

Abstract
High mobility group box 1 (HMGB1), an evolutionarily highly conserved and abundant nuclear protein also has roles within the cytoplasm and as an extracellular damage-associated molecular pattern (DAMP) molecule. Extracellular HMGB1 is the prototypic endogenous ‘danger signal’ that triggers inflammation and immunity. Recent findings suggest that posttranslational modifications dictate the cellular localization and secretion of HMGB1. HMGB1 is actively secreted from immune cells and stressed cancer cells, or passively released from necrotic cells. During cancer development or administration of therapeutic agents including chemotherapy, radiation, epigenetic drugs, oncolytic viruses, or immunotherapy, the released HMGB1 may either promote or limit cancer growth, depending on the state of progression and vascularization of the tumor. Extracellular HMGB1 enhances autophagy and promotes persistence of surviving cancer cells following initial activation. When oxidized, it chronically suppresses the immune system to promote cancer growth and progression, thereby enhancing resistance to cancer therapeutics. In its reduced form, it can facilitate and elicit innate and adaptive anti-tumor immunity, recruiting and activating immune cells, in conjunction with cytotoxic agents, particularly in early transplantable tumor models. We hypothesize that HMGB1 also functions as an epigenetic modifier, mainly through regulation of NF-kB-dependent signaling pathways, to modulate the behavior of surviving cancer cells as well as the immune cells found within the tumor microenvironment. This has significant implications for developing novel cancer therapeutics.
Keywords: Cancer, HMGB1, NF-kB signaling, activation, innate immunity, dendritic cells, CD8+ T cells, epigenetic pathways
Introduction
The high-mobility group box-1 (HMGB1) protein, also known as high-mobility group 1 (HMG-1) and amphoterin, is a highly conserved, abundant non-histone nuclear protein expressed in almost all eukaryotic cells [1]. Within the nucleus, HMGB1 modulates and ‘fluidizes’ nucleosomes and bends DNA and binds bent DNA in chromatin structure, facilitating transcription of many genes (for a review, see ref# [2]). As a component of the innate immune system, the HMGB1 protein functions as a common signal that alerts the host to stress, unscheduled cell death, and to microbial invasion [3,4]. As a result, HMGB1 is an endogenous molecule that facilitates host inflammation and the resultant immune response [5]. In response to inflammatory stimuli following infection or injury, HMGB1 is secreted by immune cells such as macrophages [6,7], natural killer (NK) cells [8], neutrophils [9] and mature dendritic cells (DC) [10], functioning as a cytokine-like molecule. Cancer cells may actively secrete HMGB1 [11-13], or passively release HMGB1 in the process of dying. Through binding to individual surface receptors, including the receptor for advanced glycation end products (RAGE) and Toll-like receptors (TLR) 2/4 on cancer and other cells, HMGB1 can activate signalling pathways, including mitogen-activated protein kinases (MAPKs) and the serine/threonine kinase AKT, which play important roles in tumor growth and inflammation [5,14]. Increased expression of HMGB1 occurs in many cancers and has functional implications. Here we will focus on the posttranslational modification, localization, and release of the HMGB1 molecule in immune cells and cancer cells, its dynamics and function during cancer treatment, and as a potential target for improved combination therapy. Interestingly, we propose that HMGB1 functions as an epigenetic modifier via epigenetic pathways to regulate surviving cancer cells and immune cells within the tumor microenvironment, thus regulating their function. This is an important factor to consider in developing combination strategies for patients with cancer.
Molecular biology of HMGB1
HMGB1 is a protein encoded by the HMGB1 gene, a member of the HMG superfamily in humans. The human HMGB1 gene contains 14 individual introns (12 gt-ag, 2 gc-ag) and five exons. In addition 16 different splice variant mRNAs and 2 unspliced forms are found. There are 6 probable alternative promoters, 2 non overlapping alternative terminal exons and 9 validated alternative polyadenylation sites. Interestingly, 799-base pairs of this gene are antisense to the ubiquitin-specific peptidase-like protein 1 (USPL1), suggesting regulated alternate expression. Both USPL1 and HMGB1 are located on chromosome 13q12 [15] (Figure 1). The gene is driven by a strong TATA box-less promoter, flanked with additional regulatory elements, including a silencer located upstream and an enhancer located in the first intron of the gene [16]. Even though expression is ubiquitous, the levels of expression can vary significantly in individual cell types presumably due to regulation of its transcription [17]. HMGB1 is often overexpressed not only in cancer cells, such as carcinomas of the breast [18], cervical [19], gastrointestinal tract [20], head and neck [21], nasopharynx [22], and lymphomas [23], but also in activated endothelial cells found within the cancer tissue [24].
Figure 1.

A schematic representation of gene structure, protein domains and their associated functions of human HMGB1. Upper Panel: The 5 exons of the gene are indicated by boxes (open for translated regions and solid for untranslated regions). Middle Panel: The human HMGB1 has 215 amino acid residues and can be divided into three domains. Box A domain at the N-terminal is required for DNA binding, Box B is also required for DNA binding, and a fragment peptide within (#80 to 123) is required for cytokine activity. Peptide #89-108 is required for TRL4 binding and peptide #150-183 for RAGE-binding. Peptide Hp91 (#91 to 108) is an immune adjuvant peptide [113]. The C tail is acidic and codes for regulatory functions. There are two nuclear localization signals (NLS1, #27-43, and NLS2, #178-184) [6]. Three cysteine residues, C23, C45 and C106 are labeled. C23 and C45 can form an intramolecular disulfite bridge, while C106 is essential for binding to TLR4. Lower Panel: Listed are main functions of four different localized forms of HMGB1. The interplay between mitochondria and two forms of HMGB1 are based on two recent studies [41,42].
At the protein level, members of the HMG superfamily are typically 25 to 30 kDa in size with homologous basic domains that bind DNA, mediated by individual HMG boxes [25]. HMGB1 is composed of three domains: two positively charged domains (A and B boxes) and a negatively charged carboxyl terminus [26,27] (Figure 1). Boxes A and B are DNA-binding domains. Intriguingly, the box B contains the cytokine-like activity that can induce macrophage secretion of additional proinflammatory cytokines [28], dependent on the cysteine 106 being nonoxidized and the more proximal cysteines at positions 23 and 45 being oxidized as the dithiol. The protein structure involved in the binding of HMGB1 with RAGE is located between amino acid residues 150 and 183 [29]. The acidic tail at the C-terminus interacts with HMG boxes and regulates HMGB1 DNA binding specificity via a unique mechanism [30,31].
HMGB1 exerts its key functions within the nucleus by binding (without sequence specificity) to the minor groove of DNA. This enables it to facilitate the assembly of nucleosomes on the chromatin and interact physically with DNA and a variety of molecules, including p53 [32], NF-κB [33], and steroid hormone receptors [34], thus regulating transcription of a number of important genes (for a review, see ref# [35]). HMGB1 is also found outside of the nucleus, within the cytoplasm following any type of cellular stress [17]. Some cells express HMGB1 on the plasma membrane as well. Membrane-associated HMGB1 controls neurite outgrowth, smooth muscle cell chemotaxis, and tumor cell metastasis [36-39]. In addition, HMGB1 can be found within mitochondria [40-42] (Figure 1).
HMGB1 is released into the extracellular space during both sterile inflammation and infection [43,44]. Immunocompetent cells also secrete HMGB1 following stimulation with other danger-associated molecular pattern (DAMP) and pathogen-associated molecular pattern (PAMP) molecules [45,46]. There is a novel role for double-stranded RNA-dependent protein kinase (PKR) in inflammasome activation and HMGB1 secretion in macrophages, offering a potential new target for intervention [47]. In addition to innate immune cells, at least three studies have demonstrated that HMGB1 can be actively secreted from cancer cells including human colon cancer [11,13] and mesothelioma [12]. HMGB1 can also be released passively from dying/stressed cancer and normal cells. Originally, it was thought that HMGB1, ATP and other inflammatory molecules were released only from necrotic, but not apoptotic cells [48]. Later studies indicated that cells dying via apoptosis could also release HMGB1 late following dissolution of an undigested cell. In a number of of cancer cell lines, HMGB1 is released following treatment with chemical inducers of cell death (staurosporine, etoposide, or camptothecin), and this release can be diminished by application of the apoptosis-inhibitor Z-VAD-fmk [49]. Thus release of HMGB1 from both apoptotic and necrotic cells can be observed [50]. Murine macrophages stimulated with lipopolysaccharide (LPS) or polyinosinic-polycytidylic acid release HMGB1 and this is correlated with the occurrence of apoptosis in these cells [51]. This observation is in part confounded by the fact that HMGB1 is actively secreted from macrophages which have taken up apoptotic cells. Autophagy also regulates selective release of HMGB1. Stressed cancer cells, in which autophagy is induced, selectively release HMGB1 without associated lysis of the cell membrane or classical necrosis [52].
Signaling pathways of HMGB1
HMGB1 can interact with other TLR ligands as well as cytokines, and activate cells through multiple cell surface receptors including TLR2, TLR4, and RAGE, functioning as a major in vivo sensor of tissue damage by eliciting inflammatory reactions, functioning much like a cytokine. HMGB1 binding to RAGE triggers Ras, PI3K and Rho activation, one of the major downstream signaling molecules being NF-kB, the latter itself inducing a number of other inflammatory molecules. HMGB1 binding to TLR2/4 triggers signaling cascades with substantial overlap, activating three signaling pathways, eventually leading to NF-kB activation. There are crosstalk and convergence between these various signaling pathways (for a review, see ref # [5,14,53]). These signaling cascades lead to activation of NF-kB, and then other proinflammatory cytokines, leukocyte adhesion molecules and angiogenic factors in both hematopoietic and endothelial cells, thus promoting inflammation in cancer and other disease states. Negative signaling pathways can also be identified. For example, inflammatory responses to DAMPs, but not to microbial PAMPs, are repressed by the interaction of CD24 and Siglect-10 in humans or Siglec-G in mice [54,55]. Interestingly, tumor-associated dendritic cells (DC) express on the cell surface high levels of another molecule, T cell immunoglobulin and mucin domain containing protein-3 (TIM-3) that binds HMGB1 and also inhibits the antitumor immunity induced by DNA vaccines and chemotherapy [56].
HMGB1 may form complexes with other proteins, bind to other receptors in synergy with endogenous and exogenous danger signals to promote inflammation, and contribute to pathogenesis or effective therapy [57,58]. In activation of human PBMC or synovial fibroblast cultures, HMGB1 on its own does not induce detectable IL-6 production. However, when the protein associates with one of several individual proinflammatory molecules (IL-1β, TLR4 ligand LPS, the TLR9 ligand CpG-ODN, or the TLR1-TLR2 ligand Pam3CSK4) it potently enhances proinflammatory cytokine production. TLR9–dependent activation by DNAcontaining immune complexes is mediated by HMGB1 and RAGE [59]. The complexes bind to TLR9 as well as to RAGE and activate pDCs and B cells in response to DNA and contribute to autoimmune pathogenesis [59]. One could take advantage of this interaction in therapeutic settings where a potent immune response is desired. Microbial CpG-DNA or its analog CpG-ODNs activate macrophages, monocytes, and DC to secrete proinflammatory cytokines, driving the Th1 response. HMGB1 can function as a CpG-ODN–binding protein, pre-associates with TLR9 in the endoplasmic reticulum-Golgi intermediate compartment, and facilitates the redistribution of TLR9 to early endosomes in response to CpG-ODN. CpG-ODN stimulates macrophages and dendritic cells to secrete HMGB1; extracellular HMGB1, in turn, accelerates the delivery of CpG-ODNs to its receptor, leading to a TLR9-dependent augmentation of IL-6, IL-12, and TNFα secretion [60], following effective recruitment of NF-κB. Interestingly, HMGB1–nucleosome complexes activate antigen presenting cells via binding to TLR2 and may crucially contribute to the pathogenesis of systemic lupus erythematosus via breaking the immunological tolerance to against nucleosomes/dsDNA [61]. HMGB1/DNA complexes could thus also serve as attractive adjuvants in vaccine strategies for patients with cancer or other diseases.
The mechanism by which HMGB1 promotes cytokine release subsequent to its interaction with TLR4 thereby promoting cell migration is still not completely understood. Inhibitors of NF-kB kinases α (IKKα) and β (IKKβ) are important for HMGB1-elicited chemotaxis of fibroblast, innate immune cells in vitro and neutrophils in vivo [62], and may play critical roles.
DAMPs are critically regulated by oxidation with terminal oxidation by myeloperoxidases as means for extinguishing the ‘danger signals’ [63-66]. Functioning as either chemoattractant or proinflammatory cytokine, HMGB1 orchestrates both processes by switching among mutually exclusive redox states [67]. Reduced cysteines make the molecule a chemoattractant, whereas a disulfide bond between the vicinal C23 and C45 cysteines makes it a proinflammatory cytokine. Further cysteine oxidation to sulfonates by reactive oxygen species abrogates both activities. In other words, HMGB1 orchestrates both key events in sterile inflammation, leukocyte recruitment and their induction to secrete inflammatory cytokines, by adopting mutually exclusive redox states. CXCL12 plays an important role in HMGB1-induced recruitment of inflammatory cells. HMGB1 and CXCL12 form a heterocomplex that acts exclusively through CXCR4 and not through other HMGB1 receptors. This heterocomplex promotes conformational rearrangements of CXCR4 from that of CXCL12 alone. Mononuclear cell recruitment in vivo into air pouches and injured muscles depends on the heterocomplex and is inhibited by AMD3100, an inhibitor of CXCR4 signaling, and glycyrrhizin, which directly binds HMGB1. Thus, both inflammatory cell recruitment and activation depend on HMGB1 [68].
Post-translational modifications dictate intracellular distribution and key functions of HMGB1
HMGB redistribution is controlled, in part, by one of several posttranslational modifications. These include acetylation [6], phosphorylation [69], methylation [9] and oxidation [70]. These changes modulate its structure, its localization and subsequent biological functions.
HMGB1 contains two nuclear localization signals (NLSs) for controlled nuclear transport [6] (Figure 1). Both acetylation and phosphorylation of the two NLSs of HMGB1 is involved in nuclear transport toward secretion. In 2003, Bianchi, Agresti and associates showed that, in all cells, HMGB1 shuttles actively between the nucleus and the cytosol. HMGB1 is acetylated extensively upon activation with LPS in monocytes and macrophages. In resting macrophages, forced hyperacetylation of the molecule leads to its translocation to the cytosol. Cytosolic HMGB1 is then concentrated by default into secretory lysosomes, and secreted when monocytic cells receive an appropriate second signal [6]. The postsynthetic acetylation of HMGB1 protein and its truncated form significantly affects its properties as an “architectural” factor - recognition of bent DNA and binding of short DNA fragments. In a study of liver ischemia/reperfusion (I/R) injury, we found that serum HMGB1 released following liver I/R in vivo is acetylated, and that hepatocytes exposed to oxidative stress in vitro also released acetylated HMGB1. They identified the histone deacetylases-1 (HDAC1) and -4 (HDAC4) as critical in regulating acetylated HMGB1 release [71]. We subsequently found that interferon regulatory factor 1 (IRF-1) mediates acetylation and release of HMGB1 via histone acetyltransferases [72]. Phosphorylation of HMGB1 dictates the dynamic shuttling between cytoplasmic and nuclear compartments [69]. Hyperphosphorylated HMGB1 relocates to the cytoplasm. In a nuclear import assay, phosphorylated HMGB1 in the cytoplasm did not enter the nucleus. The authors mutated serine residues of either or both NLSs of HMGB1 to glutamic acid to mimic a phosphorylated state and examined the binding of HMGB1 to karyopherin-α1, ae nuclear import protein for HMGB1. Substitution of serine with glutamic acid in either NLS decreased the binding with karyopherin-α1 by about 50%. Substitution, however, of both NLSs showed no binding, and HMGB1 was relocated to the cytoplasm and subsequently secreted. PKC-ζ phosphorylates HMGB1, and the phosphorylation of specific serine residues is related to enhanced HMGB1 secretion in colon cancer cells [13]. These studies establish the notion that phosphorylation and acetylation of the two NLS regions of the protein regulate the direction of transport of HMGB1.
HMGB1 can be modified by methylation as well. In neutrophils, HMGB1 is post-translationally mono-methylated at Lys42. The methylation alters the conformation of HMGB1 and weakens its DNA binding activity, causing it to become largely distributed in the cytoplasm by passive diffusion out of the nucleus [9]. This novel pathway may help explain the distribution of nuclear HMGB1 to the cytoplasm and is important for understanding how neutrophils release HMGB1 into the extracellular milieu [9].
HMGB1 is also regulated by reduction-oxidation (redox) reactions. Oxidative stress can induce a covalent disulfide bond between protein and peptide thiols that is reversible through enzymatic catalysis. HMGB1 contains three conserved cysteine (C) residues: C23 and C45 can form an intramolecular disulfide bridge, whereas C106 is unpaired and is essential for binding to TLR4 (Figure 1). The disulfide bond between C23 and C45 is a target of glutathione-dependent reduction by glutaredoxin. Cysteine targeted mutational analysis suggests that C23 and 45 in response to oxidative stress, undergo conformational changes whereas C106 appears to be critical for the nucleocytoplasmic shuttling of HMGB1 [70].
In cancer therapy with cisplatin, it is believed that HMGB1 enhances the anticancer efficacy of cisplatin by shielding platinated DNA lesions from repair. A recent study showed that C23 and C45 in the domain A form a reversible disulfide bond under mildly oxidizing conditions. The reduced domain A protein binds to a 25-bp DNA probe containing a central 1,2-d(GpG) intrastrand cross-link, the major platinum-DNA adduct, with a 10-fold greater binding affinity than the oxidized box A domain. Thus, the cellular redox environment can influence the interaction of HMGB1 with platinated DNA and suggests that the redox state of the box A domain modulates the activity of cisplatin as an anticancer drug [73]. The C106 thiol and C23-C45 disulfide bond are required for HMGB1 to induce NF-kB nuclear translocation and TNF production in macrophages [74]. Oxidation of HMGB1 occurs during liver I/R injury, and leads to attenuation of its pro-inflammatory activity after its release from cells [75]. Reduced and oxidized HMGB1 have selective roles in extracellular signaling and regulation of immune responses that are mediated by signaling through individual receptors [76]. Together, these studies demonstrate that HMGB1 posttranslational modifications are critical for the localization, DNA binding and pro-inflammatory activity of the protein (Table 1).
Table 1.
Redox regulation of HMGB1 and biological effects
| Redox states | Biological models | Biological effects | References |
|---|---|---|---|
| Native and exclusive redox states | 1. Examine its function as a cytokine or chemoattractant in different redox states; 2. “Native state” of the molecule (with C106 thiol and C23-C45 disulfite bond) is required for HMGB1 to induce nuclear NF-kB translocation and TNF production in macrophages. | 1. Reduced cysteines make it a chemoattractant; 2. A disulfite bond (C23-C45) makes a proinflammatory cytokine. 3. Full cysteine oxidation to sulfonates by ROS abrogates both activities. 4. Both irreversible oxidation and complete reduction of these cysteines inhibit TNF production. | [67] [74] |
| Oxidation | Apoptosis: HMGB1 is predominately oxidized in apoptotic cells and from primary and secondary necrotic cells. The oxidation is caspasedependent. | 1. This usually leads to immunological tolerance. 2. However, it may stimulate immune responses by scavenging or by mutating a mitochondrial caspase target protein when ROS activity was prohibited | [50] [153] |
| Oxidation and reduction | Autophagy and apoptosis: Stimuli that enhance ROS promote cytosolic translocation of HMGB1 and thereby enhance autophagic flux. HMGB1 directly interacts with the autophagy protein Beclin1 displacing Bcl-2. | 1. Reduced HMGB1 binds RAGEs, but not to TLR4, induces Beclin1-dependent autophagy and promotes tumor resistance; 2. Oxidized HMGB1 increases the cytotoxicity of the agents and induces apoptosis mediated by the caspase-9/-3 intrinsic pathway. | [82,84] |
| Mild reduction | Regeneration and remodeling of skeletal muscle: HMGB1, weakly expressed in healthy muscles, increases during regeneration in parallel with the antioxidant response in both fibers and leukocytes. | The early antioxidant response in regenerating muscle may limit HMGB1 oxidation, thus allowing successful muscle regeneration. | [154] |
| Oxidation | Prolonged ischemia and upon reperfusion: Oxidation of HMGB1 takes place and may attenuate its pro-inflammatory activity. | The results confirm that post-translational oxidation of HMGB1 attenuates its proinflammatory activity | [75] |
Relationship between autophagy, apoptosis and release of HMGB1 and immune responses
Most PAMPs and DAMPs serve as so-called ‘Signal 0s’ that bind specific receptors to promote autophagy [77]. We and others have studied how HMGB1 and other DAMPs affects autophagy and apoptosis, and conversely how they regulate HMGB1 release and function. Autophagy, or autophagocytosis, is the basic catabolic mechanism that involves degradation of unnecessary or dysfunctional cellular components through either extracellular secretion or degradation within lysosome [78]. In the context of disease, autophagy has been seen as an adaptive response to survival, however when excessive, can promote cell death and morbidity [79]. The balance between apoptosis and autophagy is important in tumor development and response to therapy. Often, tumor suppressors positively regulate autophagy, whereas oncoproteins inhibit autophagy. Autophagy in cancer can either suppress or promote tumor growth depending on the status of the cells [80,81].
Endogenous HMGB1 regulates autophagy [82]. HMGB1 and p53 form a complex that regulates the balance between tumor cell death and survival [83]. Stimuli that enhance reactive oxygen species promote cytosolic translocation of HMGB1 and thereby enhance autophagic flux. HMGB1 directly interacts with the autophagy protein Beclin1 displacing Bcl-2. Pharmacological inhibition of HMGB1 cytoplasmic translocation by agents such as ethyl pyruvate limits starvation-induced autophagy. HMGB1 is a redox-sensitive regulator of the balance between autophagy and apoptosis [84]. In cancer cells, anticancer agents enhance autophagy and apoptosis, as well as HMGB1 release. HMGB1 release may be a prosurvival signal for residual cells following various cytotoxic cancer treatments. Diminished HMGB1 leads predominantly to apoptosis and decreased autophagy in stressed cancer cells. Under these conditions, reducible HMGB1 binds to RAGE, but not to TLR4, inducing Beclin1-dependent autophagy and thereby promoting tumor resistance to a number of chemotherapeutic agents [84,85]. In contrast, oxidized HMGB1 increases the cytotoxicity of these agents and induces apoptosis mediated by the caspase-9/-3 intrinsic pathway. HMGB1 release, as well as its redox state, thus links autophagy and apoptosis, representing a suitable target when coupled with conventional tumor treatments [84].
Autophagy also regulates release of HMGB1 [52,86]. Autophagy modulates cell death by epidermal growth factor receptor-targeted diphtheria toxin (DT-EGF) lysis of cells. DT-EGF kills epithelial and glioblastoma tumor cells with similar efficiency but by different mechanisms that depend on whether the cells activate autophagy when treated with the drug. Dying cells in which autophagy is induced selectively release the immune modulator HMGB1 without causing disruption of the cell membrane or necrosis. Conversely, cells that are killed by DT-EGF where autophagy is blocked, activate caspases but retain HMGB1. These data suggest that it may be feasible to manipulate the immunogenicity of dying cells by increasing or decreasing autophagy [52].
Antineoplastic chemotherapies are particularly efficient when they elicit immunogenic cell death, thus provoking an anticancer immune response. Autophagy, which is often disabled in cancer, is dispensable for chemotherapy-induced cell death but required for its immunogenicity [87]. The key feature is that, when responding to chemotherapy, autophagy-competent, but not autophagy-deficient, cancers attracted dendritic cells and T lymphocytes into the tumor bed. Suppression of autophagy inhibits the release of ATP from dying tumor cells. Conversely, inhibition of extracellular ATP-degrading enzymes increases pericellular ATP in autophagy-deficient tumors, reestablishing recruitment of immune cells, and restoring chemotherapeutic responses but only in immunocompetent hosts. Thus, autophagy is essential for the immunogenic release of ATP from dying cells, and increased extracellular ATP concentrations improve the efficacy of antineoplastic chemotherapies in short term tumor-bearing hosts when autophagy is disabled [87]. It is important to point out that this study deals with a vaccine instead of a therapeutic tumor model and that more chronic release of HMGB1 and ATP, as occurs presumably in human tumors, more likely promotes immunosuppression.
HMGB1 promotes cancer growth and development
Six biological capacities acquired during the multistep development of cancer make up the hallmarks of cancer [88]. Unfortunately, overexpression of HMGB1 is associated with each of the six hallmarks of cancer [89] (Figure 2). HMGB1 protein serves predominantly to promote wound healing, but when excessive can cause disease [90,91]. In cancer progression and during treatment of cancer, HMGB1 signaling has also paradoxical dual effects. On the one hand, chronic release of HGMB1 promotes tumor growth and progression, enhances tumor neoangiogenesis, and promotes resistance to drug therapy (Figure 2 and 3A). On the other hand, under ideal settings such as acute release of large quantity of HMGB1 and other inflammatory molecules, it can also promote anti-tumor immunity that can promote tumor regression (Figure 3A). These yin-yang properties and functions make it an ideal therapeutic target in cancer therapy [92].
Figure 2.
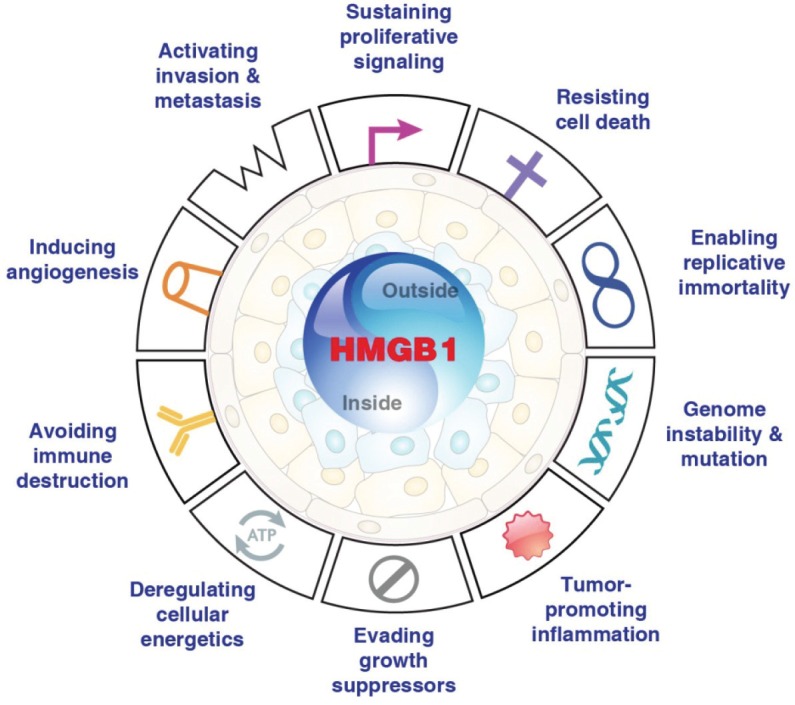
Cancer promoting properties of HMGB1. In cancer, HMGB1 dysfunction is associated with each of the six hallmarks of cancer the latter of which were summarized by Hanahan and Weinberg [88]. We have presented an earlier version of this association in our previous review [89].
Figure 3.

HMGB1 in the tumor microenvironment and hypothesis of HMGB1 functioning via NF-kB and epigenetic pathways. A. The release of HMGB1 in the tumor microenvironment. HMGB1 has yin-yang effects on cancer. Acute release of HMGB1 after cancer therapies promotes maturation of DCs through interaction with TLR4 and clonal expansion of tumor antigen-specific T cells and thus elicits antitumor responses. In contrast, persistent hypoxia in growing tumors leads to necrosis, causing chronic release of HMGB1, which promotes angiogenesis and tumor growth through the recruitment of macrophages (TAM) and endothelial precursor cells (EPC) and activation of local endothelial cells through RAGE signaling. The acutely and chronically released HMGB1 molecules are represented in red and black, indicating that they may be differently modified by redox or other mechanisms. This figure is modified from a figure by Srikrishna and Freeze [58]. B. The hypothesis of HMGB1 acting via NF-kB and epigenetic pathways to exert its long lasting effects on surviving tumor cells, immune cells and stromal cells in the tumor microenvironment. The NF-kB pathway is regulated partly via epigenetic pathways (EP) both upstream and downstream. STAT3 and NF-kB, both inflammation-regulated genes, regulate genes to promote cancer cell proliferation, survival, caner angiogenesis and metastasis. As an example to illustrate the point of role of epigenetic mechanisms, we showed that IL-6, produced and released from macrophage stimulated by HMGB1 and modulated via epigenetic mechanisms, regulates transcription of DNMT1, one of the key enzymes for DNA methylation in cancer cells. The TNF-stimulated and phosphorylated p65 binds and recruits DNMT1 to the promoter of BRMS1 gene, thus inhibits the production of BRMS1, an inhibitor of tumor metastasis.
HMGB1 promotes tumor growth. HMGB1 is over-expressed in many types of cancer [18-23]. HMGB1 is critical for survival of cancer cells and knockdown of HMGB1 results in apoptosis in prostate and gastric cancer cells [93,94]. Diminished HMGB1 promotes apoptosis and decreased autophagy in stressed colon and pancreatic cancer cells [84]. In hypoxic hepatocellular carcinoma, HMGB1 activates TLR4 and RAGE-signaling pathways to induce caspase-1 activation with subsequent production of multiple inflammatory mediators. As a result, these inflammatory signals promote invasiveness and metastases [95]. In gastric cancer, HMGB1 promotes invasiveness through enhanced MMP-9 expression [94]. In a colon cancer model, HMGB1 released from necrotic cancer cells enhances regrowth and metastasis of the remaining cancer cells via RAGE activation [96]. In malignant mesothelioma, active secretion (and passive release) of HMGB1 from cancer cells supports progression of the disease [12]. The motility, survival, and anchorage-independent growth of HMGB1-secreting malignant mesothelioma cells are inhibited in vitro by treatment with monoclonal antibodies directed against HMGB1 or RAGE. Significantly, HMGB1 inhibition diminishes the growth of mesothelioma xenografts in immunodeficient mice and extends host survival. HMGB1 also promotes drug resistance in osteosarcoma [97], and leukemia [85], and this could be attributed to HMGB1-mediated autophagy [85]. In ovarian cancer, nucleus accumbens-1 (NAC1) contributes to drug resistance. Treatment with cisplatin caused an activation of autophagy in ovarian cancer cells. Regulation of autophagy by NAC1 was mediated by HMGB1, as the functional status of NAC1 was associated with the expression, translocation and release of HMGB1 [98]. Therefore, one mechanism of HMGB1-mediated drug resistance to cisplatin is via enhanced autophagy stimulated by NAC1 in ovarian cancer.
Redox regulates interaction of HMGB1 with individual receptors and its functions. Reducible HMGB1 binds to RAGE, but not to TLR4, induces Beclin1-dependent autophagy and promotes tumor resistance to various chemotherapeutic agents. In contrast, oxidized HMGB1 increases the cytotoxicity of these agents and induces apoptosis mediated by the caspase-9/-3 intrinsic pathway [84], but may have lost its proinflammatory activity [75].
HMGB1 can also be highly immunosuppressive, particularly in the setting of absence of other TLR ligands. It actively affects the immune functions of many types of cells including DC, macrophages, NK cells, T lymphocytes, and regulatory T cells (Tregs) [3,10,99]. Recent findings suggest that the release of HMGB1 as an endogenous danger signal is important for priming an adaptive immune response that promotes malignant progression [100]. Tumorassociated DCs express on the cell surface high levels of TIM-3 that binds HMGB1, thereby inhibiting the antitumor immunity elicited by DNA vaccines and chemotherapy [56]. HMGB1 suppresses cytokine secretion and maturation of plasmacytoid DCs in response to TLR9 agonists including the hypomethylated oligodeoxy-nucleotide CpG- and DNA-containing viruses. HMGB1 inhibits secretion of several proinflammatory cytokines including IFN-α, IL-6, TNF-α, inducible protein-10, and IL-12 from pDC [101]. Knockdown of HMGB1 in tumor cells attenuates their ability to induce Tregs and leads to naturally acquired CD8 T cell- or IFN-γ–dependent tumor rejection [99]. HMGB1 directly enhances immune inhibitory functions of Treg via RAGE-mediated mechanisms and limits the number and activity of Teff. HMGB1 effects on Treg may alter immune reactivity in the setting of chronic inflammatory states such as cancer [21,102].
HMGB1 during cancer therapy
Essentially all therapeutic modalities inducing necrosis and/or apoptosis of cancer cells would lead to release of HMGB1 from cancer cells, and subsequent secretion of HMGB1 from activated immune cells. These would include, but not limit to, most types of chemotherapy, radiation, and immunotherapy. The release of HMGB1 has been demonstrated with killing of cancer cells using cytolytic cells such as NK and T cells in vitro [103], in prodrug-suicide gene therapy [104], oncolytic virotherapy [105], and epigenetic therapy [49].
The beneficial effects of HMGB1 in cytotoxic therapeutic regimens
HMGB1 sensitizes cells to cisplatin-mediated cytotoxicity by shielding its major DNA adducts from nucleotide excision repair. Treatment of steroid hormone receptors-expressing human cancer cells with the either estrogen and/or progesterone, significantly increases the potency of cisplatin and its analogue carboplatin by causing overexpression of HMGB1 [106]. Binding of HMGB1 to DNA (chromatin), could be modulated by the redox state of HMGB1. Two cysteine residues in the HMGB1 A box domain form a reversible disulfide bond under mildly oxidizing conditions. The reduced domain A protein binds to a 25-bp DNA probe containing a central 1,2-d(GpG) intrastrand cross-link, the major platinum DNA adduct, with a 10-fold greater binding affinity than the oxidized domain A. Thus, the cellular redox environment can influence the interaction of HMGB1 with the platinated DNA and suggest that the redox state of the box A domain of HMGB1 could modulate the potency of cisplatin as an anticancer drug [73]. In summary, these findings suggest that the proper combination of drugs and hormones could have potential benefit in treating hormone receptor-expressing ovarian, prostate, or breast cancers [107]. Similarly a role for HMGB1 in the deeper understanding of Vitamin D biology and therapy is in order, not just for microbial infections but also in cancer as another steroid hormone important in its biology [108].
HMGB1 plays an important role in eliciting innate and adaptive anti-tumor immunity [3,44]. The emerging concept that cancer is not just a disease of a tissue or an organ but also a host disease relies on evidence of tumor-induced immunosuppression and polymorphisms in genes involved in host protection against tumors. Many therapeutic effects require the immunoadjuvant effect of tumor cell death induced by cytotoxic anticancer agents. Zitvogel and colleagues were among the first to explore the role of immunity in conjunction with anticancer chemotherapy and radiotherapy [107]. Release of HMGB1 by dying tumor cells is mandatory to license host DCs to process and present tumor antigens. HMGB1 interacts with TLR4 on DCs, which are selectively involved in the cross-priming of antitumor T lymphocytes in vivo. A TLR4 polymorphism that limits the binding of HMGB1 to TLR4 predicts early relapse following anthracyclinebased chemotherapy in breast cancer patients [107]. In a recent study, the significance of HMGB1 release and induction anti-tumor immunity was analyzed in patients with esophageal squamous cell carcinoma (ESCC) receiving chemoradiation. The authors found that chemoradiation induces tumor antigen-specific T-cell responses, and HMGB1 production is related to clinical outcome after chemoradiation [109].
DNA alkylating agents induce sporadic cell necrosis and regression of apoptosis-deficient tumors [93]. Sporadic tumor cell necrosis is associated with extracellular release of intercellular contents including HMGB1 and subsequent recruitment of innate immune cells into tumor tissues. Although DNA alkylating therapy led to a complete tumor regression in an athymic mouse tumor xenograft model, it failed to do so in tumors deficient in HMGB1. The HMGB1-deficient tumors have an impaired ability to recruit innate immune cells including macrophages, neutrophils, and NK cells into the treated tumor tissue [93]. Suppression of innate immunity and HMGB1 using depleting Abs limits tumor regression in response to these therapies.
The epigenetic modulator apicidin has been applied in combination with the cytotoxic agent docetaxel in tumor breast cell lines characterized by different grades of invasiveness [110]. Combined treatment with apicidin and docetaxel, at minimally toxic doses, stimulates in metastatic breast cancer cells the expression of CTCF-like protein and other cancer antigens, thus favoring an antitumor immune response. Importantly, following combined exposure to these agents, metastatic cells express calreticulin on the cell surface and release considerable amounts of HMGB1, thus promoting the translation of induced cell death into effective antitumor immune responses [110].
HMGB1 mediates endogenous TLR2 activation and leads to brain tumor regression in a glioblastoma multiforme model [111]. Combined immunotherapy/conditional cytotoxic approaches that utilize adenoviral vectors (Ad) expressing Fms-like tyrosine kinase 3 ligand (Flt3L) and thymidine kinase delivered into the tumor mass lead to secretion of HMGB1. This in turn is specifically recognized by its receptor TLR2 on myeloid dendritic cells (MDC). The presence of HMGB1 attracts and activates the MDC withinin the brain which, in turn, activates T lymphocytes. The HMGB1-TLR2 axis signaling pathway thus activates anti-tumor T cell immunity and leads to brain tumor regression. Another important function of HMGB1 in immune response is that it is an endogenous immune adjuvant [112-115]. Necrotic HMGB1(-/-) cells have a reduced ability to activate APCs, and HMGB1 blockade reduces the activation induced by necrotic wild-type cell supernatants. In vivo, HMGB1 enhances the primary antibody responses to soluble antigens and transforms poorly immunogenic apoptotic lymphoma cells into efficient vaccines [112]. A short peptide derived from HMGB1 potentiates cellular immune responses to peptide antigen and cellular and humoral immune responses to protein antigen in vivo. The short peptide promotes in vivo production of the immunomodulatory cytokines, IFN-γ, TNF-α, IL-6, and IL-12 (p70), as well as antigen-specific activation of CD8+ T cells [113,114]. HMGB1 adjuvant properties enhance the adaptive effector and memory immune responses against influenza virus infection [115]. Together, these results suggest that HMGB1 could function as an adjuvant in the context of cancer vaccine to promote potent anti-cancer immunity.
HMGB1 in treatment with epigenetic drugs
Epigenetic therapy of cancer has shown very promising results in both preclinical and clinical studies [116,117]. So far four epigenetic drugs, including inhibitors of DNA methyltransferases (DNMT) and histone deacetylases (HDAC), have been approved by the FDA. They are used to treat myelodysplastic syndromes, a set of bone marrow conditions that often progress into terminal leukemias, or cutaneous T cell lymphomas. The use of epigenetic drugs to treat solid cancers has been actively pursued with limited results. Extracellular release of HMGB1 during nominal ‘apoptotic’ death has been found [49]. The HDAC inhibitor trichostatin A causes biochemical changes that promote enhanced release of HMGB1 from apoptotic cells [118]. We have applied DNMT and HDAC inhibitors to study induction of cancer/testis antigens and a transcription factor Rhox5 in cancer cells as a novel form of immunotherapy [119-121]. Epigenetic drugs such as HDAC inhibitors induce apoptosis and autophagy in cancer cells [120,122], which likely leads to the release of HMGB1. HMGB1 is released into the extracellular milieu from cancer cells treated with decitabine (Guo ZS et al., unpublished data).
HMGB1 is involved in epigenetic silencing of tumor necrosis factor alpha (TNF-α) and interleukin 1 beta (IL-1β) transcription in blood leukocytes of animals and humans after the initiation of severe systemic inflammation (SSI). In the case of endotoxin tolerance, HMGB1 and linker histone H1 couple as a component of the epigenetic complex that silences acute proinflammatory TNF-α during the assembly of heterochromatin in the severe systemic inflammation (SSI) phenotype. Depletion of HMGB1 results in dissociation of RelB, one of the five NF-kB proteins, from the promoter and partially restores TNF-α transcription [123].
HMGB1 in treatments with oncolytic viruses
Oncolytic virotherapy for cancer has shown great promise in both preclinical and clinical studies [124,125]. The death of cancer cells infected with oncolytic virus (OV) results from direct oncolysis (a mixture of apoptosis and necropoptosis), and necrotic death induced indirectly by the effects of anti-angiogenesis and hypoxia induced by the virus, or by anticancer immunity elicited by the viruses and the virus-infected cells and cytokines. As a result, the biologically active HMGB1 is released from dying cancer and endothelial cells or actively secreted from activated immune cells into the cancer tissue milieu.
Our group first reported that cancer cells infected by an oncolytic virus, in this case vaccinia virus, were induced into apoptotic/necrotic death pathways and released HMGB1 from the dying cells into the extracellular milieu [105]. Subsequently, other investigators have also observed the release of HMGB1 as well as other inflammatory signals from cancer cells infected with other OVs, such as an oncolytic adenovirus (Ad5/3-hTERT-E1A-hCD40L) [118], an oncolytic measles virus from infected melanoma cells [126], a herpes simplex virus type -2 (HSV-2) from human endometrial cancer cells [127], and with a coxsackievirus B3 [128]. Together, these studies strengthen the notion that OV exert antitumor effects through multiple mechanisms including oncolysis (necrosis), apoptosis, release of HMGB1 and other inflammatory cytokines, and induction of innate immunity and anti-tumor T cell response.
HMGB1 interacts physically and functionally with a number of viruses including OVs: 1) exogenous HMGB1 inhibits replication of human immunodeficiency virus in monocyte-derived macrophages [129]; 2) HMGB1 binds to the influenza virus nucleoprotein and promotes replication of this virus [130]; and 3) HMGB1 also regulates dengue virus (DENV) infection in human DCs [131]. DENV infection causes translocation of nuclear HMGB1 into the cytosol and secretion into the extracellular milieu. When DENV-infected DCs are co-cultured with autologous T cells, there is increased production of HMGB1 by both cell types. HMGB1 regulated TNF-α, IL-6, IL-8 and IFN-α secretion in DENVinfected DCs. As would be expected, elevated level of HMGB1 lowers the rate of DENV replication in DCs [131]. Thus, the released HMGB1 may modulate the replication and the efficacy of various OVs depending on the specific virus.
Oncolytic virotherapy itself is a form of immunotherapy [132,133]. In most cases, the positive immune effects of OV are mediated by the proimmune environment created in part by the release of immunogenic molecules, such as HMGB1, heat shock protein and IFNs. Thus, it is logical to combine OV with other forms of immunotherapy in order to further enhance its efficacy. For example, one recent study demonstrated improved therapeutic outcomes when combining an OV with a potent agonist antibody specific for the costimulatory molecule 4-1BB [134]. It is interesting to note that some most promising OVs are Ad, HSV or VV armed with GM-CSF, another immunostimulatory molecule. The HSV version, called OncoVEX GM-CSF, has shown good efficacy in cancer patients with an approximately 30% response rate against systemic disease, following local injection into accessible tumors. OncoVEX GM-CSF is currently completing a pivotal phase III trial in melanoma, and a phase III trial in head and neck cancer is also underway [135]. HMGB1 and other pro-inflammatory molecules may work in concert with GM-CSF to elicit potent anti-cancer immunity which may account for significant better clinical outcomes. Another combination strategy has been to combine OV with chemotherapeutic agents. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by IFN and HMGB1 released from the infected cells [136].
Hypothesis: HMGB1 functions via epigenetic pathways
We hypothesize that HMGB1 plays a significant role via epigenetic pathways in shaping the surviving cancer cells and immune cells within the emergent tumor microenvironment arising in the setting of chronic inflammation (Figure 3B). During cancer progression and treatment, the interplay between environment and epigenetics directs distinct inflammatory responses [137]. Cancer evolution at all stages is driven by both epigenetic abnormalities as well as genetic alterations. Alterations in DNA methylation have been observed during inflammation and inflammation-associated carcinogenesis [138]. Enzymes involved in both acetylation and methylation of histones are involved in inflammatory responses and in immunity [139,140].
One of the key molecules activated by HMGB1 signaling cascades, and a number of other TLR-ligand-initiated signaling, is the activation and nuclear localization of NF-kB [14,53]. The NF-κB/Rel family includes NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), RelB, and c-Rel. NF-kB is well connected both upstream and downstream with a number of key factors of the epigenetic pathways in cancer and immune responses [141-143]. Some recent progress further strengthens this critical connection. Constitutive NF-κB activation has causative roles in adult T cell leukemia (ATL) caused by HTLV-1 and other cancers. An epigenetic pathway is involved in Polycomb-mediated miRNA silencing and NF-κB activation [144]. MiR-31 negatively regulates the noncanonical NF-κB pathway by targeting NF-κB inducing kinase. In ATL cells, miR-31 level is epigenetically regulated, and aberrant upregulation of Polycomb proteins contribute to miR-31 downregulation in an epigenetic fashion, leading to activation of NF-κB and apoptosis resistance. Inflammation may promote metastatic disease via NF-kB and epigenetic pathways. In response to TNF in the tumor milieu, NF-κB member p65 is phosphorylated at S276 and the phosphorylated p65 recruits DNA methyltransferase 1 (DNMT-1) to the promoter and represses transcription of the tumor metastasis suppressor gene BRMS1 [145]. p65 directly recruits DNMT-1 to chromatin, resulting in promoter-specific DNA methylation and transcriptional repression of BRMS1. This highlights a new mechanism through which the inflammation-induced NF-κB can regulate metastatic disease. A number of TLR4-regulated proinflammatory genes in macrophages, also targets of HMGB1, are controlled by regulated trimethylation and demethylation of histone H4K20 located in the promoters. Signal-dependent erasure of H4K20me3 is required for effective gene activation and is achieved by NF-κB-dependent delivery of the histone demethylase PHF2 [146]. NF-kB modulates IL-6 via epigenetic pathways, which in turn affects other genes [142,147]. IL-6 and STAT3, another key inflammation-triggered signaling molecule, modulate transcription of DNMT1 in leukemia and cancer cells [148,149]. These studies clearly link epigenetic pathways with inflammation-activated NF-kB-mediated gene regulation and directly contribute to disturbed cancer biology. As for immune cells, epigenetic processes and NF-kB contribute to the development and activation of a variety of immune cells [150,151]. In addition, a number of cytokine genes in immune cells are regulated through epigenetic pathways, and accompany the activation or suppression of immune cells. The findings of these and other studies suggest that epigenetic programs from HMGB1 to NF-kB and further downstream oncogenic signaling play important roles in cancer initiation and progression. Thus, both HMGB1 and NF-kB offer ideal targets for interventions in cancer therapeutics. In this regard, it is interesting to note that both HMGB1 and NF-kB play ‘yin-yang’ roles in cancer and immunity.
Conclusions and perspectives
The majority of therapeutic regimens for cancer lead to release of HMGB1. Therefore, how to best utilize the released HMGB1 in combination therapy is one of the major questions we have addressed. In the setting of cancer arising over several years, the major role of HMGB1 is to promote a wound healing response and limit the effectiveness of immune effectors. Still, immune reactivity is a dynamic process and means to limit myeloid derived suppressor cells and Tregs in the setting of oncolytic therapy and enhancement of immune reactivity, perhaps by targeting HMGB1 cofactors or receptors is in order.
Following acute cytotoxic treatments including chemotherapy, radiation therapy, epigenetic drugs, oncolytic viruses, and immunotherapy, stressed/dying cancer cells often release HMGB1, ATP and other inflammatory molecules [152]. At the same time, activated macrophages/monocytes and other innate immune cells actively secrete HMGB1 and other inflammatory cytokines into the tumor milieu. These events could be potent signals to prime for potent anti-tumor immune responses. Any rational combinatorial therapeutic regimen should take advantage of the potential synergy of HMGB1 signaling. (1) acute release of HMGB1 to stimulate innate and adaptive immunity against cancer; and/or (2) the effect of HMGB1 to sensitize the cancer cells to certain type of chemotherapy (such as cisplatin) or other therapeutic regimens. This is a promising new direction for treatment of patients with cancer.
Acknowledgments
The authors acknowledge the financial support from National Organization of Rare Diseases grant # 707338 (ZSG) and David C. Koch Regional Therapy Cancer Center. The original research papers related to HMGB1 from our groups discussed here were supported by the National Institute of Health grants R01CA100415 (DLB) and 1P01CA101944 (MTL). We thank Dr. Gutian Xiao and Ms. Roshni Ravindranathan for critical reading of the manuscript.
Conflict of interest statement
None.
References
- 1.Vaccari T, Beltrame M, Ferrari S, Bianchi ME. Hmg4, a new member of the Hmg1/2 gene family. Genomics. 1998;49:247–252. doi: 10.1006/geno.1998.5214. [DOI] [PubMed] [Google Scholar]
- 2.Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799:101–113. doi: 10.1016/j.bbagrm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 3.Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P. HMGB1: guiding immunity from within. Trends Immunol. 2005;26:381–387. doi: 10.1016/j.it.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 4.Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, Savitsky D, Ronfani L, Akira S, Bianchi ME, Honda K, Tamura T, Kodama T, Taniguchi T. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462:99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]
- 5.Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. 2012;8:195–202. doi: 10.1038/nrrheum.2011.222. [DOI] [PubMed] [Google Scholar]
- 6.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 8.Semino C, Angelini G, Poggi A, Rubartelli A. NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood. 2005;106:609–616. doi: 10.1182/blood-2004-10-3906. [DOI] [PubMed] [Google Scholar]
- 9.Ito I, Fukazawa J, Yoshida M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem. 2007;282:16336–16344. doi: 10.1074/jbc.M608467200. [DOI] [PubMed] [Google Scholar]
- 10.Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA, Rovere-Querini P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174:7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 11.Liu S, Stolz DB, Sappington PL, Macias CA, Killeen ME, Tenhunen JJ, Delude RL, Fink MP. HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix-induced hyperpermeability of Caco-2 monolayers. Am J Physiol Cell Physiol. 2006;290:C990–999. doi: 10.1152/ajpcell.00308.2005. [DOI] [PubMed] [Google Scholar]
- 12.Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I, Pass HI, Gaudino G, Carbone M, Yang H. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 2012;72:3290–3301. doi: 10.1158/0008-5472.CAN-11-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee H, Park M, Shin N, Kim G, Kim YG, Shin JS, Kim H. High mobility group box-1 is phosphorylated by protein kinase C zeta and secreted in colon cancer cells. Biochem Biophys Res Commun. 2012;424:321–326. doi: 10.1016/j.bbrc.2012.06.116. [DOI] [PubMed] [Google Scholar]
- 14.van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1) Angiogenesis. 2008;11:91–99. doi: 10.1007/s10456-008-9093-5. [DOI] [PubMed] [Google Scholar]
- 15.Ferrari S, Finelli P, Rocchi M, Bianchi ME. The active gene that encodes human high mobility group 1 protein (HMG1) contains introns and maps to chromosome 13. Genomics. 1996;35:367–371. doi: 10.1006/geno.1996.0369. [DOI] [PubMed] [Google Scholar]
- 16.Lum HK, Lee KL. The human HMGB1 promoter is modulated by a silencer and an enhancer-containing intron. Biochim Biophys Acta. 2001;1520:79–84. doi: 10.1016/s0167-4781(01)00243-3. [DOI] [PubMed] [Google Scholar]
- 17.Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med. 2004;255:332–343. doi: 10.1111/j.1365-2796.2003.01296.x. [DOI] [PubMed] [Google Scholar]
- 18.Brezniceanu ML, Volp K, Bosser S, Solbach C, Lichter P, Joos S, Zornig M. HMGB1 inhibits cell death in yeast and mammalian cells and is abundantly expressed in human breast carcinoma. FASEB J. 2003;17:1295–1297. doi: 10.1096/fj.02-0621fje. [DOI] [PubMed] [Google Scholar]
- 19.Hao Q, Du XQ, Fu X, Tian J. [Expression and clinical significance of HMGB1 and RAGE in cervical squamous cell carcinoma] . Zhonghua Zhong Liu Za Zhi. 2008;30:292–295. [PubMed] [Google Scholar]
- 20.Choi YR, Kim H, Kang HJ, Kim NG, Kim JJ, Park KS, Paik YK, Kim HO. Overexpression of high mobility group box 1 in gastrointestinal stromal tumors with KIT mutation. Cancer Res. 2003;63:2188–2193. [PubMed] [Google Scholar]
- 21.Wild CA, Brandau S, Lotfi R, Mattheis S, Gu X, Lang S, Bergmann C. HMGB1 is overexpressed in tumor cells and promotes activity of regulatory T cells in patients with head and neck cancer. Oral Oncol. 2012;48:409–416. doi: 10.1016/j.oraloncology.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 22.Wu D, Ding Y, Wang S, Zhang Q, Liu L. Increased expression of high mobility group box 1 (HMGB1) is associated with progression and poor prognosis in human nasopharyngeal carcinoma. J Pathol. 2008;216:167–175. doi: 10.1002/path.2391. [DOI] [PubMed] [Google Scholar]
- 23.Meyer A, Staratschek-Jox A, Springwald A, Wenk H, Wolf J, Wickenhauser C, Bullerdiek J. Non-Hodgkin lymphoma expressing high levels of the danger-signalling protein HMGB1. Leuk Lymphoma. 2008;49:1184–1189. doi: 10.1080/10428190802064909. [DOI] [PubMed] [Google Scholar]
- 24.van Beijnum JR, Dings RP, van der Linden E, Zwaans BM, Ramaekers FC, Mayo KH, Griffioen AW. Gene expression of tumor angiogenesis dissected: specific targeting of colon cancer angiogenic vasculature. Blood. 2006;108:2339–2348. doi: 10.1182/blood-2006-02-004291. [DOI] [PubMed] [Google Scholar]
- 25.Laudet V, Stehelin D, Clevers H. Ancestry and diversity of the HMG box superfamily. Nucleic Acids Res. 1993;21:2493–2501. doi: 10.1093/nar/21.10.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bustin M, Lehn DA, Landsman D. Structural features of the HMG chromosomal proteins and their genes. Biochim Biophys Acta. 1990;1049:231–243. doi: 10.1016/0167-4781(90)90092-g. [DOI] [PubMed] [Google Scholar]
- 27.Weir HM, Kraulis PJ, Hill CS, Raine AR, Laue ED, Thomas JO. Structure of the HMG box motif in the B-domain of HMG1. EMBO J. 1993;12:1311–1319. doi: 10.1002/j.1460-2075.1993.tb05776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, Harris HE, Czura CJ, Wang H, Ulloa L, Warren HS, Moldawer LL, Fink MP, Andersson U, Tracey KJ, Yang H. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med. 2003;9:37–45. [PMC free article] [PubMed] [Google Scholar]
- 29.Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, Rauvala H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002;62:4805–4811. [PubMed] [Google Scholar]
- 30.Knapp S, Muller S, Digilio G, Bonaldi T, Bianchi ME, Musco G. The long acidic tail of high mobility group box 1 (HMGB1) protein forms an extended and flexible structure that interacts with specific residues within and between the HMG boxes. Biochemistry. 2004;43:11992–11997. doi: 10.1021/bi049364k. [DOI] [PubMed] [Google Scholar]
- 31.Wang Q, Zeng M, Wang W, Tang J. The HMGB1 acidic tail regulates HMGB1 DNA binding specificity by a unique mechanism. Biochem Biophys Res Commun. 2007;360:14–19. doi: 10.1016/j.bbrc.2007.05.130. [DOI] [PubMed] [Google Scholar]
- 32.Imamura T, Izumi H, Nagatani G, Ise T, Nomoto M, Iwamoto Y, Kohno K. Interaction with p53 enhances binding of cisplatin-modified DNA by high mobility group 1 protein. J Biol Chem. 2001;276:7534–7540. doi: 10.1074/jbc.M008143200. [DOI] [PubMed] [Google Scholar]
- 33.Brickman JM, Adam M, Ptashne M. Interactions between an HMG-1 protein and members of the Rel family. Proc Natl Acad Sci U S A. 1999;96:10679–10683. doi: 10.1073/pnas.96.19.10679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Onate SA, Prendergast P, Wagner JP, Nissen M, Reeves R, Pettijohn DE, Edwards DP. The DNA-bending protein HMG-1 enhances progesterone receptor binding to its target DNA sequences. Mol Cell Biol. 1994;14:3376–3391. doi: 10.1128/mcb.14.5.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev. 2005;15:496–506. doi: 10.1016/j.gde.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 36.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 37.Salmivirta M, Rauvala H, Elenius K, Jalkanen M. Neurite growth-promoting protein (amphoterin, p30) binds syndecan. Exp Cell Res. 1992;200:444–451. doi: 10.1016/0014-4827(92)90194-d. [DOI] [PubMed] [Google Scholar]
- 38.Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007;220:35–46. doi: 10.1111/j.1600-065X.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- 39.Rauvala H, Rouhiainen A. RAGE as a receptor of HMGB1 (Amphoterin): roles in health and disease. Curr Mol Med. 2007;7:725–734. doi: 10.2174/156652407783220750. [DOI] [PubMed] [Google Scholar]
- 40.Stumbo AC, Cortez E, Rodrigues CA, Henriques MG, Porto LC, Barbosa HS, Carvalho L. Mitochondrial localization of non-histone protein HMGB1 during human endothelial cell-Toxoplasma gondii infection. Cell Biol Int. 2008;32:235–238. doi: 10.1016/j.cellbi.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 41.Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B, Zeh HJ 3rd, Lotze MT. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011;13:701–711. doi: 10.1016/j.cmet.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gdynia G, Keith M, Kopitz J, Bergmann M, Fassl A, Weber AN, George J, Kees T, Zentgraf HW, Wiestler OD, Schirmacher P, Roth W. Danger signaling protein HMGB1 induces a distinct form of cell death accompanied by formation of giant mitochondria. Cancer Res. 2010;70:8558–8568. doi: 10.1158/0008-5472.CAN-10-0204. [DOI] [PubMed] [Google Scholar]
- 43.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 44.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD, Dixit VM. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–4392. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 47.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, Ting JP, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 49.Bell CW, Jiang W, Reich CF 3rd, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291:C1318–1325. doi: 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- 50.Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang W, Pisetsky DS. The role of IFN-alpha and nitric oxide in the release of HMGB1 by RAW 264.7 cells stimulated with polyinosinic-polycytidylic acid or lipopolysaccharide. J Immunol. 2006;177:3337–3343. doi: 10.4049/jimmunol.177.5.3337. [DOI] [PubMed] [Google Scholar]
- 52.Thorburn J, Horita H, Redzic J, Hansen K, Frankel AE, Thorburn A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009;16:175–183. doi: 10.1038/cdd.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 54.Chen GY, Chen X, King S, Cavassani KA, Cheng J, Zheng X, Cao H, Yu H, Qu J, Fang D, Wu W, Bai XF, Liu JQ, Woodiga SA, Chen C, Sun L, Hogaboam CM, Kunkel SL, Zheng P, Liu Y. Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nat Biotechnol. 2011;29:428–435. doi: 10.1038/nbt.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. 2009;323:1722–1725. doi: 10.1126/science.1168988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, Hirashima M, Uede T, Takaoka A, Yagita H, Jinushi M. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13:832–842. doi: 10.1038/ni.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hreggvidsdottir HS, Ostberg T, Wahamaa H, Schierbeck H, Aveberger AC, Klevenvall L, Palmblad K, Ottosson L, Andersson U, Harris HE. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol. 2009;86:655–662. doi: 10.1189/jlb.0908548. [DOI] [PubMed] [Google Scholar]
- 58.Srikrishna G, Freeze HH. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia. 2009;11:615–628. doi: 10.1593/neo.09284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 60.Ivanov S, Dragoi AM, Wang X, Dallacosta C, Louten J, Musco G, Sitia G, Yap GS, Wan Y, Biron CA, Bianchi ME, Wang H, Chu WM. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 2007;110:1970–1981. doi: 10.1182/blood-2006-09-044776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, Kalden JR, Schett G, Rovere-Querini P, Herrmann M, Voll RE. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–3018. doi: 10.1084/jem.20081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Penzo M, Molteni R, Suda T, Samaniego S, Raucci A, Habiel DM, Miller F, Jiang HP, Li J, Pardi R, Palumbo R, Olivotto E, Kew RR, Bianchi ME, Marcu KB. Inhibitor of NF-kappa B kinases alpha and beta are both essential for high mobility group box 1-mediated chemotaxis [corrected] . J Immunol. 2010;184:4497–4509. doi: 10.4049/jimmunol.0903131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lotfi R, Herzog GI, DeMarco RA, Beer-Stolz D, Lee JJ, Rubartelli A, Schrezenmeier H, Lotze MT. Eosinophils oxidize damage-associated molecular pattern molecules derived from stressed cells. J Immunol. 2009;183:5023–5031. doi: 10.4049/jimmunol.0900504. [DOI] [PubMed] [Google Scholar]
- 64.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, Devera ME, Liang X, Tor M, Billiar T. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 65.Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28:429–436. doi: 10.1016/j.it.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 66.Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, Lotze MT. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res. 2007;13:2836–2848. doi: 10.1158/1078-0432.CCR-06-1953. [DOI] [PubMed] [Google Scholar]
- 67.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A, Thelen M, Varani L, Mellado M, Proudfoot A, Bianchi ME, Uguccioni M. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. 2012;209:551–563. doi: 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177:7889–7897. doi: 10.4049/jimmunol.177.11.7889. [DOI] [PubMed] [Google Scholar]
- 70.Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312:3526–3538. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 71.Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, Klune JR, Zlotnicki J, Billiar T, Tsung A. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 2010;285:39888–39897. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dhupar R, Klune JR, Evankovich J, Cardinal J, Zhang M, Ross M, Murase N, Geller DA, Billiar TR, Tsung A. Interferon regulatory factor 1 mediates acetylation and release of high mobility group box 1 from hepatocytes during murine liver ischemia-reperfusion injury. Shock. 2011;35:293–301. doi: 10.1097/SHK.0b013e3181f6aab0. [DOI] [PubMed] [Google Scholar]
- 73.Park S, Lippard SJ. Redox state-dependent interaction of HMGB1 and cisplatin-modified DNA. Biochemistry. 2011;50:2567–2574. doi: 10.1021/bi2000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, Al-Abed Y, Andersson U, Tracey KJ, Antoine DJ. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1) Mol Med. 2012;18:250–259. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 75.Liu A, Fang H, Dirsch O, Jin H, Dahmen U. Oxidation of HMGB1 causes attenuation of its pro-inflammatory activity and occurs during liver ischemia and reperfusion. PLoS One. 2012;7:e35379. doi: 10.1371/journal.pone.0035379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tang D, Kang R, Zeh HJ 3rd, Lotze MT. Highmobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011;14:1315–1335. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 79.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.White EJ, Martin V, Liu JL, Klein SR, Piya S, Gomez-Manzano C, Fueyo J, Jiang H. Autophagy regulation in cancer development and therapy. Am J Cancer Res. 2011;1:362–372. [PMC free article] [PubMed] [Google Scholar]
- 81.Xiao G. Autophagy and NF-kappaB: fight for fate. Cytokine Growth Factor Rev. 2007;18:233–243. doi: 10.1016/j.cytogfr.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ 3rd, Lotze MT. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Livesey K, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC, Zhang L, Manfredi JJ, Zeh HJ, Li L, Lotze M, Tang D. p53/HMGB1 Complexes Regulate Autophagy and Apoptosis. Cancer Res. 2012 Apr 15;72:1996–2005. doi: 10.1158/0008-5472.CAN-11-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, Zeh HJ, Lotze MT. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299–5310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Lotze MT, Tang D, Cao L. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia. 2011;25:23–31. doi: 10.1038/leu.2010.225. [DOI] [PubMed] [Google Scholar]
- 86.Thorburn J, Frankel AE, Thorburn A. Regulation of HMGB1 release by autophagy. Autophagy. 2009;5:247–249. doi: 10.4161/auto.5.2.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, Vacchelli E, Galluzzi L, Ghiringhelli F, di Virgilio F, Zitvogel L, Kroemer G. Autophagydependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 88.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 89.Tang D, Kang R, Zeh HJ 3rd, Lotze MT. Highmobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ulloa L, Messmer D. High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev. 2006;17:189–201. doi: 10.1016/j.cytogfr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 91.Campana L, Bosurgi L, Rovere-Querini P. HMGB1: a two-headed signal regulating tumor progression and immunity. Curr Opin Immunol. 2008;20:518–523. doi: 10.1016/j.coi.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 92.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 93.Guerriero JL, Ditsworth D, Catanzaro JM, Sabino G, Furie MB, Kew RR, Crawford HC, Zong WX. DNA alkylating therapy induces tumor regression through an HMGB1-mediated activation of innate immunity. J Immunol. 2011;186:3517–3526. doi: 10.4049/jimmunol.1003267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Song B, Song WG, Li ZJ, Xu ZF, Wang XW, Wang CX, Liu J. Effect of HMGB1 silencing on cell proliferation, invasion and apoptosis of MGC-803 gastric cancer cells. Cell Biochem Funct. 2012;30:11–17. doi: 10.1002/cbf.1811. [DOI] [PubMed] [Google Scholar]
- 95.Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH, Monga SP, Geller DA, Lotze MT, Tsung A. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012;55:1863–1875. doi: 10.1002/hep.25572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luo Y, Chihara Y, Fujimoto K, Sasahira T, Kuwada M, Fujiwara R, Fujii K, Ohmori H, Kuniyasu H. High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived chemotherapy. Eur J Cancer. 2012 Oct 3; doi: 10.1016/j.ejca.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 97.Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R, Vernon P, Cao L, Tang D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 2012;72:230–238. doi: 10.1158/0008-5472.CAN-11-2001. [DOI] [PubMed] [Google Scholar]
- 98.Zhang Y, Cheng Y, Ren X, Zhang L, Yap KL, Wu H, Patel R, Liu D, Qin ZH, Shih IM, Yang JM. NAC1 modulates sensitivity of ovarian cancer cells to cisplatin by altering the HMGB1-mediated autophagic response. Oncogene. 2012;31:1055–1064. doi: 10.1038/onc.2011.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu Z, Falo LD Jr, You Z. Knockdown of HMGB1 in tumor cells attenuates their ability to induce regulatory T cells and uncovers naturally acquired CD8 T cell-dependent antitumor immunity. J Immunol. 2011;187:118–125. doi: 10.4049/jimmunol.1003378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.He Y, Zha J, Wang Y, Liu W, Yang X, Yu P. Tissue damage-associated ‘danger signals’ influence T cell responses that promote the progression of pre-neoplasia to cancer. Cancer Res. 2013;73:1–11. doi: 10.1158/0008-5472.CAN-12-2704. [DOI] [PubMed] [Google Scholar]
- 101.Popovic PJ, DeMarco R, Lotze MT, Winikoff SE, Bartlett DL, Krieg AM, Guo ZS, Brown CK, Tracey KJ, Zeh HJ 3rd. High mobility group B1 protein suppresses the human plasmacytoid dendritic cell response to TLR9 agonists. J Immunol. 2006;177:8701–8707. doi: 10.4049/jimmunol.177.12.8701. [DOI] [PubMed] [Google Scholar]
- 102.Wild CA, Bergmann C, Fritz G, Schuler P, Hoffmann TK, Lotfi R, Westendorf A, Brandau S, Lang S. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int Immunol. 2012;24:485–494. doi: 10.1093/intimm/dxs051. [DOI] [PubMed] [Google Scholar]
- 103.Dong Xda E, Ito N, Lotze MT, Demarco RA, Popovic P, Shand SH, Watkins S, Winikoff S, Brown CK, Bartlett DL, Zeh HJ 3rd. High mobility group box I (HMGB1) release from tumor cells after treatment: implications for development of targeted chemoimmunotherapy. J Immunother. 2007;30:596–606. doi: 10.1097/CJI.0b013e31804efc76. [DOI] [PubMed] [Google Scholar]
- 104.Candolfi M, Yagiz K, Foulad D, Alzadeh GE, Tesarfreund M, Muhammad AK, Puntel M, Kroeger KM, Liu C, Lee S, Curtin JF, King GD, Lerner J, Sato K, Mineharu Y, Xiong W, Lowenstein PR, Castro MG. Release of HMGB1 in response to proapoptotic glioma killing strategies: efficacy and neurotoxicity. Clin Cancer Res. 2009;15:4401–4414. doi: 10.1158/1078-0432.CCR-09-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guo ZS, Naik A, O’Malley ME, Popovic P, Demarco R, Hu Y, Yin X, Yang S, Zeh HJ, Moss B, Lotze MT, Bartlett DL. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005;65:9991–9998. doi: 10.1158/0008-5472.CAN-05-1630. [DOI] [PubMed] [Google Scholar]
- 106.He Q, Liang CH, Lippard SJ. Steroid hormones induce HMG1 overexpression and sensitize breast cancer cells to cisplatin and carboplatin. Proc Natl Acad Sci U S A. 2000;97:5768–5772. doi: 10.1073/pnas.100108697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 108.Jo EK. Innate immunity to mycobacteria: vitamin D and autophagy. Cell Microbiol. 2010;12:1026–1035. doi: 10.1111/j.1462-5822.2010.01491.x. [DOI] [PubMed] [Google Scholar]
- 109.Suzuki Y, Mimura K, Yoshimoto Y, Watanabe M, Ohkubo Y, Izawa S, Murata K, Fujii H, Nakano T, Kono K. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 2012;72:3967–3976. doi: 10.1158/0008-5472.CAN-12-0851. [DOI] [PubMed] [Google Scholar]
- 110.Buoncervello M, Borghi P, Romagnoli G, Spadaro F, Belardelli F, Toschi E, Gabriele L. Apicidin and docetaxel combination treatment drives CTCFL expression and HMGB1 release acting as potential antitumor immune response inducers in metastatic breast cancer cells. Neoplasia. 2012;14:855–867. doi: 10.1593/neo.121020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C, Muhammad AK, Clark MC, Arditi M, Comin-Anduix B, Ribas A, Lowenstein PR, Castro MG. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, Dumitriu IE, Muller S, Iannacone M, Traversari C, Bianchi ME, Manfredi AA. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5:825–830. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Telusma G, Datta S, Mihajlov I, Ma W, Li J, Yang H, Newman W, Messmer BT, Minev B, Schmidt-Wolf IG, Tracey KJ, Chiorazzi N, Messmer D. Dendritic cell activating peptides induce distinct cytokine profiles. Int Immunol. 2006;18:1563–1573. doi: 10.1093/intimm/dxl089. [DOI] [PubMed] [Google Scholar]
- 114.Saenz R, Souza Cda S, Huang CT, Larsson M, Esener S, Messmer D. HMGB1-derived peptide acts as adjuvant inducing immune responses to peptide and protein antigen. Vaccine. 2010;28:7556–7562. doi: 10.1016/j.vaccine.2010.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fagone P, Shedlock DJ, Bao H, Kawalekar OU, Yan J, Gupta D, Morrow MP, Patel A, Kobinger GP, Muthumani K, Weiner DB. Molecular adjuvant HMGB1 enhances anti-influenza immunity during DNA vaccination. Gene Ther. 2011;18:1070–1077. doi: 10.1038/gt.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Popovic R, Licht JD. Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov. 2012;2:405–413. doi: 10.1158/2159-8290.CD-12-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yu XD, Guo ZS. Epigenetic drugs for cancer treatment and prevention: mechanisms of action. Biomol Concepts. 2010;1:239–252. doi: 10.1515/bmc.2010.020. [DOI] [PubMed] [Google Scholar]
- 118.Diaconu I, Cerullo V, Hirvinen ML, Escutenaire S, Ugolini M, Pesonen SK, Bramante S, Parviainen S, Kanerva A, Loskog AS, Eliopoulos AG, Pesonen S, Hemminki A. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012;72:2327–2338. doi: 10.1158/0008-5472.CAN-11-2975. [DOI] [PubMed] [Google Scholar]
- 119.Guo ZS, Hong JA, Irvine KR, Chen GA, Spiess PJ, Liu Y, Zeng G, Wunderlich JR, Nguyen DM, Restifo NP, Schrump DS. De novo induction of a cancer/testis antigen by 5-aza-2’-deoxycytidine augments adoptive immunotherapy in a murine tumor model. Cancer Res. 2006;66:1105–1113. doi: 10.1158/0008-5472.CAN-05-3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li Q, Bartlett DL, Gorry MC, O’Malley ME, Guo ZS. Three epigenetic drugs up-regulate homeobox gene Rhox5 in cancer cells through overlapping and distinct molecular mechanisms. Mol Pharmacol. 2009;76:1072–1081. doi: 10.1124/mol.109.056291. [DOI] [PubMed] [Google Scholar]
- 121.Li Q, O’Malley ME, Bartlett DL, Guo ZS. Homeobox gene Rhox5 is regulated by epigenetic mechanisms in cancer and stem cells and promotes cancer growth. Mol Cancer. 2011;10:63. doi: 10.1186/1476-4598-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2004;101:18030–18035. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.El Gazzar M, Yoza BK, Chen X, Garcia BA, Young NL, McCall CE. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol Cell Biol. 2009;29:1959–1971. doi: 10.1128/MCB.01862-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–231. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Donnelly OG, Errington-Mais F, Steele L, Hadac E, Jennings V, Scott K, Peach H, Phillips RM, Bond J, Pandha H, Harrington K, Vile R, Russell S, Selby P, Melcher AA. Measles virus causes immunogenic cell death in human melanoma. Gene Ther. 2013;20:7–15. doi: 10.1038/gt.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Borde C, Barnay-Verdier S, Gaillard C, Hocini H, Marechal V, Gozlan J. Stepwise release of biologically active HMGB1 during HSV-2 infection. PLoS One. 2011;6:e16145. doi: 10.1371/journal.pone.0016145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, Okazaki T, Marumoto T, Takahashi A, Takayama K, Nakanishi Y, Shimizu H, Tani K. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012;72:2609–2621. doi: 10.1158/0008-5472.CAN-11-3185. [DOI] [PubMed] [Google Scholar]
- 129.Nowak P, Barqasho B, Treutiger CJ, Harris HE, Tracey KJ, Andersson J, Sonnerborg A. HMGB1 activates replication of latent HIV-1 in a monocytic cell-line, but inhibits HIV-1 replication in primary macrophages. Cytokine. 2006;34:17–23. doi: 10.1016/j.cyto.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 130.Moisy D, Avilov SV, Jacob Y, Laoide BM, Ge X, Baudin F, Naffakh N, Jestin JL. HMGB1 protein binds to influenza virus nucleoprotein and promotes viral replication. J Virol. 2012;86:9122–9133. doi: 10.1128/JVI.00789-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kamau E, Takhampunya R, Li T, Kelly E, Peachman KK, Lynch JA, Sun P, Palmer DR. Dengue virus infection promotes translocation of high mobility group box 1 protein from the nucleus to the cytosol in dendritic cells, upregulates cytokine production and modulates virus replication. J Gen Virol. 2009;90:1827–1835. doi: 10.1099/vir.0.009027-0. [DOI] [PubMed] [Google Scholar]
- 132.Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA, Errington F. Oncolytic viruses: a novel form of immunotherapy. Expert Rev Anticancer Ther. 2008;8:1581–1588. doi: 10.1586/14737140.8.10.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thorne SH. Immunotherapeutic potential of oncolytic vaccinia virus. Immunol Res. 2011;50:286–293. doi: 10.1007/s12026-011-8211-4. [DOI] [PubMed] [Google Scholar]
- 134.John LB, Howland LJ, Flynn JK, West AC, Devaud C, Duong CM, Stewart TJ, Westwood JA, Guo ZS, Bartlett DL, Smyth MJ, Kershaw MH, Darcy PK. Oncolytic virus and anti-4-1BB combination therapy elicits strong anti-tumor immunity against established cancer. Cancer Res. 2012;72:1651–1660. doi: 10.1158/0008-5472.CAN-11-2788. [DOI] [PubMed] [Google Scholar]
- 135.Kaufman HL, Bines SD. OPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GMCSF for unresectable stage III or IV melanoma. Future Oncol. 2010;6:941–949. doi: 10.2217/fon.10.66. [DOI] [PubMed] [Google Scholar]
- 136.Huang B, Sikorski R, Kirn DH, Thorne SH. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. 2011;18:164–172. doi: 10.1038/gt.2010.121. [DOI] [PubMed] [Google Scholar]
- 137.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hartnett L, Egan LJ. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis. 2012;33:723–731. doi: 10.1093/carcin/bgs006. [DOI] [PubMed] [Google Scholar]
- 139.Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011;32:335–343. doi: 10.1016/j.it.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 140.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, Mosley J, Ramirez-Molina C, Rowland P, Schofield CJ, Sheppard RJ, Smith JE, Swales C, Tanner R, Thomas P, Tumber A, Drewes G, Oppermann U, Patel DJ, Lee K, Wilson DM. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vanden Berghe W, Ndlovu MN, Hoya-Arias R, Dijsselbloem N, Gerlo S, Haegeman G. Keeping up NF-kappaB appearances: epigenetic control of immunity or inflammation-triggered epigenetics. Biochem Pharmacol. 2006;72:1114–1131. doi: 10.1016/j.bcp.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 142.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xiao G, Fu J. NF-kappaB and cancer: a paradigm of Yin-Yang. Am J Cancer Res. 2011;1:192–221. [PMC free article] [PubMed] [Google Scholar]
- 144.Yamagishi M, Nakano K, Miyake A, Yamochi T, Kagami Y, Tsutsumi A, Matsuda Y, Sato-Otsubo A, Muto S, Utsunomiya A, Yamaguchi K, Uchimaru K, Ogawa S, Watanabe T. Polycomb-mediated loss of miR-31 activates NIK-dependent NF-kappaB pathway in adult T cell leukemia and other cancers. Cancer Cell. 2012;21:121–135. doi: 10.1016/j.ccr.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 145.Liu Y, Mayo MW, Nagji AS, Smith PW, Ramsey CS, Li D, Jones DR. Phosphorylation of RelA/p65 promotes DNMT-1 recruitment to chromatin and represses transcription of the tumor metastasis suppressor gene BRMS1. Oncogene. 2012;31:1143–1154. doi: 10.1038/onc.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, Boutros M, Perrimon N, Rosenfeld MG, Glass CK. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol Cell. 2012;48:28–38. doi: 10.1016/j.molcel.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Meng F, Wehbe-Janek H, Henson R, Smith H, Patel T. Epigenetic regulation of microRNA-370 by interleukin-6 in malignant human cholangiocytes. Oncogene. 2008;27:378–386. doi: 10.1038/sj.onc.1210648. [DOI] [PubMed] [Google Scholar]
- 148.Hodge DR, Xiao W, Clausen PA, Heidecker G, Szyf M, Farrar WL. Interleukin-6 regulation of the human DNA methyltransferase (HDNMT) gene in human erythroleukemia cells. J Biol Chem. 2001;276:39508–39511. doi: 10.1074/jbc.C100343200. [DOI] [PubMed] [Google Scholar]
- 149.Zhang Q, Wang HY, Woetmann A, Raghunath PN, Odum N, Wasik MA. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood. 2006;108:1058–1064. doi: 10.1182/blood-2005-08-007377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Reiner SL. Epigenetic control in the immune response. Hum Mol Genet. 2005;14:R41–46. doi: 10.1093/hmg/ddi115. [DOI] [PubMed] [Google Scholar]
- 151.Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9:91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- 152.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 153.Urbonaviciute V, Meister S, Furnrohr BG, Frey B, Guckel E, Schett G, Herrmann M, Voll RE. Oxidation of the alarmin high-mobility group box 1 protein (HMGB1) during apoptosis. Autoimmunity. 2009;42:305–307. doi: 10.1080/08916930902831803. [DOI] [PubMed] [Google Scholar]
- 154.Vezzoli M, Castellani P, Corna G, Castiglioni A, Bosurgi L, Monno A, Brunelli S, Manfredi AA, Rubartelli A, Rovere-Querini P. High-mobility group box 1 release and redox regulation accompany regeneration and remodeling of skeletal muscle. Antioxid Redox Signal. 2011;15:2161–2174. doi: 10.1089/ars.2010.3341. [DOI] [PubMed] [Google Scholar]