

Abstract
A major challenge in the future development of cancer therapeutics is the identification of biological targets and pathways, and the subsequent design of molecules to combat the drug-resistant cells hiding in virtually all cancers. This therapeutic approach is justified based upon the limited advances in cancer cures over the past 30 years, despite the development of many novel chemotherapies and earlier detection, which often fail due to drug resistance. Among the various targets to overcome tumor resistance are the DNA repair systems that can reverse the cytotoxicity of many clinically used DNA-damaging agents. Some progress has already been made but much remains to be done. We explore some components of the DNA-repair process, which are involved in repair of alkylation damage of DNA, as targets for the development of novel and effective molecules designed to improve the efficacy of existing anticancer drugs.
Cancer resistance to drugs
The war on cancer has gone on around the world for more than 40 years, with the expenditure of billions of US dollars. As a result, the age-adjusted cancer death rate has been reduced by approximately 12% for both males and females between 1975 and 2008, with some major improvements in colon, breast and some hematological cancers [201,202]. A significant fraction of the overall reduction in death rates, specifically since the late 1980s, is linked to reduced lung cancer deaths due to reduced tobacco consumption. There are also improvements in 5-year survival rates for a number of important cancers, which reflect earlier detection and better treatments. Still, widespread cures remain elusive despite increased cancer screening, using improved methods for early diagnosis and an increase in the number and nature of the therapies to treat the different diseases that we call cancer. It has long been known that cancer is multiple diseases with multiple etiologies, involving genetic and epigenetic events, and that a single therapeutic agent would not be sufficient to lower cancer mortality [1]. However, the lack of significant progress in the development of approaches to eliminate cancer in patients rather than incrementally increase survival, would suggest a basic flaw in our therapeutic strategies, and a disconnect between the clinic and the laboratory. A classic example of the success and failure of cancer therapy is illustrated by trastuzumab, which targets the HER2 receptor that is over-expressed in a significant percentage of breast cancers and induces a cytostatic G1 arrest [2]. There is an initial excellent response to trastuzumab but, eventually, the disease reoccurs [3]. There are several mechanisms responsible for the development of resistance:
Reduced trastuzumab binding due to mutations in the HER2 gene that lead to a truncated, but active, receptor [4];
Upregulation of downstream signaling (e.g., loss of PTEN or increase in PI3K/Akt activity) that compensates for reduced receptor activation [5];
Compensation due to alternative signaling pathways involving other members of the HER family of receptors and other receptors (e.g., IGF-1R [6]);
Impaired immune-mediated activity [7].
Many other effective drugs eventually become ineffective, due to similar issues related to mutations in the binding domain that they target and epigenetic changes that activate alternative compensating signaling pathways.
For cytostatic compounds, which do not kill tumor cells and that continually need to be administered, cancer cell survival due to resistance appears to be inevitable owing to genetic diversity in the tumor mass and selection pressure for epigenetic changes. However, the same scenario occurs even for cytotoxic DNA-alkylating therapies [8,9]. Hence, the development of strategies to block resistance should be developed upfront rather than to adopting a passive ‘wait and watch’ approach [10–12]. We discuss below the role of DNA repair in tumor resistance to alkylating agents and the progress in developing agents that inhibit enzymes involved in the different pathways of repair.
DNA-damaging drugs
The history of anticancer therapies began with the development of World War I mustard gas agents [13] that at low doses had therapeutic activity due to the covalent modification of DNA. The first effective molecule clinically used in the treatment of cancer was the nitrogen mustard, bis(2-chloroethylamine) (Figure 1). The nitrogen mustards and related analogues, for example, bis(2-chloroethyl)-N-nitrosourea [14] and cyclophosphamide [15], react with DNA in all cells that they reach to produce complex arrays of monofunctional and bifunctional lesions that qualitatively and quantitatively differ with the different drugs [16]. The bifunctional lesions include intra- and inter-strand cross-links. The latter are extremely cytotoxic since they prevent the DNA strand separation that is required for replication by DNA polymerases and transcription by RNA polymerases. However, if not repaired, the monofunctional and intrastrand crosslink lesions generated by these compounds are also cytotoxic based upon their ability to block the processivity of DNA polymerases. There are a number of anticancer agents that only produce monofunctional lesions. Regardless, toxicity due to alkylation damage generally requires cell division, which is one reason why tumor cells (assuming that they are replicating) are selectively more sensitive to DNA-damaging agents than most noncancer cells. Over the years, numerous DNA-modifying drugs with superior specificity and bioavailability have been developed (Figure 2). Therefore, this class of compounds continues to constitute an important tool in the treatment of many cancers, despite efforts to produce mechanistically based noncytotoxic antineoplastic drugs.
Figure 1.

Initial nitrogen mustard-based anticancer drug (mechlorethamine) and some clinically used analogues.
Figure 2.

Classes of clinically used alkylating anticancer drugs: nitrosourea, triazene and sulfonate ester.
The major limitation of DNA-damaging drugs is that they are generally cytotoxic in any cell type that is rapidly dividing; for example, tumor cells, epithelial cells in the GI tract and the hematopoietic cells in bone marrow. Accordingly, the difference between a therapeutic and toxic dose can be small and dosing must be carefully monitored. A second important drawback of DNA-alkylating agents is that they are mutagenic as well as cytotoxic; therefore, they increase the risk of secondary cancers derived from the initial round of therapy [17–20]. Efforts to restrict DNA alkylation to the formation of lesions that are cytotoxic but marginally mutagenic have been reported but none of these compounds have yet made it into clinical trials [21–24].
Tumor resistance to alkylating agents
In addition to the undesirable side effects mentioned above, tumors can develop resistance to specific types of genotoxic insults via a number of mechanisms, including the upregulation of gene products that enhance DNA-repair capacity. This mode of resistance is in addition to the more generic multidrug resistance pathways involving increased expression of transporters (e.g., P-glycoprotein MRP1/ABCC1) [25–28] and higher levels of cellular nucleophiles that can scavenge the reactive intermediates generated from this class of antineoplastic drugs [29,30]. There is experimental evidence using genetic manipulations that reducing the expression of many, albeit not all (e.g., mismatch repair [MMR]) DNA-repair proteins makes cells and animals more sensitive to DNA-damaging agents [31–41]. Conversely, increasing repair protein expression can confer resistance [42,43]. Therefore, DNA-repair pathways afford valid targets to potentiate clinically useful chemotherapeutic agents and to block cellular resistance to the same drugs.
It is worth some discussion on whether the strategy to potentiate DNA-damaging agents to overcome resistance represents a rational approach to drug design. The genetic deletion of many DNA-repair proteins shows virtually no phenotype, so inhibitors of specific repair targets are not expected to be toxic in the absence of a DNA-damaging drug. There are notable exceptions to this; for example, APE-1 and polymerase β (Pol β) (see below). Clearly, the significant off-target toxicities associated with antineoplastic agents may also be affected by any agent that diminishes DNA repair. However, it has been proposed that the overexpression of repair enzymes in some tumors may indicate that these proteins are critical to tumor growth or survival, which may make these cells selectively sensitive to repair inhibitors, even in the absence of alkylating agents [44]. It is also important to determine the effect of repair inhibitors on tumor stem cells, which are critical to the survival and expansion of the tumor [45], and how repair inhibitors in combination with alkylating agents alter the sensitivity of these cells. The role of DNA-repair pathways in tumor stem cells has been explored with some reports suggesting that human-induced pluripotent cells have enhanced levels of DNA-repair proteins, including those involved in repair of double-strand breaks [46]. A similar effect of DNA repair has been suggested for hematopoietic stem cells [47]. Embryonic stem cells are thought to be sensitive to DNA damage and undergo apoptosis more rapidly because of genotoxic insults [48]. Similarly, murine embryonic stem cells show higher repair activity than the corresponding fibroblast cells [49]. While the question is still not resolved, the inhibition of specific repair pathways may selectively induce tumor cells to undergo apoptosis.
Below, we discuss the major pathways associated with the repair of DNA damage induced by anticancer agents, as well as some of the many proteins that can be targeted to increase the efficacy of existing antineoplastic drugs. We have focused specifically on the proteins that are enzymatically involved in the repair of lesions. As mentioned above, it is reassuring that the genetic ablation of many of these repair proteins has no obvious phenotype, except when animals or cells are challenged with DNA-damaging agents. It is important to note that the interaction between unrepaired DNA lesions and the biological pathways that they eventually trigger is not discussed in any detail (Figure 3). Clearly, tumor cells may respond very differently to lesions due to differences in, for example, cell cycle checkpoints and apoptotic pathways, and there are already small-molecule inhibitors that target some of these pathways. With the development of DNA-repair inhibitors and research to understand how they can be used in combination with drugs that target these pathways, it may be possible to develop a systems biology approach to selectively kill tumor cells and eliminate resistance.
Figure 3. Biological effects of DNA damage and DNA repair.

DNA damage blocks processivity of DNA polymerases, which induces cell cycle arrest due to the formation of stalled replication forks. DNA repair can occur during cell cycle arrest, which is a survival pathway, but persistent stalled replication forks eventually give rise to single-strand and double-strand breaks that lead to cell death by apoptosis. Persistent lesions can result in mutations and possibly secondary cancers due to error prone repair or translesion synthesis by low-fidelity polymerases. DNA repair can also lead to cell death due to overactivation of PARP, which results in depletion of cellular levels of ATP. The processes in green are desired effects for successful chemotherapy and red are undesirable effects.
Potential drug targets to overcome
Resistance to DNA damage
The four major routes to repair DNA damage induced by alkylating anticancer agents are discussed below (Figure 4). The determination of the predominant pathway in response to DNA damage depends on the type and extent of damage but, in most cases, there is some overlap because of the diversity of DNA lesions that are generated by DNA-alkylating agents. This review focuses on proteins that are enzymatically involved in the removal of DNA damage created by anticancer drugs. There are more than 100 other proteins that are associated with DNA-repair pathways and each may constitute a viable target for inhibitors designed to sensitize cells to DNA-damaging drugs.
Figure 4. The four major pathways for the repair of DNA damaged by anticancer drugs.

BER: in the initial step the adducted base is excised off the DNA backbone, leaving an abasic site that is sequentially processed by a number of enzymes (see Figure 5). NER: the lesion, which generally has a helix distorting effect, is initially recognized by proteins that then recruit endonucleases that cut a segment out of DNA including the lesion. The gap is filled in by DNA polymerase and the ends ligated. Direct reversal: the alkyl lesion is directly transferred from the DNA nucleobases to the repair protein in a one-step suicide reaction. ICL repair: in the initial step the crosslink is recognized by enzymes associated with NER and then one end of the crosslink is unhooked by endonucleases. The remaining lesion (shown in the figure) is then bypassed by a translesion polymerase. The remaining monofunctional lesion is then processed as in NER. If the repair occurs during DNA replication, potential errors made during the error-prone bypass step can be corrected by homologous recombination in which a strand from the sister chromatid serves as an undamaged template.
BER: Base excision repair; ICL: Interstrand crosslink; NER: Nucleotide excision repair; X: Monofunctional or crosslinked lesion.
Base excision repair
Base excision repair (BER) removes specific types of damaged bases from DNA. The specificity is a function of the glycosylase protein that initially ‘recognizes’ the lesion and then excises it off the DNA backbone. There are 11 human DNA-repair glycosylases associated with BER but only a few are involved in recognition of lesions caused by anticancer drugs. After the initial stage of excision to afford an abasic site, the remaining enzymes and steps in the BER pathway are universal; that is, it does not apparently matter how the abasic site originated. Therefore, specificity for inhibition of the repair of drug-generated DNA lesions has to be achieved at the glycosylase stage of BER.
DNA glycosylases
As the name suggests, the BER pathway involves the initial recognition and removal of a single modified base by a DNA glycosylase (Figure 5). There are two distinct classes of DNA glycosylases; monofunctional, which cleave the modified base off the DNA leaving an abasic lesion, and bifunctional, which both remove the lesion and then excise the DNA at the abasic site [50]. While many of the DNA glycosylases repair lesions that predominately form from endogenous reactions of cellular metabolites with DNA, for example 8-oxoguanine and 5-hydroxycytosine, which are generated by the production of reactive oxygen species [51,52], there are proteins that efficiently remove some of the alkylation lesions produced by anticancer drugs. Specifically, N-methylpurine-DNA glycosylase (MPG) efficiently excises N3-alkyladenine and N7-alkylguanine adducts from DNA, although it also processes hypoxanthine, which is derived from the deamination of adenine [53,54], and 1,N6-ethenoadenine, which is created from the reaction of adenine with lipid peroxidation byproducts [54]. The N3-alkylA adduct blocks DNA replication and is cytotoxic [55,56], while N7-methylG appears innocuous to polymerization [57]. The N3-alkylA lesion is generated in significant amounts by many DNA-alkylating drugs, so its efficient removal by MPG constitutes a mode of resistance. In fact, the repair of N3-alkylA lesions is normally so efficient and rapid in cells and animals that the lesion can only be isolated using high concentrations of alkylating agent and at short incubation times. However, in high-dose chemotherapy, or in cells with low MPG levels, the cytotoxicity of the lesion becomes apparent [57,58].
Figure 5. Short patch base excision repair involving sequential processing of lesion by N-methylpurine-DNA glycosylase, APE-1, Pol β and DNA ligase.

In long patch repair (not shown), the polymerase fills in the gap and displaces a number of bases (2–10) leaving a dangling flap, which is digested by FEN-1.
MPG: N-methylpurine-DNA glycosylase.
MPG inhibitors
The crystal structure of MPG in the presence of DNA and a hypoxanthine substrate is shown in Figure 6 [59]. There are relatively few reports on small-molecule inhibitors of the glycosylase [60]; the most active inhibitors in this study were Trp-P-1 and 2-thioxanthine (Figure 7) with the latter having an IC50 of 76 μM against human MPG. Both compounds clearly resemble potential substrates for MPG and presumably bind in the enzyme’s active site. A number of other groups have conducted extensive screening, without success, to identify potent and selective MPG inhibitors. The lack of more potent glycosylase inhibitors is probably because these proteins have electropositive surfaces that nonspecifically electrostatically interact with DNA through salt bridges to the phosphate backbone while they scan for damaged bases. The higher proportion of disordered regions in glycosylases, which may contribute to initial steps of recognition and binding [61,62], also pose a challenge to structure-based rational drug design, due to implied changes in global topology of the glycosylase that are not yet well understood. In addition, the active site, which is buried in the protein, is relatively small. The modified bases are excised off the deoxyribose ring upon extrusion from the base pair stack into the active site, which is a scenario common to all DNA glycosylases. A tyrosine residue stacks in the void in the DNA left by the lesion when it enters the active site. The modified bases make relatively few enthalpic interactions with the active site, which sterically blocks nonsubstrate bases from entering. This prevents the inadvertent excision of canonical bases and the formation of AP sites. In fact, overexpression of MPG is a weak mutator phenotype, presumably due to an increase in the background level removal of natural bases [63]. A potential alternative to reduce BER-mediated repair is to interfere with the displacement of the glycosylase from the DNA after the base excision reaction (Figure 5). However, exactly how the displacement of the glycosylase by the next enzyme in the BER pathway (i.e., APE-1; Figure 5) occurs is not well understood, but blocking this step would involve interfering with a protein–protein interaction. Because the molecular interactions between the different glycosylases and APE-1 have not been determined, and because of the challenges in targeting protein–protein binding sites with small-molecule inhibitors, there has been no reported effort to block the interaction between the different proteins in the BER pathway.
Figure 6. N-methylpurine-DNA glycosylase.
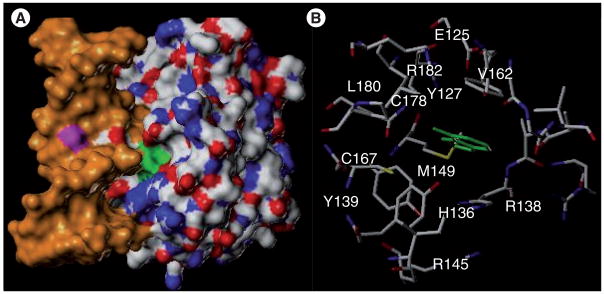
(A) Structure of human N-methylpurine-DNA glycosylase in the presence of DNA (orange) and the extrahelical hypoxanthine modification (green) buried in the enzyme’s active site. (B) View of hypoxanthine (green) substrate in active site of N-methylpurine-DNA glycosylase.
Adapted from PDB entry: 1EWN [58].
Figure 7.
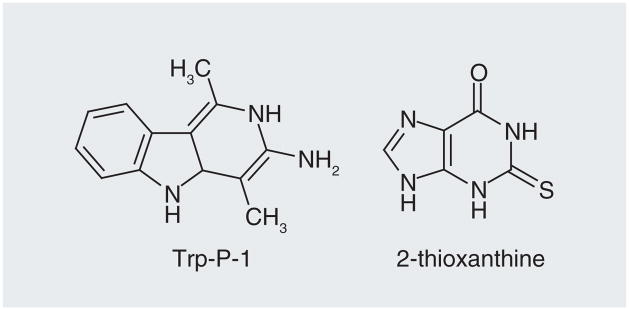
Small-molecule inhibitors of N-methylpurine-DNA glycosylase.
AP endonuclease
All of the DNA glycosylases, including MPG, produce an abasic site as a result of their initial excision of damaged bases (Figure 5). An abasic site, which has no coding information, can also be formed by ‘spontaneous’ hydrolysis of an unmodified base. The rates of ‘spontaneous’ depurination/depyrimidination at near neutral pH and physiological temperature are quite slow. However, the formation of a charged nucleic acid base due to alkylation on a ring nitrogen greatly accelerates the hydrolysis of the glycosidic C-N bond [64]. It is estimated that there are 10,000 abasic sites formed per day per cell without any treatment [65,66]. Apparently for this reason, neither animals nor cells can survive without the main enzyme (APE-1) that repairs abasic sites [67]. The lethality of APE-1 knockouts has been linked to loss of the repair activity and the mechanism of cell death appears to involve apoptosis [68–70]. There is also evidence that APE-1 expression can be induced by genotoxic agents, including cancer drugs [10]. The lethality of clinically used anticancer treatments can be enhanced by a temporal decrease in APE-1 using antisense technology [35,71,72]. Therefore, temporarily decreasing APE-1 activity could be an important adjuvant to DNA-damaging antineoplastic agents. In addition to its endonuclease activity, APE-1 also plays a role as a redox co-factor for a number of transcription factors [73–75]. The cellular effects of this redox activity are still not fully understood [76].
APE-1 inhibitors
The structure in the region around the active site of hAPE-1 with DNA containing a stable tetrahydrofuran abasic site is shown in Figure 8A [77]. As is the case with the binding of many of the repair enzymes, the DNA molecule is significantly distorted from linearity, the lesion is extrahelical and the protein introduces a side chain (Arg-177 for APE-1) into the void left in the base stack. Based upon the structures of the protein without and with DNA containing a lesion, rearrangement of this arginine residue into the DNA stack is a major structural change that takes place in the protein when APE-1 binds to DNA with a substrate. Therefore, the molecular modeling to design active site inhibitors of APE-1 requires some attention to the dynamic movement of the loop with the arginine side chain. The enzymatic center of APE-1, where a bound Mg2+ activates a water molecule for the nucleophilic attack on the 5′-phosphate, lies in a pocket that is deep inside the protein and is lined with residues that can form H-bonds, π-cation and hydrophobic interactions with potential inhibitor ligands (Figure 8). Most of the nonlesion interactions between the protein and DNA are salt bridges with the phosphate backbone on both strands in the vicinity of the lesion.
Figure 8. Human APE-1 protein.
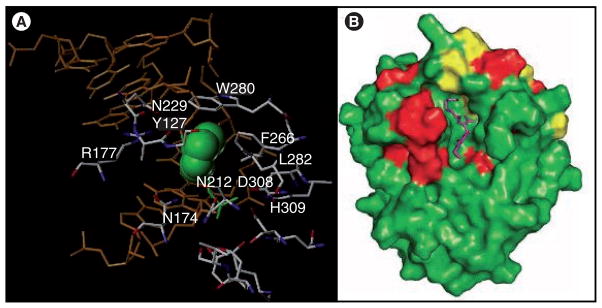
(A) The amino acids that flank the enzymatic site of APE-1 are shown along with the DNA (orange) and the abasic site (green) adapted from PDB entry 1DEW [76]. (B) The proposed binding of E3330 (purple; see Figure 9 for structure) to APE-1 based on NMR studies (red and yellow represent amino acid residues whose chemical shifts change by >0.01 and >0.02 ppm, respectively, when E3330 is present) [84].
A number of small-molecule inhibitors of APE-1 have been reported (Figure 9 shows some examples along with their IC50 values) [78–82]. Many of the compounds identified are based on dianionic molecules (e.g., 3-CCPPA and 2-BBIDA) that may mimic the diphosphate linkage that is embedded in the DNA substrate structure [78]. In a search for APE-1 inhibitors to enhance the cytotoxicity of alkylating compounds in melanoma and glioma cells, a number of structurally related compounds were identified, including ‘compound 4’ (Figure 9) [79]. The molecule has low micromolar activity against APE-1 and enhances the cytotoxicity of both methyl methanesulfonate and temozolomide in glioma and melanoma cell lines. However, the level of abasic sites detected in the cells, which should be a biomarker of APE-1 inhibition, did not indicate any synergistic effect between ‘compound 4’ and the alkylating agent, methyl methanesulfonate. Both the APE-1 inhibitor and methyl methanesulfonate alone increased the number of abasic sites by approximately 2.7-fold but together the increase was only fourfold. This raises the question regarding the mechanism of action for this compound. A benzo[b][1,8]naphthyridine (Figure 9; AR03) molecule, which was isolated after a large screening effort, in vitro showed a 2-μM IC50 against APE-1 activity and some potentiation when used with temozolomide and methyl methanesulfonate in cell culture [80]. Included in this class of APE-1 inhibitors is a series of arylstibinic acids (Figure 9; compounds 13755 and 13743), which although potent at nanomolar concentrations in biochemical experiments, lacked activity in cells [81]. Lucanthone, which inhibits APE-1/REF-1 activity and binds to the protein [82], also interacts with other cellular targets, including DNA via intercalation [83], so the mechanism of action remains uncertain. E3330 (Figure 9), which inhibits APE-1 endonuclease activity at low micromolar concentrations, was originally identified as a specific inhibitor of APE-1 redox activity [84]. However, NMR experiments have recently shown that the quinone binds to the active site of APE-1 (Figure 8B) and it also blocks the endonuclease activity [85]. We have confirmed that E3330 is an endonuclease inhibitor in two biochemical assays with an IC50 in the low micromolar range [Srinivasan A, Gold B, unpublished data]. Treatment of T98G glioma cells with E3330 also caused a modest increase in the number of abasic sites based on an aldehyde colorimetric assay [Srinivasan A, Gold B, unpublished data]. Because of these recent results, it has been suggested that E3330 bound in the catalytic site associated with endonuclease activity also blocks the binding of transcription factors to APE-1. If this overlap of activities is common, it may be difficult to discover the mechanism(s) of action of small-molecule APE-1 inhibitors. Regardless, there is much interest in developing APE-1 inhibitors for clinical use. Another approach to block APE-1 activity is to chemically modify the AP site using compounds such as methoxyamine. Methoxyamine forms an imine with the aldehyde group in the ring-open form of the AP site (Figure 10), which prevents the APE-1-mediated cleavage of the AP site [40,86]. Although methoxy-amine will react with any aldehyde (or ketone), it is being clinically evaluated in a Phase I trial as an adjunct therapy in combination with the DNA-methylating agent temozolomide (Figure 2). The fact that this simple reactive compound, which has no structural specificity for abasic sites, is being tested in humans illustrates the desperate need to block resistance to DNA-alkylating agents.
Figure 9.

APE-1 inhibitors and their reported in vitro IC50 values.
Figure 10.

Abasic lesion; cyclic and ring-opened form.
The interactions of APE-1 with other repair proteins, such as DNA glycosylases, Pol β and PARP, are also topics of interest for medicinal chemistry. APE is proposed to associate with MPG and in doing so influences base excision turnover [87,88]. The association of APE with Pol β is expected, considering that the latter enzyme follows the phosphodiesterase activity of APE [89]. PARP has been known to compete with APE for binding to the same APE-cleaved BER intermediate [90]. The activities of Flap endonuclease and DNA ligase are also proposed to be coordinated by APE activity [91]. While all these interactions are of general academic interest to understand repair pathways and the interacting domains/surfaces, they also provide unique opportunities to design small molecules aimed at blocking these interactions so that the substrate ‘hand-off ‘ does not proceed, thereby arresting the process of lesion repair.
Pol β
After APE-1 has removed the abasic lesion, the remaining break in the DNA backbone, with 3′-hydroxyl and 5′-deoxyribose-phosphate termini, is processed by Pol β. In short patch repair, the enzyme trims the 5′-deoxyribose terminus to a phosphate via its lyase activity and inserts the appropriate complementary base into the vacant position via its polymerase activity (Figure 5). In long patch repair, the enzyme performs a strand-displacing synthesis, leaving an extended nucleotide flap that is degraded by FEN1 endonuclease [92]. Mice deficient in Pol β die at birth and show neurogenic developmental deficiencies [93]. However, Pol β null cells are viable, although sensitive to DNA-alkylating agents [94]. Overexpression of Pol β has been observed in a number of cancers but it is not certain that it is required for tumor maintenance [95]. In addition, a variety of human tumors often express mutated forms of Pol β, including variants that have reduced BER activity or reduced fidelity [96]. Some of the mutants appear to be dominant negative in that they maintain their DNA-binding affinity but are defective in the repair functionality [97]. The crystal structures of Pol β with a variety of DNA substrates have been solved [98].
Pol β inhibitors
Because of the sensitivity of Pol β-defective cells to alkylating agents, there have been several reports of molecules that inhibit both the lyase and/or polymerase activities of Pol β (Figure 5). The natural products oleanolic acid, edgeworin, harbinatic acid and myristinin A (Figure 11) have low micromolar activity in biochemical assays, little toxicity on their own and weakly potentiate bleomycin cytotoxicity [99–101]. Using the crystal structure as a guide (Figure 12) [98], Jaiswal et al. more recently identified a small-molecule inhibitor (NSC666715) of Pol β that dramatically potentiates the cytotoxicity of temozolomide in vitro and in vivo, and even had an effect on MMR-deficient cells [102]. Cells that are defective in MMR are generally resistant to temozolomide (see below). NSC666715 interacts with Pol β in such a way that it blocks both short and long patch repair but does not affect other BER proteins (i.e., APE-1 and ligase). Another molecule identified by the same research group that is effective at inhibiting Pol β is the deoxynucleotide analogue NSC124854 (Figure 11) [103]. It causes a concentration-dependent decrease in the strand-displacement synthesis activity of Pol β with an IC50 value of 5.3 μM [103]. Both NSC666715 and NCS124854 are proposed to bind in the same region on Pol β based upon molecular modeling studies (Figure 12). This region of Pol β is where the tumor suppressor adenomatous polyposis coli (APC) protein binds [104]. APC expression is induced by DNA-alkylating agents [105]. Mutations in the APC gene occur early in the neoplastic transformation of the colon [106], and APC blocks Pol β-mediated BER pathways and increases the sensitivity of cells in vitro to alkylation-induced DNA damage [107]. As expected, APC gene mutant cells become more resistant to alkylating treatment [108].
Figure 11. Inhibitors of DNA polymerase b and their reported IC50 values in parentheses.

Different assays were used to generate the IC50 values.
Figure 12. Models of inhibitors docked with Pol β.
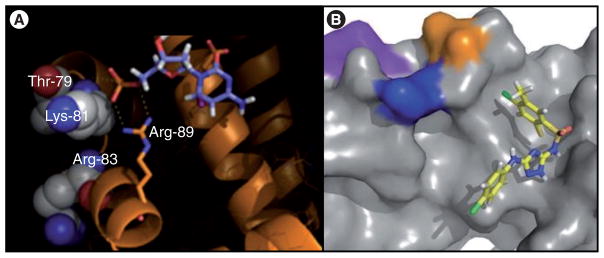
(A) NSC124854 (Figure 11) docked to Pol β via salt bridge between phosphate and Arg residue and hydrophobic contacts with heterocycle [102]; (B) NSC666715 docked in the same region of the protein (Lys-81: blue; Thr-79: orange; Arg-83: purple) [101].
NSC124854 was evaluated in a xenograft model of colorectal cancer employing MMR-deficient and -proficient cell lines in the absence and presence of the methylating agent temozolomide (20 mg/kg). This is well below the maximum tolerated dose of temozolomide. NSC124854 was given intraperitoneally at 10 mg/kg, which is tenfold lower than the in vitro IC50 dose [103]. NSC124854 and temozolomide were given for 5 consecutive days and the mice maintained for approximately 6 weeks. Both compounds independently reduced in vivo tumor growth, with NSC124854 unexpectedly being more effective than temozolomide. Both compounds in combination lowered the IC50 of temozolomide in cell culture experiments. However, there was no synergistic effect when the two molecules were combined in the animal tumor model experiment. The observation that NSC124854 was effective by itself in the mouse xenograft model in the absence of alkylating agent implies that the molecule may have other targets or that the formation of abasic sites in the tumor line may require a high constitute level of BER. Clearly, the challenges of using two drugs simultaneously in vivo will require additional studies.
FEN-1
FEN-1, a protein required for normal development [109,110], plays an important role in the long patch BER of alkylation damage DNA in addition to the cleavage of Okazaki fragments that arise during replication.
FEN-1 inhibitors
In order to potentiate the therapeutic efficacy of DNA-damaging drugs, a series of nanomolar FEN-1 inhibitors, which have some specificity for this endonuclease, have been identified [111]. These inhibitors have a 3-hydroxydihydropyrimidine-2,4(1H,3H ) -dione core and IC50 values in the nanomolar range. The compounds also increase the toxicity of methyl methanesulfonate in cell culture. However, the one compound assayed in cells did not appear to show a clear dose-dependent effect.
DNA ligase
The last enzymatic step in BER (Figure 5), as well as in other DNA-repair pathways, is the ligation of the 5′-phosphate and 3′-hydroxy groups at the nick (i.e., single strand break) by ligase-I or ligase-III associated with XRCC1. The XRCC1 appears to act as a scaffold that facilitates the repair process but does not perform an enzymatic function. The crystal structure of ligase-I in complex with a nicked DNA template shows that the enzyme distorts the DNA and exposes the nick that is the target of ligation [112]. The DNA-binding domain of the protein completely encircles the DNA and stabilizes the distorted structure that is required to join the break.
DNA-ligase inhibitors
Small molecules were initially screened in silico using the crystal structure (Figure 13) [112]. The target for binding was the surface on the DNA-binding domain of the protein. Molecules identified in the screen were then tested in a DNA-joining assay and in binding assays using a nonligatable template. Four low micromolar inhibitors are shown in Figure 14 [113]. Although all of the compounds inhibited ligase activity, their mechanisms of action varied [114]. Compound 197, which was the most active, is a dicarboxylic acid and is reminiscent of the APE-1 inhibitors that mimic the DNA backbone (Figure 9). Compounds 67 and 189 were competitive inhibitors and cytotoxic. Compound 82 was a noncompetitive inhibitor and cytostatic. The cytostatic activity was attributed to activation of a G1-S cell cycle checkpoint. A notable observation was that compounds 67 and 189 markedly enhanced the toxicity of the DNA-methylating agent, methyl methanesulfonate, and ionizing radiation in tumor cells but not in ‘normal’ breast epithelial cells. The authors measured the levels of ligase-I activity in the cancer and normal cells and found them to be higher in the former, as previously observed in other cancers [115]. The question has been raised whether the tumor selectivity of these inhibitors can be attributed mainly to overexpression of ligase-I in tumor cells. Whether high ligase-I activity is required for these tumors is not clear, but the data indicate that the amount of DNA ligase-I enzyme in malignant tumors is higher than that in benign normal tissues and peripheral blood lymphocytes. The level of DNA ligase-I in human tumors grown in nude mice was also shown to be very high. Therefore, the selective activity of the molecules in tumor cells makes this a potentially exciting target for additional medicinal chemistry.
Figure 13. DNA Ligase-1.
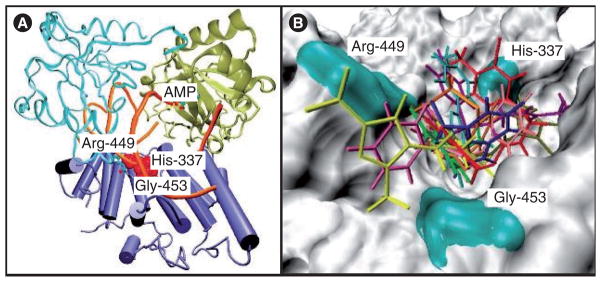
(A) Structure of the DNA-binding domain of DNA ligase-I with the predicted binding site in red and residues His-337, Arg-449 and Gly-453 that constitute the potential binding pocket. (B) Predicted docking of some of the ligase inhibitors (Figure 14) with the DNA-binding domain [112].
Figure 14.

DNA human ligase inhibitors against ligase-I and -III with in vitro IC50 values (μM) in parasentheses.
PARP-1
The final protein that we will discuss in connection with BER is PARP-1; although PARP actually does not play a direct enzymatic role in repair. The PARP proteins are nuclear enzymes that bind as homodimers to both single- and double-strand breaks [116–119]. This binding activates its catalytic activity that transfers ADP-ribose subunits derived from NAD+ onto a number of protein substrates. Depending on the level of strand breaks, this process can deplete cells of NAD+, which results in low ATP levels and rapid cell death by necrosis [120,121]. In PARP null mice or depleted cells, the death pathway switches from necrosis to apoptosis [122–124]. Regardless of its role in BER, the development of small-molecule inhibitors of PARP has received significant attention since they can produce a build up of toxic single-strand breaks by trapping PARP on the break site [125]. There is also a synthetic lethality generated by PARP inhibitors in tumors that express defective mutant BRCA tumor suppressor proteins [126,127]. The BRCA proteins are involved in one pathway for repair of DNA double-strand breaks by homologous recombination [128–130]. These toxic lesions persist in PARP-inhibited BRCA-mutant cells. The synthetic lethal effect induced by PARP inhibitors in BRCA-mutated cells is the focus of clinical trials, since PARP inhibitors have virtually no toxicity in BRCA wild-type cells.
PARP-1 inhibitors
There has been a significant effort in the development of PARP inhibitors [131–134], and there are 88 clinical trials listed that have been completed or are ongoing [203]. Some examples of the inhibitors are shown in Figure 15. Sanofi’s iniparib (BSI-201) is presently in a Phase I clinical trial in combination with temozolomide for the treatment of newly diagnosed malignant glioma and in an ongoing Phase III trial in combination with gemcitabine and carboplatin treatment in patients with previously untreated stage IV squamous non-small-cell lung cancer. However, an iniparib Phase III trial in triple negative (negative for estrogen receptor, progesterone receptor and HER2) breast cancer did not reach its goal [204]. Another PARP inhibitor, olaparib (AZD2281), is being used in combination with carboplatin for refractory breast and ovarian cancer in a Phase I trial [205]. However, AstraZeneca does not plan on pursuing this PARP inhibitor in hereditary BRCA-1 and -2-positive breast cancers, and will focus their efforts on ovarian cancer. Pfizer’s rucaparib (PF-01367338) molecule is in a Phase II trial with carboplatin for the treatment of advanced breast and ovarian tumors [206]. Regardless of the less-than-spectacular results to date, based on the number of PARP-1 inhibitors that are under development, it seems likely that even more effective and selective drugs will soon be in clinical trials. Only then will the clinical value of PARP inhibitors be determined.
Figure 15.

Nanomolar inhibitors of PARP-1 activity.
Nucleotide excision repair
Global nucleotide excision repair (NER) is fundamentally different from BER in that it lacks the same level of structure-based specificity, and repair involves excision of a long stretch of DNA containing the lesion by the coordinated action of multiple enzymes; more than 15 have been assigned a role in NER (Figure 4). The key element in the initiation of NER is the disruption of the canonical Watson–Crick helix, due to lesions that distort DNA. Most alkylating agents do not yield adducts that are substrates for NER. However, there are notable exceptions, including the cisplatin drugs that generate intrastrand crosslinks, protein–DNA crosslinks and mono-functional DNA lesions [135]. For the cisplatin intrastrand lesions, NER excises a fragment that contains both modified bases, leaving a gap that is eventually filled in by polymerase. One mechanism associated with resistance to cisplatin drugs is overexpression of NER proteins, specifically ERCC1-XPF [136], which acts as a single-stranded endonuclease in excising the stretch of DNA containing the damage. This effect on cisplatin toxicity was confirmed using antisense technology to decrease the levels of ERCC1 [137]. NER is also involved in a transcription-coupled process that allows RNA synthesis to proceed at lesion-halting modifications.
Inhibitors
A fluorinated epipodophylloid molecule, F-11782 (Figure 16), which inhibits topoisomerases I and II, is also an inhibitor of NER [138]. F-11782 inhibits the incision step in repair and the target may involve the ERCC1-XPF or XPG endonuclease activity associated with NER removal of the strand containing the damage. It was suggested that F-11782, in combination with DNA cross-linking agents would be a candidate for future clinical trials. Small-molecule inhibitors have also been reported for XPA (Figure 16), which is a factor associated with initial recognition of DNA damage in the NER pathway [139]. It is of interest that the molecules have a dianionic flavor similar to other repair inhibitors, including those that target APE-1 (Figure 9) and ligase (Figure 14). It may well be that these small dianions with different linker regions structurally mimic the DNA diphosphate backbone, electrostatically interact with DNA via salt bridges and physically block access of the specific repair protein to DNA. It would be interesting to determine if some of the molecules identified in a screen for XPA inhibitors would also inhibit APE-1.
Figure 16. Small-molecule inhibitors of nucleotide excision repair.

(A) Structure of nucleotide excision repair inhibitor F-11782 (formulated as the N-methyl-D-glucamine salt) and XPA inhibitors (B & C).
Another NER target that has attracted interest is replication protein A, which is necessary for formation of the pre-incision complex that is required for NER removal of damaged DNA [140]. The repair of cisplatin intrastrand cross-links was shown to be inhibited by gemcitabine (Figure 17), which has a number of mechanisms of action, and the use of gemcitabine has been explored in a carboplatin Phase II trial in platinum-resistant ovarian cancer [141].
Figure 17.
Gemcitabine, which inhibits repair of cisplatin intrastrand crosslinks.
Interstrand crosslink repair
Interstrand crosslinks (ICL) formed from bifunctional alkylating agents (e.g., nitrogen mustards, cisplatin, nitrosoureas and mito-mycin C) physically prevent the complementary DNA strands from separating and are extremely toxic. However, even these lesions can be repaired by an excision reaction on both sides of one of the crosslinked bases to afford an unhooked intermediate, translesion synthesis past the unhooked intermediate by specialized polymerases, NER nonremoval of the unhooked monofunctional adduct and homologous recombination (Figure 18) [142–144]. As expected, the sensitivity to crosslinking drugs has been correlated with the efficiency of ICL repair [145].
Figure 18. Pathway for interstrand crosslink repair.

(A) The interstrand crosslink represents a block to polymerization; (B) one end of the crosslink is excised to yield a double strand break terminus, a gap and a lesion connected to the excised DNA; (C) the remaining unhooked monofunctional lesion is bypassed by translesion synthesis polymerase; (D) the remaining unhooked lesion is excised by nucleotide excision repair proteins; (E) polymerase fills in the gap in the DNA (F) homologous recombination is used to provide a template for DNA synthesis; (G) after resolution of the Holiday junction the DNA is fully repaired.
Inhibitors
At this time, there have been no reports on small-molecule inhibitors that selectively affect ICL repair. Because many of the same proteins associated with NER are involved in ICL repair, some of the inhibitors developed for NER may also be effective against ICL repair. Finally, the unique translesion synthesis polymerases that can bypass damaged bases, albeit often with reduced fidelity, constitute another potential target to inhibit ICL repair.
Direct reversal repair
Direct reversal repair by alkylguanine DNA-alkyltransferase (AGT) is highly specific for the repair of O6-alkylguanine lesions, which are commonly produced during exposure to alkylating agent chemotherapy by the nitrosoureas and triazenes (Figure 2). The toxicity of this type of monofunctional lesion requires functional MMR that performs a futile cycle of removing and replacing a natural base opposite the O6-alkylG lesion. Eventually, this process leads to strand breaks and cell death by apoptosis [146]. It should be noted that some O6-alkylG lesions, such as those formed from chloroethylating agents, also exert their toxicity via DNA crosslinking [147]. Tumors that are tolerant to drugs that produce O6-alkylguaine lesions (e.g., temozolomide) are often defective in MMR and/or overexpress the AGT protein, which transfers the alkyl group from the imidate ester lesion onto the protein in a suicide reaction with a turnover number of unity. The question has been raised whether tumors are tolerant of temozolomide because they become defective in MMR or whether they are tolerant because they are MMR defective. This is a chicken-or-egg question. However, it is likely that MMR-defective cells exist in any sizable tumor mass and they may be selected for by the chemotherapy treatment with temozolomide.
Inhibitors
There are many inhibitors of the AGT protein and they are all based on analogues that closely resemble the imidate ester substrate; for example, 2-amino-6-[(4-bromo-2-thienyl) methoxy]-9H-purine (lomeguatrib) (Figure 19), O6-benzylguanine and related O6-alkylguanine derivatives [148]. There are 32 clinical trials listed for O6-benzylguanine [207]. In all of these studies, the inhibitors are being used as an adjuvant to a DNA alkylating drug; for example, temozolomide and carmustine [149]. An example of the combination of AGT inhibitor with an alkylating agent is the clinical trial with 2-amino-6-[(4-bromo-2-thienyl)methoxy]-9H-purine in combination with temozolomide in metastatic colon cancer patients [150]. In spite of the trial obtaining the goal of depleting AGT activity, no beneficial response was observed. The lack of response in this trial could be attributed to the decision to use temozolomide to treat colon cancer and/or the presence of MMR-deficient cells in the tumors that are resistant to the toxicity of 6-methylgua-nine lesions produced by temozolomide. Another reason for the lack of efficacy in this trial, and probably for many others, is the late stage of the cancer in the enrolled patients. It is conceivable that early-stage cancers with less genetic diversity may be more sensitive to specific therapies than advanced cancers that are genetically and epigenetically more heterogeneous.
Figure 19.

Inhibitors of O6- alkylguanine alkyltransferase.
In terms of adverse effects, the main difference between temozolomide without and with 2-amino-6-[(4-bromo-2-thienyl)methoxy]-9H-purine was a more pronounced myelosuppressive. This raises the issue of specificity; if there is no differentiation between cancer cells and other proliferating cells, the difference in the therapeutic index will remain constant. To combat this problem, clinical trials have explored the use of autologous infusion of AGT-transduced hematopoietic progenitors into patients with gliomas [207]. This would provide the sensitization of tumors to alkylating agents but protect the hematopoietic system.
At this time, there are no viable gene therapy strategies to restore MMR activity, which is required for tumor cells to be sensitive to O6-alkylguanine adducts formed by many DNA-alkylating agents.
Future perspective: targeting DNA-repair pathways
We have presented an overview of many of the enzymes that can be targeted to directly interfere with normal DNA repair in order to potentiate existing DNA-damaging anticancer drugs and to overcome tumor resistance that results from the upregulation of tumor cell DNA-repair capacity. In addition, there are a large number of potential protein targets that do not directly participate in DNA repair but transmit the signals induced in cells as a consequence of DNA damage, which eventually leads to cell growth arrest and/or death. These are also attractive drug targets.
The rationale for combining DNA-damaging drugs with molecules that will specifically inhibit the repair of the DNA lesions that the drugs generate appears well founded. There are numerous in vitro and in vivo experiments in which proteins have been modulated genetically or by antisense or siRNA approaches, which demonstrate the potentiation of anticancer drugs by compromising DNA repair. Moreover, the deletion of many DNA-repair proteins does not cause toxicity in untreated cells or create a discernable phenotype in unchallenged null animals. Therefore, many of the molecular target that have been structurally characterized, which we discussed, have been validated and there are well-defined biochemical and cellular assays, animal models and biomarkers to evaluate new inhibitor compounds. The ongoing clinical trials of several DNA-repair inhibitors in combination with alkylating drugs will provide additional insight into the potential of the approach, as well as the limitations. In this regard, the development of biomarkers to establish the appropriate patient population to target and the beneficial effect of the inhibitors is paramount. Unfortunately, the late-stage cancer patients enrolled in many of the clinical trials may confound an objective analysis of the clinical efficacy of the compounds.
The design of drugs to target DNA-repair proteins involves several significant technical challenges. Many of these enzymes electrostatically associate with DNA via basic amino acid sidechains that weakly and nonspecifically interact with the polyanionic phosphate backbone. While this allows the proteins that initially ‘find’ damaged bases to conduct an efficient 1D search, the requirement for numerous electrostatic contacts is not an attractive strategy in small-molecule drug design. In addition, the active sites for many repair proteins are relatively small and buried deep inside the protein and are lined with amino acid residues that present limited potential for selective stabilizing enthalpic interactions. Regardless, compounds with greater than micromolar IC50 values have been identified for many repair targets. Some of the compounds show the predicted synergistic cytotoxic effect when used with drugs that produce the relevant DNA lesions. In some cases, but not all, the appropriate biomarkers indicate that the mechanism of action is indeed related to DNA repair. Based upon many of the reported structures, converting these compounds into drugs will require a significant amount of medicinal chemistry engineering.
The ultimate challenge for the development of therapeutic molecules that target DNA repair is not a new one. It is the ability to selectively differentiate between tumor cells and normal dividing cells. There is already evidence in the clinical combination of DNA-methylating agents and AGT inhibitors that this problem will need to be addressed in order to achieve an increase in the therapeutic index of existing DNA-damaging drugs. Related to this issue of selectivity, research needs to be done to catalog the susceptibility of tumor stem cells to DNA-repair inhibitors since this is the critical cell population that must be eliminated for successful therapy.
Clearly, the upregulation of DNA-repair pathways through genetic and/or epigenetic changes is only one mechanism that allows tumor cells to avoid the toxicity of DNA-damaging agents. Regardless of the challenges and the potential pitfalls, the need to develop approaches to overcome drug resistance remains absolutely critical to improving cancer survival.
Executive summary.
Background
The development of drugs to prevent or limit tumor cell resistance is critical to improve long-term cancer survival.
Cancer resistance
Many anticancer drugs initially work very effectively but eventually fail, because tumors reoccur due to drug resistance.
Rather than the continuous cycle of developing new drugs, including those that specifically target cancers via nontoxic mechanism, and watching them fail due to resistance, we need to predict potential pathways for resistance and develop drugs to block them.
Resistant cells may not be readily observable due to their number, but it should be assumed that they are present in tumors and this should be reflected in treatment therapies.
Tumor resistance to DNA damage
The efficient repair of the DNA damage induced by genotoxic drugs (and ionizing radiation) by populations of cells within a tumor is a druggable target to address drug resistance.
The targets to inhibit DNA repair should be chosen to reflect the nature of the lesions induced by the drugs used in chemotherapy and the potential resistance pathways.
DNA repair inhibitors
The screening of existing chemical libraries has already provided many interesting candidate molecules that efficiently and selectively inhibit DNA repair, including some that act synergistically with DNA-damaging drugs. One example of this is the development of PARP inhibitors that create a synthetic lethal combination in breast tumor cells lacking functional BRCA-1 or -2. Unfortunately, nontarget tissue (e.g., hematopoietic system) is also sensitized to the drugs so the issue of specificity must be addressed.
Key Terms
- Resistance
Ability of selected cancer cells to survive drug treatment
- DNA repair
Error free or error prone processing of DNA damage
- Polymerase β
Key polymerase enzyme in base excision repair pathway
- DNA damage
Lesions in DNA generated by anticancer drugs
Footnotes
For reprint orders, please contact reprints@future-science.com
Financial disclosure
The authors would like to thank the NIH for support of some of the research mentioned in this manuscript. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
- 1.Weinberg RA. The Biology of Cancer. Garland Science; New York, NY, USA: 2006. [Google Scholar]
- 2.Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol. 2007;18:977–984. doi: 10.1093/annonc/mdl475. [DOI] [PubMed] [Google Scholar]
- 3.Fiszman GL, Jasnis MA. Molecular mechanisms of trastuzumab resistance in HER2 overexpressing breast cancer. Int J Breast Cancer. 2011;2011:352182. doi: 10.4061/2011/352182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molina MA, Sáez R, Ramsey EE, et al. NH2-terminal truncated HER-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clin Cancer Res. 2002;8:347–353. [PubMed] [Google Scholar]
- 5.Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002;62:4132–4141. [PubMed] [Google Scholar]
- 6.Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-non growth factor-I receptor signaling and resistance to transtuzumab (herceptin) J Natl Cancer Inst. 2001;93:1852–1857. doi: 10.1093/jnci/93.24.1852. [DOI] [PubMed] [Google Scholar]
- 7.Barok M, Isola J, Pályi-Krekk Z, et al. Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance. Mol Cancer Ther. 2007;6:2065–2072. doi: 10.1158/1535-7163.MCT-06-0766. [DOI] [PubMed] [Google Scholar]
- 8.Boeckmann L, Nickel AC, Kuschal C, et al. Temozolomide chemoresistance heterogeneity in melanoma with different treatment regimens: DNA damage accumulation contribution. Melanoma Res. 2011;21:206–216. doi: 10.1097/CMR.0b013e328345af95. [DOI] [PubMed] [Google Scholar]
- 9.Beier D, Schulz JB, Beier CP. Chemoresistance of glioblastoma cancer stem cells – much more complex than expected. Mol Cancer. 2011;10:128–138. doi: 10.1186/1476-4598-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madhusudan S, Middleton MR. The emerging role of DNA repair proteins as predictive, prognostic and therapeutic targets in cancer. Cancer Treat Rev. 2005;31:603–617. doi: 10.1016/j.ctrv.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Adhikari S, Choudhury S, Mitra PS, Dubash JJ, Sajankila SP, Roy R. Targeting base excision repair for chemosensitization. Anticancer Agents Med Chem. 2008;8:351–357. doi: 10.2174/187152008784220366. [DOI] [PubMed] [Google Scholar]
- 12.Jalal S, Earley JN, Turchi JJ. DNA repair: from genome maintenance to biomarker and therapeutic target. Clin Cancer Res. 2011;17:6973–6984. doi: 10.1158/1078-0432.CCR-11-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghabili K, Agutter PS, Ghanei M, Ansarin K, Panahi Y, Shoja MM. Sulfur mustard toxicity: history, chemistry, pharmacokinetics, and pharmacodynamics. Crit Rev Toxicol. 2011;41:384–403. doi: 10.3109/10408444.2010.541224. [DOI] [PubMed] [Google Scholar]
- 14.Kohn KW. Interstrand cross-linking of DNA by 1,3-bis(2-chloroethyl)-1-nitrosourea and other 1-(2-haloethyl)-1-nitrosoureas. Cancer Res. 1977;37:1450–1454. [PubMed] [Google Scholar]
- 15.Foye LV, Jr, Chapman CG, Willett FM, Adams WS. A preliminary study of a new alkylating agent. Cyclophosphamide. Cancer Chemother Rep. 1977;6:39–40. [PubMed] [Google Scholar]
- 16.Tokuda K, Bodell WJ. Cytotoxicity and sister chromatid exchanges in 9L cells treated with monofunctional and bifunctional nitrogen mustards. Carcinogenesis. 1987;8:1697–1701. doi: 10.1093/carcin/8.11.1697. [DOI] [PubMed] [Google Scholar]
- 17.Shrivastav N, Li D, Essigmann JM. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 2010;31:59–70. doi: 10.1093/carcin/bgp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchesi F, Turriziani M, Tortorelli G, Avvisati G, Torino F, De Vecchis L. Triazene compounds: mechanism of action and related DNA repair systems. Pharmacol Res. 2007;56:275–287. doi: 10.1016/j.phrs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Hijiya N, Hudson MM, Lensing S, et al. Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. J Am Med Assoc. 2007;297:1207–1215. doi: 10.1001/jama.297.11.1207. [DOI] [PubMed] [Google Scholar]
- 20.Hijiya N, Ness KK, Ribeiro RC, Hudson MM. Acute leukemia as a secondary malignancy in children and adolescents: current findings and issues. Cancer. 2009;115:23–35. doi: 10.1002/cncr.23988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inga A, Chen F-X, Monti P, et al. N-(2-chloroethy)-N-nitrosourea tethered to lexitropsin induces minor groove lesions at the p53 cDNA which are more cytotoxic than mutagenic. Cancer Res. 1999;59:689–695. [PubMed] [Google Scholar]
- 22.Monti PA, Campomenosi P, Iannone R, et al. Influences of base excision repair defects on the lethality and mutagenesis induced by Melex, a sequence selective N3-adenine methylating agent. J Biol Chem. 2002;277:28663–28668. doi: 10.1074/jbc.M203384200. [DOI] [PubMed] [Google Scholar]
- 23.Gold B, Fronza G. The biological effects of N3-methyladenine. J Cell Biochem. 2004;91:250–257. doi: 10.1002/jcb.10698. [DOI] [PubMed] [Google Scholar]
- 24.Bobola MS, Varadarajan S, Smith NW, et al. Human glioma cell sensitivity to the sequence-specific alkylating agent methyl-lexitropsin. Clin Cancer Res. 2007;13:612–620. doi: 10.1158/1078-0432.CCR-06-1127. [DOI] [PubMed] [Google Scholar]
- 25.Türk D, Szakács G. Relevance of multidrug resistance in the age of targeted therapy. Curr Opin Drug Discov Devel. 2009;12:246–252. [PubMed] [Google Scholar]
- 26.Baumert C, Hilgeroth A. Recent advances in the development of P-gp inhibitors. Anticancer Agents Med Chem. 2009;9:415–436. doi: 10.2174/1871520610909040415. [DOI] [PubMed] [Google Scholar]
- 27.Gangemi R, Paleari L, Orengo AM, et al. Cancer stem cells: a new paradigm for understanding tumor growth and progression and drug resistance. Curr Med Chem. 2009;16:1688–1703. doi: 10.2174/092986709788186147. [DOI] [PubMed] [Google Scholar]
- 28.Baguley BC. Multiple drug resistance mechanisms in cancer. Mol Biotechnol. 2010;46:308–316. doi: 10.1007/s12033-010-9321-2. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Powell KL, Thames HD, MacLeod MC. Detoxication of sulfur half-mustards by nucleophilic scavengers: robust activity of thiopurines. Chem Res Toxicol. 2010;23:488–496. doi: 10.1021/tx900190j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abel EL, Bubel JD, Simper MS, et al. Protection against 2-chloroethyl ethyl sulfide (CEES)-induced cytotoxicity in human keratinocytes by an inducer of the glutathione detoxification pathway. Toxicol Appl Pharmacol. 2010;255:176–183. doi: 10.1016/j.taap.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 31.Sobol RW, Watson DE, Nakamura J, et al. Mutations associated with base excision repair deficiency and methylation-induced genotoxic stress. Proc Natl Acad Sci USA. 2002;99:6860–6865. doi: 10.1073/pnas.092662499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bobola MS, Finn LS, Ellenbogen RG, et al. Apurinic/apyrimidinic endonuclease activity is associated with response to radiation and chemotherapy in medulloblastoma and primitive neuroectodermal tumors. Clin Cancer Res. 2005;11:7405–7414. doi: 10.1158/1078-0432.CCR-05-1068. [DOI] [PubMed] [Google Scholar]
- 33.Ono Y, Furuta T, Ohmoto T, Akiyama K, Seki S. Stable expression in rat glioma cells of sense and antisense nucleic acids to a human multifunctional DNA repair enzyme, APEX nuclease. Mutat Res. 1994;315:55–63. doi: 10.1016/0921-8777(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 34.Walker LJ, Craig RB, Harris AL, Hickson ID. A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress. Nucleic Acids Res. 1994;22:4884–4889. doi: 10.1093/nar/22.23.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silber JR, Bobola MS, Blank A, et al. The apurinic/apyrimidinic endonuclease activity of Ape1/Ref-1 contributes to human glioma cell resistance to alkylating agents and is elevated by oxidative stress. Clin Cancer Res. 2003;8:3008–3018. [PubMed] [Google Scholar]
- 36▪.Liu L, Gerson SL. Therapeutic impact of methoxyamine: blocking repair of abasic sites in the base excision repair pathway. Curr Opin Investig Drugs. 2004;5:623–627. Discusses the exploitation of methoxyamine, which is in clinical trials, to inhibit base excision repair in cancer chemotherapy. [PubMed] [Google Scholar]
- 37.Engelward BP, Weeda G, Wyatt MD, et al. Base excision repair deficient mice lacking the Aag DNA glycosylase. Proc Natl Acad Sci USA. 1997;94:13087–13092. doi: 10.1073/pnas.94.24.13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wirtz S, Nagel G, Eshkind L, Neurath MF, Samson LD, Kaina B. Both base excision repair and O6-methylguanine-DNA methyltransferase protect against methylation-induced colon carcinogenesis. Carcinogenesis. 2010;31:2111–2117. doi: 10.1093/carcin/bgq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kisby GE, Olivas A, Park T, et al. DNA repair modulates the vulnerability of the developing brain to alkylating agents. DNA Repair. 2009;8:400–412. doi: 10.1016/j.dnarep.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu L, Yan L, Donze JR, Gerson SL. Blockage of abasic site repair enhances antitumor efficacy of 1,3-bis-(2-chloroethyl)-1-nitrosourea in colon tumor xenografts. Mol Cancer Ther. 2003;2:1061–1066. [PubMed] [Google Scholar]
- 41.Kruse V, Rottey S, De Backer O, Van Belle S, Cocquyt V, Denys H. PARP inhibitors in oncology: a new synthetic lethal approach to cancer therapy. Acta Clin Belg. 2011;66:2–9. doi: 10.2143/ACB.66.1.2062507. [DOI] [PubMed] [Google Scholar]
- 42.Rinne M, Caldwell D, Kelley MR. Transient adenoviral N-methylpurine DNA glycosylase overexpression imparts chemotherapeutic sensitivity to human breast cancer cells. Mol Cancer Ther. 2004;3:955–967. [PubMed] [Google Scholar]
- 43.Trivedi RN, Wang XH, Jelezcova E, Goellner EM, Tang JB, Sobol RW. Human methyl purine DNA glycosylase and DNA polymerase beta expression collectively predict sensitivity to temozolomide. Mol Pharmacol. 2008;74:505–516. doi: 10.1124/mol.108.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarasin A, Kauffmann A. Overexpression of DNA repair genes is associated with metastasis: a new hypothesis. Mutat Res. 2008;659:49–55. doi: 10.1016/j.mrrev.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 45.Kauffmann A, Rosselli F, Lazar V, et al. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene. 2008;27:565–573. doi: 10.1038/sj.onc.1210700. [DOI] [PubMed] [Google Scholar]
- 46.Fan J, Robert C, Jang YY, et al. Human induced pluripotent cells resemble embryonic stem cells demonstrating enhanced levels of DNA repair and efficacy of nonhomologous end-joining. Mutat Res. 2011;713:8–17. doi: 10.1016/j.mrfmmm.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niedernhofer LJ. DNA repair is crucial for maintaining hematopoietic stem cell function. DNA Repair. 2008;7:523–529. doi: 10.1016/j.dnarep.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tichy ED, Stambrook PJ. DNA repair in murine embryonic stem cells and differentiated cells. Exp Cell Res. 2008;314:1929–1936. doi: 10.1016/j.yexcr.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tichy ED, Liang L, Deng L, et al. Mismatch and base excision repair proficiency in murine embryonic stem cells. DNA Repair. 2011;10:445–451. doi: 10.1016/j.dnarep.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477:7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 52.Martinez GR, Loureiro AP, Marques SA, et al. Oxidative and alkylating damage in DNA. Mutat Res. 2003;544:115–127. doi: 10.1016/j.mrrev.2003.05.005. [DOI] [PubMed] [Google Scholar]
- 53.Kow YW. Repair of deaminated bases in DNA. Free Radic Biol Med. 2002;33:886–893. doi: 10.1016/s0891-5849(02)00902-4. [DOI] [PubMed] [Google Scholar]
- 54.Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002;181–182:219–222. doi: 10.1016/s0300-483x(02)00448-1. [DOI] [PubMed] [Google Scholar]
- 55.Fronza G, Gold B. The biological effects of N3-methyladenine. J Cell Biochem. 2004;91:250–257. doi: 10.1002/jcb.10698. [DOI] [PubMed] [Google Scholar]
- 56.Larson K, Sahm J, Shenkar R, Strauss B. Methylation-induced blocks to in vitro DNA replication. Mutat Res. 1985;150:77–84. doi: 10.1016/0027-5107(85)90103-4. [DOI] [PubMed] [Google Scholar]
- 57.Paik J, Duncan T, Lindahl T, Sedgwick B. Sensitization of human carcinoma cells to alkylating agents by small interfering RNA suppression of 3-alkyladenine-DNA glycosylase. Cancer Res. 2005;65:10472–10477. doi: 10.1158/0008-5472.CAN-05-1495. [DOI] [PubMed] [Google Scholar]
- 58.Bobola MS, Varadarajan S, Smith NW, et al. Human glioma cell sensitivity to the sequence-specific alkylating agent methyl-lexitropsin. Clin Cancer Res. 2007;13:612–620. doi: 10.1158/1078-0432.CCR-06-1127. [DOI] [PubMed] [Google Scholar]
- 59.Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci USA. 2000;97:13573–13578. doi: 10.1073/pnas.97.25.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Speina E, Cieśla JM, Graziewicz MA, Laval J, Kazimierczuk Z, Tudek B. Inhibition of DNA repair glycosylases by base analogs and tryptophan pyrolysate, Trp-P-1. Acta Biochim Pol. 2005;52:167–178. [PubMed] [Google Scholar]
- 61.Hegde ML, Hazra TK, Mitra S. Functions of disordered regions in mammalian early base excision repair proteins. Cell Mol Life Sci. 2010;67:3573–3587. doi: 10.1007/s00018-010-0485-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Setser JW, Lingaraju GM, Davis CA, Samson LD, Drennan CL. Searching for DNA lesions: structural evidence for lower- and higher-affinity DNA binding conformations of human alkyladenine DNA glycosylase. Biochemistry. 2012;51:382–390. doi: 10.1021/bi201484k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klapacz J, Lingaraju GM, Guo HH, et al. Frameshift mutagenesis and microsatellite instability induced by human alkyladenine DNA glycosylase. Mol Cell. 2010;37:843–853. doi: 10.1016/j.molcel.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fujii T, Saito T, Nakasaka T. Purines. XXXIV 3-methyladenosine and 3-methyl-2′-deoxyadenosine: their synthesis, glycosidic hydrolysis, and ring fission. Chem Pharm Bull. 1989;37:2601–2609. [Google Scholar]
- 65.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 66.Nakamura J, Swenberg JA. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999;59:2522–2526. [PubMed] [Google Scholar]
- 67.Xanthoudakis S, Curran T. Redox regulation of AP-1: a link between transcription factor signaling and DNA repair. Adv Exp Med Biol. 1996;387:69–75. [PubMed] [Google Scholar]
- 68.Chang IY, Kim JN, Jun JY, et al. Repression of apurinic/apyrimidinic endonuclease by p53-dependent apoptosis in hydronephrosis-induced rat kidney. Free Radic Res. 2011;45:728–734. doi: 10.3109/10715762.2011.574289. [DOI] [PubMed] [Google Scholar]
- 69.Fung H, Demple B. Distinct roles of Ape1 protein in the repair of DNA damage induced by ionizing radiation or bleomycin. J Biol Chem. 2011;286:4968–4977. doi: 10.1074/jbc.M110.146498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fung H, Demple B. Overexpression of APE-1 makes cells resistant to alkylating agents. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 71.McNeill DR, Wilson DM. III A dominant-negative form of the major human abasic endonuclease enhances cellular sensitivity to laboratory and clinical DNA-damaging agents. Mol Cancer Res. 2007;5:61–70. doi: 10.1158/1541-7786.MCR-06-0329. [DOI] [PubMed] [Google Scholar]
- 72.Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008;68:6425–6434. doi: 10.1158/0008-5472.CAN-08-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 74.Fritz G. Human APE/Ref-1 protein. Int J Biochem Cell Biol. 2000;32:925–929. doi: 10.1016/s1357-2725(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 75.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal. 2009;11:601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Su D, Delaplane S, Luo M, et al. Interactions of apurinic/apyrimidinic endonuclease with a redox inhibitor: evidence for an alternate conformation of the enzyme. Biochemistry. 2010;50:82–92. doi: 10.1021/bi101248s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 78.Mohammed MZ, Vyjayanti VN, Laughton CA, et al. Development and evaluation of human AP endonuclease inhibitors in melanoma and glioma cell lines. Br J Cancer. 2011;104:653–663. doi: 10.1038/sj.bjc.6606058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zawahir Z, Dayam R, Deng J, Pereira C, Neamati N. Pharmacophore guided discovery of small-molecule human apurinic/apyrimidinic endonuclease 1 inhibitors. J Med Chem. 2009;52:20–32. doi: 10.1021/jm800739m. [DOI] [PubMed] [Google Scholar]
- 80.Bapat A, Glass LS, Luo M, et al. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J Pharmacol Exp Ther. 2010;334:988–998. doi: 10.1124/jpet.110.169128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Seiple LA, Cardellina JH, II, Akee R, Stivers JT. Potent inhibition of human apurinic/apyrimidinic endonuclease 1 by arylstibonic acids. Mol Pharmacol. 2008;73:669–677. doi: 10.1124/mol.107.042622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Luo M, Kelley MR. Inhibition of the human apurinic/apyrimidinic endonuclease (APE1) repair activity and sensitization of breast cancer cells to DNA alkylating agents with lucanthone. Anticancer Res. 2004;24:2127–2134. [PubMed] [Google Scholar]
- 83.Bailly C, Waring MJ. Preferential intercalation at AT sequences in DNA by lucanthone, hycanthone, and indazole analogs. A footprinting study. Biochemistry. 1993;32:5985–5993. doi: 10.1021/bi00074a009. [DOI] [PubMed] [Google Scholar]
- 84.Nyland RL, Luo M, Kelley MR, Borch RF. Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/Ref-1) J Med Chem. 2010;53:1200–1210. doi: 10.1021/jm9014857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Manvilla BA, Wauchope O, Seley-Radtke KL, Drohat AC. NMR studies reveal an unexpected binding site for a redox inhibitor of AP endonuclease 1. Biochemistry. 2011;50:10540–10549. doi: 10.1021/bi201071g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu L, Gerson SL. Therapeutic impact of methoxyamine: blocking repair of abasic sites in the base excision repair pathway. Curr Opin Investig Drugs. 2004;5:623–627. [PubMed] [Google Scholar]
- 87.Baldwin MR, O’Brien PJ. Human AP endonuclease 1 stimulates multiple-turnover base excision by alkyladenine DNA glycosylase. Biochemistry. 2009;48:6022–6033. doi: 10.1021/bi900517y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fitzgerald ME, Drohat AC. Coordinating the initial steps of base excision repair. Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J Biol Chem. 2008;283:32680–32690. doi: 10.1074/jbc.M805504200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abyzov A, Uzun A, Strauss PR, Ilyin VA. An AP endonuclease 1-DNA polymerase beta complex: theoretical prediction of interacting surfaces. PLoS Comput Biol. 2008;4 doi: 10.1371/journal.pcbi.1000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cistulli C, Lavrik OI, Prasad R, Hou E, Wilson SH. AP endonuclease and poly(ADP-ribose) polymerase-1 interact with the same base excision repair intermediate. DNA Repair. 2004;3:581–591. doi: 10.1016/j.dnarep.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 91.Ranalli TA, Tom S, Bambara RA. AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase I activity in long patch base excision repair. J Biol Chem. 2002;277:41715–41724. doi: 10.1074/jbc.M207207200. [DOI] [PubMed] [Google Scholar]
- 92.Prasad R, Beard WA, Batra VK, Liu Y, Shock DD, Wilson SH. A review of recent experiments on step-to-step ‘hand-off ‘ of the DNA intermediates in mammalian base excision repair pathways. Mol Biol (Mosk) 2011;45:586–600. [PMC free article] [PubMed] [Google Scholar]
- 93.Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 2000;19:1397–1404. doi: 10.1093/emboj/19.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sobol RW, Horton JK, Kühn R, et al. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 95.Canitrot Y, Cazaux C, Fréchet M, et al. Overexpression of DNA polymerase beta in cell results in a mutator phenotype and a decreased sensitivity to anticancer drugs. Proc Natl Acad Sci USA. 1996;95:12586–12590. doi: 10.1073/pnas.95.21.12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]
- 97.Bhattacharyya N, Banerjee S. A variant of DNA polymerase beta acts as a dominant negative mutant. Proc Natl Acad Sci USA. 1997;94:10324–10329. doi: 10.1073/pnas.94.19.10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 99.Deng JZ, Starck SR, Hecht SM, Ijames CF, Hemling ME. Harbinatic acid, a novel and potent DNA polymerase beta inhibitor from Hardwickia binata. J Nat Prod. 1999;62:1000–1002. doi: 10.1021/np990099r. [DOI] [PubMed] [Google Scholar]
- 100.Gao Z, Maloney DJ, Dedkova LM, Hecht SM. Inhibitors of DNA polymerase beta: activity and mechanism. Bioorg Med Chem. 2008;16:4331–4340. doi: 10.1016/j.bmc.2008.02.071. [DOI] [PubMed] [Google Scholar]
- 101.Maloney DJ, Deng JZ, Starck SR, Gao Z, Hecht SM. (+)-myristinin A, a naturally occurring DNA polymerase beta inhibitor and potent DNA-damaging agent. J Am Chem Soc. 2005;127:4140–4141. doi: 10.1021/ja042727j. [DOI] [PubMed] [Google Scholar]
- 102.Jaiswal AS, Banerjee S, Panda HA, et al. A novel inhibitor of DNA polymerase β enhances the ability of temozolomide to impair the growth of colon cancer cells. Mol Cancer Res. 2009;7:1973–1983. doi: 10.1158/1541-7786.MCR-09-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jaiswal AS, Banerjee S, Aneja R, Sarkar FH, Ostrov DA, Narayan S. DNA polymerase β as a novel target for chemotherapeutic intervention of colorectal cancer. PLoS ONE. 2011;6:e16691. doi: 10.1371/journal.pone.0016691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Balusu R, Jaiswal AS, Armas ML, Kundu CN, Bloom LB, Narayan S. Structure/function analysis of the interaction of adenomatous polyposis coli with DNA polymerase beta and its implications for base excision repair. Biochemistry. 2007;46:13961–13974. doi: 10.1021/bi701632e. [DOI] [PubMed] [Google Scholar]
- 105.Narayan S, Jaiswal AS. Activation of adenomatous polyposis coli (APC) gene expression by the DNA-alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine requires p53. J Biol Chem. 1997;272:30619–30622. doi: 10.1074/jbc.272.49.30619. [DOI] [PubMed] [Google Scholar]
- 106.Markowitz SD, Bertagnolli MM. Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jaiswal AS, Narayan S. A novel function of adenomatous polyposis coli (APC) in regulating DNA repair. Cancer Lett. 2008;271:272–280. doi: 10.1016/j.canlet.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kundu CN, Balusu R, Jaiswal AS, Narayan S. Adenomatous polyposis coli-mediated hypersensitivity of mouse embryonic fibroblast cell lines to methylmethane sulfonate treatment: implication of base excision repair pathways. Carcinogenesis. 2007;28:2089–2095. doi: 10.1093/carcin/bgm125. [DOI] [PubMed] [Google Scholar]
- 109.Kucherlapati M, Yang K, Kuraguchi M, et al. Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc Natl Acad Sci USA. 2002;99:9924–9929. doi: 10.1073/pnas.152321699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Larsen E, Gran C, Saether BE, Seeberg E, Klungland A. Proliferation failure and gamma radiation sensitivity of Fen1 null mutant mice at the blastocyst stage. Mol Cell Biol. 2003;23:5346–5353. doi: 10.1128/MCB.23.15.5346-5353.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tumey LN, Bom D, Huck B, et al. The identification and optimization of a N-hydroxy urea series of flap endonuclease 1 inhibitors. Bioorg Med Chem Lett. 2005;15:277–281. doi: 10.1016/j.bmcl.2004.10.086. [DOI] [PubMed] [Google Scholar]
- 112.Pascal JM, O’Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432:473–478. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- 113.Zhong S, Chen X, Zhu X, et al. Identification and validation of human DNA ligase inhibitors using computer-aided drug design. J Med Chem. 2008;51:4553–4562. doi: 10.1021/jm8001668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen X, Zhong S, Zhu X, et al. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008;68:3169–3177. doi: 10.1158/0008-5472.CAN-07-6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sun D, Urrabaz R, Nguyen M, et al. Elevated expression of DNA ligase I in human cancers. Clin Cancer Res. 2001;7:4143–4148. [PubMed] [Google Scholar]
- 116.Khodyreva SN, Prasad R, Ilina ES, et al. Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1) Proc Natl Acad Sci USA. 2010;107:22090–22095. doi: 10.1073/pnas.1009182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Eustermann S, Videler H, Yang JC, et al. The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger. J Mol Biol. 2011;407:149–170. doi: 10.1016/j.jmb.2011.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhu G, Chang P, Lippard SJ. Recognition of platinum-DNA damage by poly(ADP-ribose) polymerase-1. Biochemistry. 2010;49:6177–6183. doi: 10.1021/bi100775t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huambachano O, Herrera F, Rancourt A, Satoh MS. Double-stranded DNA binding domain of poly(ADP-ribose) polymerase-1 and molecular insight into the regulation of its activity. J Biol Chem. 2011;286:7149–7160. doi: 10.1074/jbc.M110.175190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 121.Herceg Z, Wang Z-Q. Failure of poly(ADP-ribose) polymerase cleavage by caspases leads to induction of necrosis and enhanced apoptosis. Mol Cell Biol. 1999;19:5124–5133. doi: 10.1128/mcb.19.7.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Masutani M, Suzuki H, Kamada N, et al. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc Natl Acad Sci USA. 1999;96:2301–2304. doi: 10.1073/pnas.96.5.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pieper AA, Brat DJ, Krug DK, et al. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci USA. 1999;96:3059–3064. doi: 10.1073/pnas.96.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tentori L, Balduzzi A, Portarena I, et al. Poly (ADP-ribose) polymerase inhibitor increases apoptosis and reduces necrosis induced by a DNA minor groove binding methyl sulfonate ester. Cell Death Differ. 2001;8:817–828. doi: 10.1038/sj.cdd.4400863. [DOI] [PubMed] [Google Scholar]
- 125▪.Ström CE, Johansson F, Uhlén M, Szigyarto CA, Erixon K, Helleday T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011;39:3166–3175. doi: 10.1093/nar/gkq1241. Describes the first appreciation of the synthetic lethality between BRCA mutants and inhibition of PARP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 127.Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Curr Opin Pharmacol. 2008;8:363–369. doi: 10.1016/j.coph.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 128.Lee H, Trainer AH, Friedman LS, et al. Involvement of BRCA2 in DNA repair. Mol Cell. 1998:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- 129.Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res. 2005;3:531–539. doi: 10.1158/1541-7786.MCR-05-0192. [DOI] [PubMed] [Google Scholar]
- 130.Karran P. A double strand break repair in mammalian cells. Curr Opin Genet Dev. 2000;10:144–150. doi: 10.1016/s0959-437x(00)00069-1. [DOI] [PubMed] [Google Scholar]
- 131.Graziania G, Szabób C. Clinical perspectives of PARP inhibitors. Pharmacol Res. 2005;52:109–118. doi: 10.1016/j.phrs.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 132.Telli ML, Ford JM. PARP inhibitors in breast cancer. Clin Adv Hematol Oncol. 2010;8:629–635. [PubMed] [Google Scholar]
- 133.Yap TA, Sandhu SK, Carden CP, de Bono JS. Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J Clin. 2011;61:31–49. doi: 10.3322/caac.20095. [DOI] [PubMed] [Google Scholar]
- 134.Plummer R, Jones C, Middleton M, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14:7917–7923. doi: 10.1158/1078-0432.CCR-08-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jamieson ER, Lippard SJ. Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 136.Ferry KV, Hamilton TC, Johnson SW. Increased nucleotide excision repair in cisplatin-resistant ovarian cancer cells: role of ercc1–xpf. Biochem Pharmacol. 2000;60:1305–1313. doi: 10.1016/s0006-2952(00)00441-x. [DOI] [PubMed] [Google Scholar]
- 137.Selvakumaran M, Pisarcik DA, Bao R, Yeung AT, Hamilton TC. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003;63:1311–1131. [PubMed] [Google Scholar]
- 138.Barret J-M, Cadou M, Hill BT. Inhibition of nucleotide excision repair and sensitisation of cells to DNA cross-linking anticancer drugs by F 11782, a novel fluorinated epipodophylloid. Biochem Pharmacol. 2002;63:251–258. doi: 10.1016/s0006-2952(01)00835-8. [DOI] [PubMed] [Google Scholar]
- 139.Neher TM, Shuck SC, Liu J, Zhang J-T, Turchi JJ. Identification of novel small molecule inhibitors of the XPA protein using in silico based screening. ACS Chem Biol. 2010;5:953–965. doi: 10.1021/cb1000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Neher TM, Bodenmiller D, Fitch RW, Jalal SI, Turchi JJ. Novel irreversible small molecule inhibitors of replication protein a display single-agent activity and synergize with cisplatin. Mol Cancer Ther. 2011;10:1796–1806. doi: 10.1158/1535-7163.MCT-11-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ledermann JA, Gabra H, Jayson GC, et al. Inhibition of carboplatin-induced DNA interstrand cross-link repair by gemcitabine in patients receiving these drugs for platinum-resistant ovarian cancer. Clin Cancer Res. 2010;16:4899–4905. doi: 10.1158/1078-0432.CCR-10-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nojima K, Hochegger H, Saberi A, et al. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res. 2005;65:11704–11711. doi: 10.1158/0008-5472.CAN-05-1214. [DOI] [PubMed] [Google Scholar]
- 144.Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 145.Spanswick VJ, Craddock C, Sekhar M, et al. Repair of DNA interstrand crosslinks as a mechanism of clinical resistance to melphalan in multiple myeloma. Blood. 2002;100:224–229. doi: 10.1182/blood.v100.1.224. [DOI] [PubMed] [Google Scholar]
- 146.Bignami M, O’Driscoll M, Aquilina G, Karran P. Unmasking a killer: DNA O(6)-methylguanine and the cytotoxicity of methylating agents. Mutat Res. 2000;462:71–82. doi: 10.1016/s1383-5742(00)00016-8. [DOI] [PubMed] [Google Scholar]
- 147.Kohn KW. Interstrand crosslinking of DNA by 1,3-bis(2-chloroethyl)-1-nitrosourea and other 1-(2-haloethyl)-1-nitrosoureas. Cancer Res. 1977;37:1450–1454. [PubMed] [Google Scholar]
- 148▪.Baer JC, Freeman AA, Newlands ES, Watson AJ, Rafferty JA, Margison GP. Depletion of O6-alkylguanine-DNA alkyltransferase correlates with potentiation of temozolomide and CCNU toxicity in human tumour cells. Br J Cancer. 1993;67:1299–1302. doi: 10.1038/bjc.1993.241. Details a completed clinical trial using an inhibitor of the direct reversal repair enzyme (alkylguanine DNA alkyltransferase on the efficacy of temozolomide and the problems that were observed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ranson M, Middleton MR, Bridgewater J, et al. Lomeguatrib, a potent inhibitor of O6-alkylguanine-DNA-alkyltransferase: Phase I safety, pharmacodynamic, and pharmacokinetic trial and evaluation in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2006;12:1577–1584. doi: 10.1158/1078-0432.CCR-05-2198. [DOI] [PubMed] [Google Scholar]
- 150.Khan OA, Ranson M, Michael M, et al. A Phase II trial of lomeguatrib and temozolomide in metastatic colorectal cancer. Br J Cancer. 2008;98:1614–1618. doi: 10.1038/sj.bjc.6604366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- 201.American Cancer Society. Cancer facts and figures. 2011 www.cancer.org/Research/CancerFactsFigures/CancerFactsFigures/2011-most-requested-tables-and-figures.
- 202.NCI. Finding cancer statistics. www.cancer.gov/statistics/finding.
- 203.ClinicalTrials.gov. PARP studies. http://clinicaltrials.gov/ct2/results?term=PARP&pg=1.
- 204.ClinicalTrials.gov. Study to assess the safety and tolerability of a PARP inhibitor in combination with carboplatin and/or paclitaxel. www.clinicaltrials.gov/ct2/show/NCT00516724.
- 205.ClinicalTrials.gov. AZD2281 and carboplatin in treating patients with BRCA1/BRCA2-associated, hereditary, or triple negative metastatic or unresectable breast cancer or ovarian epithelial cancer. www.clinicaltrials.gov/ct2/show/NCT00647062.
- 206.ClinicalTrials.gov. Rucaparib (CO-338; formally called AG-014699 or PF-0136738) in treating patients with locally advanced or metastatic breast cancer or advanced ovarian cancer. www.clinicaltrials.gov/ct2/show/NCT00664781.
- 207.ClinicalTrials.gov. Benzylguanine studies. http://clinicaltrials.gov/ct2/results?term=benzylguanine.