fat promotes obesity and enhances the risk for the development of type 2 diabetes and cardiovascular disease (1–5). It has been proposed that oxidative stress may serve as the causal link between diet-induced obesity and cardiovascular disease (6, 7). Hearts from rodents fed a high fat diet display increased indices of lipid and protein oxidation as well as increased markers of apoptosis (8–11). Several different mechanisms of free radical production have been implicated in obesity, including the release of pro-inflammatory adipokines (12), elevated NAD(P)H oxidase activity (13), and increased mitochondrial hydrogen peroxide (H2O2) production (14, 15). However, the overall role of oxidative stress in the pathobiology of obesity and high dietary fat is not well understood.
Although free radicals are capable of damaging lipids, DNA, and proteins, it is important to note that pro-oxidant species such as H2O2 also act as signaling molecules (16–18). Furthermore, an extensive network of endogenous antioxidant enzymes and molecules exists to preclude oxidative damage while maintaining an environment conducive to redox regulation. If high dietary fat induces oxidative stress, it is logical to assume that the antioxidant network will mount a compensatory response to preserve redox homeostasis. Assessing the response of the antioxidant network to a high fat diet would provide valuable insight into the nature and source of oxidative stress associated with high dietary fat and offer targeted therapeutic options for the accompanying diseases. However, little has been reported on the antioxidant network response to high dietary fat, particularly in the heart. What is known is largely limited to the measurement of superoxide dismutase, glutathione peroxidase, and catalase (9, 10, 19, 20). Given that there are a number of antioxidant enzymes residing in multiple cellular locations, and catalyzing distinct reactions, it is important that a comprehensive proteomic and temporal assessment of the antioxidant response be performed.
The goal of the experiments described here was to determine the effect of high dietary fat on cardiac antioxidant protein expression. We utilized C57BL/6J mice fed a high fat diet (60% kcal from fat) for varying durations, with mice fed a low fat diet (10% kcal from fat) serving as controls. Mice consuming the high fat chow develop obesity, hyperinsulinemia, hyperglycemia, and hypertension (21). Characterization of the antioxidant response using Western blot analysis is both logistically challenging given the large number of targets and not suited for rigorous quantification of changes in protein content. Therefore, a high throughput quantitative mass spectrometry-based method was developed to analyze a large fraction of the antioxidant network in a single experiment with the use of liquid chromatography-tandem mass spectrometry with selected reaction monitoring (SRM)2 (22). In the SRM experiments, a series of collision-induced dissociation reactions were monitored for a subset of peptides, produced by trypsin digestion, specifically found in each target protein of interest. Key aspects of the SRM approach include high selectivity for the proteins analyzed, direct quantitation of protein concentration, as well as a degree of multiplexing that measures many proteins in a single experiment (22).
Using this quantitative proteomics approach, we found that, within 1 day, high dietary fat invokes a cardiac antioxidant response that is specific to the H2O2-metabolizing enzyme catalase. This work also shows that catalase is present within mouse cardiac mitochondria and that mitochondrial catalase content and activity increase in response to a high fat diet, consistent with elevated mitochondrial H2O2 production. Furthermore, these data show that 1 day of fasting also induces a quantitatively similar and selective increase in catalase expression and activity. These opposing nutritional states have in common increased levels of circulating triglycerides and increased delivery of free fatty acids to the heart (9, 23–25). It has previously been demonstrated that, in each case, this results in enhanced fatty acid oxidation (24–26) presumably by both peroxisomes and mitochondria. Given that increased reliance on fatty acid oxidation promotes peroxisomal and mitochondrial H2O2 production, up-regulation of catalase may represent an important adaptive antioxidant response designed to accommodate changes in energetic substrate availability. We believe that this unique up-regulation of catalase is a means to prevent oxidative damage from toxic levels of H2O2 without perturbing H2O2-mediated signaling within the physiological range.
EXPERIMENTAL PROCEDURES
Animals, Dietary Model, and Sample Preparation
For studies with wild-type animals, 6-week-old male C57BL6/J mice (The Jackson Laboratory) were fed either a high fat diet or a control low fat diet (60% versus 10% total kilocalories from lard) purchased from Research Diets (D12492, D12450B, and D12450J). Previously described NFE2L2−/− (Nrf2) global knock-out mice (27) were obtained from Dr. Scott Plafker at the Oklahoma Medical Research Foundation, and CAT−/− global knock-out mice (28) were acquired from Dr. Eugene Chen at the University of Michigan. PPARA−/− global knock-out mice were purchased from The Jackson Laboratory (stock 008154). At the designated time points, mice were sacrificed by cervical dislocation, and the hearts removed for analysis. Approximately 10 mg of heart tissue was removed from the apex for RNA extraction as described below. The remaining tissue was homogenized in buffer A (buffer A: 10 mm MOPS, 1 mm EDTA, 210 mm mannitol, 70 mm sucrose, pH 7.4). These whole heart lysates were cleared by centrifugation at 550 × g for 5 min and decanted through cheesecloth. Protein concentrations were determined using the BCA Protein Assay Reagent (Thermo Scientific) according to manufacturer's protocol. All experiments were approved by the Oklahoma Medical Research Foundation Institutional Animal Care and Use Committee.
Mass Spectrometry Analysis (22)
Quantitative proteomics was used to determine changes in the expression of the antioxidant proteins in all samples. For these assays, 60-μg amounts of whole heart lysates were mixed with 8 pmol of bovine serum albumin (BSA) as an internal standard and 50 μl of 10% SDS. The samples were heated at 80 °C for 15 min before precipitating the proteins in 80% acetone overnight at −20 °C. The protein pellet was dissolved in 60 μl of sample buffer and a 20-μl aliquot containing 20 μg of protein run 1.5 cm into a 12.5% SDS-polyacrylamide gel. The gel was fixed and stained with GelCode Blue (Pierce). For each sample, the entire 1.5-cm lane was cut out of the gel and divided coarsely. The gel pieces were washed to remove the stain, reduced with DTT, alkylated with iodoacetamide, and digested with 1 μg of trypsin overnight at room temperature. The peptides produced in the digest were extracted with 50% methanol, 10% formic acid in water. The extract was evaporated to dryness and reconstituted in 150 μl of 1% acetic acid in water for analysis.
The samples were analyzed using SRM with a triple quadrupole mass spectrometer (ThermoScientific TSQ Vantage) configured with a splitless capillary column HPLC system (Eksigent). The samples (10-μl aliquots) were injected onto a 10 cm × 75 μm inner diameter column packed with a C18 reversed phase material (Phenomenex, Jupiter C18). The column was eluted at 160 nl/min with a 30-min linear gradient of acetonitrile in 0.1% formic acid. The SRM program was constructed using the program Pinpoint (ThermoScientific) to contain a total of ∼400 collision-induced dissociation reactions for ∼100 peptides. These peptides represented 25 target proteins, two housekeeping proteins (heat shock protein 1 and voltage-dependent anion-selective channel protein 1), and the BSA internal standard. Each peptide was monitored in a 5-min window centered around the known elution time of that peptide.
The data were processed using the program Pinpoint, which aligned the various collision-induced dissociation reactions monitored for each peptide and determined the chromatographic peak areas. The response for each protein was taken as the total response for all peptides monitored. Changes in the relative abundance of the proteins were determined by normalization to the BSA internal standard, with confirmation by normalization to the housekeeping proteins. The amount of catalase in each sample was determined based on its ratio to the BSA internal standard according to the best flyer approach (29).
Western Blot Analysis
Whole heart lysates were immunoblotted with antibodies for catalase (Santa Cruz Biotechnology) as well as the E1 subunit of α-ketoglutarate dehydrogenase (Biosynthesis), which served as a loading control. Protein samples were incubated for 10 min at 70 °C in the presence of 70 mm SDS, 100 mm DTT, and 50 μm MG132, subjected to electrophoresis using a NuPAGE 10% BisTris gel (Invitrogen), and transferred to PVDF (Bio-Rad). Antibody treatments were carried out in 10 mm K2HPO4, 150 mm NaCl, 0.05% Tween 20, and 5% nonfat dry milk. Horseradish peroxidase-conjugated secondary antibodies (Pierce) were visualized with using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) according to manufacturer's protocol.
Catalase Enzyme Activity
Catalase enzyme activity was determined by measuring the rate of H2O2 consumption via absorbance at 240 nm (30). Briefly, activity assays were carried out with 40 μg of protein in 1 ml of 10 mm MOPS, 10 mm H2O2, pH 7.4. The amount of catalase present in each sample, determined by the quantitative proteomics described above, was used to calculate catalase-specific activity as H2O2 consumed per min/pmol of catalase protein.
Mitochondrial H2O2 Production
Cardiac mitochondria were pelleted from whole heart lysates via centrifugation at 10,000 × g for 10 min at 4 °C. The supernatant was discarded and the mitochondrial pellets resuspended in ice-cold buffer A. For the assay, isolated mitochondria (200 μg of protein equivalent) were incubated at room temperature in a 2-ml reaction in buffer B (buffer B: 10 mm MOPS, 210 mm mannitol, 70 mm sucrose, 5 mm K2HPO4, 0.05% BSA, 100 mm malate, 20 μm Amplex red, 4 units of horseradish peroxidase, pH 7.4) with either 25 μm palmitoylcarnitine or 100 μm pyruvate as respiration substrate. The amount of H2O2 produced was measured as the rate of resorufin accumulation determined by fluorescence (excitation 571 nm/emission 582 nm).
Density Gradient Purification of Cardiac Mitochondria
Mitochondria were isolated as described above and further purified by differential centrifugation using a self-generating Percoll gradient (GE Healthcare). Isolated mitochondria were layered onto a 40% isotonic Percoll solution and centrifuged at 66,000 × g for 40 min at 4 °C. The purified mitochondria formed a single distinct layer within the gradient that was collected and diluted in 12 ml of ice-cold buffer A. The purified mitochondria were then separated from any residual Percoll by pelleting at 10,000 × g for 15 min at 4 °C. This final purified mitochondrial pellet was resuspended in ice-cold buffer A. The peroxisome-enriched pellets were obtained from the mitochondrially depleted whole heart lysate by centrifugation at 20,000 × g for 90 min at 4 °C. The resulting peroxisome-enriched pellet was resuspended in ice-cold buffer A.
Immunogold Electron Microscopy
The electron microscopy experiments were carried out at the Oklahoma Medical Research Foundation Imaging Facility using established procedures. Briefly, heart tissue samples were fixed overnight in 4% EM grade paraformaldehyde, dehydrated in ethanol, and embedded in LR White. Blocks were cured by UV light exposure at 4 °C. Sections, 100-nm thick, were placed on square mesh nickel grids. Immunogold labeling was performed serially in droplets of the following solutions: hydration in PBS with 2% BSA for 5 min, blocking buffer for 30 min (PBS, 1% BSA, 3% donkey serum, 0.1% cold water fish skin gelatin), catalase antibody for 1.5 h (PBS, 1% BSA, 3% donkey serum), PBS rinses three times for 5 min each, secondary antibody for 1 h (0.05% BSA), PBS rinses three times for 5 min each, water rinses two times for 1 min each. Immunogold labeled tissues were counter-stained by serial exposure to droplets of Sato's Lead for 5 min, saturated uranyl acetate for 30 min, Sato's Lead for 15 min, and water rinses four times for 1 min each. Images at ×5,000 to 25,000 magnification were collected using a Hitachi H-7600 transmission electron microscope.
Quantitative RT-PCR
The apex of the heart (about 10 mg) was snap-frozen in liquid nitrogen at harvest and stored at −80 °C. RNA was extracted using Tripure (Roche Applied Science) according to the manufacturer's protocol, and concentration and 260/280 nm ratio (≥1.8 for all samples) were measured by absorbance on a NanoDrop 2000 UV-visible spectrophotometer (ThermoScientific). RNA (1 μg) was converted to cDNA, a 20-μl final volume, using QuantiTect reverse transcription kit (Qiagen). Quantitative PCR was performed on a CFX96 thermocycler (Bio-Rad). Reactions consisted of 1 μl of cDNA, 125 nm final concentrations of each primer, and iQ SYBR Green Supermix (Bio-Rad) in a total volume of 20 μl. Technical duplicates were performed for all samples. Relative transcript expression ratios were calculated using CFX Manager software, version 2.1 (Bio-Rad), and statistics were generated with REST2009 software (31). Catalase transcript levels were normalized to three reference genes (GAPDH, SDHA, and HSP90ABI) determined to be stable between dietary conditions using geNorm analysis (32). The respective cDNA was amplified with the following primers: CAT (89.1% efficiency) 181-bp product, forward, 5′-agcgaccagatgaagcagtg-3′, and reverse, 5′-tccgctctctgtcaaagtgtg-3′; ACOX1 (93.0% efficiency) 283-bp product, forward, 5′-taacttcctcactcgaagcca-3′, and reverse, 5′-agttccatgacccatctctgtc-3′; GAPDH (78.5% efficiency) 136-bp product, forward, 5′-atgttccagtatgactccactc-3′, and reverse, 5′-ggcctcaccccatttgatgt-3′; SDHA (85.5% efficiency) 106-bp product, forward,: 5′-ggaacactccaaaaacagacct-3′, and reverse, 5′-ccaccactgggtattgagtagaa-3′; HSP90ABI (90.5% efficiency) 102-bp product, forward, 5′-tcaaacaaggagattttcctccg-3′, and reverse, 5′-actgtccaacttagaagggtc-3′.
RESULTS
Diet-induced Obesity
To investigate the endogenous antioxidant response to high dietary fat and the obesity produced by such a diet, C57BL/6J mice were placed on either a high fat diet or a low fat control diet that gave 60 or 10% of total kilocalories from lard, respectively. The mice were maintained on the respective diets for 2, 20, or 30 weeks to evaluate the effects of diet duration. The animals eating the high fat diet had significantly increased body weights, compared with low fat control diet animals, after as little as 2 weeks (27 ± 3 g versus 23 ± 2 g, p < 0.001, n = 11). Further increases were evident at 20 weeks (45 ± 4 g versus 31 ± 3 g, p < 0.001, n = 14) and 30 weeks (53 ± 6 g versus 33 ± 3 g, p < 0.001, n = 5). This pattern of weight gain is consistent with other reports using this model of diet-induced obesity (33).
Catalase Expression Is Induced by High Fat Feeding
The endogenous antioxidant response in the heart to this high fat diet was measured using a quantitative proteomics approach (22). Analysis of whole heart lysates revealed that, of 16 major antioxidant proteins measured, the H2O2-metabolizing enzyme catalase was the only observed enzyme induced by 30 weeks of high fat feeding (Fig. 1A). The increase in catalase detected by quantitative mass spectrometry was also seen by Western blot with densitometry analysis (Fig. 1, B and C). This unique response was also observed after 2 weeks and 20 weeks on the high fat diet (data not shown). Compared with low fat diet controls, catalase protein content in the heart was significantly increased by 38% at 2 weeks (p < 0.01, n = 11), 48% at 20 weeks (p < 0.001, n = 14),and 82% at 30 weeks (p < 0.001, n = 5) (Fig. 2A). Thus, the catalase induction is specific and contrasts with the notable lack of an increase in the expression of any other assayed enzymes involved in either the consumption of H2O2 (the glutathione peroxidases and peroxiredoxins) or the production of H2O2 (the superoxide dismutases).
FIGURE 1.
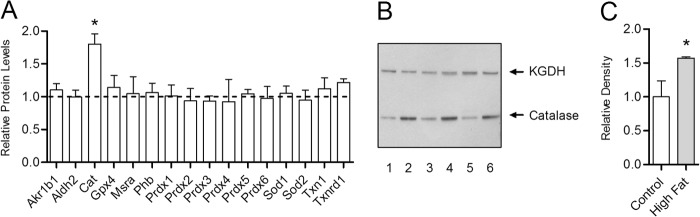
Antioxidant network response to a high fat diet within the heart is specific to catalase. The relative difference in protein expression between high fat and low fat fed control mice was determined for enzymes of the cardiac antioxidant network by quantitative mass spectrometry with selected reaction monitoring. A, antioxidant protein expression in mice fed a high fat diet for 30 weeks relative to low fat fed controls (dashed line). The abbreviations used are aldo-keto reductase family 1 Member B1 (Akr1b1), aldehyde dehydrogenase (Aldh2), catalase (Cat), glutathione peroxidase (Gpx), methionine sulfoxide reductase A (Mrsa), prohibitin (Phb), peroxiredoxin (Prdx), superoxide dismutase (Sod), thioredoxin (Txn), and thioredoxin reductase (Txnrd). Values are presented as the mean ± S.D. (n = 5), where * indicates a significant increase (2-tailed t test) in high fat-fed mice versus low fat-fed controls (p < 0.05). B, Western blot analysis of whole heart lysates from mice fed high fat diet (lanes 2, 4, and 6) or low fat control diet (lanes 1, 3, and 5) for 30 weeks with antibodies for catalase and the E1 subunit of α-ketoglutarate dehydrogenase (KGDH), which served as a loading control. C, densitometry analysis of catalase Western blot band intensities in mice fed high fat diet for 30 weeks (closed bar) relative to low fat-fed controls (open bar).
FIGURE 2.

Catalase protein concentration and enzymatic activity are increased in the hearts of high fat-fed mice. Catalase protein concentration and activity were determined by quantitative mass spectrometry with selected reaction monitoring and measuring the rate of H2O2 clearance, respectively, within whole heart lysates from mice maintained on either high fat or low fat control diet for 2, 20, and 30 weeks. A, catalase protein concentration in whole heart lysates from high fat-fed mice (closed bars) relative to low fat-fed controls (open bars). B and C, total catalase activity (H2O2 consumed per mg of total protein) in wild-type (B) and catalase knock-out mice (C) fed a high fat diet compared with low fat fed controls. D, specific catalase activity (H2O2 consumption per pmol of catalase) in high fat-fed mice compared with low fat-fed controls. Values are presented as the mean ± S.D. (n ≥5), where * indicates a significant increase (2-tailed t test) in high fat diet versus low fat-fed controls within each diet duration (p < 0.01).
Catalase activity was measured to determine whether increased catalase protein content translates into elevated enzyme activity in these hearts. Total catalase activity, measured as H2O2 consumption per mg of total protein, was significantly elevated in mice fed the high fat diet at all three time points, compared with the low fat diet controls (Fig. 2B). The magnitude of the increase in catalase activity was relatively constant at all three time points, i.e. ∼40%. The catalase inhibitors sodium cyanide, sodium azide, and 3-amino-1,2,4-triazole all blocked the H2O2 consumption in the assay (data not shown). Finally, in a parallel 2-week high fat diet experiment, catalase global knock-out mice showed no appreciable level of catalase activity (Fig. 2C). It is therefore clear that the increase in H2O2 metabolism seen in the hearts of animals on the high fat diet is due to catalase activity as opposed to any other enzymatic or nonenzymatic process.
The quantitation of catalase amounts in the quantitative proteomics experiments makes it possible to calculate specific activity. This calculation revealed no change in catalase specific activity at 2 and 20 weeks on the high fat diet (Fig. 2D) with a modest but statistically significant decrease seen at 30 weeks (−19%, p < 0.01, n = 5). It is important to note that, overall, catalase specific activity declined significantly with age on both the high fat diet and the low fat control diet. However, it appears the heart is capable of compensating for the age-dependent decline in specific activity by increasing catalase protein content to maintain a relatively constant level of total catalase activity throughout the 30-week experiment in both diet groups (Fig. 2, A and B).
As shown in Table 1, the low fat diet used for these experiments offsets the reduction of lard content with increased sucrose content. The use of this high sucrose diet as the low fat control raises the possibility that the differences in catalase expression and activity seen in Fig. 2 are due to a down-regulation driven by the high sucrose rather than an up-regulation by the high fat diet. Therefore, to determine whether high dietary sucrose affects catalase activity, total catalase activity was measured in mice fed an alternative low fat control diet where cornstarch, rather than sucrose, is used to offset the reduced fat content. Cornstarch is a complex carbohydrate and as such does not induce the postprandial blood glucose spike observed following sucrose consumption. After 4 weeks on diet, total catalase activity in the cornstarch-enriched low fat diet-fed mice was not significantly different from mice fed the sucrose-enriched low fat control diet (Table 1, p = 0.625, n = 5). These data indicate that the observed changes in catalase protein content and activity are increased due to the high dietary fat.
TABLE 1.
Catalase induction by high fat feeding is due to increased fat content
Two different control diets are used to probe potential effects of the carbohydrate needed to compensate for the calorie content of the high fat diet (HF). The sucrose content in the low fat chow was increased to offset the reduced level of fat. The respective control diets are either low fat/low sucrose (LF/LS) or low fat/high sucrose (LF/HS). Catalase total activity (micromoles of H2O2/min/mg) was determined in mice fed a low fat chow where the fat calories are offset by increased cornstarch (LF/LS). Values are presented as the mean ± S.D. (n = 5).
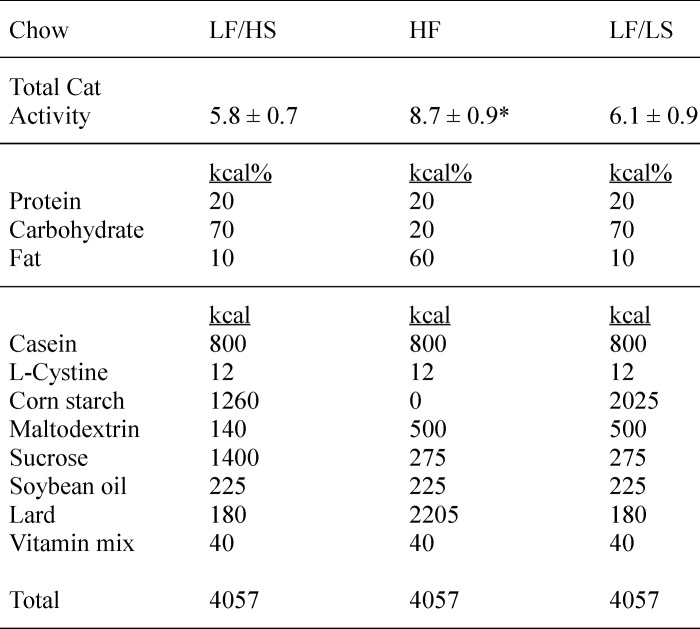
* This indicates a significant increase (2-tailed t test) in high fat-fed mice compared with mice fed either low fat control diet (p < 0.01).
Catalase Induction by the High Fat Diet Occurs Prior to Onset of Obesity
The persistent nature and relatively constant magnitude of the increased total catalase activity seen as early as 2 weeks on the high fat diet prompted us to determine the exact onset of this response and, in particular, whether this response was related to the weight gain or the high fat diet itself. Fig. 3 shows that catalase protein content is significantly increased, relative to controls, in mice fed a high fat diet for 1 day. Total catalase activity was also increased following 1 day of the high fat feeding (Fig. 3B) with no significant change in catalase specific activity (data not shown). The magnitude by which total catalase activity is increased in mice fed a high fat diet throughout the 1- to 3-day experiments, ∼40%, was similar to that observed for the 2- to 30-week experiments described above (Fig. 2). These observations strongly suggest that catalase induction is an important component of an immediate antioxidant response to a high fat diet and that elevated dietary fat increases H2O2 production within heart tissue.
FIGURE 3.
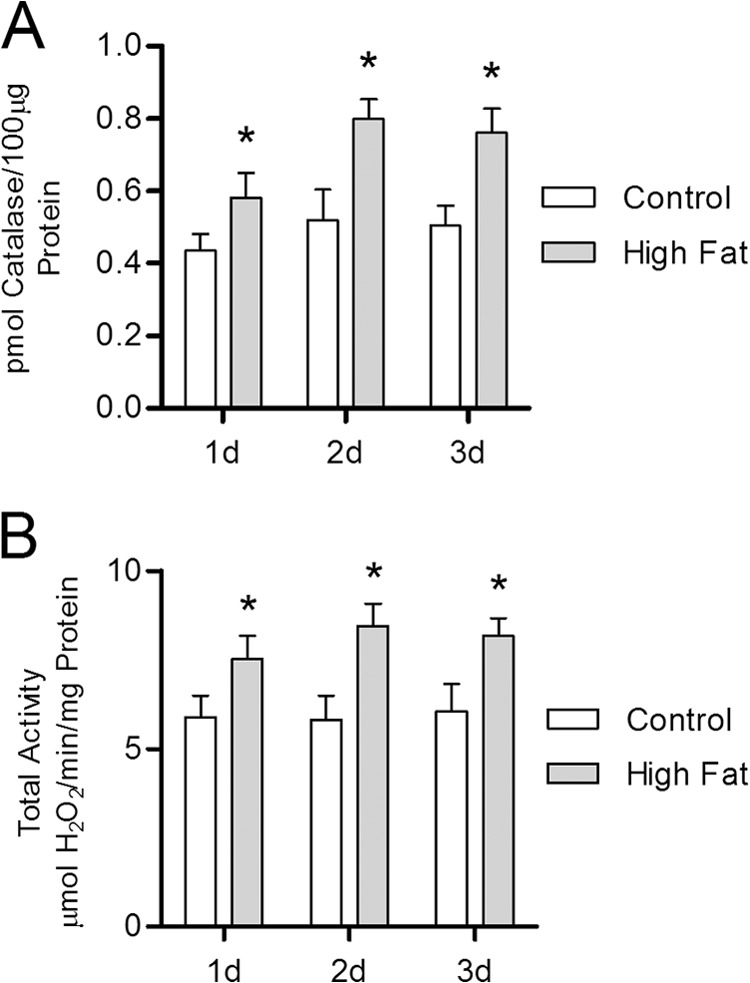
Catalase induction is immediately responsive to high fat feeding. Catalase protein concentration and activity were determined by quantitative mass spectrometry and measuring the rate of H2O2 clearance via absorbance spectrophotometry, respectively, within whole heart lysates from mice maintained on either high fat or low fat control diet for 1–3 days (d). A, catalase protein concentration in whole heart lysates from high fat-fed mice (closed bars) relative to low fat-fed controls (open bars). B, total catalase activity (H2O2 consumed per mg of total protein) in high fat-fed mice compared with low fat-fed controls. Values are presented as the mean ± S.D. (n ≥5), where * indicates a significant increase (2-tailed t test) in high fat-fed mice versus low fat-fed controls within each diet duration (p < 0.01).
Both Peroxisomes and Mitochondria Are Potential Sources of Increased H2O2 Production during High Fat Feeding
The organelles chiefly responsible for fatty acid oxidation are the peroxisomes and mitochondria. Peroxisomal fatty acid oxidation is largely restricted to very long chain fatty acids and is a potential contributor to increased H2O2 production within the heart in response to a high fat diet. The enzyme ACOX1 performs the first step in this process in a reaction that directly produces H2O2 during the removal of the α- and β-carbons from the acyl-CoA ester. Mice consuming high fat diet for periods of 1–3 days all displayed increased ACOX1 protein amounts in the heart (+31, +30, and +33%, respectively, p < 0.05, n = 5) compared with low fat fed controls (Fig. 4A). These data implicate ACOX1-mediated fatty acid oxidation as a potential source of increased H2O2 production induced by high dietary fat.
FIGURE 4.

Both peroxisomes and mitochondria are potential sources of increased H2O2 production during high fat feeding. Protein concentrations of ACOX1 and ACADVL were determined by mass spectrometry analysis of whole heart lysates from mice maintained on either a high fat or a low fat control diet. Rate of H2O2 production was determined using isolated cardiac mitochondria. A and B, ACOX1 and ACADVL protein concentration, as determined by mass spectrometry, in mice maintained on either a high fat (closed bars) or a low fat control (open bars) diet for 1–3 days (d). C, representative trace of H2O2 production (see under “Experimental Procedures”) from mitochondria, isolated from low fat-fed mice, respiring on either palmitoylcarnitine or pyruvate. D, mean rate of H2O2 production, during pyruvate- (Pry) (open bar) or palmitoylcarnitine (PC) (closed bar)-supported respiration, from mitochondria isolated from mice maintained on a low fat diet for 2 weeks. Values are presented as the mean ± S.D. (n ≥5), where * indicates a significant increase (2-tailed t test) in high fat-fed mice versus low fat-fed controls within each diet duration (p < 0.01).
Mitochondria also generate H2O2 as a by-product during energetic substrate oxidation to produce cellular energy. In contrast to ACOX1, the enzyme acyl-coenzyme A dehydrogenase very long chain (ACADVL), which catalyzes the first step in mitochondrial oxidation of very long chain fatty acids, does not directly produce H2O2 and is not induced by 1 week of high fat feeding (Fig. 4B). Although there are no changes in ACADVL protein levels, the rate of H2O2 production from rodent skeletal muscle mitochondria is known to be influenced by the energetic substrate used to support respiration (34, 35). Respiring cardiac mitochondria isolated from control mice produce 23% more H2O2 (p < 0.01, n = 6) using palmitoylcarnitine as substrate versus pyruvate (Fig. 4, C and D). The elevation in H2O2 production is likely due to increased entry of electrons into the ubiquinone pool of the electron transport chain during fatty acid oxidation (15), thereby increasing the half-life of reduced components in complex I and producing superoxide anion that is rapidly converted to H2O2 by superoxide dismutase. High levels of dietary fat also depress glycolysis in the heart through the Randle cycle leading to increased dependence on fatty acid oxidation for energy production (36). As such, increased fatty acid oxidation driven by a high fat diet likely enhances cardiac mitochondrial H2O2 production.
Mitochondrial Catalase Content and Activity Are Elevated by High Dietary Fat
Despite possession of a peroxisomal targeting sequence, catalase has been shown to reside within rat cardiac mitochondria (37). Given that both mitochondria and peroxisomes produce H2O2 during fatty acid oxidation, it was important to determine the subcellular site of high fat diet-induced catalase expression to gain insight into the potential physiological significance. Cardiac mitochondrial and peroxisomal fractions were purified from mice fed either a high fat or low fat control diet for 3 days. Western blot analysis was used to assess the purity of each fraction and showed both that the subcellular fractions were free of cytosolic proteins and that there was no cross-contamination between the mitochondrial and peroxisomal compartments (Fig. 5A). Quantitative mass spectrometric analyses and enzyme activity assays were then used to measure the amount of catalase in each compartment. These quantitative assays showed that peroxisomal catalase content and total activity did not change in response to high dietary fat (Fig. 5, B and C). However, mitochondria isolated from high fat-fed mice had both increased catalase content (+49%, p < 0.05, n = 5) and total activity (+50%, p < 0.05, n = 5) compared with low fat-fed controls (Fig. 5, D and E). The presence and increase in catalase protein within the mitochondria of high fat-fed versus low fat-fed control mice was directly demonstrated by electron microscopy with immunogold staining (Fig. 5F). Although peroxisomes have a 3-fold greater catalase protein and total activity per μg of protein than mitochondria, it is important to note that peroxisomes are relatively scarce in the heart (38). Mitochondria, however, are highly abundant due to the large energy demand of the heart and display an increase in catalase content that is reflective of the increase observed in whole heart lysates. Together, these data suggest that the bulk of increased catalase content occurs within the mitochondrial fraction that is consistent with enhanced mitochondrial H2O2 production during high fat feeding.
FIGURE 5.

High fat feeding increases catalase content and activity within cardiac mitochondria. Catalase content and activity were determined in peroxisomes, and mitochondria were isolated from mice maintained on a high fat or low fat control diet for 3 days. A, Western blot analysis of total (Tot), mitochondrial (Mito), and peroxisome-enriched (Perox) fractions isolated from animals fed either a high fat diet (HF) or a low fat control diet (C). The relative protein levels of SOD1 (cytosolic), SOD2 (mitochondrial), and ABCD3 (peroxisomal) were assayed to determine purity of the mitochondrial fraction. B and C, analysis of the peroxisomal fraction to determine catalase protein via mass spectrometry (B) as well as catalase total activity (C) in mice maintained on high fat diet (closed bars) compared with low fat-fed controls (open bars). D and E, analysis of purified mitochondria to determine catalase protein via mass spectrometry (D) as well as catalase total activity (E) in mice maintained on high fat diet compared with low fat-fed controls. F, representative electron micrograph images of ultrathin sections of mitochondria (M) within heart tissue (at ×5,000 and 25,000 magnification). Catalase was visualized via immunogold labeling (arrows). The left-most section was treated with secondary antibody only to illustrate the lack of nonspecific binding by the catalase primary antibody. Values are presented as the mean ± S.D. (n = 5), where * indicates a significant increase (2-tailed t test) in high fat-fed mice compared with low fat-fed controls (p < 0.05).
High Fat Diet Increases Catalase mRNA Levels
Catalase mRNA levels were determined in mice fed a high fat diet relative to low fat-fed controls to determine whether the catalase response to a high fat diet results from increased transcription. Catalase mRNA content was significantly elevated following 24 h on the high fat diet, compared with controls, and remained elevated throughout 30 weeks on diet (Fig. 6A). These data demonstrate that the catalase induction triggered by high fat feeding is at least partly regulated at the transcription level.
FIGURE 6.

High fat diet increases catalase mRNA in NFE2L2−/− and PPARA−/− knock-out mice. The level of catalase mRNA was determined using quantitative RT-PCR analysis of RNA isolated from the heart tissue of wild-type, NFE2L2−/−, and PPARA−/− mice. A, catalase mRNA levels in wild-type mice maintained on high fat diet (closed bars) from 1 day to 30 weeks relative to low fat-fed controls (open bars). B–D, relative catalase mRNA levels (B), protein concentration (C), and total activity (D) were determined in wild-type, NFE2L2−/−, and PPARA−/− mice fed either a high fat or low fat control diet for 1 week. E, ACOX1 mRNA levels in wild-type or PPARA−/− knock-out mice maintained on high fat diet from 1 week relative to low fat fed controls. Values are presented as the mean ± S.E. (catalase mRNA, Bio-Rad CFX manager software version 2.1) or mean ± S.D. (catalase concentration and total activity, 2-tailed t test) where * indicates a significant difference between high fat- and low fat-fed control mice within each mouse strain, and † indicates a significant difference between wild-type and knock-out mouse strain within each dietary condition (p < 0.05, n ≥4).
We therefore sought to identify transcription factors that contribute to catalase induction. Nrf2 is a stress-response transcription factor implicated in the regulation of catalase in response to oxidative stress (39). To assess the role of Nrf2 in the catalase response to high dietary fat, wild-type and NFE2L2−/− global knock-out mice were analyzed after 1 week on the high fat diet. NFE2L2−/− mice fed the high fat diet had significantly increased catalase mRNA, catalase protein concentration, and total catalase activity (Fig. 6, B–D, p < 0.05, n ≥4) relative to controls. No differences were seen in the total catalase activity between NFE2L2−/− and wild-type mice in either dietary condition. This lack of an effect shows that Nrf2 does not mediate the induction of catalase in response to high dietary fat.
Pparα is a lipid-sensitive nuclear receptor that acts as a transcription factor to drive the expression of several target genes, including ACOX1, in response to conditions that increase lipid availability. In light of increased ACOX1 mRNA levels in the hearts of mice consuming a high fat diet (Fig. 6E), the role of Pparα in the induction of catalase by high fat feeding was assessed using PPARA−/− global knock-out mice. The loss of Pparα likely leads to a general reduction in the overall level of fatty acid oxidation based on the significant decrease in ACOX1 mRNA in low fat fed PPARA−/− mice (p < 0.05, n = 5) compared with wild-type (Fig. 6E). Nevertheless, PPARA−/− mice fed a high fat diet for 1 week displayed a significant increase in catalase mRNA, protein concentration, and total activity (p < 0.05, n = 5) relative to low fat fed controls (Fig. 6, B–D). Furthermore, the percent increase in catalase total activity induced by high dietary fat in PPARA−/− mice is nearly identical to that observed in NFE2L2−/− and wild-type mice (+47, +56, and +48%, respectively). However, PPARA−/− mice possess significantly lower levels of catalase protein and total activity compared with wild-type mice in both high fat fed (−40 and −32%, respectively) and control animals (−41 and −29%, respectively). These data indicate that although Pparα may contribute to the basal levels of catalase content and activity, it is not required for the induction of catalase by high dietary fat.
Fasting Uniquely Up-regulates Catalase
The results linking a high fat diet with increased catalase expression and activity, an increase in ACOX1 expression, and the increased H2O2 production by mitochondria respiring on palmitoylcarnitine rather than pyruvate strongly implicate high lipid availability as a driving force. If increased lipid availability drives catalase induction then alternative metabolic states that increase circulating lipid levels should also induce catalase expression. One such metabolic state is fasting. In a fasted state, the heart becomes more reliant on fatty acid oxidation for energy production due to increased levels of circulating glucagon and lipids (24). Experiments using a 1-day fast gave a unique increase in catalase protein concentration (+44%, p < 0.001, n = 5) compared with control animals fed a low fat diet (Fig. 7A). Total catalase activity and mRNA levels were also elevated in the fasted mice (p < 0.05, n = 5) compared with the fed controls (Fig. 7, B and C) with no change in specific activity (data not shown). Additionally, this fasting state produced an increase, relative to low fat fed controls, in heart ACOX1 protein concentration (Fig. 7D, +50%, p < 0.05, n = 5). The magnitude of this increase was significantly greater than the increase seen in mice fed a high fat diet for the same 1 day (+31%, p < 0.05, n = 5). Together, the congruence of the high fat diet and fasting data is consistent with lipid availability triggering a specific and selective induction of catalase.
FIGURE 7.
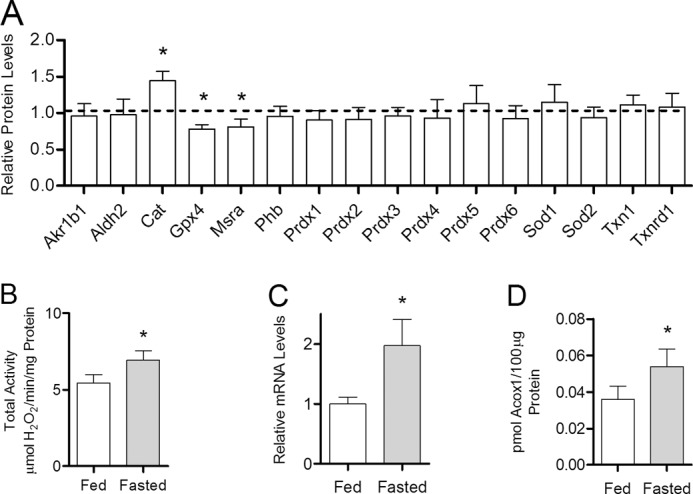
Catalase is uniquely up-regulated by fasting. Cardiac catalase protein concentration, activity, and mRNA levels were determined in mice either fed low fat control diet or fasted for 1 day. A, quantitative mass spectrometric analysis of antioxidant protein expression in mice fasted for 1 day relative to low fat-fed controls (dashed line). The abbreviations used are: aldo-keto reductase family 1 member B1 (Akr1b1), aldehyde dehydrogenase (Aldh2), catalase (Cat), glutathione peroxidase (Gpx), methionine sulfoxide reductase A (Mrsa), prohibitin (Phb), peroxiredoxin (Prdx), superoxide dismutase (Sod), thioredoxin (Txn), thioredoxin reductase (Txnrd). B, total catalase activity (H2O2 consumed per mg of total protein) in low fat-fed control mice (open bar) compared with mice fasted for 1 day (closed bar). C, catalase mRNA levels in mice maintained on low fat control diet or fasted for 1 day. D, ACOX1 protein concentration determined by quantitative mass spectrometry in mice fed low fat control diet or fasted for 1 day. Values are presented as the mean ± S.E. (catalase mRNA, Bio-Rad CFX manager software version 2.1) or mean ± S.D. (protein concentration and activity, 2-tailed t test), where * indicates a significant difference between low fat-fed and fasted mice (p < 0.05, n = 5).
DISCUSSION
In this study, we measured the effect of a high fat diet on the endogenous antioxidant enzyme system in the heart. Using a novel quantitative proteomics method, based on LC-tandem MS with SRM, we have simultaneously quantified changes in 16 members of the antioxidant enzyme network in mice fed a high fat diet. We found that catalase was selectively induced within 24 h, and catalase total activity persisted at a relatively constant magnitude throughout 30 weeks of high fat feeding. Moreover, the same selective and rapid induction of catalase was observed in mice fasted for 1 day. Together, these data suggest that both high fat feeding and fasting increase H2O2 production within the heart, which is countered by the specific induction of catalase.
Hydrogen peroxide can damage biological systems by a number of mechanisms. For example, Fenton chemistry converts H2O2 into the highly reactive hydroxyl radical that is capable of damaging proteins, lipids, and DNA. H2O2 is also a substrate for the enzyme myeloperoxidase, which uses H2O2 to generate other damaging molecules such a hypochlorous acid (40). It is also clear that H2O2 functions as a signaling molecule through transient oxidation of proteins such as the protein-tyrosine phosphatases PTP1B and PTEN, which negatively regulate insulin sensitivity (41–43). Low concentrations of H2O2 can also have a proliferative effect, whereas high concentrations are cytotoxic (44). As such, H2O2-consuming enzymes such as the glutathione peroxidases, peroxiredoxins, and catalase should be readily and selectively regulated to limit toxic levels of H2O2 accumulation while permitting H2O2-mediated cell signaling. The low binding affinity of catalase for H2O2, which is well below the micromolar binding affinity of glutathione peroxidases and peroxiredoxins, may underlie the specificity of catalase induction by high fat diet (45–47). This difference makes catalase ideal for consuming toxic levels of H2O2 while allowing the glutathione peroxidases and peroxiredoxins to manipulate the lower subtoxic amounts of H2O2 needed for signaling. Proper coordination between these enzymes is critical given that subtle changes in cellular H2O2 concentration can exert both positive and negative effects. For example, treatment of hepatocytes with 1 μm H2O2 has been shown to increase insulin sensitivity, whereas treatment with 5–10 μm H2O2 promotes insulin resistance via inhibition of PTP1B and activation of the stress-responsive protein c-Jun N-terminal kinase (48). Based on low binding affinity, we hypothesize that the main role of catalase in the maintenance of intracellular H2O2 concentration is largely to protect against H2O2-induced oxidative damage.
Overexpression of catalase in transgenic mice confers protection against conditions that increase fatty acid oxidation. Whole heart overexpression of catalase reduces malondialdehyde protein modification and preserves fractal shortening in models of type 1 and 2 diabetes (49). Of particular importance to this study, mice that overexpress mitochondrial targeted catalase (mCat) are protected from high fat diet-induced insulin resistance in skeletal muscle and displayed decreased mitochondrial H2O2 production (14). The data from transgenic mice coupled with H2O2 concentration-dependent regulation of insulin sensitivity in hepatocytes support H2O2 as a key regulator of insulin sensitivity. Why then do mice capable of selectively increasing catalase expression and activity in response to high dietary fat still become insulin-resistant (21)? One explanation is that the level of catalase activity in mitochondria from mCat mice is 50-fold higher than wild type (50) compared with the ∼50% increase we observe for the endogenous catalase response. Although the binding affinity of catalase is low, the Vmax is near the rate of diffusion. Thus, massive overexpression of catalase could eliminate the H2O2 needed to drive skeletal muscle insulin resistance in response to high dietary fat.
The data from mCat mice suggest that increased mitochondrial H2O2 production induced by high fat feeding promotes insulin resistance. High dietary fat is known to increase fatty acid oxidation for energy production via the Randle cycle, and we and others have shown that mitochondria produce more H2O2 when oxidizing fatty acids versus pyruvate derived from glycolysis (35). We demonstrate here for the first time that catalase is present and is increased within mouse cardiac mitochondria following exposure to a high fat diet. The increase in mitochondrial catalase (∼50%) is consistent with a response designed to protect mitochondria rather than prevent insulin resistance as observed in mCat mice. Very long chain fatty acids present in the high fat chow are oxidized within peroxisomes where β-oxidation is initiated by ACOX1 activity that directly produces H2O2. The observed increase in ACOX1 expression within 1 day suggests that high dietary fat enhances peroxisomal fatty acid oxidation. The excess H2O2 produced by peroxisomes is likely to be consumed by the high concentration of catalase present within peroxisomes. However, peroxisomal β-oxidation is incomplete, and the shortened fatty acids are able to be exported for uptake and complete oxidation by the mitochondria in a manner similar to pristanic acid (51), potentially enhancing mitochondrial H2O2 production. Together, these data are consistent with both peroxisomes and mitochondria cooperatively affecting the cellular redox environment via increased fatty acid oxidation during high fat feeding.
A question that emerges from our findings is what serves as the stimulus for the induction of catalase in response to high dietary fat, increased fatty acid delivery, or increased H2O2 production? Two candidate transcription factors that are sensitive to either oxidative stress (Nrf2) or lipid levels (Pparα) were explored in our work. Nrf2-dependent regulation of catalase has been observed in mouse cardiomyocytes challenged with 3H-1,2-dithiole-3-thione, which induces various antioxidants and phase 2 enzymes, including catalase (39). Other work from our laboratory has found that Nrf2 siRNA reduces the increase in catalase expression seen in macrophages treated with oxidized low density lipoprotein (22). Similarly, a functional Ppar-response element has been found within the rat catalase promoter (52). However, no differences were seen in the response of increased catalase protein expression or activity induced by high dietary fat between wild-type, NFE2L2−/−, and PPARA−/− mice clearly arguing against the involvement of these transcription factors. The overall reduction in catalase concentration seen in PPARA−/− global knock-out mice may arise from a general reduction in fatty acid oxidation, which suggests that although PPARA−/− does not regulate the catalase response to high dietary fat the amount of catalase present is linked to the level of fatty acid oxidation.
The requirement of c-Jun N-terminal kinase (JNK) for development of insulin resistance in response to high dietary fat may provide the first clue as to the regulatory mechanism responsible for catalase induction. JNK1 knock-out mice are resistant to high fat diet-induced increases in body weight, blood glucose levels, and insulin resistance (53). Low micromolar H2O2 treatment of mouse embryonic fibroblasts activates the small GTPase Ral leading to JNK-dependent phosphorylation of transcription factor forkhead box protein 4 (FOXO4) thereby promoting nuclear translocation and up-regulation of target genes (54). FOXO3-dependent catalase induction has been demonstrated in response to oxidative stress-mediated hypertrophy of rat cardiomyocytes (55). Additionally, treatment of rat cardiomyocytes with insulin leads to increased phosphorylation, ubiquitination, and eventual down-regulation of FOXO1 (56). Altogether, these data suggest that JNK activation by H2O2 promotes insulin resistance and FOXO-mediated transactivation of antioxidant genes, including catalase. Therefore, H2O2 production from peroxisomes or mitochondria during high fat feeding may serve both to induce catalase expression while promoting insulin resistance through a JNK/FOXO-dependent mechanism. Although this idea needs to be tested, any potential regulation of catalase by FOXO family members is potentially complex given that these transcription factors, like Nrf2 and Pparα, typically up-regulate gene sets rather than individual antioxidant enzymes. The existence of multiple FOXO isoforms and a variety of co-transactivators with which they interact may provide the specificity required for the unique induction of catalase by high dietary fat.
In summary, we have characterized the cardiac antioxidant response to a high fat diet and identified catalase as an important component of the immediate antioxidant response. Furthermore, we found that fasting elicited a similar catalase-specific response. Ad libitum consumption of high dietary fat and fasting are fundamentally different nutritional states; however, each is characterized by elevated plasma levels and oxidation of fatty acids (9, 23–26). Thus, our findings are consistent with elevated circulating fatty acid levels promoting H2O2 production from organelles that oxidize fatty acids. The coupling of fatty acid oxidation with the production of H2O2, a signaling molecule known to influence insulin sensitivity and thereby energetic fuel utilization, place both the mitochondria and peroxisomes in an ideal position to sense and communicate dietary composition. The rapid induction of catalase suggests that this enzyme is critical for the heart's ability to accommodate increased fatty acid oxidation for energy production, during either high fat feeding or fasting, by preventing oxidative damage while not perturbing H2O2-mediated signaling. From this perspective, short term metabolic adaptations such as insulin resistance may be an entirely appropriate response to a high fat diet that favors utilization of lipid rather than carbohydrate to ensure complete clearance of potentially toxic lipid intermediates. Although these changes may reflect an evolutionarily conserved adaptation to cycles of fasting and feeding, chronic consumption of excess calories leads to obesity and a metabolic syndrome characterized by chronic dyslipidemia. In this scenario, the well intended response needed for short term challenges is transformed into the unwanted long term metabolic abnormality of chronically elevated blood glucose levels that drive downstream complications such as diabetes and cardiovascular disease.
Acknowledgments
We thank Melinda West for work with the care of the animals used in this study and Caroline Kinter for work on the quantitative mass spectrometry analyses. We also thank the Imaging Core, supported by the Oklahoma Medical Research Foundation, for their services in generating the immunogold labeling data. We thank Dr. Eugene Chen (University of Michigan) for providing the catalase knock-out mice.
This work was supported, in whole or in part, by National Institutes of Health Grants AG016339 (to L. I. S.) and GM092900 from NIGMS (to S. M. P.). This work was also supported by the Oklahoma Medical Research Foundation.
- SRM
- selected reaction monitoring
- ACADVL
- acyl-coenzyme A dehydrogenase very long chain
- ACOX1
- acyl-coenzyme A oxidase 1 palmitoyl
- FOXO
- forkhead box protein
- mCat
- mitochondrial specific overexpression of catalase
- Ppar
- peroxisome proliferator-activated receptor
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Nrf2
- nuclear factor E2-related factor 2.
REFERENCES
- 1. Fox C. S., Coady S., Sorlie P. D., D'Agostino R. B., Sr., Pencina M. J., Vasan R. S., Meigs J. B., Levy D., Savage P. J. (2007) Increasing cardiovascular disease burden due to diabetes mellitus. The Framingham Heart Study. Circulation 115, 1544–1550 [DOI] [PubMed] [Google Scholar]
- 2. Boudina S., Abel E. D. (2007) Diabetic cardiomyopathy revisited. Circulation 115, 3213–3223 [DOI] [PubMed] [Google Scholar]
- 3. Kenchaiah S., Evans J. C., Levy D., Wilson P. W., Benjamin E. J., Larson M. G., Kannel W. B., Vasan R. S. (2002) Obesity and the risk of heart failure. N. Engl. J. Med. 347, 305–313 [DOI] [PubMed] [Google Scholar]
- 4. Hubert H. B., Feinleib M., McNamara P. M., Castelli W. P. (1983) Obesity as an independent risk factor for cardiovascular disease. A 26-year follow-up of participants in the Framingham Heart Study. Circulation 67, 968–977 [DOI] [PubMed] [Google Scholar]
- 5. Abel E. D., Litwin S. E., Sweeney G. (2008) Cardiac remodeling in obesity. Physiol. Rev. 88, 389–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsutsui H., Ide T., Kinugawa S. (2006) Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid. Redox Signal. 8, 1737–1744 [DOI] [PubMed] [Google Scholar]
- 7. Penckofer S., Schwertz D., Florczak K. (2002) Oxidative stress and cardiovascular disease in type 2 diabetes. The role of antioxidants and pro-oxidants. J. Cardiovasc. Nurs. 16, 68–85 [DOI] [PubMed] [Google Scholar]
- 8. Wang H.-T., Liu C.-F., Tsai T.-H., Chen Y.-L., Chang H.-W., Tsai C.-Y., Leu S., Zhen Y.-Y., Chai H.-T., Chung S.-Y., Chua S., Yen C.-H., Yip H.-K. (2012) Effect of obesity reduction on preservation of heart function and attenuation of left ventricular remodeling, oxidative stress and inflammation in obese mice. J. Transl. Med. 10, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bhandari U., Kumar V., Khanna N., Panda B. P. (2011) The effect of high fat diet-induced obesity on cardiovascular toxicity in Wistar albino rats. Hum. Exp. Toxicol. 30, 1313–1321 [DOI] [PubMed] [Google Scholar]
- 10. Noeman S. A., Hamooda H. E., Baalash A. A. (2011) Biochemical study of oxidative stress markers in the liver, kidney, and heart of high fat diet-induced obesity in rats. Diabetol. Metab. Syndr. 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fang C. X., Dong F., Thomas D. P., Ma H., He L., Ren J. (2008) Hypertrophic cardiomyopathy in high fat diet-induced obesity. Role of suppression of forkhead transcription factor and atrophy gene transcription. Am. J. Physiol. Heart Circ. Physiol. 295, H1206–H1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fernández-Sánchez A., Madrigal-Santillán E., Bautista M., Esquivel-Soto J., Morales-González A., Esquivel-Chirino C., Durante-Montiel I., Sánchez-Rivera G., Valadez-Vega C., Morales-González J. A. (2011) Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 12, 3117–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sonta T., Inoguchi T., Tsubouchi H., Sekiguchi N., Kobayashi K., Matsumoto S., Utsumi H., Nawata H. (2004) Evidence for contribution of vascular NAD(P)H oxidase to increased oxidative stress in animal models of diabetes and obesity. Free Radic. Biol. Med. 37, 115–123 [DOI] [PubMed] [Google Scholar]
- 14. Anderson E. J., Lustig M. E., Boyle K. E., Woodlief T. L., Kane D. A., Lin C. T., Price J. W., 3rd, Kang L., Rabinovitch P. S., Szeto H. H., Houmard J. A., Cortright R. N., Wasserman D. H., Neufer P. D. (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 119, 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fisher-Wellman K. H., Neufer P. D. (2012) Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 23, 142–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McLain A. L., Szweda P. A., Szweda L. I. (2011) α-Ketoglutarate dehydrogenase. A mitochondrial redox sensor. Free Radic. Res. 45, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Veal E. A., Day A. M., Morgan B. A. (2007) Hydrogen peroxide sensing and signaling. Mol. Cell 26, 1–14 [DOI] [PubMed] [Google Scholar]
- 18. Rhee S. G. (2006) Cell signaling. H2O2, a necessary evil for cell signaling. Science 312, 1882–1883 [DOI] [PubMed] [Google Scholar]
- 19. Anderson E. J., Thayne K., Harris M., Carraway K., Shaikh S. R. (2012) Aldehyde stress and up-regulation of Nrf2-mediated antioxidant systems accompany functional adaptations in cardiac mitochondria from mice fed n-3 polyunsaturated fatty acids. Biochem. J. 441, 359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vincent H. K., Powers S. K., Dirks A. J., Scarpace P. J. (2001) Mechanism for obesity-induced increase in myocardial lipid peroxidation. Int. J. Obes. Relat. Metab. Disord. 25, 378–388 [DOI] [PubMed] [Google Scholar]
- 21. Surwit R. S., Kuhn C. M., Cochrane C., McCubbin J. A., Feinglos M. N. (1988) Diet-induced type II diabetes in C57BL/6J mice. Diabetes 37, 1163–1167 [DOI] [PubMed] [Google Scholar]
- 22. Kinter C. S., Lundie J. M., Patel H., Rindler P. M., Szweda L. I., Kinter M. (2012) A quantitative proteomic profile of the Nrf2-mediated antioxidant response of macrophages to oxidized LDL determined by multiplexed selected reaction monitoring. PLoS ONE 7, e50016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Orfali K. A., Fryer L. G., Holness M. J., Sugden M. C. (1993) Long-term regulation of pyruvate dehydrogenase kinase by high fat feeding. Experiments in vivo and in cultured cardiomyocytes. FEBS Lett. 336, 501–505 [DOI] [PubMed] [Google Scholar]
- 24. Horton T. J., Hill J. O. (2001) Prolonged fasting significantly changes nutrient oxidation and glucose tolerance after a normal mixed meal. J. Appl. Physiol. 90, 155–163 [DOI] [PubMed] [Google Scholar]
- 25. Lopaschuk G. D., Ussher J. R., Folmes C. D., Jaswal J. S., Stanley W. C. (2010) Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90, 207–258 [DOI] [PubMed] [Google Scholar]
- 26. Wright J. J., Kim J., Buchanan J., Boudina S., Sena S., Bakirtzi K., Ilkun O., Theobald H. A., Cooksey R. C., Kandror K. V., Abel E. D. (2009) Mechanisms for increased myocardial fatty acid utilization following short-term high fat feeding. Cardiovasc. Res. 82, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chan K., Lu R., Chang J. C., Kan Y. W. (1996) NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. U.S.A. 93, 13943–13948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ho Y. S., Xiong Y., Ma W., Spector A., Ho D. S. (2004) Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem. 279, 32804–32812 [DOI] [PubMed] [Google Scholar]
- 29. Ludwig C., Claassen M., Schmidt A., Aebersold R. (November 20, 2012) Mol. Cell. Proteomics 10.1074/mcp.M111.013987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aebi H. (1984) Catalase in vitro. Methods Enzymol. 105, 121–126 [DOI] [PubMed] [Google Scholar]
- 31. Pfaffl M. W., Horgan G. W., Dempfle L. (2002) Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Griffin T. M., Huebner J. L., Kraus V. B., Yan Z., Guilak F. (2012) Induction of osteoarthritis and metabolic inflammation by a very high fat diet in mice: effects of short-term exercise. Arthritis Rheum. 64, 443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seifert E. L., Estey C., Xuan J. Y., Harper M. E. (2010) Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 285, 5748–5758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anderson E. J., Yamazaki H., Neufer P. D. (2007) Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J. Biol. Chem. 282, 31257–31266 [DOI] [PubMed] [Google Scholar]
- 36. Randle P. J., Newsholme E. A., Garland P. B. (1964) Regulation of glucose uptake by muscle. 8. Effects of fatty acids, ketone bodies and pyruvate, and of alloxan-diabetes and starvation, on the uptake and metabolic fate of glucose in rat heart and diaphragm muscles. Biochem. J. 93, 652–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Radi R., Turrens J. F., Chang L. Y., Bush K. M., Crapo J. D., Freeman B. A. (1991) Detection of catalase in rat heart mitochondria. J. Biol. Chem. 266, 22028–22034 [PubMed] [Google Scholar]
- 38. De Craemer D., Vamecq J., Roels F., Vallée L., Pauwels M., Van den Branden C. (1994) Peroxisomes in liver, heart, and kidney of mice fed a commercial fish oil preparation. Original data and review on peroxisomal changes induced by high fat diets. J. Lipid Res. 35, 1241–1250 [PubMed] [Google Scholar]
- 39. Zhu H., Jia Z., Misra B. R., Zhang L., Cao Z., Yamamoto M., Trush M. A., Misra H. P., Li Y. (2008) Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovasc. Toxicol. 8, 71–85 [DOI] [PubMed] [Google Scholar]
- 40. Hazen S. L., Heinecke J. W. (1997) 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Invest. 99, 2075–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Salmeen A., Andersen J. N., Myers M. P., Meng T. C., Hinks J. A., Tonks N. K., Barford D. (2003) Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769–773 [DOI] [PubMed] [Google Scholar]
- 42. Wijesekara N., Konrad D., Eweida M., Jefferies C., Liadis N., Giacca A., Crackower M., Suzuki A., Mak T. W., Kahn C. R., Klip A., Woo M. (2005) Muscle-specific Pten deletion protects against insulin resistance and diabetes. Mol. Cell. Biol. 25, 1135–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Elchebly M., Payette P., Michaliszyn E., Cromlish W., Collins S., Loy A. L., Normandin D., Cheng A., Himms-Hagen J., Chan C. C., Ramachandran C., Gresser M. J., Tremblay M. L., Kennedy B. P. (1999) Increased insulin sensitivity and obesity resistance in mice lacking the protein-tyrosine phosphatase-1B gene. Science 283, 1544–1548 [DOI] [PubMed] [Google Scholar]
- 44. Loo A. E., Ho R., Halliwell B. (2011) Mechanism of hydrogen peroxide-induced keratinocyte migration in a scratch-wound model. Free Radic. Biol. Med. 51, 884–892 [DOI] [PubMed] [Google Scholar]
- 45. Nicholls P., Fita I., Loewen P. (2001) Enzymology and structure of catalases. Adv. Inorg. Chem. 51, 51–106 [Google Scholar]
- 46. Gaetani G. F., Ferraris A. M., Rolfo M., Mangerini R., Arena S., Kirkman H. N. (1996) Predominant role of catalase in the disposal of hydrogen peroxide within human erythrocytes. Blood 87, 1595–1599 [PubMed] [Google Scholar]
- 47. Manta B., Hugo M., Ortiz C., Ferrer-Sueta G., Trujillo M., Denicola A. (2009) The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch. Biochem. Biophys. 484, 146–154 [DOI] [PubMed] [Google Scholar]
- 48. Iwakami S., Misu H., Takeda T., Sugimori M., Matsugo S., Kaneko S., Takamura T. (2011) Concentration-dependent dual effects of hydrogen peroxide on insulin signal transduction in H4IIEC hepatocytes. PLoS ONE 6, e27401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ye G., Metreveli N. S., Donthi R. V., Xia S., Xu M., Carlson E. C., Epstein P. N. (2004) Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 53, 1336–1343 [DOI] [PubMed] [Google Scholar]
- 50. Schriner S. E., Linford N. J., Martin G. M., Treuting P., Ogburn C. E., Emond M., Coskun P. E., Ladiges W., Wolf N., Van Remmen H., Wallace D. C., Rabinovitch P. S. (2005) Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308, 1909–1911 [DOI] [PubMed] [Google Scholar]
- 51. Verhoeven N. M., Roe D. S., Kok R. M., Wanders R. J., Jakobs C., Roe C. R. (1998) Phytanic acid and pristanic acid are oxidized by sequential peroxisomal and mitochondrial reactions in cultured fibroblasts. J. Lipid Res. 39, 66–74 [PubMed] [Google Scholar]
- 52. Girnun G. D., Domann F. E., Moore S. A., Robbins M. E. (2002) Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol. Endocrinol. 16, 2793–2801 [DOI] [PubMed] [Google Scholar]
- 53. Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., Hotamisligil G. S. (2002) A central role for JNK in obesity and insulin resistance. Nature 420, 333–336 [DOI] [PubMed] [Google Scholar]
- 54. Essers M. A., Weijzen S., de Vries-Smits A. M., Saarloos I., de Ruiter N. D., Bos J. L., Burgering B. M. (2004) FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 23, 4802–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tan W.-Q., Wang K., Lv D.-Y., Li P.-F. (2008) Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. J. Biol. Chem. 283, 29730–29739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu T. J., Lai H. C., Ting C. T., Wang P. H. (2007) Bidirectional regulation of upstream IGF-I/insulin receptor signaling and downstream FOXO1 in cardiomyocytes. J. Endocrinol. 192, 149–158 [DOI] [PubMed] [Google Scholar]