

Abstract
MicroRNAs (miRNAs) are short, noncoding RNAs that modulate genes posttranscriptionally. Frequent gains and losses of miRNA genes have been reported to occur during evolution. However, little is known systematically about the periods of evolutionary origin of the present miRNA gene repertoire of an extant mammalian species. Thus, in this study, we estimated the evolutionary periods during which each of 1,433 present human miRNA genes originated within 15 periods, from human to platypus–human common ancestral branch and a class “conserved beyond theria,” primarily using multiple genome alignments of 38 species, plus the pairwise genome alignments of five species. The results showed two peak periods in which the human miRNA genes originated at significantly accelerated rates. The most accelerated rate appeared in the period of the initial phase of hominoid lineage, and the second appeared shortly before Laurasiatherian divergence. Approximately 53% of the present human miRNA genes have originated within the simian lineage to human. In particular, approximately 28% originated within the hominoid lineage. The early phase of placental mammal radiation comprises approximately 28%, while no more than 15% of human miRNAs have been conserved beyond placental mammals. We also clearly showed a general trend, in which the miRNA expression level decreases as the miRNA becomes younger. Intriguingly, amid this decreasing trend of expression, we found one significant rise in the expression level that corresponded to the initial phase of the hominoid lineage, suggesting that increased functional acquisitions of miRNAs originated at this particular period.
Keywords: microRNA, gene gain, gene loss, regulatory network
Introduction
MicroRNAs (miRNAs) act as trans-regulators by posttranscriptionally modulating mRNAs through base-complementarity of the miRNA seed sequences (5′ nucleotides 2 to 7 of a mature miRNA) to 3′ untranslated regions (UTRs) of mRNAs (Ambros 2004; Bartel 2004, 2009). There are more than 1,500 genes encoding human miRNAs that are registered in miRBase (Griffiths-Jones et al. 2008; Kozomara and Griffiths-Jones 2011), which is comparable with the number of known human transcription factors (TFs) (Vaquerizas 2009). A characteristic difference between miRNAs and TFs, as trans-regulators, is that miRNA genes undergo fast turnover of gains and losses during evolution. This fast turnover has been studied by the small RNA deep sequencing approach (Lu et al. 2008) and by computational analyses on Drosophila species (Nozawa et al. 2010). For mammals, the miRNA gene expansions specific to primates (Bentwich et al. 2005; Zhang et al. 2007, 2008) and those specific to mouse (Li et al. 2010; Lehnert et al. 2011; Zheng et al. 2011) have been studied; however, little has been elucidated on the evolutionary periods of origin within mammalian evolution for the entire miRNA gene repertoire currently known for an extant species.
Thus, the aim of this study was to estimate the period of origin of each of the extant human miRNA genes, and thereby determine which evolutionary periods have had critical influences on the present repertoire of human miRNA genes. We focused on the human miRNA gene set because of availability of 1) extensive annotations on the genome and miRNAs for human, 2) human-anchored synteny-oriented multiple genome alignments, and 3) various genome sequences that correspond to fine-scaled evolutionary positions within the primate lineage.
For each of the extant human miRNA genes, we examined the human-anchored multiple alignment and its corresponding species tree to identify the common ancestral node at which the miRNA precursor first appeared. This approach has three characteristics. First, we focused on the way in which the miRNA gene repertoire has accumulated to comprise the present gene set; therefore, we did not focus on bona fide gains and losses over short time spans in the past. Second, available inter-genome synteny information was taken into account; thus, we did not simply depend on homology searches, which often fail to identify the correct orthology. Third, we adopted a criterion that the seed sequence of either of the two mature miRNAs should exactly match the orthologous precursor miRNAs, in addition to the often-used threshold of miRNA precursor secondary structure folding energy. The exact seed match constraint was introduced such that the miRNA of interest would be more likely to maintain its functionality and affect the same group of target mRNAs as that of the present human miRNAs.
To incorporate synteny information, we primarily used synteny-oriented Ensembl Enredo–Pecan–Orteus (EPO) 35-way placental mammal (Paten et al. 2008; Flicek et al. 2012) and Pecan 19-way amniote multiple alignments (Dewey 2007). We prioritized the EPO 35-way over Pecan 19-way alignments because the former comprises a larger number of species that are evolutionarily closer to each other and covers a larger span of the human genome than the latter. Thus, the EPO 35-way alignments were expected to be more accurate and informative. The pecan 19-way alignments were used to identify the origins of human miRNAs when the origins were estimated to be in the placental mammal root branch (or earlier) by the EPO 35-way alignments. This was because the Pecan 19-way alignments include Metatherian, Prototherian, and avian species, as well as 13 placental mammal species that are shared with the EPO 35-way alignments. Only in cases where the EPO 35-way and Pecan 19-way alignments did not solve the period of origin did we examine pairwise genome alignments. In the species tree, the edge from human to the common ancestral branch between human and prototheria (i.e., platypus) was partitioned into 15 evolutionary periods by the divergences of species/clades whose genomes have been sequenced (fig. 1). In total, we examined the genome sequences of 43 species (fig. 2). We report here that drastic alterations in the rate of origination of human miRNA genes showed two remarkably accelerated peaks.
Fig. 1.
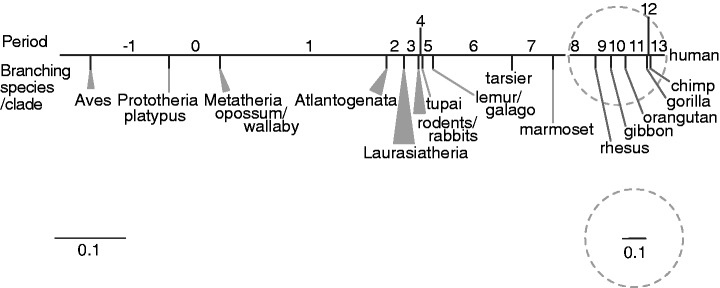
Schematic diagram of the relationships among periods and divergences. A horizontal line stands for an edge connecting human and the root of the species tree. Period numbers are indicated above and along the edge. Diverging species or clades are shown below the edge. The divided edge lengths indicating the intervals of the periods are proportional to the neutral distance. The part of the edge in a dashed gray circle is enlarged 3-fold.
Fig. 2.

Cladogram of 43 species used. A thick line drawn from the upper right to the left stands for the edge connecting human and the root of the cladogram. On this edge, 16 internal nodes are located (nodes −3 through node 12) corresponding to each divergence of a single species or clade. The leaf node, human, is regarded as node 13. A period X in figure 1 corresponds to an interval between node X and node X−1. The 43 species are each numbered 1 through 43, indicated with italicized numbers.
Results
Period of Origin for Each Human Precursor miRNA
We prioritized the synteny-oriented multiple alignments; therefore, we first examined the EPO 35-way alignment that included 35 placental mammalian genome sequences. We specified the period of origin within period 2 to 13 for 968 human precursor miRNAs in the mammalian evolution after Atlantogenatan divergence (fig. 2 and the workflow chart of fig. 3). In addition, 15 human-specific duplicates could be identified, because the multiple human genome segments that included human precursor miRNAs were aligned in the same slice of the EPO 35-way alignment. The Pecan 19-way alignment was then examined to assign the remaining 450 human precursor miRNAs to any of the periods –1, 0, 1 or the class “conserved beyond theria.” Using this method, periods of origin for 238 human precursor miRNAs could be assigned.
Fig. 3.

Flowchart of the processes for estimating the periods of origin. The number of human precursor miRNAs resulting from the each corresponding step is indicated in parentheses.
For the 238 human miRNAs, the concordance of presence or absence of each of the functional miRNAs estimated as discussed earlier was examined between Pecan and EPO alignments, to assess the validity of using the Pecan 19-way alignments as an extension of the EPO-35 way alignments. The 2 multiple alignments included 13 eutherian species in common, in which 8 species were based on high-coverage genomes. The concordance was generally high; 83% for the high-coverage genome species, and 79% for all the EPO-Pecan-shared species. Therefore, it is generally valid to use Pecan 19-way as extension of the EPO 35-way alignments. Only three miRNAs (hsa-mir-217, hsa-mir-152, and hsa-mir-30a) were below 50% of the concordance for both the high-coverage and the all-shared species. They were each indicated with a note in supplementary table S1, Supplementary Material online.
For the remaining 212 human precursor miRNAs, pairwise genome alignments of human to opossum, wallaby, platypus, chicken, and zebrafish were examined to identify the corresponding functional orthologs to permit their periods of origin to be assigned to periods −1, 0, 1 and the class “conserved beyond theria.” As a result, we successfully specified the period of origin of all 1,433 human precursor miRNAs to the 15 periods or to “conserved beyond theria.” All the estimates are provided in the order of chromosomal locations in supplementary table S1, Supplementary Material online.
Number of Present Human miRNAs that Originated in Each Period
As shown in figure 4a and b, the numbers of present human miRNAs revealed a remarkably skewed distribution in terms of their origins among the periods. Sixteen percentage of human miRNAs originated during the eutherian root branch (period 1), which was the highest number in any one period. The early eutherian radiation phase (periods 1 and 2) included the origins of approximately 28% of the human miRNA repertoire.
Fig. 4.

Graphs showing the fraction and the rate of human miRNA originations by period. The percent of human miRNA genes that originated during each period is shown in a bar chart (a) in which the x axis stands for the neutral evolutionary distance from human. Above each vertical bar, the corresponding period number is shown. Each vertical bar is located at the mid point of the corresponding period of origin. The relationship of each bar with the divergence of a species or clade is indicated in a schematic drawing (b). Graph (c) shows the rate of origination of human miRNAs for each period, in which the y axis indicates the percent of the miRNA that originated during each period, with a scale of per 0.01 neutral substitution/site.
After divergence of tarsiers, the numbers of human miRNAs originating in the Simiformes (or simian) lineage through to human remarkably increased. More than one-half (approximately 53%) of the present human miRNAs originated within the simian lineage (i.e., periods 7–13), In particular, more than a quarter (28%) of the human miRNAs originated within the hominoid lineage (periods 9–13). Approximately 81% of the present human miRNA repertoire originated either in the initial placental mammal radiation phase or after the initial phase of simian lineage. On the other hand, the conserved miRNAs beyond eutheria comprised no more than 15% of the human miRNAs.
Rate of the Origination of the Present Human miRNA along the Periods
The rate of origination of the human miRNAs for each period was computed as the fraction of the human miRNAs that originated per neutral distance of the period. The result showed that the rate of human miRNA origination had two outstanding accelerated peaks (fig. 4c). The largest accelerated peak was a particularly sharp one corresponding to the initial internal branch of the hominoid lineage (i.e., period 9), preceding the gibbon divergence. The rate of origination of the human miRNA repertoire increased during period 9 to approximately 260 miRNA originating per 0.01 neutral substitution/site. The number of miRNA that originated within period 9 was 187, and the neutral branch length ascending from node 9 was 0.0072; therefore, (187/0.0072) × 0.01 makes approximately 260. This result suggests that the initial phase of the hominoid lineage predominantly fostered the miRNA genes that form the present human miRNA repertoire. The second most accelerated peak of the miRNA origination rate was in period 2, which corresponds to the internal branch preceding the Laurasiatherian divergence in the early phase of placental mammal radiation. The rate increased to 72 miRNA originations per 0.01 neutral substitution/site. This result suggests that the human miRNAs that originated during the early phase of the placental mammal evolution contributed much to the present human miRNA repertoire.
Expression Levels Decrease as miRNAs Become Younger, Except for the Period of Accelerated miRNA Origination in Hominoid Lineage
To determine the relationship between the period of origin and the expression level of the human miRNAs, we examined the expressed clone counts in a mammalian gene expression atlas (Landgraf et al. 2007). We used only the clone count data that were derived from normal human tissues. For each period, we computed the mean of the expressed clone counts of miRNAs that originated in the particular period and regarded it as the expression level.
In figure 5, the expression levels of the human miRNAs whose origins were classified as periods 0 through 13 are plotted, except for period 4, which lacked the corresponding precursor miRNAs in the Landgraf’s data set, together with the expression levels of the human miRNAs classified as “conserved beyond theria.” The plot shows a general trend in which the human miRNAs’ expression levels decreased as the miRNAs’ period of origin become closer to the present. The expression level of the human miRNAs conserved beyond theria was higher than the level of any other periods of origin.
Fig. 5.

Expression levels of human miRNAs that originated during each period. The plot shows the mean expression levels of human miRNAs in normal human tissues along the periods during which the particular miRNAs originated. The y axis of the plot is indicated on a logarithmic scale. On the plot, the left-most dot stands for the expression level of the human miRNAs conserved beyond theria. The other dots indicate the expression levels of human miRNAs that originated during period 0 to period 13, except for period 4, which lacks the corresponding expression data. On the top of the plot, the period number corresponding to each dot is indicated. An arrow points to the dot during period 9. In the plot, the dots for periods 12 and 13 are placed beneath the bottom scale line, because the expressed clone counts are 0 for these periods. The x axis is proportional to the neutral branch length from human, above which each dot is located at the mid point of the corresponding period of origin.
Amid this general decreasing trend, however, the plots revealed a significant paradoxical rise during periods 8 and 9, with a peak during period 9. The expressed clone count of miRNAs that originated during period 8 was significantly higher than that of period 7 (P < 0.04, t-test), and the clone count that originated during period 9 was significantly higher than that of period 10 (P < 0.02, t-test). Notably, period 9 coincides with the estimated peak rate of origination of human miRNAs. From this, we suggest that during period 9, that is, the initial phase of the hominoid lineage, a larger number of miRNAs rapidly became highly expressed, probably because of a high rate of functional acquisitions of the particular miRNAs.
Periods of Origin of Human miRNA Clusters
The feasibility of the results was assessed in comparison to published findings by focusing on four miRNA large clusters on three distinct human chromosomes. A dense array of 46 miRNAs from hsa-mir-512-1 to hsa-mir-519a-2 on chromosome 19 (fig. 6a), termed C19MC, constitutes a large primate-specific miRNA cluster spanning approximately 100 kb (Bentwich et al. 2005). The C19MC is an imprinted domain (Zhang et al. 2008; Bortolin-Cavaille et al. 2009), which has been reported to be involved in tumorigenesis (Flor and Bullerdiek 2012; Fornari et al. 2012). With regard to the primate specificity of the origin of this cluster, our result consistently showed that the human miRNAs in the C19MC originated in the common ancestral nodes between human and primates, except for two miRNAs: hsa-mir-512-1 and hsa-mir-498 (fig. 6b). Notably, our estimates showed that most of them originated within the period immediately preceding the common ancestor between human and gibbon (period 9).
Fig. 6.

Human miRNA gene clusters and their estimated periods of origin. Periods of origin (y axis) of the human miRNA genes are shown (blue vertical bars) along the chromosomal physical location (x axis) and along the order of miRNA genes within the chromosome (red vertical bars) for human chromosomes 19 (a and b), 14 (c and d), and X (e and f), respectively. Ticks beside bar charts indicate period numbers in the decreasing order from the bottom, and the top tick indicates an origin beyond theria. In the bar charts (b, d, and f), each miRNA gene has the same width on the x axis; therefore, the regions of densely clustered miRNA genes results in enlarged width. A square bracket in the panel indicates each miRNA cluster and the connected arrow points to the physical location on the chromosome. In (f), the two contiguous clusters are indicated separately with a gray and a black square bracket.
Human chromosome 14 harbors another miRNA cluster (fig. 6c) that comprises 41 miRNA genes, hsa-mir-379 to hsa-mir-656, in an imprinted DLK–DIO3 region (Cavaille et al. 2002; Glazov et al. 2008). This cluster of miRNAs has been reported to have emerged in the early eutherian radiation by tandem duplication (Glazov et al. 2008). Our results supported the view that all of the miRNAs in this cluster originated in the early eutherian radiation phase (periods 1 and 2) (fig. 6d). Thirty-eight out of the 41 human miRNAs originated in the eutherian root branch (period 1), and the remaining three miRNAs originated in the branch between Atlantogenatan and Laurasiatherian divergences (period 2). Consistent with the findings by Glazov et al., our analysis found no extra-eutherian orthologs to the miRNAs in this cluster. Our results further suggested the possibility that a wider range of eutherian species have functional miRNA orthologs to humans than the range of species that Glazov et al. reported.
On the human X chromosome, there are two known miRNA clusters, as shown in figure 6e. The first cluster comprises hsa-mir-890, 888, 892a, 892b, and 891a. These miRNAs have been reported to exist in humans to marmosets, caused by a duplication that occurred in the primate-lineage (Li et al. 2010). Our results were consistent with this view: all the miRNAs in this cluster originated within the primate lineage (periods 8 and 9) (fig. 6f, indicated by a gray square bracket). The second cluster on the human X chromosome spans 95 kb and includes 15 miRNAs (hsa-mir-513c to hsa-mir-514a-3). It has been reported that most of the miRNA genes in this cluster are conserved within primates, but some miRNA genes are conserved in mouse and dog (Bentwich et al. 2005; Zhang et al. 2007). Our analysis also showed that the human miRNAs in this cluster have mixed periods of origin, with five appearing after marmoset divergence and the rest in the early eutherian radiation phase (fig. 6f, indicated by a black square bracket).
As mentioned earlier, our systematic analyses provided consistent estimates of the periods of origin for four large, representative human miRNA clusters that have been previously studied. Thus, these comparative results support the feasibility of our method for estimating the periods of origin of the human miRNA repertoire.
Simulation Assessments Show that the Two Accelerated-Rate Peaks Are Robust
In this study, each evolutionary period was defined by the divergence of a species or a clade that includes multiple species. Some clades include many species; for example, the Laurasiatherian clade comprises 12 species. It is expected that clades with more species have higher chances of detecting orthologs than those of single-species clades or small clades. To assess the influence of the clades with many species on the estimation of the periods of origin of human miRNA genes, we randomly chose one species’ genome sequence from three large multi-species clades in the EPO 35-way alignment (i.e., Atlantogenatan, Laurasiatherian, and rodents/rabbits clades). The period of origin was then estimated in the same way as the original data, with one exception: no distinction was made between high and low coverage sequences. This exception was introduced because only one species genome from each clade was used in this simulation; therefore, the condition in the original procedure that demanded that the ortholog should be identified in two or more species’ genomes for low-coverage genomes became meaningless.
The simulation result (fig. 7a) showed that the number of human miRNA origins assigned to period 1 decreased, while the number assigned to periods 2 and 3 increased. As shown in figure 7b, these alterations in the assignments of the periods of origin resulted in an increase in the peak of the rate of human miRNA origination during period 2; the other sharp peak during period 9 remained intact.
Fig. 7.

Effects of multi-species clades on the estimates of the periods of origin of human miRNA genes. The results of simulations in which one species’ genome sequence is randomly chosen from the multi-species clades are shown. A graph (a) shows the fraction of the origin of human miRNA genes, and a graph (b) shows the rate of the human miRNA origination during periods 1 (left) through 13 (right). Black lines indicate the result of the original data set and a red line indicates the result of the simulation data set. Each error bar stands for the 1 SD. obtained by the simulation. For plots (a) and (b), the x axis each is proportional to the neutral distance from human, above which each dot is located at the mid point of the origination period. The period number corresponding to each dot is indicated on the top of each plot. The two accelerated-rate peaks were reproduced by simulation.
The Atlantogenatan clade comprised only low-coverage genomes; however, the Laurasiatherian and rodents/rabbits clades included four and two high-coverage genomes, respectively. Therefore, it is feasible that a single genome choice from each of the clades operated in favor of the Laurasiatherian and rodents/rabbits clades in the detection of orthologous miRNA genes. Consequently, the orthologs that failed to be detected in the Atlantogenatan clade in the simulation were recaptured in the Laurasiatherian and rodents/rabbits clades. Furthermore, the clades containing many species are evolutionarily more remote from human; thus, those large clades could complement the low detection rates of orthologs caused by the long distances from human. These series of simulations suggest that the strategy of demanding ortholog detection in two or more species for the low-coverage genomes in multi-species clades played an effective role in the reliable estimation of period of origin of the human miRNA genes.
Assessment of the Robustness of the Results Against Possible Inaccuracies in the miRBase Annotation
The miRBase we utilized as a source of materials is the primary online repository for all miRNA sequences and annotation, including a rapidly growing number of new miRNA data derived from, for example, deep-sequencing approaches. Some of such deposited miRNAs may be unsupported by relatively low-confidence evidence (Kozomara and Griffiths-Jones 2011; Tarver et al 2012).
Therefore, to assess the extent of robustness of our present results against possible inaccuracies in the miRBase annotation, we applied two additional conditions to the retrieval procedure of human miRNAs (hereafter referred to as conditions 1 and 2). Condition 1: We tightened the threshold of the folding energy of precursor miRNA secondary structure (<−20.5 kcal/mol) to obtain secure human miRNAs in terms of structure. We adopted the threshold value as a 95 percentile value over all the 1,523 human precursor miRNAs. Condition 2: In addition to condition 1, we retrieved only human miRNAs that each had two annotated mature miRNAs in human, known as miR (mature miRNA) and miR* (star sequence), which are derived from both the 5- and 3-prime stem regions of the corresponding precursor miRNA. Evidence of both miR and miR* is one of the key criteria for high-confidence miRNAs (Kozomara and Griffiths-Jones 2011; Tarver et al. 2012).
Condition 1 (folding energy threshold of −20.5 kcal/mol) resulted in the exclusion of 73 out of the 1,444 human precursor miRNAs (N = 1,371). Condition 2 (condition 1 plus presence of both miR and miR* in human) excluded 897 out of 1,444 human precursor miRNAs (N = 547). Thus, applying condition 2 was so stringent as to leave no more than 38% of the human precursor miRNAs used in the original analysis (supplementary table S2, Supplementary Material online).
Under conditions 1 and 2, as shown in figure 8a, the pattern of skewed distribution of the number of present human miRNAs along the period of their origins was basically unchanged from the result under the original conditions. Along therian evolution, the largest number of human miRNAs originated during the eutherian root branch (period 1) followed by period 2 under every condition. The second largest numbers of human miRNA gains were observed during periods 7–9; although under condition 2, numbers of human miRNA origins during periods 8–13 were relatively reduced. The correspondence of the period to the species/clade divergence is indicated figure 8b.
Fig. 8.

Comparison among miRNA sets under different conditions on the fraction and the rate of human miRNA originations by period. The percent of human miRNA genes that originated during each period is shown in a line chart (a), in which the x axis stands for the neutral evolutionary distance from human. A red line stands for the percent miRNA origination under the original condition (precursor folding energy <−13 kcal/mol); a green line for that under condition 1 (precursor folding energy <−20.5 kcal/mol); a purple line for that under condition 2 (precursor folding energy <−20.5 kcal/mol plus presence of both miR and miR*). Above the line chart, the corresponding period number is shown. Each vertex is located at the midpoint of the corresponding period of origin. The relationship of each vertex with the divergence of a species or clade is indicated in a schematic drawing (b). Graph (c) shows the rate of origination of human miRNAs for each period, in which the y axis indicates the percent of the miRNA that originated during each period, with a scale of per 0.01 neutral substitution/site. The line colors are the same as those in (a), respectively.
The rate of origination of the human miRNAs clearly showed the two accelerated peaks (fig. 8c), consistent with the result under the original conditions; the largest sharp peak appeared in the initial branch of the hominoid lineage (period 9), and second largest peak appeared during period 2. The plots matched to one another consistently under every condition; except for the reduction of the rates of periods 10–13 under condition 2.
The relationship of human miRNA expression levels with the period of miRNA origin is shown in figure 9, with a comparison of the results under the original conditions with the results under the conditions 1 and 2. The general trend of lower expression level for younger human miRNAs was consistently reproduced under conditions 1 and 2. The paradoxical rise in the expression level during periods 8 and 9 identified under the original conditions was also clearly shown under conditions 1 and 2.
Fig. 9.

Comparison among miRNA sets under different conditions on expression levels of human miRNAs originated during each period. Plots show the mean expression levels of human miRNAs in normal human tissues along the periods during which the particular miRNAs originated. The y axis of plot is indicated on a logarithmic scale. On each plot, the left-most dot stands for the expression level of the human miRNAs conserved beyond theria. The other dots indicate the expression levels of human miRNAs that originated during period 0 to period 13, except for period 4, which lacks the corresponding expression data. Red dots and lines stand for the expression levels under the original condition (precursor folding energy <−13 kcal/mol); green for those under condition 1 (precursor folding energy <−20.5 kcal/mol); purple for those under condition 2 (precursor folding energy <−20.5 kcal/mol plus presence of both miR and miR*). On the top of the plots, the period number corresponding to each dot is indicated. The dots for periods 12 and 13 are placed beneath the bottom scale line because the expressed clone counts are 0 for these periods. The x axis is proportional to the neutral branch length from human, above which each dot is located at the midpoint of the corresponding period of origin.
Based on the reproducibility of the main findings that were assessed under the two-step stringent conditions 1 and 2, we confirmed that the results and the methods in the present study were robust against the possible inaccuracy in the annotation of the miRBase.
Discussion
In this study, we have elucidated the distribution of the evolutionary periods of origin for 1,433 extant human miRNA genes among 15 periods that represent the internal branches placed from human to the common ancestral branch of prototheria (platypus) with human. The estimates demonstrated that the rate of origination of the present human miRNAs was skewed, with two remarkably accelerated peaks during mammalian evolution. The largest accelerated peak was in the initial phase of hominoid lineage and the second largest corresponded to the early radiation phase of the placental mammals.
We also demonstrated that the expression levels of human miRNAs in human normal tissues generally decrease as the miRNAs become younger. Furthermore, this study illustrated this trend with a finer resolution than the previous studies that noted the same trend, and which used the small RNA deep sequencing approaches (Berezikov et al. 2006; Lu et al. 2008) and the sequence and database analyses approaches (Liang and Li 2009).
The newly emerged miRNAs could affect or even perturb the established regulatory networks, even though their expressions were very weak. For the newly emerged miRNAs to survive, they needed to acquire a certain function, even though the function is trivial. The function may not be novel but only a redundancy-making role bridging over a well-established function (Iwama et al. 2011). Redundancy in function would be more likely, particularly for new precursor miRNAs that yield the same mature miRNAs. Subsequently, some of the new functional miRNAs could increase their expression levels, which could permit their further survival. In this scenario, for the new miRNAs to join the miRNA repertoire at an accelerated rate, loosening of the constraint at the regulatory network level may be an underlying condition. Under such a condition, there would be more chances of nascent miRNAs acquiring new functions at a high rate. Consistent with this theory, we revealed a significant increase in the expression levels of miRNAs that originated during the most accelerated-rate period, that is, the initial phase of the hominoid lineage, amid a decreasing trend of the expression for younger miRNAs. This seemingly paradoxical rise in expression levels suggests an increased rate of miRNA functional acquisition.
Therefore, the initial phase of the hominoid lineage and the early eutherian radiation phase could have extensive impacts on the present human gene regulatory networks. It seems likely that many miRNA genes that originated during these periods would have a tendency to gain functions. We could not, however, detect significant increases in the expression levels of miRNAs that originated during the initial phase of eutherian radiation, which could be reasoned as follows.
In the eutherian radiation phase, period 2 spanned from ∼104.7 Ma (Atlantogenatan divergence) through ∼97.4 Ma (Laurasiatherian divergence) (Steiper and Young 2009; Kumar and Hedges 2011). Period 9, on the other hand, spanned from ∼29.6 Ma (rhesus macaque divergence) through ∼18.8 Ma (gibbon divergence) (Marphy and Eizirk 2009; Kumar and Hedges 2011). There is a large gap in the depth of evolution. It has been estimated that 60 My is generally required for a small fraction of new miRNAs to be stably expressed and integrated into the transcriptome of an organism, with the subsequent gradual increase in their expressions (Lu et al. 2008). This long process is considered the main factor causing the general decreasing trend for the younger miRNAs. The effect of the long process would still be weak for period 9, but strong enough during period 2 to mask alterations in expression that might be related to the accelerated acquisition of functional miRNAs. However, the influence of the accelerated miRNA origination might be indicated as the less steep decline in the expressions of periods 1–2 relative to the decline of periods 3–7 (fig. 5).
The estimates of the periods of origin of the human miRNA gene set were validated by 1) comparing our findings with previously published results on the origins of four large human miRNA clusters, and 2) by the simulation analyses that assessed the bias caused by large clades. Our criteria, that is, prioritizing synteny-oriented multiple alignments over pairwise alignments, the exact seed sequence must match to assign an ortholog, and that orthologs should be detected in at least two genome sequences if they both are low-coverage ones, effectively enhanced the reliability of the estimated periods of origin of the human miRNA gene set. The estimated periods of origin of 1,433 human miRNAs also facilitate the miRNA target prediction by limiting the range of species to search for the conservation of mRNAs’ 3′-UTRs.
Materials and Methods
Human miRNA Information
The miRNA data files, miRNA.dat and hsa.gff, of miRBase release 18 were downloaded from ftp://mirbase.org/pub/mirbase/18/ (last accessed 29 November 2012). Human genome coordinates for human miRNA precursors were retrieved from the hsa.gff file. The coordinates of mature miRNAs and their seed sequences were computed by integrating the information of the miRNA.dat file. The seed sequence was defined as nucleotides 2–7 from the 5′-end of each mature miRNA sequence. Four miRNA precursors, that is, hsa-mir-1273e, hsa-mir-3155b, hsa-mir-4482-2, and hsa-mir-941-2 in the miRNA.dat file were not present in the hsa.gff file, and were therefore omitted from further analyses. The hsa.gff file included 1,523 human precursor miRNA entries.
Multiple Genome Alignments, the Species Trees, and Neutral Branch Lengths
For eutherians, the EPO 35-way multiple alignment was downloaded as a set of Ensembl Multi Format (EMF) flat files, together with the species tree, from ftp.ensembl.org/pub/release-65/emf/ensembl-compara/epo_35_eutherian/ (last accessed 29 November 2012). The provided species tree with neutral branch lengths had been computed using four-fold degenerated sites by programs msa_view (Hubisz et al. 2011) and phyloFit (Siepel and Haussler 2004; Siepel et al. 2005), under the general reversible model (Yang 1997). Ensembl and UCSC use this same species tree for their alignments. (Please also refer to http://genomewiki.ucsc.edu/index.php/Human/hg19/GRCh37_46-way_multiple_alignment#Multiple_Trees; last accessed 29 November 2012). For the low-coverage genomes, this alignment pipeline adopts BlastZ-net to map the low-coverage genome alignments to EPO alignments of high coverage genomes (Dewey 2007). To examine the conservation of precursor miRNAs beyond eutherian species, a set of EMF flat files of the Pecan 19-way amniota vertebrate multiple alignment and its corresponding species tree were downloaded from ftp.ensembl.org/pub/release-65/emf/ensembl-compara/pecan_19_amniota/ (last accessed 29 November 2012). This alignment pipeline adopts Mercator (Dewey 2007) to obtain the synteny map, and the syntenic regions were aligned by Pecan. We used the alignment blocks and the species tree of six species that were not included in the EPO 35-way (Gallus gallus, Meleagris gallopavo, Taeniopygia guttata, Anolis carolinensis, Monodelphis domestica, and Ornithorhynchus anatinus).
Excising Alignment Slices Corresponding to Human Precursor miRNAs
EPO 35-way and Pecan 19-way alignments comprise 58,159 and 11,282 alignment blocks, respectively, most of which are discontinuous with the neighboring blocks on the human genome coordinates. From the EPO 35-way alignment blocks, we excised the alignment slices that corresponded to the genome coordinates of each human precursor miRNA, 1) if the entire stretch of the human precursor miRNA was included in a single alignment block, or 2) if it was included in two neighboring alignment blocks with one gap of, at most, two-nucleotides or less. This process ruled out 46 human precursor miRNAs for which informative alignment blocks were not available (N = 1477). Subsequently, we selected the alignment slices in which a single aligned sequence was assigned to every non-human species. In this regard, however, we accepted alignment slices whose non-human additional aligned sequences each 1) comprised an all-gap region or 2) included 10 or less nucleotides in the sequence. These criteria excluded 33 entries (N = 1,444). Every sequence that corresponded to each of the 1,444 human precursor miRNA stretches was excised from the alignment blocks as an alignment slice.
Criteria of Functional Orthologs of Human miRNAs
In this study, we defined each non-human precursor miRNA as functional based on the RNA secondary structure folding energy and the exact seed sequence match to that of the human sequence, rather than simply based on the homology of the entire miRNA precursor sequence. First, RNAfold (version 2.0.2) (Hofacker et al. 1994) (http://www.tbi.univie.ac.at/∼ivo/RNA/; last accessed 29 November 2012) was used to compute the folding energy of each of 1,523 human precursor miRNAs with the default settings, and obtained the 99.5 percentile value of −12.5 kcal/mol. Accordingly, <−13 kcal/mol was regarded as the threshold of a miRNA precursor being functional for any species examined. Eleven human precursor miRNAs were excluded that were above the threshold (N = 1,433). For each excised non-human precursor miRNA candidate sequence, two criteria were applied to define it as a functional precursor miRNA orthologous to human: 1) the folding energy was <−13 kcal/mol, and 2) the seed sequence of either of the two mature miRNAs of a precursor miRNA should exactly match the orthologous precursor miRNAs. If a single mature miRNA was assigned to the human miRNA precursor, the seed sequence should be identical between human and the examined species.
Pairwise Genome Alignments
For the human miRNAs, only when no multiple alignment blocks were available (N = 266), we used the Ensembl BlastzNet pairwise genome alignments of human to opossum, wallaby, platypus, and chicken, through COMPARA API (Stabenau et al. 2004) at Ensembl. For all the 266 human miRNAs, the pairwise alignments were available for all four species. Ensembl BlastzNet alignments include only avians as non-therian species; therefore, to further confirm the sequence conservation, the UCSC human–zebrafish pairwise genome alignments were downloaded from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/vsDanRer7/axtNet/ (last accessed 29 November 2012). The UCSC human-zebrafish alignments were available for all the 266 miRNAs. Then, the aligned stretches exactly corresponding to the coordinates of each of the human miRNA precursors were excised.
Estimation of the Period of Origin Using EPO 35-Way
In the species tree, 17 nodes were placed on the edge that connects the human terminal node and the root of the tree, each of which corresponded to a divergence of a single species or a clade (fig. 2). We termed each as node X (−3 ≤ X ≤ 13). Node 13 stood for the terminal node, human, and a neighboring ancestral node on the human-root edge was assigned a number of X − 1. A node X and a node X − 1 define a period X (−1 ≤ X ≤ 13). Each of the 43 species was numbered as i (1 ≤ i ≤ 43), as indicated in figure 2. A parsimony method was adopted. For each human precursor miRNA, m, 1) if a species i contains a functional ortholog, we denoted 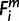 = 1, 2) if either of the ortholog’s seeds does not exactly match to that of human,
= 1, 2) if either of the ortholog’s seeds does not exactly match to that of human, 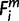 = −1, and 3) if a species i contains no ortholog,
= −1, and 3) if a species i contains no ortholog, 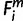 = 0.
= 0.
Initially, the EPO 35-way multiple alignment that includes 35 species was prioritized (1 ≤ i ≤ 35), and then the high-coverage genomes were prioritized (ihigh ∈ {1, 2, 3, 4, 6, 7, 12, 13, 19, 22, 26, 28}) over low-coverage ones (ilow ∈ {5, 8, 9, 10, 11, 14, 15, 16, 17, 18, 20, 21, 23, 24, 25, 27, 29, 30, 31, 32, 33, 34, 35}). For each miRNA, m, whose 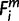 = 1 (i ∈ ihigh), the node that connects species i and human was denoted as
= 1 (i ∈ ihigh), the node that connects species i and human was denoted as 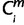 , and assigned the node number to
, and assigned the node number to 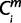 (=X). For each m, when the species i is a one-species clade to node X (i ∈ 1 through 8 and 11); if
(=X). For each m, when the species i is a one-species clade to node X (i ∈ 1 through 8 and 11); if 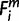 = 1, then
= 1, then 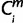 = X. For the case of a multi-species clade connecting to node X, because ihighs were prioritized over ilows, if one i (i ∈ ihigh) suffices
= X. For the case of a multi-species clade connecting to node X, because ihighs were prioritized over ilows, if one i (i ∈ ihigh) suffices 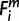 = 1, then
= 1, then 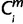 = X. For is (i ∈ ilow) that were members of a multi-species clade, only if
= X. For is (i ∈ ilow) that were members of a multi-species clade, only if 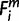 = X held for two or more species is in the clade (i ∈ ilow), then
= X held for two or more species is in the clade (i ∈ ilow), then 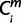 = X. For each miRNA m, the minimum
= X. For each miRNA m, the minimum 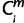 over is (i.e., closest to the root of the tree) was the value of the originating node Om. If 2 ≤ Om ≤ 13, we assigned Om to the period of origin X for the miRNA, m.
over is (i.e., closest to the root of the tree) was the value of the originating node Om. If 2 ≤ Om ≤ 13, we assigned Om to the period of origin X for the miRNA, m.
Estimation of the Period of Origin by Pecan 19-Way and Pairwise Alignments
Each m whose Om is 1 by the EPO 35-way alignment, it remained uncertain in whether the period of origin X is assigned 1 or less (i.e., more ancient in origin). For these miRNAs, ms, to specify the period of origin at more ancestral common nodes, we used the Pecan 19-way alignment for the species iPecan (37 ≤ iPecan ≤ 42). We prioritized the Pecan 19-way alignment over pairwise alignments because the former is based on inter-genome synteny. Using iPecan, for each m whose 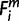 = 1 (i ∈ iPecans), then
= 1 (i ∈ iPecans), then 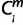 = X where a node X (−2 ≤ X ≤ 0) was the nearest common node between species i and human. The minimum
= X where a node X (−2 ≤ X ≤ 0) was the nearest common node between species i and human. The minimum 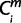 over is defines the Om; thus, the period of origin X was assigned Om (−2 ≤ X ≤ 0). Only for miRNAs, ms, whose Pecan 19-way alignment was uninformative, we used pairwise alignments for species ipairwises (ipairwise ∈ 36, 37, 38, 42, 43). For the miRNAs, ms, whose
over is defines the Om; thus, the period of origin X was assigned Om (−2 ≤ X ≤ 0). Only for miRNAs, ms, whose Pecan 19-way alignment was uninformative, we used pairwise alignments for species ipairwises (ipairwise ∈ 36, 37, 38, 42, 43). For the miRNAs, ms, whose 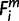 = 1 (i ∈ ipairwises), then
= 1 (i ∈ ipairwises), then 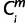 = X where a node X (−3 ≤ X ≤ 0) was the nearest common node between species i and human. The minimum
= X where a node X (−3 ≤ X ≤ 0) was the nearest common node between species i and human. The minimum 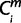 over is defined the originating node Om. If the Om is 0 or −1, then for the m, the period of origin X was assigned Om. Otherwise, if the Om is −2 or −3, then for the m, the period of origin was defined as “conserved beyond theria.”
over is defined the originating node Om. If the Om is 0 or −1, then for the m, the period of origin X was assigned Om. Otherwise, if the Om is −2 or −3, then for the m, the period of origin was defined as “conserved beyond theria.”
Human-Specific Duplications
Out of the alignment blocks for the 1,444 precursor miRNAs, 36 alignment blocks included multiple human genome segments. These are candidates for human-specific duplications detected by the EPO 35-way multiple alignment. Therefore, the genome location of each of the additional human multiply aligned sequences was checked to determine whether they corresponded to the location of the other human precursor miRNAs. In this way, if such human duplicated precursor miRNAs were detected, a further check for duplicated precursor miRNAs that have one-to-one correspondence to those of chimpanzee was performed.
Human miRNA Expression Data Set
We downloaded the human miRNA expression data set (Landgraf et al. 2007) from http://www.cell.com/supplemental/S0092-8674(07)00604-6 (last accessed 29 November 2012). The data set contained the relationship between human precursor miRNAs and the clone counts expressed in both normal and malignant tissues. Every expressed clone count of each miRNA that unambiguously corresponded to a single human precursor miRNA in our data set was retrieved. Subsequently, the expressed clone counts derived only from normal human tissues were retrieved. For each period of origin, the retrieved expressed clone counts of the miRNAs that originated during the period were summed, and then divided by the number of precursor miRNAs of the particular period of origin to obtain the mean expression level.
Simulation Assessments of Effect of Clades With Many Species
A matrix was compiled that represented presence (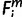 = 1) or absence (
= 1) or absence (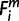 = 0 or −1) of an ortholog within the EPO 35 species (1 ≤ i ≤ 35) for each of the 1,444 human miRNA precursors. Each row of the matrix stood for a human miRNA, each column for a species. Out of the 1,444 miRNAs, every human miRNA whose precursor’s secondary structure folding energy is −13 kcal/mol or higher was excluded from this simulation.
= 0 or −1) of an ortholog within the EPO 35 species (1 ≤ i ≤ 35) for each of the 1,444 human miRNA precursors. Each row of the matrix stood for a human miRNA, each column for a species. Out of the 1,444 miRNAs, every human miRNA whose precursor’s secondary structure folding energy is −13 kcal/mol or higher was excluded from this simulation.
To assess the effect of the clades that comprise many species, we randomly chose one species’ genome from each clade. From the Atlantogenatan clade (31 ≤ i ≤ 35), from the Laurasiatherian clade (19 ≤ i ≤ 30) and from the rodents/rabbits clade (12 ≤ i ≤ 18), we randomly chose one species for each clade; thus, the recompiled matrix for simulation comprised 13 columns (or species). Subsequently, we monitored the change in the number of miRNAs whose origin was assigned to each period. The period of origin was estimated in the same way as used for the original matrix. However, this time, each clade had only one genome; therefore, regardless of whether the chosen genome had high or low coverage; if 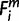 = 1, then
= 1, then 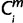 = X for each miRNA, m. The minimum
= X for each miRNA, m. The minimum 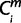 over is defined the originating node Om (13 ≤ Om ≤ 2). Thus, the number of miRNAs whose periods of origin were among 2–13 was obtained for the simulated matrix. The miRNAs, ms, whose Om = 1 include the miRNAs that originated at period 1 and during more ancestral periods. In this simulation, the number of miRNAs originating at period 1 was obtained by subtracting the number of miRNAs originating at periods 0 and before in the original result from the number of ms whose Om = 1. This series of processes was repeated 10,000 times.
over is defined the originating node Om (13 ≤ Om ≤ 2). Thus, the number of miRNAs whose periods of origin were among 2–13 was obtained for the simulated matrix. The miRNAs, ms, whose Om = 1 include the miRNAs that originated at period 1 and during more ancestral periods. In this simulation, the number of miRNAs originating at period 1 was obtained by subtracting the number of miRNAs originating at periods 0 and before in the original result from the number of ms whose Om = 1. This series of processes was repeated 10,000 times.
Supplementary Material
Supplementary tables S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work was supported by JSPS KAKENHI Grant Number 23570273 from Japan Society for the Promotion of Science (JSPS).
References
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentwich I, Avniel A, Karov Y, et al. (13 co-authors) Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- Berezikov E, Thuemmler F, van Laake LW, Kondova I, Bontrop R, Cuppen E, Plasterk RH. Diversity of microRNAs in human and chimpanzee brain. Nat Genet. 2006;38:1375–1377. doi: 10.1038/ng1914. [DOI] [PubMed] [Google Scholar]
- Bortolin-Cavaille ML, Dance M, Weber M, Cavaille J. C19MC microRNAs are processed from introns of large Pol-II, non-protein-coding transcripts. Nucleic Acids Res. 2009;37:3464–3473. doi: 10.1093/nar/gkp205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaille J, Seitz H, Paulsen M, Ferguson-Smith AC, Bachellerie JP. Identification of tandemly-repeated C/D snoRNA genes at the imprinted human 14q32 domain reminiscent of those at the Prader-Willi/Angelman syndrome region. Hum Mol Genet. 2002;11:1527–1538. doi: 10.1093/hmg/11.13.1527. [DOI] [PubMed] [Google Scholar]
- Dewey CN. Aligning multiple whole genomes with mercator and mavid. Methods Mol Biol. 2007;395:221–236. doi: 10.1007/978-1-59745-514-5_14. [DOI] [PubMed] [Google Scholar]
- Flicek P, Amode R, Barrell D, et al. (57 co-authors) Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor I, Bullerdiek J. The dark side of a success story: microRNAs of the C19MC cluster in human tumours. J Pathol. 2012;227:270–274. doi: 10.1002/path.4014. [DOI] [PubMed] [Google Scholar]
- Fornari F, Milazzo M, Chieco P, et al. (15 co-authors) In hepatocellular carcinoma miR-519d is upregulated by p53 and DNA hypomethylation and targets CDKN1A/p21, PTEN, AKT3 and TIMP2. J Pathol. 2012;227:275–285. doi: 10.1002/path.3995. [DOI] [PubMed] [Google Scholar]
- Glazov EA, McWilliam S, Barris WC, Dalrymple BP. Origin, evolution, and biological role of miRNA cluster in DLK-DIO3 genomic region in placental mammals. Mol Biol Evol. 2008;25:939–948. doi: 10.1093/molbev/msn045. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL, Fontana W, Stadler PF, Bonhoeffer S, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 1994;125:167–188. [Google Scholar]
- Hubisz MJ, Pollard KS, Siepel A. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform. 2011;12:41–51. doi: 10.1093/bib/bbq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwama H, Murao K, Imachi H, Ishida T. MicroRNA networks alter to conform to transcription factor networks adding redundancy and reducing the repertoire of target genes for coordinated regulation. Mol Biol Evol. 2011;28:639–646. doi: 10.1093/molbev/msq231. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Hedges SB. TimeTree2: species divergence times on the iPhone. Bioinformatics. 2011;27:2023–2024. doi: 10.1093/bioinformatics/btr315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, et al. (51 co-authors) A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnert S, Kapitonov V, Thilakarathne PJ, Schuit FC. Modeling the asymmetric evolution of a mouse and rat-specific microRNA gene cluster intron 10 of the Sfmbt2 gene. BMC Genomics. 2011;12:257. doi: 10.1186/1471-2164-12-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Liu Y, Dong D, Zhang Z. Evolution of an X-linked primate-specific micro RNA cluster. Mol Biol Evol. 2010;27:671–683. doi: 10.1093/molbev/msp284. [DOI] [PubMed] [Google Scholar]
- Liang H, Li WH. Lowly expressed human microRNA genes evolve rapidly. Mol Biol Evol. 2009;26:1195–1198. doi: 10.1093/molbev/msp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Shen Y, Wu Q, Kumar S, He B, Shi S, Carthew RW, Wang SM, Wu CI. The birth and death of microRNA genes in Drosophila. Nat Genet. 2008;40:351–355. doi: 10.1038/ng.73. [DOI] [PubMed] [Google Scholar]
- Marphy WJ, Eizirk E. Placental mammals (Eutheria) In: Hedges SB, Kumar S, editors. The timetree of life. New York: Oxford University Press; 2009. pp. 471–474. [Google Scholar]
- Nozawa M, Miura S, Nei M. Origins and evolution of microRNA genes in Drosophila species. Genome Biol Evol. 2010;2:180–189. doi: 10.1093/gbe/evq009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paten B, Herrero J, Beal K, Fitzgerald S, Birney E. Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 2008;18:1814–1828. doi: 10.1101/gr.076554.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, et al. (16 co-authors) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Haussler D. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol Biol Evol. 2004;21:468–488. doi: 10.1093/molbev/msh039. [DOI] [PubMed] [Google Scholar]
- Stabenau A, McVicker G, Melsopp C, Proctor G, Clamp M, Birney E. The Ensembl core software libraries. Genome Res. 2004;14:929–933. doi: 10.1101/gr.1857204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiper ME, Young NM. Primates (Primates) In: Hedges SB, Kumar S, editors. The timetree of life. New York: Oxford University Press; 2009. pp. 482–486. [Google Scholar]
- Tarver JE, Donoghue PC, Peterson KJ. Do miRNAs have a deep evolutionary history? Bioessays. 2012;34:857–866. doi: 10.1002/bies.201200055. [DOI] [PubMed] [Google Scholar]
- Vaquerizas JM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Zhang R, Peng Y, Wang W, Su B. Rapid evolution of an X-linked microRNA cluster in primates. Genome Res. 2007;17:612–617. doi: 10.1101/gr.6146507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Wang YQ, Su B. Molecular evolution of a primate-specific microRNA family. Mol Biol Evol. 2008;25:1493–1502. doi: 10.1093/molbev/msn094. [DOI] [PubMed] [Google Scholar]
- Zheng GX, Ravi A, Gould GM, Burge CB, Sharp PA. Genome-wide impact of a recently expanded microRNA cluster in mouse. Proc Natl Acad Sci U S A. 2011;108:15804–15809. doi: 10.1073/pnas.1112772108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.