

Abstract
Aging and exposure to environmental toxins including MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) are strong risk factors for developing Parkinson's disease (PD), a common neurologic disorder characterized by selective degeneration of midbrain dopaminergic (DAergic) neurons and astrogliosis. Aging and PD impair the subventricular zone (SVZ), one of the most important brain regions for adult neurogenesis. Because inflammation and oxidative stress are the hallmarks of aging and PD, we investigated the nature, timing, and signaling mechanisms contributing to aging-induced SVZ stem/neuroprogenitor cell (NPC) inhibition in aging male mice and attempted to determine to what extent manipulation of these pathways produces a functional response in the outcome of MPTP-induced DAergic toxicity. We herein reveal an imbalance of Nrf2-driven antioxidant/anti-inflammatory genes, such as Heme oxygenase1 in the SVZ niche, starting by middle age, amplified upon neurotoxin treatment and associated with an exacerbated proinflammatory SVZ microenvironment converging to dysregulate the Wingless-type MMTV integration site (Wnt)/β-catenin signaling, a key regulatory pathway for adult NPCs. In vitro experiments using coculture paradigms uncovered aged microglial proinflammatory mediators as critical inhibitors of NPC proliferative potential. We also found that interruption of PI3K (phosphatidylinositol3-kinase)/Akt and the Wnt/Fzd/β-catenin signaling cascades, which switch glycogen synthase kinase 3β (GSK-3β) activation on and off, were causally related to the impairment of SVZ-NPCs. Moreover, a synergy between dysfunctional microglia of aging mice and MPTP exposure further inhibited astrocyte proneurogenic properties, including the expression of key Wnts components. Last, pharmacological activation/antagonism studies in vivo and in vitro suggest the potential that aged SVZ manipulation is associated with DAergic functional recovery.
Introduction
Aging and exposure to the neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) are critical risk factors for developing Parkinson's disease (PD), a common neurodegenerative disorder of unknown causes, characterized by progressive loss of midbrain dopaminergic (DAergic) neurons (Langston et al., 1999; Olanow et al., 2003; Hindle, 2010). While host genetics accounting for <10% of cases, environmental factors also can affect disease onset and/or progression (Warner and Schapira, 2003; Morale et al., 2008). Inflammation and oxidative stress in particular are important hallmarks of aging and PD development (Olanow et al., 2003; Chen et al., 2005; Marchetti and Abbracchio, 2005; Gao and Hong, 2008; Gao et al., 2008; McGeer and McGeer, 2008; Hirsch and Hunot, 2009; Przedborski, 2010; Marchetti et al., 2011). One mechanism by which cells combat oxidative/inflammatory insults is through the redox-sensitive transcription factor Nrf2, which is activated by oxidants to induce the expression of antioxidant, anti-inflammatory, and cytoprotective genes, such as Heme oxygenase1 (Hmox) (Chen et al., 2009; Surh et al., 2009; Bitar and Al-Mulla, 2011). With age, however, antioxidant self-defense response is reduced (Suh et al., 2004; Shih and Yen, 2007), while microglia become “primed” (i.e., capable of adopting a potent neurotoxic phenotype) (Henry et al., 2009; Njie et al., 2012). Importantly, aging reduces the degree of DAergic neuron plasticity (Ricaurte et al., 1987a,b; Ho and Blum, 1998; Collier et al., 2007), at least in part as a result of a reduced or limited neurogenesis, with causes and mechanisms not fully elucidated.
Indeed, with the process of aging and PD, the neurogenic potential is dramatically reduced in the subventricular zone (SVZ) of the lateral wall of the lateral ventricles (Tropepe et al., 1997; Enwere et al., 2004; Höglinger et al., 2004; Maslov et al., 2004; Luo et al., 2006; Ahlenius et al., 2009). Here, stem/neuroprogenitor cells (NPCs) are in intimate contact with surrounding glia, forming the so-called “stem cell niche” (Lim and Alvarez-Buylla, 1999; Alvarez-Buylla et al., 2001; Song et al., 2002; Kazanis, 2009), where neurotransmitters, different growth/neurotrophic factors, morphogens, nitric oxide (NO), cytokines, and key components of the Wingless-type MMTV integration site (Wnt)/β-catenin signaling pathway contribute to SVZ regulation (Höglinger et al., 2004; Estrada and Murillo-Carretero, 2005; Butovsky et al., 2006; Ziv et al., 2006; Adachi et al., 2007; Borta and Höglinger, 2007; Kalani et al., 2008; Pluchino et al., 2008; Ziv and Schwartz, 2008; Ekdahl et al., 2009; O'Keeffe et al., 2009; Young et al., 2011; Zhang et al., 2011).
Recently, we uncovered an active and concerted role of reactive astrocytes and microglia in the remodeling of the SVZ niche upon MPTP/1-methyl-4-phenylpyridinium ion injury, and we found that this role is at least in part regulated by cross talk between inflammation and Wnt/β-catenin signaling cascades (L'Episcopo et al., 2012). Here, we highlight a candidate role for Nrf2/Hmox axis imbalance in aging SVZ and demonstrate that aging-induced dysfunctional astrocyte–microglia cross talk (the first hit) acts as a key driver of SVZ impairment through reduced Nrf2-mediated SVZ adaptive response to neurotoxin exposure (the second hit), with harmful consequences for SVZ cell homeostasis. Synergy between “primed”/dysfunctional microglia of aging mice and MPTP exposure further inhibits astrocyte proneurogenic properties, including the expression of key Wnts components. Hence, the interruption of two pivotal signaling cascades, the PI3K (phosphatidylinositol3-kinase)/Akt and the Wnt/Fzd/β-catenin, which switch GSK-3β activation on and off, were causally related to SVZ neurogenic impairment. Importantly, pharmacological activation/antagonism studies, in vivo and in vitro, suggest that aged SVZ manipulation is potentially associated with DAergic functional recovery.
Materials and Methods
Mice and treatments.
Male C57BL/6 mice of 2–5, 8–10, and 22–24 months of age (Charles River, Calco) received n = 4 intraperitoneal injections of vehicle (saline) or MPTP-HCl (Sigma-Aldrich) dissolved in saline, 2 h apart in 1 d, at the dose of 20 mg/kg−1 free base for young mice and 10 mg-kg−1 free base for aging mice, respectively. These doses were selected based on titration studies that produced comparable depletions of DAergic endpoints in both striatum (Str) and substantia nigra pars compacta (SNpc) in young and older mice, without causing toxicity (Table 1). MPTP was handled in accordance with the reported guidelines (Jackson-Lewis and Przedborski, 2007). At the indicated days post-MPTP treatment (dpt), mice were given bromodeoxyuridine (BrdU, 50 mg/kg−1, injected 4×, 2 h apart) and killed 2 h after the last injection. All animal studies were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals (NIH), and approved by the Institutional Animal Care and Use Committee guidelines. All surgeries were performed under anesthesia with all efforts made to minimize suffering.
Table 1.
Dose–response effect of MPTP-induced DAergic toxicity in young and middle-aged mice
| Analyses | MPTP doses (n = 4 in 1 d) |
|||
|---|---|---|---|---|
| −MPTP | 20 mg/kg−1 | 15 mg/kg−1 | 10 mg/kg−1 | |
| Young control mice | ||||
| Striatal DA (pm/mg protein) | 405 ± 28 | 127 ± 12* | 170 ± 10* | 218 ± 19* |
| DA uptake (% of ct) | 100 ± 14 | 29 ± 10* | 42 ± 8* | 54 ± 9* |
| SNpc TH+ neurons (% of ct) | 100 ± 12 | 38 ± 8* | 48 ± 8* | 60 ± 10* |
| Middle-aged mice | ||||
| Striatal DA (pm/mg protein) | 390 ± 35 | 75 ± 10* | 125 ± 9* | |
| DA uptake (% of ct) | 100 ± 8 | 15 ± 8* | 27 ± 6* | |
| SNpc TH+ neurons (% of ct) | 100 ± 10 | 26 ± 8* | 37 ± 7* | |
Young (2–5 months) mice received n = 4 intraperitoneal injections of vehicle (saline, 10 ml/kg) or MPTP-HCl at the indicated doses (mg/kg−1 free base; Sigma-Aldrich) dissolved in saline, 2 h apart in one day (80, 60, and 40 mg/kg−1 cumulative). Middle-aged (8–10 months) mice received only the 15 and 10 mg/kg−1 doses. The evaluation point was set 5 dpt, based on previous studies (L'Episcopo et al., 2011a). No mortality was observed in young mice while survival rate was reduced in middle-aged mice at 15 mg/kg−1. For each set of analyses, five mice were used for the indicated time points. The brains processed as indicated for striatal DA concentration by HPLC analysis, for synaptosomial high-affinity [3H] dopamine (DA) uptake in striatum (total high-affinity and mazindol noninhibitable), and for TH+ neuron number in the SNpc, and values expressed as percentages of controls (−MPTP = 100). Differences were analyzed by ANOVA followed by Newman–Keuls test, and considered significant when p < 0.05. *p < 0.05 versus −MPTP. The doses of 20 and 10 mg/kg−1 leading to comparable loss of DAergic endpoints in young and ageing mice, respectively, were then selected for all subsequent experiments.
Experimental design.
The study included both in vivo and in vitro experiments. In vivo studies, performed in young and aging mice, both in basal condition and at different time points (tps) following MPTP injection as above (6–8 mice/age group/tp), were conducted two times, and quantification of the different parameters studied for each series of analyses performed in n ≥ 6. For studies in SVZ tissues and stem/NPCs isolated from mice of the indicated age groups and tps following saline or MPTP exposure (7–8 mice/age group/tp), the freshly isolated SVZ and NPCs were processed as described for either real-time PCR or Western blotting (n ≥ 3 individual determinations). For in vitro studies in NPC cultures derived from young and aging mice as above, the NPC–glial cocultures and astrocyte–microglial coculture paradigms were performed in triplicates, and at least three independent cultures were used for quantifications.
Immunohistochemistry.
After mice were deeply anesthetized and transcardially perfused, the brains were carefully removed and stored at −80°C until further analyses. Criostatic coronal sections (14 μm thick) were collected, mounted on poly-l-lysine-coated slides and processed with preabsorbed primary antibodies. Serial coronal sections (14 μm thick), containing the SVZ (0.74, 0.5, 0.14, and 0.02 mm anterior to bregma) (Paxinos and Watson, 1997) were collected and mounted on poly-l-lysine-coated slides. The following preabsorbed primary antibodies previously characterized in brain-tissue sections and cell cultures (Gennuso et al., 2004; Morale et al., 2004; L'Episcopo et al., 2010b, 2011a, 2011b, 2011c, 2012) were used: mouse anti-BrdU (1:200; Sigma-Aldrich); goat anti-doublecortin (anti-DCX, 1:400; Santa Cruz Biotechnology); mouse anti-glial fibrillary acidic protein (anti-GFAP,1:500, Sigma-Aldrich); rabbit anti-GFAP (Dako, Cytomation); rabbit anti-epidermal growth factor receptor (anti-EGF-R, 1:200, Millipore Bioscience Research Reagents), goat anti-heme oxygenase 1 (anti-Hmox, 1:150, Santa Cruz Biotechnology); goat anti-ionized calcium-binding adapter molecule 1 (anti-IBA1, 1:200, Novus Biologicals); rat anti-dopamine transporter (anti-DAT, 1: 500, Millipore Bioscience Research Reagents); mouse anti-tyrosine hydroxylase (anti-TH, Boehringer Mannheim), mouse anti-neuron-specific nuclear protein (anti-NeuN,1: 500, United States Biological); rabbit anti-cleaved Caspase3 (1:200, Cell Signaling Technology); rabbit anti-β-catenin (1:200, Abcam).
Visualization of incorporated BrdU requires DNA denaturation performed by incubating the sections in HCl for 30 min at 65°C. After overnight incubation, sections were rinsed and incubated in darkness for 2 h with CY3/FITC-conjugated donkey anti-mouse, donkey anti-rabbit, and donkey anti-goat antibodies (1:100/300; Jackson ImmunoResearch), mounted on glass slides and coverslipped with glycerol-based mounting medium. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) or propidium iodine (PI) in mounting medium (Vector Laboratory). For TH+ neuron cell counting, cresyl violet was used to visualize Nissl substance. In all of these protocols, blanks were processed as for experimental samples except that the primary antibodies were replaced with PBS.
Apoptotic DNA damage was identified in cells already immunostained with anti-GFAP or with anti-NeuN by applying the TUNEL technique (Gennuso et al., 2004), with negative and positive controls according to the manufacturer's instructions (TACS TdT in situ apoptosis detection kit, R&D Systems). Confocal laser scanning microscopy was applied on a minimum of four different SVZ sections/brain, in 6 mice/experimental group and at the indicated tps, using a 20×, 40×, and 100× objective (Gennuso et al., 2004).
Microscopical analysis.
All assessments were performed by a blinded observer. Immunostaining was examined along the entire lateral wall of the lateral ventricle, from the dorsolateral aspect to the level of the anterior commisure, using a Leica LCS-SPE confocal microscope equipped with image analysis software (Leica Microsystems), using 20×, 40×, and 100× (oil) immersion objectives (L'Episcopo et al., 2010b, 2011a,b, 2012) using a semiautomatic stereology system (Mercator, Explora Nova). Briefly, four coronal sections throughout the SVZ, located 0.74, 0.5, 0.14, and 0.02 mm anterior to bregma (Paxinos and Watson, 1997), were analyzed bilaterally. Estimations of the different phenotypic markers (cell numbers/mm3 SVZ, neurons per unit volume) in SVZ was performed using the 20× objective and conventional stereological methods (Gundersen and Jensen, 1987).
Image analysis of Hmox immunofluorescent staining in SVZ.
For Hmox fluorescence intensity assessment and colocalization with the astrocyte marker, GFAP, SVZ brain sections labeled by immunofluorescence were visualized and analyzed using a Leica LCS-SPE confocal microscope, as above (Leica Lasertechnik), equipped with an argon–krypton laser using 10×, 20×, 40×, and 100× oil-immersion objectives (Gennuso et al., 2004). Pinhole was set at 1.0–1.2 for optical sections of 0.5–0.6 μm. Single lower-power scans were followed by 16–22 serial optical sections in 6 fields/section using the same settings. The average fluorescence (mean ± SD) intensity (pixel) in SVZ areas was measured throughout the stack. To estimate Hmox fluorescence localized in SVZ astrocytes, sections double-stained with anti-Hmox and anti-GFAP were acquired separately with the FITC and CY3 filters, and the intensity of Hmox fluorescence was determined in GFAP-positive areas in the SVZ, and corrected for background measured in areas devoid of cells (Gennuso et al., 2004). Additionally, the percentages of Hmox+/GFAP+ cells out of the total GFAP+ cells were estimated in each condition, in ≥100 cells, read from at least four SVZ sections/brain, in 6 mice/experimental group, and results expressed as mean ± SD. 3D reconstruction from z-series were used to verify colocalization in the x–y, y–z, and x–z planes.
NPC isolation from SVZ of young and aged mice.
Animals were deeply anesthetized with halothane and killed by cervical dislocation. Brain coronal sections from young and aging mice treated with vehicle or MPTP were taken from 2 mm from the anterior pole of the brain, excluding the optic tracts, and 3 mm posterior to the previous cut, at the indicated time intervals (3, 21, and 65 dpt). The SVZs were then dissected out under a microscope (Pluchino et al., 2003, 2005; L'Episcopo et al., 2011a,b, 2012). Dissected tissue was transferred to Earle's balanced salt solution (Invitrogen) containing 1 mg/ml papain (27 U/mg; Sigma-Aldrich), 0.2 mg/ml cysteine (Sigma-Aldrich), and 0.2 mg/ml EDTA (Sigma-Aldrich), and incubated for 45 min at 37°C on a rocking platform. Tissues were then transferred to DMEM-F-12 medium (1:1 v/v; Invitrogen) containing 0.7 mg/ml ovomucoid (Sigma-Aldrich) and mechanically dissociated. After digestion, the number of viable cells was determined by trypan blue (Sigma-Aldrich) exclusion and cells processed for cell culture as described below.
Cell culture conditions.
The isolated cells were plated in 24-well uncoated plates (0.5 ml/well, Corning) at 8000 cells/cm2 in neurosphere growth medium (DMEM/F12 containing 2 mm l-glutamine, 0.6% glucose, 0.1 mg/ml apo-transferrin, 0.025 mg/ml insulin, 9.6 μg/ml putrescin, 6.3 ng/ml progesterone, 5.2 ng/ml Na selenite, 2 μg/ml heparin) supplemented with epidermal growth factor (EGF) (20 ng/ml) and fibroblast growth factor (FGF-II) (10 ng/ml) (proliferative medium), as described previously (Gritti et al., 2002; Pluchino et al., 2003, 2008). Seven days after plating, primary spheres were mechanically dissociated into single cells and plated at a final density of 1 × 105 cells/cm2 on poly-d-lysine-coated 24-well plates and processed for proliferation and differentiation assays. Additionally, we extended in vitro culturing of neurospheres in the presence of FGF-II and EGF for 10–12 passages of amplification. Proliferative capacity was studied in neurospheres exposed to proliferative medium for 3 DIV, by addition of the nucleotide analog BrdU (5 μm) at 2 DIV and the cells fixed after 24 h at 3 DIV. For differentiation studies, neurospheres cultured in proliferation medium for 48–72 h were then shifted in differentiation medium consisting of a 1:1 mixture of F12 and MEM, without growth factors, containing HEPES and glutamine, with N2 supplements (Invitrogen), with 1% fetal calf serum (Pluchino et al., 2008). Neurospheres were let to differentiate for 5–10 DIV in the absence or the presence of the different treatments/coculture paradigms, as described.
Glial cell cultures from young and aging mice.
Mixed glial cell cultures were obtained from mouse Str at postnatal day 2 and at 2, 10, and 24 months, and cultured as described previously (L'Episcopo et al., 2011a,b, 2012). The astrocyte cultures obtained after separation of microglia [>95% of the cells were GFAP immunoreactive (IR) astrocytes] and the enriched microglial (>95% of the cells were IBA1-IR microglia) monolayers were rinsed with sterile PBS and replated at a final density of 0.4–0.6 × 105 cells/cm2 in poly-d-lysine (10 μg/ml)-coated 6-well, 12-well, or 24-well plates, or in insert membranes (0.4 μm polyethylene terephthalate) for indirect coculture (BD Biosciences).
NPC–glial cocultures.
Glial monolayers of the different ages were cocultured with either young or aged NPCs in both direct (allowing for cell-to-cell contacts) and indirect coculture paradigms. As a control (ct-insert), a nonglial preparation (i.e., neuronal GT1–7 cells) was used (L'Episcopo et al., 2011a). For proliferation studies, NPCs cultured as above were layered on the top of the glial cell monolayers. After 24 h, the nucleotide analog BrdU (5 μm) was added and the cells fixed after 24–48 h. For differentiation studies, NPCs were cultured in differentiation medium as above, and cells fixed at 5 DIV. The cells were then processed for fluorescent immunocytochemistry as described. For the indirect coculture paradigm, the inserts containing the glial monolayers or the ct-inserts were added on the top of the young or aged NPCs (L'Episcopo et al., 2011a, 2012). These inserts allowed diffusion of factors from the glia monolayer to the NPCs and vice versa, without direct contact between cells (L'Episcopo et al., 2011a, 2012). NPC survival was estimated by cell counting and determination of Caspase3 activity.
Cell treatments.
The IBA1+ cells acutely isolated from mouse brain at different ages were cultured for 24–48 h in the absence or the presence of the NO-donating cyclo-oxygenase inhibitor, HCT1026 [2-fluoro-α-methyl(1,1′-biphenyl)-4-acetic-4-(nitrooxy)butyl ester]. Pilot experiments were performed with increasing doses (5–25 μm) of HCT1026 and the dose of 10 μm was selected for its ability to reverse exacerbated ROS and inducible nitric oxide synthase-derived NO production (Marchetti et al., 2002), without causing microglial cell toxicity (Table 2).
Table 2.
Effect of HCT1026 on ageing-induced increased reactive microglial cell markers and survival assays
| Treatments | DCFH-DA (% of control) | Nitrite (% of control) | MTT (% of control) | Caspase3 (% of control) |
|---|---|---|---|---|
| Plus PBS | 220 ± 22 | 205 ± 18 | 95 ± 10 | 105 ± 12 |
| Plus HCT1026 | ||||
| 5 μm | 170 ± 24* | 156 ± 15* | 100 ± 9 | 98 ± 10 |
| 10 μm | 105 ± 14* | 110 ± 10* | 102 ± 12 | 100 ± 14 |
| 25 μm | 90 ± 12* | 87 ± 11* | 95 ± 8 | 110 ± 8 |
Microglial (IBA1+) cells acutely isolated from young (control) and aging mouse brain were cultured for 24–48 h in the presence of the NO-donating NSAID, HCT1026 [2-fluoro-α-methyl(1,1'-biphenyl)-4-acetic-4-(nitrooxy)butyl ester] or phosphate buffered saline (PBS), as described. Experiments were performed with the freshly prepared IBA1+ cells cultured in the presence of increasing doses of HCT1026 (5–25 μm) applied after plating. The cells were processed 24–48 h after plating. Part of the cultures were processed for intracellular ROS using the redox membrane-permeant probe 2',7'-dichlorofluorescein diacetate (DCFH-DA, 50 μm, added for 1 h at 37°C), and cells viewed under the confocal microscope (Gennuso et al. 2004). Measurement of iNOS-derived NO was carried out in cell-free supernatant using Griess reagent (Marchetti et al. 2002; Morale et al. 2004; L'Episcopo et al. 2011c, 2012). Cell survival was determined by the MTT and Caspase3 assays. Results are expressed as percent changes over control (young microglial treated with PBS = 100). *p < 0.05 versus PBS.
RNA extraction, reverse transcription and real-time PCR.
RNA extraction was performed in freshly isolated SVZ tissue, in acutely isolated SVZ cells (7–8 mice/age group/tp), and in cell cultures under the different treatment conditions, as previously detailed (Pluchino et al., 2008; L'Episcopo et al., 2011a,b, 2012). Briefly, the tissue/cell samples were homogenized in 1 ml of QIAzol Lysis Reagent (#79306, Qiagen) using a rotor-stator homogenizer. Total RNA was isolated from homogenized samples using RNeasy Lipid Tissue Kit (#74804, Qiagen) including Dnase digestion. At the end, RNA samples were redissolved in 30 μl of RNase-free water and their concentrations were determined spectrophotometrically by A260 (Nanodrop-ND 1000), and the cDNA was synthesized from 2 μg of total RNA using the Retroscript Kit (Ambion). After purification using a QIAquick PCR Purification kit (Qiagen), 250 ng of cDNA were used for real-time PCR using predeveloped Taqman Assay Reagents (Applied Biosystems). Real-time quantitative PCR was performed with Step One Detection System (Applied Biosystems) according to the manufacturer's protocol, using the TaqMan Universal PCR master mix (#4304437). For each sample, we designed a duplicate assay and β-actin was used exclusively as the housekeeping gene. The assay IDs were as follows: Nrf2, Mm00477784_m1; Hmox1, Mm00516005_m1; Hmox2, Mm00468922_m1; Nos2, Mm00440485_m1; gp91PHOX, Mm01287743_m1; TNFα, Mm00443258_m1; Fzd-1, Mm00445405_s1; Fzd-3, Mm00445423_m1; Fzd-4, Mm00433382_m1; Fzd-5, Mm03053323_s1; Fzd-6, Mm00433383_m1; Fzd-7, Mm01255614_s1; Fzd-8, Mm00433419_s1; Fzd-9, Mm01206511_s1; β-catenin, Mm00483039_m1; Axin2, Mm00443610_m1; Wnt1, Mm00810320_s1. We used the housekeeping gene, β-actin, as normalizer and embrionic mouse brain as calibrator. Results are expressed as arbitrary units or as n-fold induction over levels from cells not exposed to MPTP (−MPTP levels). Real-time PCR determinations were repeated three times independently.
Western blot analysis.
Protein extracts were prepared from SVZ tissue/cells isolated from saline or MPTP mice (n = 6–8 mice/age group/tp) and from cell cultures (run in triplicates) within the different experimental groups, as described previously (L'Episcopo et al., 2010b, 2011a,b,c). The samples were loaded into a 9–12% SDS-polyacrilamide gel and separated by electrophoresis for 3 h at 100 V. Proteins were transferred to polyvinylidene difluoride membrane (GE Healthcare) for 1.5 h at 300 mA. The following primary antibodies were used: Akt (rabbit polyclonal, 1:1000, Cell Signaling Technology); Akt phospho-Ser473 (rabbit polyclonal, 1:1000, Cell Signaling Technology); GSK-3β phospho-Tyr216 (rabbit polyclonal, 1:500, Abcam); GSK-3β (rabbit polyclonal, 1:200), β-catenin (rabbit polyclonal, 1:200), Fzd-1 (goat polyclonal, 1:1000), and Hmox (goat polyclonal, 1:200), all from Santa Cruz Biotechnology; β-actin (1:1000, Cell Signaling Technology). Blots were first probed for phosphorylated protein, followed by stripping in Restore Plus Western Blot Stripping Buffer (Thermo Scientific/Pierce Biotechnology) and reprobing for the corresponding unphosphorylated proteins, or for β-actin as a loading control. The bands from the Western blots were densitometrically visualized and the signals quantified on x-ray film using ImageQuantity One, the data subjected to statistical ANOVA. Western blot measurements were repeated three times independently.
Caspase3 activity.
The cells were lysed in ice-cold lysis buffer containing 25 mm HEPES, 5 mm EDTA, 1 mm EGTA, 5 mm MgCl2, 5 mm dithiothreitol (DTT), 1 mm phenylmethylsulfonyl fluoride, and 10 μg/ml each of pepstatin and leupeptin, pH 7.5. The cells were left for 20 min on ice and then sonicated. The lysate was centrifuged for 20 min at 10,000 × g and the supernatant was quickly frozen in a methanol dry-ice bath and stored at −80°C. Lysates (30 μg protein) were incubated at 37°C in a buffer containing 25 mm HEPES, pH 7.5, 10% sucrose, 0.1 3-[(3-cholamido propyl) dimethyl ammonio]-1-propanesulphonate, and 10 mm DTT with the fluorogenic substrate N-acetyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethylcoumarin (DEVD-AFC, 15 μm in dimethylsulfoxide; Calbiochem System Products), and quantification of DEVD-like fluorescent signal assessed in luminescence-spectrophotometer (excitation, 400 nm; emission, 505 nm) (L'Episcopo et al., 2011b, 2012). Enzymatic activity is expressed as arbitrary fluorescent units.
Mitochondrial activity with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
The colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to measure mitochondrial functionality in glial cells (Gennuso et al., 2004). Briefly, cells were incubated with 0.25 mg/ml MTT for 3 h at 37°C, and mitochondrial enzyme activity was measured in culture supernatants in a spectrophotometer (Molecular Devices) at 570 nm, with a reference wavelength of 630 nm. Results are expressed as percentage changes of control (young microglial preparation treated with PBS).
In vivo treatment of middle-aged mice with HCT1026.
Middle-aged mice received the NO-donating derivative of flurbiprofen, HCT1026 (kindly provided by NicOx), which belongs to a novel class of nonsteroidal anti-inflammatory drugs (Keeble and Moore, 2002) endowed with additional anti-inflammatory activity and strongly reduced side effects, administrated in the diet (190 ppm in the diet or 30 mg/kg−1 per day per animal) (Furlan et al., 2004; Bernardo et al., 2005; L'Episcopo et al., 2010b, 2011c). Control mice received a control diet (plain Teklad 2018 chow). Three weeks after introducing HCT1026, some mice were killed and their brains processed for different determinations. Meanwhile other mice were challenged with either saline or MPTP, and killed at different time points, as indicated, and the brains processed for SVZ isolation. For immunohistochemistry, groups of mice were anesthetized and transcardially perfused (n = 6 mice/tp/group). For neurochemical and Western blot analyses (n = 5 mice/tp/group), the brains were rapidly dissected and stored at −80°C (see Figs. 10, 11).
Figure 10.
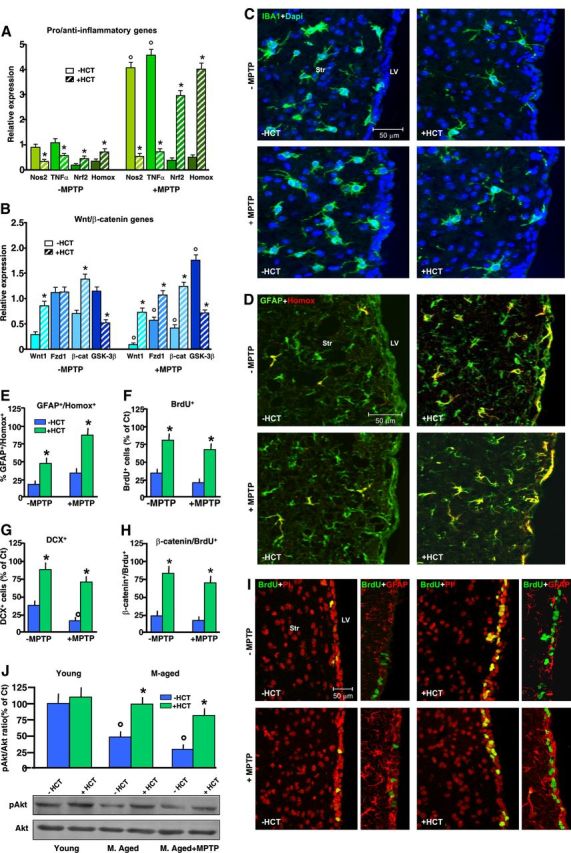
HCT1026-induced normalization of redox balance in aging SVZ reverses neurogenic impairment via Nrf2/Akt/Wnt/Fzd1/β-catenin signaling activation. A, After middle-aged mice were fed with a control (Ct) or HCT1026 (HCT) diet for 3 weeks, some mice were killed and their brains processed for either SVZ isolation and gene/protein expression analyses, or immunohistochemistry, while other mice were challenged with either saline or MPTP and killed at both early and later time upon treatment, as indicated. A, B, Real-time PCR analyses for Nos2, TNF-α, Nrf2, and Hmox (A); and Wnt1, Fzd-1, β-catenin, and GSK-3β (B) in SVZ samples. Changes in mRNA levels are expressed as n-fold induction over saline in mice of the different groups. The results obtained in basal conditions (−MPTP) and 24 h post-MPTP (means ± SEM of 3 independent experiments) are shown. Differences analyzed as described and considered significant when p < 0.05. *p < 0.05 versus −HCT1026; °p < 0.05 versus −MPTP. C, D, Representative images of IBA+ cells (green), counterstained with the nuclear marker, DAPI (blue) (C), and dual labeling of GFAP+ cells (green) with Hmox (red) in middle-aged mice fed with a Ct or HCT1026 diet for 3 weeks, and 3 dpt after saline or MPTP challenge both in the absence or the presence of HCT1026 diet (D). E–H, Changes in percentages of GFAP+/Hmox+ (E), BrdU+ (F), DCX+ (G), and β-catenin+/BrdU+ cells (H, mean ± SD). I, Representative images of BrdU+ cells (green), counterstained with the nuclear marker, PI (red), and dual labeling of GFAP+ cells (red) with BrdU (green) in middle-aged mice fed with a Ct or HCT1026 diet for 3 weeks, and 3 dpt after saline or MPTP challenge both in the absence or the presence of HCT1026 diet. J, Western blot analysis of phospho-Akt-Ser473 in protein extracts from SVZ cells. Densitometric values depicted as ratio of the phosphorylated form over total amount of Akt (p-Akt expressed as percentage of young control). Differences analyzed as above. *p < 0.05 versus −HCT1026; °p < 0.05 versus −MPTP.
Figure 11.

HCT1026-induced aged-SVZ rescue promotes DAergic neuroprotection upon MPTP challenge. Determination of DAergic endpoints in middle-age (8 months) mice fed with a control (ct) or HCT1026 diet for 3 weeks (−MPTP), and 7 and 65 d after saline or MPTP challenge, in the continuous presence of either a control or HCT diet. A, B, Fluorescence intensity values (means ± SEM) expressed as percentage (%) of saline. DAT (A) and TH (B) staining. Image analysis by confocal laser microscopy in Str shows a significant neuroprotection against MPTP in HCT1026-fed mice compared with mice fed with a control diet at both time points. C, Representative confocal images of DAT (red) and TH (green) immunofluorescent staining in Str of middle-aged mice 7 d after MPTP in the absence or the presence of HCT1026 are shown. D, E, Specific high-affinity neuronal DA uptake in Str (D) and TH+ neuron numbers in the SNpc (E) show a significant degree of neuroprotection in HCT1026-fed mice challenged with MPTP, compared with mice fed with a control diet. Differences were analyzed by ANOVA followed by Newman–Keuls test, and considered significant when p < 0.05. *p < 0.05 versus −HCT1026.
Dopaminergic endpoints in Str and ventral midbrain.
High-affinity [3H] dopamine (DA) up-take was determined in synaptosomes obtained from left and right striata, as described previously (Morale et al., 2004; L'Episcopo et al., 2010b, 2011a,b,c). Specific high-affinity neuronal DA uptake is expressed as fentomoles of DA uptake per microgram of protein minus the fentomoles of mazindol uptake. Values are represented as percentage changes in dopamine uptake versus control. Striatal DA was determined by HPLC according to Morale et al. (2004) in striata of both sides weighed, pooled, and homogenized in 1% meta-H3PO4 containing 1 mm EDTA. After centrifugation (17,500 × g for 10 min at 4°C), the supernatant was filtered and immediately injected into the HPLC system for DA determination. The loss of TH+ SNpc neurons was determined by serial section analysis of the total number of TH+ cells counted through the entire rostro-caudal axis of the murine SNpc (bregma coordinates: −2.92, −3.08, −3.16, −3.20, −3.40, and −3.52) according to Franklin and Paxinos (1997) (L'Episcopo et al., 2010b, 2011a,b). Cell counting was done in both sides of the brain for each animal, and then right and left values were added to generate a total DA SNpc neuron count. TH+ neurons were scored as positive only if their cell-body image included well defined nuclear counterstaining. Estimates of total TH+-stained and cresyl-violet-stained neurons in the SNpc were calculated using Abercrombie's correction (Abercrombie, 1946). DAT-fluorescence and TH-fluorescence intensity (FI) in Str was assessed in n = 3 coronal sections at three levels (bregma coordinates: +0.5, +0.86, and 1.1 mm, respectively) of caudate–putamen, in n = 6 mice/group/time, with DAT-FI and TH-FI above a fixed threshold using the corpus callosum for background subtraction, and changes in average FI (mean ± SD) expressed as percentage of saline-injected controls (L'Episcopo et al., 2010b, 2011a,b,c, 2012).
Data analysis.
Statistical significance between means ± SEM was analyzed by a two-way ANOVA, and Student's t test for paired or unpaired data. Experimental series performed on different days were compared by the Student–Newman–Keuls t test. A value of p < 0.05 was considered to be statistically significant.
Results
Aging-induced SVZ impairment is exacerbated in MPTP-induced nigrostriatal DAergic toxicity with no recovery
We first correlated, in vivo, spatiotemporal changes in proliferation and neuroblast formation in the SVZ of middle-aged (8–10 months) and aged (22–24 months) compared with young (2–5 months) mice (Fig. 1). We used DCX, a microtubule-associated protein for type-A cells and EGF-R mainly localized in rapidly cycling transit amplifying cells (so-called C cells) (Doetsch et al., 1997, 1999, 2002; García-Verdugo et al., 1998; Höglinger et al., 2004). In accordance with previous studies (Enwere et al., 2004; Maslov et al., 2004; Luo et al., 2006; Ahlenius et al., 2009), the process of aging is accompanied by a marked decrease in the total number of BrdU+ cells, DCX+ neuroblasts, and EGF-R+ cells. As observed by Luo et al. (2006), a major reduction in BrdU+ cells occurs already by middle age, and a further, albeit smaller decline in BrdU+ cells is observed in aged mice (Fig. 1A–F). Likewise, the formation of DCX+ neuroblasts was markedly inhibited by middle age, with a further decrease measured in aged SVZ (Fig. 1B–D,F), in line with previous reports (Luo et al., 2006; Ahlenius et al., 2009). As far as the C-cell compartment is concerned, we confirmed a decrease in EGF-R+ cells starting by middle age on (Fig. 1A–F), in good agreement with previous reports showing diminished C-cell numbers (Luo et al., 2006) and reduced immunoreactivity and expression levels of EGF-R in aged SVZ (Enwere et al., 2004), thereby supporting the idea that SVZ neurogenic impairment is an early event in mice.
Figure 1.
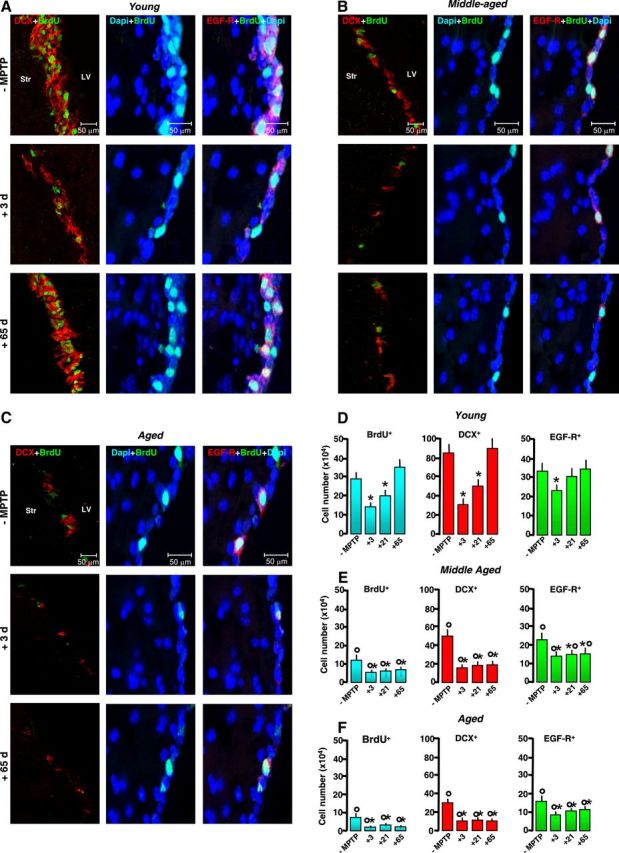
Aging-induced SVZ neurogenic impairment is exacerbated by MPTP exposure in vivo. Young (2–5 months), middle-aged (8–10 months), and aged (22–24 months) mice received saline or MPTP (7–8 mice/tp). At the indicated (+3, +21, and +65) days, mice were given BrdU. A–C, Representative confocal images of dual labeling of DCX+ neuroblasts (red) with BrdU (green); BrdU+ (green) cells counterstained with the nuclear marker, DAPI (blue); EGF-R+ cells (red) with DAPI (blue); and EGF-R+ (red) with BrdU+ (green) and DAPI (blue) in young (A), middle-aged (B), and aged (C) mice before exposure to MPTP and 3 and 65 d after exposure to MPTP. SVZ cell proliferation, neuroblast formation, and EGF-R immunoreactivity are decreased by middle age on. The capacity of young mice to recover from SVZ impairment after MPTP exposure is lost with age. D–F, Comparison of proliferation as assessed by number of BrdU+ cells; neuroblast formation as assessed by DCX; transit amplifying cell number as assessed by EGF-R, in young (D), middle-aged (E), and aged (F) SVZ, in basal conditions and upon MPTP challenge. Means ± SEM (n ≥ 6). Differences analyzed by ANOVA followed by Newman–Keuls test, and considered significant when p < 0.05. *p < 0.05 versus −MPTP; °p < 0.05 versus young within each respective treatment group.
Histopathological and neurochemical analyses of different nigrostriatal DAergic endpoints in young and middle-aged mice were next performed to verify whether changes in nigrostriatal DAergic innervation (Borta and Höglinger, 2007; Young et al., 2011) might be implicated in SVZ impairment already apparent by middle age (Fig. 2). However, we did not find significant changes in DAT-immunofluorescent (IF) reaction in Str (Fig. 2A–C), in striatal DA (Fig. 2D), in high-affinity synapotosomial DA uptake (a very sensitive quantitative parameter of striatal DAergic functionality) (Fig. 2E), and in the number of DAergic cell bodies in SNpc (Fig. 2F), indicating that, in addition to nigrostriatal DAergic influence, other factors may contribute to SVZ impairment starting by middle age.
Figure 2.

Aging-induced failure to recover from nigrostriatal DAergic toxicity upon MPTP exposure in vivo. Young (2–5 months) and middle-aged (8–10 months) mice received saline or MPTP (n = 6/tp). At the indicated days, the mice were killed and the brain processed for the determination of different DAergic endpoints in Str. For immunohistochemical analyses, the mice were deeply anesthetized and transcardially perfused, and their the brains were processed as described for Str and SNpc determinations. A, B, Representative confocal images of DAT (red) in Str of young (A) and middle-aged (B) mice after saline (−MPTP) injection and at 3 and 65 tps after MPTP injection. C–F, DAT fluorescence intensity image analysis (C); HPLC analysis of striatal DA (D), specific high-affinity neuronal DA uptake in Str (E), and TH+ neuron number in SNpc (F) in young and middle-aged mice at the indicated dpt. Differences were analyzed by ANOVA followed by Newman–Keuls test, and considered significant when p < 0.05. *p < 0.05 versus −MPTP. No significant difference is observed between young and middle-aged mice in basal conditions. MPTP induced a comparable DAergic impairment 3 and 7 dpt in both age groups, but only younger mice exhibit a substantial recovery from MPTP by 65 dpt.
We next investigated the effect of MPTP-induced DAergic toxicity on SVZ impairment during aging. Given that aging increases DAergic neuron vulnerability, the doses of MPTP administration were adjusted to obtain a comparable nigrostriatal DAergic toxicity so that young and aging mice were exposed to the same strength of stimuli (Table 1, Fig. 2A–F). In aging mice, MPTP induced a further significant inhibition of BrdU+ cells lasting until 65 dpt (i.e., for the remaining lifespan of aged mice) (Fig. 1B,C,E,F). These effects on proliferation were associated with a persistent inhibition of DCX+ neuroblast production. By contrast, in younger mice, we observed a transient SVZ impairment 3 dpt, next followed by a full recovery starting 21 dpt (Fig. 1A,D), supporting previous results (Höglinger et al., 2004; L'Episcopo et al., 2012) and underscoring the plasticity of young as opposed to old SVZ response to MPTP injury. As previously recognized (Ricaurte et al., 1987a,b; Ho and Blum, 1998; L'Episcopo et al., 2011a), we found that young adult mice did show a robust nigrostriatal DAergic recovery (Fig. 2A–F). By contrast, aging mice did not recover from MPTP-induced nigrostriatal histopathological and neurochemical impairment for the entire duration of the study both at striatal and SNpc levels (Fig. 2A–F).
Together, these results indicated that SVZ neurogenic deficit is already established by middle age in the absence of significant changes in the studied nigrostriatal DAergic endpoints. In addition, MPTP magnified aging-induced SVZ impairment associated with failure to recover from SVZ and nigrostrial DAergic injury, prompting us to verify changes in SVZ-NPC proliferative potential, in vitro.
Aging “primes” SVZ cells to neurotoxin-induced long-lasting neurogenic impairment in vitro
Studying the self-renewal of NPCs acutely isolated from young mice at different time points after saline or MPTP treatment, we recently found that, between 24 h and 14 dpt, primary neurospheres from MPTP-exposed mice did show significant early impairment in their growth rate, which, however, was completely recovered after expansion in vitro in the presence of FGF-II and EGF (L'Episcopo et al., 2012). We thus explored potential changes in proliferation and neuron differentiation properties of NPCs acutely isolated from the SVZ of aging mice both 3 and 65 d post-MPTP, corresponding to SVZ impairment and recovery observed in young mice. When primary neurospheres from young mice SVZ were established as described, and grown in proliferative medium, they express the stem cell marker, nestin, and a certain percentage incorporate the proliferative marker, BrdU (Fig. 3A,D). When shifted in differentiation medium and stained after 5 DIV, a certain proportion of cells expressed the neuronal marker, Tuj1 (red), out of the total number of DAPI-stained nuclei (Fig. 3A,E). By contrast, NPCs from middle-aged (Fig. 3B,D) and aged (Fig. 3C,D) mice exhibited significantly decreased BrdU incorporation and Tuj1+ cell formation (Fig. 3B,C,E), thus supporting impairment of proliferative capacity observed in vivo. The effect of treatment in vivo with MPTP was both age-dependent and time-dependent. Hence, in primary neurospheres from young MPTP mice, both proliferation and neuron differentiation capacity were decreased 3 dpt, but fully recovered to pre-MPTP levels by 65 dpt (Fig. 3A,D,E). By contrast, in primary neurospheres from middle-aged and aged MPTP mice, the proliferative capacity was further reduced at either 3 or 65 d post-MPTP (Fig. 3B–D), corroborating the in vivo data. In addition, when middle-aged and aged NPCs were let to differentiate for 5 DIV and then stained with the neuronal marker, Tuj1, regardless of isolation time after MPTP, they exhibited a further significant decrease in percentage of Tuj1+ neurons, compared with their younger counterparts (Fig. 3B,C,E), in keeping with our in vivo findings of diminished proliferation potential and neuroblast formation with no recovery of old compared with young MPTP mice (Fig. 1). Finally, using the fluorogenic substrate DEVD-AFC to measure Caspase3-like activity as death marker, we observed increased DEVD-AFC signal in NPCs isolated from aged MPTP mice compared with younger counterparts (Fig. 3H), indicating that aging and MPTP exposure sharply reduced NPC survival capacity.
Figure 3.
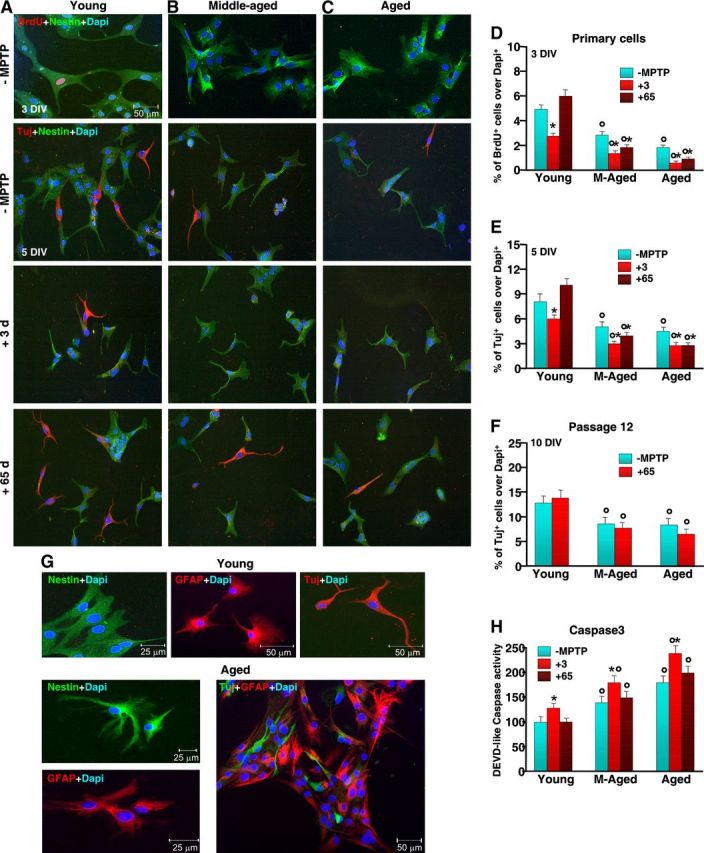
Aging primes SVZ cells to neurotoxin-induced long-lasting impairment of proliferative potential and neuroblast formation in vitro. NPCs were acutely isolated from the SVZ of 2–5-month-old (A), 8–10-month-old (B) and 22–24-month-old (C) mice, at both 3 and 65 d (7–8 mice/tp) after saline or MPTP injection, and cells processed as described. Proliferation (D) and neuron differentiation were studied in primary neurospheres (E) and after expansion for 12 passages, in vitro (F). A–C, Representative images comparing primary NPCs isolated from young (A), middle-aged (B), and aged (C) naive (−MPTP) mice and 3 and 65 dpt. Dual labeling with nestin (green) and BrdU (red) or nestin (green) and Tuj1 (red) counterstained with the nuclear marker, DAPI (blue) shows reduced BrdU+ and Tuj1+ cells with age and failure to recover upon exposure to MPTP (B, C), compared with young NPCs (A). D–F, NPC proliferation (D) as assessed by BrdU at 3 DIV; neuron differentiation, as assessed by Tuj1, after 5 DIV in primary cells (E); and neuron differentiation, as assessed by Tuj1, after 10 DIV, in expanded cells (F). G, Representative images of NPC isolated from young and aged mice after in vitro expansion. The cells were let to differentiate and stained with nestin (green), GFAP (red), Tuj1 (red), or with GFAP (red) and Tuj1 (green), counterstained with DAPI (blue). H, Comparison of cell death as assessed by Caspase3-like activity using the fluorogenic substrate DEVD-AFC as a function of age and MPTP. Results expressed as percentage changes relative to control (young −MPTP). *p < 0.05 versus −MPTP; °p < 0.05 versus young, within each respective group. The means ± SEM from three individual experiments is shown.
We then evaluated the differentiation potential of NPCs isolated from aged MPTP mice when expanded for 12 passages in the presence of FGF-II and EGF (Fig. 3F,G). After withdrawal of growth factors and addition of serum and N2 supplements to the culture medium, the cells were let to differentiate for 5–10 DIV and stained with Tuj1 for neurons and GFAP for astrocytes. After 5 DIV, we found that young and aged NPCs expressed nestin, Tuj1, and GFAP (Fig. 3G). However in aged NPCs, we observed higher percentages of GFAP+ cells. By 10 DIV in differentiation medium, we found increased neuron production in all age groups, in accord with previous findings (Ahlenius et al., 2009), supporting the idea that aged SVZ cells are still capable of responding to extrinsic factors, albeit with a lower efficiency compared with younger NPCs (Fig. 3F). Together, these results suggested that a double hit, aging and exposure to environmental toxins in vivo, can synergize in a negative fashion to reduce NPC survival, proliferation, and neuron differentiation capacity in vitro, but extending in vitro culturing in the presence of FGF-II and EGF can rescue neurospheres from aged-MPTP mice, indicating that both the nature of exogenous factors and/or changes in NPC responsiveness to environmental stimuli may in part account for SVZ cell impairment with age.
Aging downregulates Nrf2-driven-Hmox adaptive SVZ response to MPTP in the face of exaggerated inflammation
We next examined the changes in the redox/inflammatory SVZ response as a function of aging and MPTP exposure (Figs. 4, 5). The transcription factor Nrf2, a chief redox master regulator, binds to antioxidant response element (ARE) to induce antioxidant and phase II detoxification enzymes, such as Hmox, a key mediator of cellular adaptive (i.e., antioxidant and anti-inflammatory) response (Chen et al., 2009; Surh et al., 2009; Bitar and Al-Mulla, 2011). Using quantitative real-time PCR, we identified Nrf2 and Hmox transcripts in SVZ tissue freshly derived from young saline-treated mice, while lower basal Nrf2 and Hmox mRNA levels were measured in SVZ from middle-age and aged mice (Fig. 4A,B). Time course studies performed in response to MPTP challenge revealed a different response in young as opposed to aging mice. Hence, in young mice we observed a very early (3–6-12 h post-MPTP) Nrf2-Hmox upregulation, with maximal increase by 24 h (Fig. 4A,B) and an initial decrease starting 3 dpt (data not shown). By contrast, aging mice failed to upregulate both Nrf2 and Hmox transcripts at all time intervals studied (Fig. 4A,B). Aging-induced decreased Nrf2-Hmox response was further confirmed using Western blotting (wb, Fig. 4C,D), and fluorescence immunohistochemistry (Fig. 4E) showed upregulation of Hmox protein in response to MPTP in young as opposed to aging SVZ (Fig. 4C–E).
Figure 4.
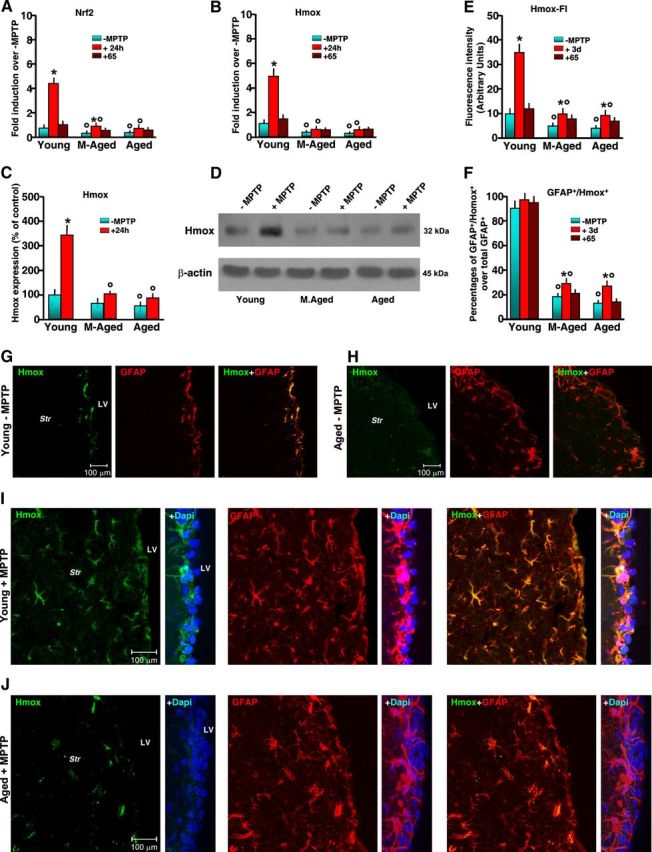
Aging inhibits Nrf2-mediated Hmox response in SVZ upon MPTP exposure. Mice of the different age groups treated with saline or MPTP were killed at the indicated time points, and freshly derived SVZ tissues were processed for real-time PCR or Western blot analyses. For immunohistochemical determinations, the mice were deeply anesthetized and transcardially perfused, and the brains were processed as described. A, B, Quantification of real-time PCR data using specific primers for Nrf2 and Hmox in SVZ samples (6 mice/tp) before treatment, 24 h after treatment, and 65 dpt. Changes in mRNA levels are expressed as n-fold induction over saline. Means ± SEM of three individual determinations are shown. Differences were analyzed by ANOVA followed by Newman–Keuls test, and considered significant when p < 0.05. *p < 0.05 versus saline; °p < 0.05 versus young within each experimental group. C, D, Western blotting of Hmox in protein extracts of SVZ isolated from mice of the different groups. The bands were densitometrically quantified on x-ray film using ImageQuantity One. Data from experimental bands were normalized to β-actin, and values expressed as percentage of control (−MPTP). Differences were analyzed as above; *p < 0.05 versus saline; °p < 0.05 versus young within each experimental group. E, F, Hmox fluorescence intensity (FI) (E) and changes in percentages of GFAP+/Hmox+ out of the total GFAP+ cells in SVZ (means ± SEM) assessed in sections double-stained with anti-Hmox and anti-GFAP (F), as detailed in Materials and Methods. *p < 0.05 versus saline; °p < 0.05 versus young within each experimental group. G, H, Representative confocal images showing Hmox-IF (green), GFAP-IF (red), and Hmox plus GFAP (merge) in young (G) and aged SVZ (H) showing reduced Hmox-IF in aged SVZ in basal conditions. I, J, Representative confocal images showing Hmox-IF (green), with DAPI (blue) counterstaining; GFAP-IF (red) with DAPI; Hmox plus GFAP (merge) with DAPI in young (I) and aged SVZ (J) showing reduced Hmox-IF in aged SVZ and striatal astrocytes in response to MPTP.
Figure 5.
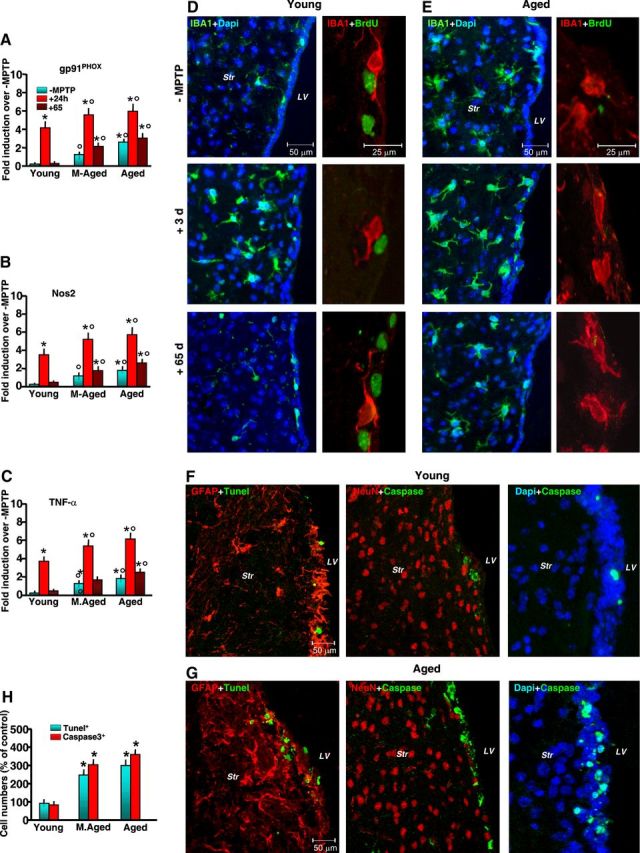
Aging exacerbates gpPhox, Nos2, and TNF-α mRNAs associated with microglial activation and cell-death marker expression in SVZ in response to MPTP. A–C, Comparison of quantified real-time PCR data using specific primers for gp91Phox, Nos2, and TNF-α in SVZ samples (6 mice/tp) from young, middle-aged, and aging mice in the absence or in the presence of MPTP treatment. The means ± SEM from three individual experiments are shown. Differences were analyzed by ANOVA followed by Newman–Keuls test, and considered significant when p < 0.05. *p < 0.05 versus saline; °p < 0.05 versus young within each experimental group. D, E, Representative confocal images showing dual staining with IBA1 (green) and DAPI (blue), and with IBA1 (red) and BrdU (green), in young (D) and aged (E) mice without (−MPTP) and 3 and 65 dpt. F–H, Dual localization of the death marker TUNEL (green) with GFAP (red), Caspase3 (green) with NeuN (red), and Caspase3 (green) with DAPI (blue) in young (F) and aged (G) mice show increased TUNEL+ and Caspase3+ cells and percentages of TUNEL+ and Caspase3+ cells (6 mice/tp, mean ± SD) in SVZ of aging compared with younger mice (H). *p < 0.05 versus young.
Earlier and more recent studies pointed to astrocytes as central players in Nrf2-Hmox induction following different types of brain insult, including MPTP exposure (Fernandez-Gonzalez et al., 2000; Lee et al., 2003; Chen et al., 2009). We then used dual staining with the astrocytic cell markers GFAP+ (red) and Hmox (green), coupled to confocal laser scanning microscopy, to localize Hmox in SVZ astrocytes (Fig. 4F–J). In basal conditions, we observed a reduced Hmox-IF signal in aging compared with younger SVZ counterparts (Fig. 4G,H). In accordance with previous studies, we found that MPTP significantly increased Hmox in striatal astrocytes of young mice, and further documented increased Hmox-IF in SVZ astrocytes (Fig. 4F,G,I). By contrast, aging mice failed to upregulate Hmox-IF (Fig. 4F,H,J), thus supporting PCR and wb analyses. It should be stressed that MPTP is recognized to increase GFAP+ cell density both in young and aging mice (Ho and Blum, 1998; Morale et al., 2004; Marchetti et al., 2005a,b), but percentages of GFAP+/Hmox+ cells out of the total GFAP+ cells were reduced in aging as opposed to younger mice (Fig. 4F,I,J). Together, these results indicated that aging and MPTP acted in synergy to reduce Nrf2-driven Hmox response in SVZ at mRNA and protein levels. By contrast, the mRNA transcript of the pivotal mediator of microglial ROS generation, gp91Phox (Gao et al., 2003; Zhang et al., 2007), was significantly higher in aging SVZ compared with younger counterparts. In addition, upon MPTP exposure, gp91Phox, was sharply upregulated in middle-aged and aged mice starting from 3 to 24 h on, compared with younger mice (Fig. 5A). Likewise, Nos2 (inducible nitric oxide synthase) mRNA was greater in aging mice compared with young adult mice, with a further increase detected after MPTP exposure (Fig. 5B). Importantly, such increase was long-lasting in aging mice, whereas a return to pre-MPTP condition was observed in younger counterparts. Finally, mRNA levels of a major proinflammatory (Th1) cytokine, TNF-α, which is critically involved in inflammation-dependent DAergic degeneration, were significantly increased in aging SVZ, a phenomenon amplified by MPTP exposure (Fig. 5C). In keeping with these results, aging induced ameboid-shaped IBA1+ reactive microglia within the Str bordering the SVZ (Fig. 5D,E). Dual staining with BrdU and IBA1 further supported reduced proliferation of SVZ cells in aging mice exposed to MPTP compared with their younger counterparts (Fig. 5D,E). Importantly, while MPTP-induced increased microglial reaction 3 dpt returned back to pre-MPTP levels in young mice, aging mice still exhibited a long-lasting and highly activated microglial phenotype up to 65 dpt (Fig. 5D,E). That an exacerbated SVZ microenvironment might contribute to impair SVZ cell homeostasis was further indicated by increased death cell markers, such as TUNEL-IF and Caspase3-IF signals in SVZ of aged compared with younger mice (Fig. 5F–H), thus corroborating the in vitro findings showing decreased NPC survival in aged SVZ cells (Fig. 3H).
Together, these data revealed that, starting by middle age on, SVZ redox/inflammatory balance is disrupted. Exposure to MPTP further exacerbated microglial proinflammatory phenotype at the expense of the antioxidant/anti-inflammatory response, likely contributing to impair astrocyte-NPC homeostasis in aged SVZ.
“Aged” microglia impair young NPCs while “young” microglia override neurogenic inhibition of aged NPCs: pharmacological modulation in vitro
We thus addressed the distinct roles of young and aged microglia using a controlled in vitro environment, where it is easy to manipulate signaling pathways and measure the resulting effects on specific cell populations. NPCs derived from young and aged SVZs were then cocultured with either young or aged microglia. We used both direct (allowing cell-to-cell contacts) and indirect (allowing diffusion of factors from the glia monolayer to the NPCs and vice versa) coculture paradigms, as previously established (L'Episcopo et al., 2011b, 2012). Microglial cells were isolated (>95% IBA1+ microglia) from 2-d-old mice brain and from young adult (2 months old), middle-aged (8–10 months old), and aged (24 months old) mice, and young and aged NPCs were layered on the top (Fig. 6). At the indicated time intervals, the cultures were processed for either NPC proliferation and differentiation or Caspase3-like activity analyses. As observed in Figure 6A,C, direct coculture of young NPCs with purified microglia markedly influenced neurogenesis as a function of age: exposure to 2-d-old and 2-month-old microglia increased the proportion of BrdU+ and Map2a+ out of the DAPI+ nuclei, compared with NPCs exposed to a nonspecific insert. Dual localization of the microglial marker IBA1 and Map2a revealed the significant effect of 2-d-old and 2-month-old microglia in direct coculture with young NPCs, as shown by the sharp increase of Map2a+ cell number, as opposed to NPCs grown alone (compare Figs. 3A–C, 6A,B). On the other hand, exposure of young NPCs to aged microglia significantly decreased BrdU+ and Map2a+ cells (Fig. 6A,C), indicating that the aging process switches microglia from a neurogenesis-promoting to neurogenesis-inhibitory phenotype. We next compared the effect of microglial age on proliferative potential of NPC isolated from aging mice (Fig. 6B,D). Neurospheres isolated from aging mice cultured as described were layered on the top of microglia monolayers of different ages, as above. Interestingly enough, dual staining with IBA1 and Map2a clearly revealed the ability of 2-d-old and 2-month-old microglia to significantly increase the number of Map2a+ cells compared with NPCs grown alone (Figs. 3A–C, 6B,D). Hence, the percentage of both Map2a+ and BrdU+ cells was increased to levels measured in young NPCs cocultured with young microglia (Fig. 6C,D), thus implying that the responsiveness of aged NPCs to exogenous factors is not impaired. By contrast, 24-month-old microglia reduced NPC proliferative potential and the formation of Map2a+ cells (Fig. 6B,D), thus supporting the idea that microglial age, and not NPC age, is critical for directing beneficial or harmful effects on NPCs. In keeping with this finding, microglial monolayers isolated from MPTP mice of increasing age further inhibited both BrdU incorporation and Map2a production (data not shown). Finally, DEVD-like fluorescent signal was significantly increased in NPCs exposed to microglial inserts from aged-MPTP mice compared with younger counterparts, supporting synergy between age and MPTP exposure in microglial-induced decreased NPC survival (Figs. 3H, 6E).
Figure 6.
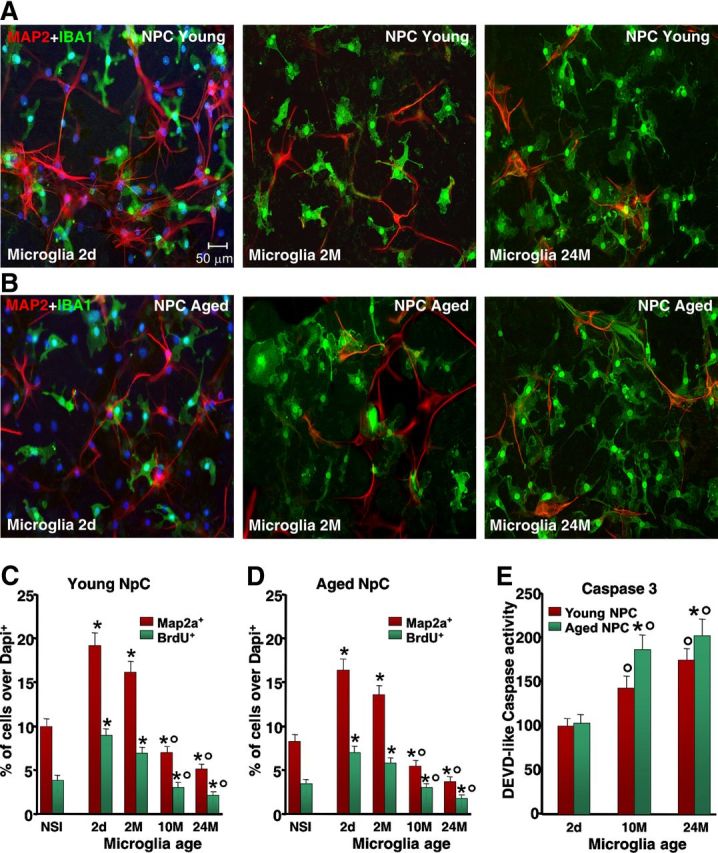
Effect of microglial age in the proliferation, neuron differentiation, and Caspase3 activation of NPCs derived from young, middle-aged, and aged SVZ. NPCs were cocultured with purified microglia acutely isolated from mice of the indicated ages, or with a nonspecific insert. A, B, Comparison of representative confocal images of dual staining with IBA1 (green) and Map2a (red) counterstained with DAPI (blue) in young NPCs cocultured with microglia of 2 d (2d), 2 months (2M), or 24 months (24M) (A); or aged NPCs cocultured with 2-d-old (2d), 2-month-old (2M), or 24-month-old (24M) microglia (B). C, D, Differences in NPC proliferation and neuron differentiation assessed with Map2a in young (C) and aged (D) NPCs. E, Caspase3-like activity, assessed with the fluorogenic substrate DEVD-AFC in NPCs cocultured with microglia with the indirect coculture paradigm, shows increased activity as a function of microglia age. Results are expressed as percentage changes relative to controls. Three independent cultures were used for quantification. *p < 0.05 versus nonspecific insert; °p < 0.05 versus 2-d-old and 2-month-old microglia. Caspase3-like activity was increased in young and aged NPCs cocultured with aged microglia. NSI, nonspecific insert.
Given our recent work establishing microglial-derived ROS and reactive nitrogen species (RNS) as candidate inhibitory mediators in MPTP-induced NPC neurogenic impairment of young mice (L'Episcopo et al., 2012), we next used the nonsteroidal anti-inflammatory NO-donating drug, HCT1026, previously shown to mitigate microglial activation in aging mice, in vivo and in vitro, and to protect nigrostriatal DAergic neurons in both young, middle-aged, and aged PD rodent models (see L'Episcopo et al., 2010a,b, 2011a, 2012). Pilot experiments with aged-microglia monolayers from MPTP mice were performed to establish the HCT1026 dose (10 μm) capable to efficiently downregulate the exacerbated Phox-derived ROS and Nos2-derived RNS levels of aged microglia, without influencing microglial cell viability (Table 2). After 24 h, the glial inserts were rinsed extensively and placed on the top of young or aged NPCs in the indirect coculture paradigm. When young (Fig. 7A,C,D) or aged (Fig. 7B,E,F) NPC were exposed to HCT1026-treated aged microglia inserts, numbers of both BrdU-expressing and Map2a-expressing NPCs were significantly increased, thereby indicating that the HCT1026-induced switch of aged microglia phenotype may have a rescue effect on NPC proliferative potential and Map2a+ neuron production.
Figure 7.
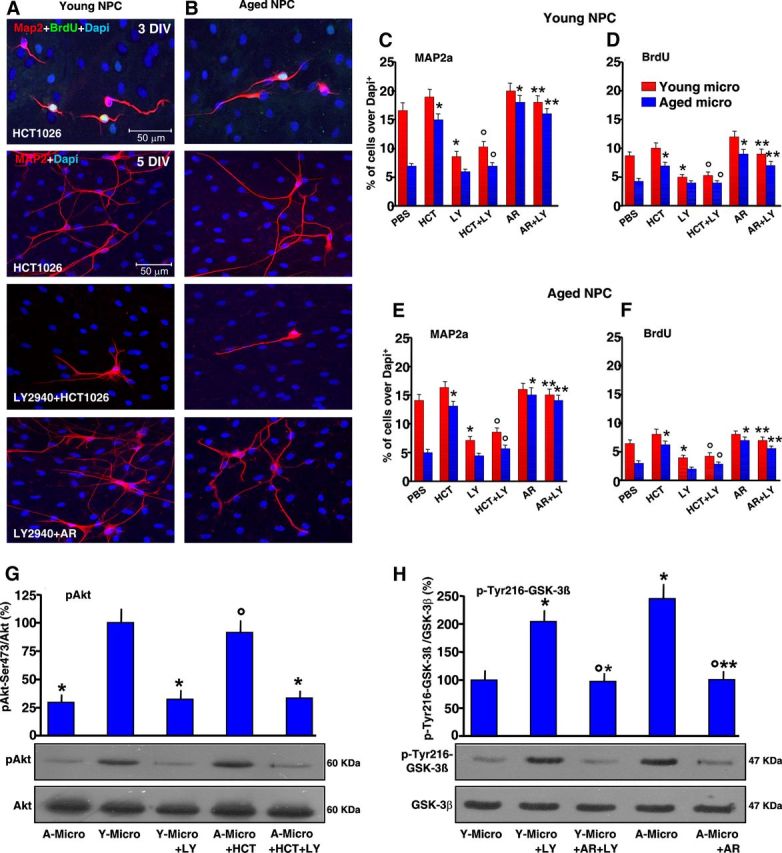
HCT1026 reverses aged-microglia-induced NPC neurogenic impairment via PI-3K/Akt signaling activation. Microglia monolayers from aged mice were treated with HCT1026 (10 μm) for 24 h and layered on the top of young (A, C, D) or aged (B, E, F) NPCs, in the indirect coculture paradigm, for assessment of proliferation with BrdU, and neuron differentiation with Map2a. The effect of young-microglia or aged-microglia HCT1026 was tested in the absence or the presence of the PI-3K inhibitor, LY294002 (LY, 10 μm). The effect of GSK-3β antagonist, AR-AO14418 (AR, 5 μm) was tested on LY294002-treated cocultures. A, B, Representative images showing young (A) or aged (B) NPCs cocultured with aged microglial inserts pretreated with HCT1026, showing Map2a (red) production and BrdU (green) incorporation, an effect reversed by LY294002. Inhibition of GSK-3β with AR counteracts LY294002-induced inhibition. C–F, Map2a+ cell percentages and BrdU incorporation in young (C, D) and aged (E, F) NPCs cocultured with either young or aged microglia, as indicated. *p < 0.05 versus PBS,°p < 0.05 versus HCT1026, **p < 0.05 versus LY294002. G, H, Western blotting of phospho-Akt-Ser473 and phospho-Tyr216GSK-3β in protein extracts from NPCs. The bands were densitometrically quantified on x-ray film using ImageQuantity One. Densitometric values, depicted as ratio of the phosphorylated form over total amount of the specific proteins, are plotted for p-Akt (G) and p-GSK-3β (H), expressed as percentage of control (Y-Micro). The means ± SEM of three individual determinations are presented. *p < 0.05 versus young microglia, °p < 0.05 versus LY, **p < 0.05 versus aged microglia. A-Micro, aged microglia; Y-Micro, young microglia.
Phosphatidylinositol 3-kinase/Akt is involved in young microglial-induced and HCT1026-induced neurogenic effects in vitro
We next addressed the signaling pathways involved in inflammatory microglial NPC modulation, and focused on the phosphatidylinositol3-kinase (PI3K)/Akt pathway. PI3K promotes the phosphorylation and activation of Akt (also known as protein kinase B). By making use of phosphorylating-dependent mechanisms, Akt can inhibit apoptosis induced by several stimuli in a multitude of cell types. Of importance, the PI3K/Akt pathway mediates the effect of various neurogenic growth factors (Ojeda et al., 2011). To investigate a potential contributory role of PI3K/Akt signaling cascade, young and aged NPCs were cocultured with young microglia, as described, both in the absence and in the presence of the PI-3K inhibitor, LY294002, at a concentration (10 μm) unable per se to influence cell viability. Inhibition of PI-3K significantly reduced the capacity of young microglia to increase Map2a+ neuron production (Fig. 7C) and BrdU incorporation (Fig. 7D) from young NPCs, implicating the requirement of PI-3K activation in microglial neurogenic effects. In addition, LY294002 reversed microglial-induced aged NPC rescue (Fig. 7E,F), indicated by the reduced BrdU+ and Map2a+ cell production.
We next investigated whether PI3K signaling is involved in HCT1026-mediated effects. When young and aged NPCs exposed to HCT1026-treated aged-microglial inserts were treated with LY294002, we observed a significant decrease of both neuron differentiation (Fig. 7A–C,E) and proliferation (Fig. 7D,F) compared with cocultures without LY294002, indicating failure to reverse the harmful effects of old glia when PI-3K was inhibited. Western blotting (Fig. 7G) supported the idea that PI-3K-induced Akt phosphorylation (pAkt) was indeed involved in beneficial effect of young or HCT1026-treated old glia. As observed, lower pAkt protein levels were measured in NPC cocultured with aged microglia compared with pAkt measured in NPCs cocultured with young microglia and HCT1026-treated aged microglia. Conversely, LY294002 reversed pAkt protein increase induced by coculture with young microglia or with aged HCT1026-treated microglia (Fig. 7G). Together, these data revealed that the PI-3K signaling pathway is required for the neurogenesis-promoting and/or neurogenesis-protective effects of young microglia. In addition, the PI-3K signaling cascade appeared a necessary pathway in producing HCT1026-mediated effects.
Glycogen synthase kinase-3β is a downstream effector of PI-3K/Akt-mediated microglial effects
The PI-3K/Akt pathway is known to inhibit glycogen synthase kinase 3β (GSK-3β), in turn associated with the activation of cell adaptive and survival pathways in different types of cells (Kim and Snider, 2011). By contrast, GSK-3β activation by phosphorylation of the tyrosine 216 residue (p-Tyr216) located in the kinase domain, is implicated in oxidative-stress-induced cell death mechanisms (see Duka et al., 2009; Petit-Paitel et al., 2009; L'Episcopo et al., 2011a,b).
We next studied the involvement of GSK-3β in LY294002-induced NPC impairment by testing the ability of the selective GSK-3β antagonist, AR-AO14418 (AR, 5 μm, Osakada et al., 2007; L'Episcopo et al., 2011b,c, 2012) to override LY294002-induced inhibition of young microglia. In this experimental paradigm, AR was coadministrated with LY294002 in young and aged NPCs cocultured with microglia in the indirect coculture paradigm, and NPCs were processed after 24 h. In young NPC–young microglia cocultures, AR did not modify the parameters studied, whereas coadministration of AR and LY294002 significantly increased the number of BrdU and Map2a+ cells compared with cocultures treated with LY294002 or AR alone (Fig. 7C–F). The possible implication of GSK-3β as a downstream mediator of PI-3K/Akt-induced effects was next studied by Western blotting. As observed (Fig. 7H), LY294002 increased pTyr216-GSK-3β, the active form of GSK-3β protein, in young NPCs cocultured with young microglia compared with PBS-treated coculture, supporting the idea that the failure of PI-3K to phosphorylate Akt might result in active GSK-3β upregulation. Likewise, coculture with aged microglia resulted in increased pTyr216-GSK-3β protein, likely suggesting antagonism of PI3-Akt by aged microglia. By contrast, GSK-3β-specific inhibition with AR efficiently reversed active GSK-3β protein increase in LY294002-treated cocultures with young microglia, and fully reverted aged microglia-induced increased pTyr216-GSK-3β protein (Fig. 7H).
Aging and MPTP-induced reduced SVZ sensitivity to “canonical” Wnt/β-catenin signaling
Because the Wnt/β-catenin signaling cascade is an attractive convergence point for interactions of multiple signaling molecules in NPCs (Kalani et al., 2008; Kuwabara et al., 2009; L'Episcopo et al., 2011a,b; Munji et al., 2011; Piccin and Morshead, 2011; Zhang et al., 2011), we next addressed whether the age-dependent imbalanced redox/inflammatory status of the SVZ microenvironment might influence the prototypical components of Wnt/β-catenin pathway in aging SVZ.
Frizzled receptors
The first step in Wnt signaling involves binding to the seven-pass transmembrane receptors of the Fzd family. Using real-time PCR and specific Fzd primers (Fig. 8A), we found that young-adult SVZ-NPCs harbor most frizzled receptors (Fzds), but Fzd-1 was the most abundant within the transcripts studied. On the other hand, while NPCs from aged SVZ exhibit small changes in Fzd transcript levels, after MPTP challenge, a significant decrease in Fzd-1 was observed in aging but not young NPCs. In keeping with this result, Western blotting shows a sharp Fzd-1 downregulation only in MPTP-aged NPCs (Fig. 8C), indicating that neurotoxin exposure may impair aged NPC ability to respond to Fzd-1 ligands.
Figure 8.
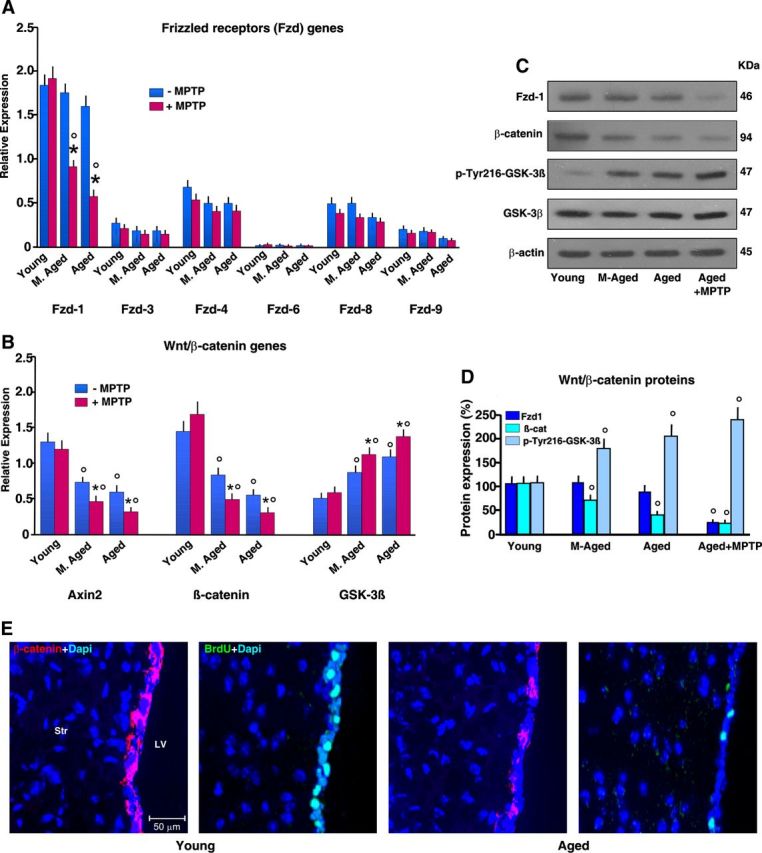
Effect of age on key components of Wnt/β-catenin signaling in saline-treated and MPTP-treated mice. After mice treated with saline or MPTP were killed 65 dpt (8 mice/tp), the SVZ was isolated and processed for real-time PCR. A, B, Comparison of Frizzled (A, Fzd-1–Fzd-9) and Wnt/β-catenin (B, β-catenin, Axin2, and GSK-3β) gene expression in SVZ from mice of the different age groups. Changes in mRNA levels expressed as relative expression (arbitrary units). Aging and neurotoxin exposure differentially modulate Fzds expression (A). Only Fzd-1 is downregulated in aging mice upon exposure to MPTP (A). β-Catenin and Axin2 transcript levels are decreased with age, in the face of upregulated GSK-3β (B). *p < 0.05 versus −MPTP; °p < 0.05 versus young within each experimental group. C, D, Western blots of Fzd-1, β-catenin, and pGSK-3β. The bands were densitometrically quantified on x-ray film using ImageQuantity One. Densitometric values of Fzd-1 and β-catenin were normalized to β-actin, p-GSK-3β, total GSK-β, and values expressed as percentage changes compared with control (young NPC). The means ± SEM of three individual determinations is shown. °p < 0.05 versus young. E, Comparison of representative images showing β-catenin+ (red) or BrdU (green) cells counterstained with DAPI (blue) within the SVZ of young and aged mice showing loss of β-catenin+ and BrdU+ cells with age.
β-Catenin, Axin2, GSK-3β
It is recognized that Fzd-1 provides the best discrimination of the Wnt/β-catenin versus the Wnt/Ca+2 pathway (Gordon and Nusse, 2006). The hallmark of “canonical” Wnt/β-catenin pathway is the stabilization of cytosolic β-catenin (Aberle et al., 1997). Importantly, Wnt signaling inhibits GSK-3β activity, thus increasing the amount of the transcriptional activator, β-catenin. As observed, young NPCs express β-catenin, both at mRNA and protein levels (Fig. 8B,C). In keeping with this finding, using specific primers for Axin2, a direct Wnt target induced by Wnt/β-catenin activation (Jho et al., 2002), we found Axin2 expression in young NPCs supporting Wnt/β-catenin signaling activity in SVZ cells of young-adult mice (Adachi et al., 2007; L'Episcopo et al., 2012). In stark contrast, both aging and neurotoxin exposure exerted a synergic inhibition of canonical Wnt/β-catenin activation in SVZ cells, as reflected by decreased β-catenin and Axin2 transcript levels, associated with a sharp upregulation of GSK-3β (Fig. 8B). Accordingly, immunoblotting (Fig. 8C,D) supported aging-induced inhibition of β-catenin, which was associated with increased pTyr216-GSK-3β protein levels, suggesting activation of GSK-3β under these conditions. In keeping with these results, immunohistochemistry showed reduction of β-catenin-IF (red) cells associated with reduced BrdU (green) incorporation in the SVZ of aged compared with younger mice (Fig. 8E), thus corroborating real-time PCR data, and supporting aging-induced dysregulation of Wnt/β-catenin in SVZ.
Aging impairs astrocyte proneurogenic effects: involvement of Wnt1 and dysfunctional microglia
The observed downregulation of Wnt/β-catenin signaling with age could be the result of different mechanisms, including astrocyte dysfunction. Indeed, compelling evidence indicates that neural progenitors in neurogenic areas are in intimate contact with astrocytes that help generate an instructive niche for promoting neurogenesis (Lim and Alvarez-Buylla, 1999; Lie et al., 2005; Jiao et al., 2008). Here, direct coculture of young NPCs with 2-d-old or 2-month-old Str astrocytes sharply increased the percentage of BrdU-expressing and Map2a-expressing cells, compared with young NPCs grown without astrocytes or with a nonspecific insert (Fig. 9A,E,F). In stark contrast, aged Str astrocytes decreased the percentages of both BrdU− and Map2a+ neurons, and reduced Map2a+ process length in coculture with either young or old NPCs (Fig. 9A,B,G,H). Additionally, young but not aged astrocytes promoted old NPC rescue (Fig. 9A,B,G,H). The contribution of Wnt/β-catenin signaling was investigated using cysteine-rich domain Fzd-1 (CRD-Fzd-1) (1 μg/ml), which blocks the effects of Wnt's ligands that bind to Fzd-1 (L'Episcopo et al., 2011c). Accordingly, when young and aged NPCs were treated with Fzd-1-CRD and then exposed to young Str astrocyte monolayers, astrocyte-induced increased NPC proliferative potential and Map2a+ neuron production were significantly reduced (Fig. 9C,E–H). On the other hand, exposure of young and old NPCs to aged astrocytes in the presence of Fzd-1-CRD produced only a slight reduction in BrdU incorporation and neuron differentiation (Fig. 9G,H), thereby indicating that with age, the astrocyte-derived Fzd-1 ligands may decline. Real-time PCR analysis was next performed to verify changes in Wnt's expression (Fig. 9D). In analogy to our previous findings, we detected Wnt1 transcripts in Str astrocytes. However, Wnt1 expression levels were sharply downregulated in aged astrocytes (Fig. 9D). Interestingly enough, when young astrocytes were exposed to aged microglia in the indirect coculture paradigm, we observed a marked loss of Wnt1 expression, whereas astrocyte exposure to young microglia or HCT1026-treated aging microglia significantly upregulated Wnt1 transcript levels and efficiently reversed aging astrocyte Wnt1 decline (Fig. 9D), suggesting that aged microglial pro-oxidant/proinflammatory mediators disrupt astrocyte-mediated Wnt/β-catenin signaling, likely via dysfunctional astrocyte–microglia cross talk. In keeping with this finding, direct activation of Wnt/β-catenin signaling by treatment of aged astrocyte–NPC cocultures with AR (5 nm) resulted in a significant rescue, revealed by increased proportion of BrdU+ and Map2a+ cells (Fig. 9E–H).
Figure 9.
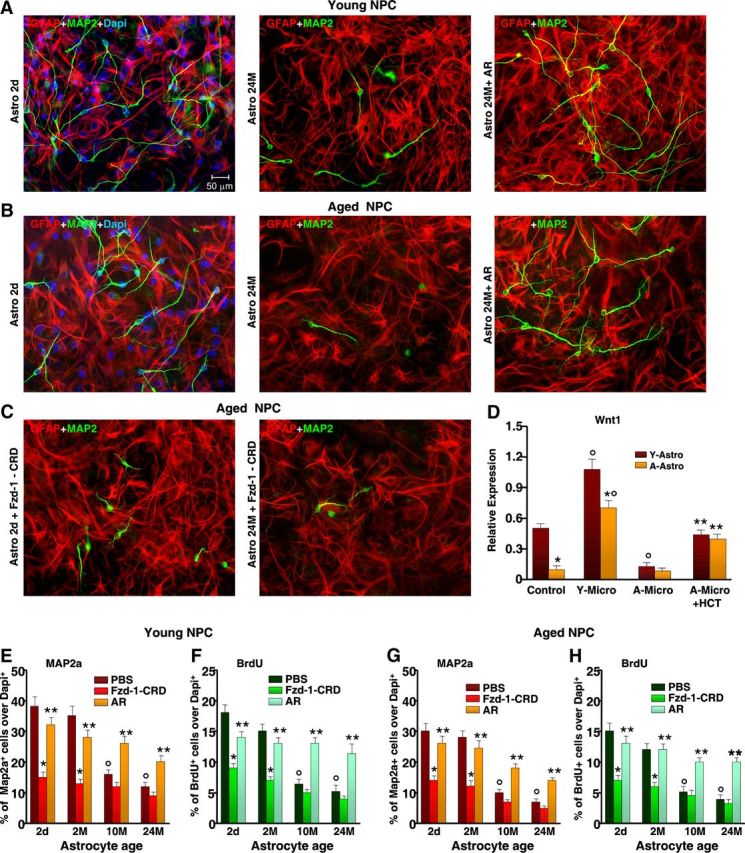
Age impairs astrocyte proneurogenic capacities and Wnt expression: modulatory role of microglia. A, B, Astrocytes were acutely isolated from young [2-d-old (2d) and 2-month-old (2M)] and aged [10-month-old (10M) and 24-month-old (24M)] mice, and NPCs from either young (A) or old (B) mice were layered on the top of astrocyte monolayers. Proliferation/differentiation were measured using BrdU and Map2a. A–C, Representative confocal images of dual staining with the astrocytic marker, GFAP (red) and Map2a (green) counterstained with DAPI (blue) revealing the robust neurogenic effect of young astrocytes when cocultured with either young (A) or aged (B) NPCs, whereas old astrocytes loose their neurogenic promoting capacities (A, B). Antagonism of astrocyte-derived Fzd-1 ligands with the CRD of Fzd-1 (C) sharply inhibits young astrocyte (red)-induced Map2a (green) neuron production. D, Real-time PCR using specific Wnt1 primers as a function of astrocyte age and astrocyte exposure to either young microglia, aging microglia, or aging HCT1026 microglia, using the indirect coculture paradigm. Note the sharp decline of Wnt1 expression in aging astrocytes or after exposure of young astrocytes (Y-Astro) to aging microglia, while aging HCT1026 microglia (A-Micro + HCT) reverses Wnt1 downregulation. The means ± SEM of three individual experiments is shown. *p < 0.05 versus young astrocytes; °p < 0.05 versus control, **p < 0.05 versus aging microglia. E–H, Comparison of proliferation and differentiation in young (E, F) and aged (G, H) NPCs as a function of astrocyte age. The effects of antagonism of Fzd-1 ligands or Wnt/β-catenin activation with AR are illustrated. *p < 0.05 versus PBS; ** versus Fzd-1-CRD; ° p < 0.05 versus 2-d-old and 2-month-old astrocytes. A-Astro, aging astrocytes.
Normalization of Nrf2 axis by HCT1026 in vivo rescues SVZ impairment associated with nigrostriatal DAergic restoration: PI3K/Akt–Wnt/β-catenin cooperation
Together, these results are in line with recent findings supporting the implication of the immune system in the modulation of adult neurogenesis in the injured brain (Ekdahl et al., 2003, 2009; Monje et al., 2003; Butovsky et al., 2006; Pluchino et al., 2008; Ziv and Schwartz, 2008; Martino et al., 2011; Tepavčević et al., 2011; Cusimano et al., 2012; Ekdahl, 2012; L'Episcopo et al., 2012), which prompted us to address the ability of HCT1026 to rescue SVZ neurogenic impairment of middle-aged mice and verify to what extent pharmacological manipulation of these age-related SVZ pathways influences the functional response in the outcome of MPTP-induced PD. The NO-donating derivative of flurbiprofen, HCT1026, is a mixed cyclooxygenase (COX1/COX2) inhibitor endowed with a safe profile and additional immunomodulatory properties (Furlan et al., 2004; Bernardo et al., 2005; Idris et al., 2009; L'Episcopo et al., 2010b, 2011c). Middle-age (8 months old) mice were fed with a control or HCT1026 diet for 3 weeks, at which time they were challenged with either saline or MPTP in the continuous presence of either a control or HCT1026 diet. At different time intervals (1–65 dpt), groups of mice were killed for histochemical analyses, neurochemical analyses, gene expression analyses, or immunoblotting (Fig. 10).
Treatment of aging mice with HCT1026 resulted in a substantial downregulation of microglial pro-oxidant and proinflammatory mediators in SVZ, including Nos2 and TNF-α, and a prevention of MPTP-induced upregulation of inflammatory mRNA species, as opposed to aged mice fed with a control diet (Fig. 10A). Normalization of redox/inflammatory balance in aged mice treated with HCT1026 resulted in a robust upregulation of Nrf2 and Hmox transcripts, early (+1 dpt) upon MPTP challenge, confirming that HCT1026 induced upregulation of SVZ anti-inflammatory response (Fig. 10A). These results of gene expression were associated with a decreased stage-4 microglial cell reactivity in HCT1026 mice (Fig. 10C) and with a significant upregulation of GFAP+/Hmox+ cells, especially upon MPTP exposure (3 dpt, Fig. 10D,E). Importantly, HCT1026-induced mitigation of the harmful SVZ microenvironment of aging mice resulted in increased Wnt1 and β-catenin mRNAs and reversed GSK-3β upregulated gene expression in SVZ both in basal condition and after MPTP challenge, as revealed by real-time PCR analysis (Fig. 10B), thus supporting in vitro findings. Concerning SVZ proliferation and DCX+ neuroblast formation (Fig. 10F–I), BrdU and PI counterstaining indicated reversal of aging-induced reduced BrdU+/PI+-nuclei within the SVZ niche and prevention of MPTP-induced SVZ impairment of HCT1026-fed mice, compared with middle-aged mice fed with a control diet (Fig. 10F,I). In keeping with these findings, dual staining with BrdU and β-catenin showed reversal of aging-induced decreased BrdU+/β-catenin+ cells and prevention of MPTP-induced loss of both markers in HCT1026 as opposed to control both without and after MPTP challenge (Fig. 10H). Likewise, HCT1026 efficiently counteracted aging and MPTP-induced decreased percentages of DCX+ neuroblasts as opposed to mice fed with a control diet (Fig. 10G). Of importance, these effects were associated with HCT1026-induced reversal of pAkt downregulation in SVZ of aged, MPTP-exposed mice (Fig. 10J), thus supporting in vitro findings.
Next, HCT1026-induced SVZ rescue in aging mice was correlated with MPTP-induced DAergic toxicity (Fig. 11). The temporal analysis of different DAergic endpoints both at striatal and SNpc levels indicated a significant neuroprotection in aged SVZ-rescued mice upon MPTP challenge, as revealed by the significant increase in DAT-IF and TH-IF in Str (Fig. 11A–C), high-affinity synaptosomial DA uptake (Fig. 11D), and TH+ neuronal cell bodies in SNpc (Fig. 11E). Together, these results suggested a temporal link between the early normalization of SVZ redox/inflammatory balance of aging SVZ niche and Akt/Wnt/β-catenin upregulation associated with increased proliferation and neuroblast formation, and correlated to DAergic neuroprotection (Fig. 12).
Figure 12.
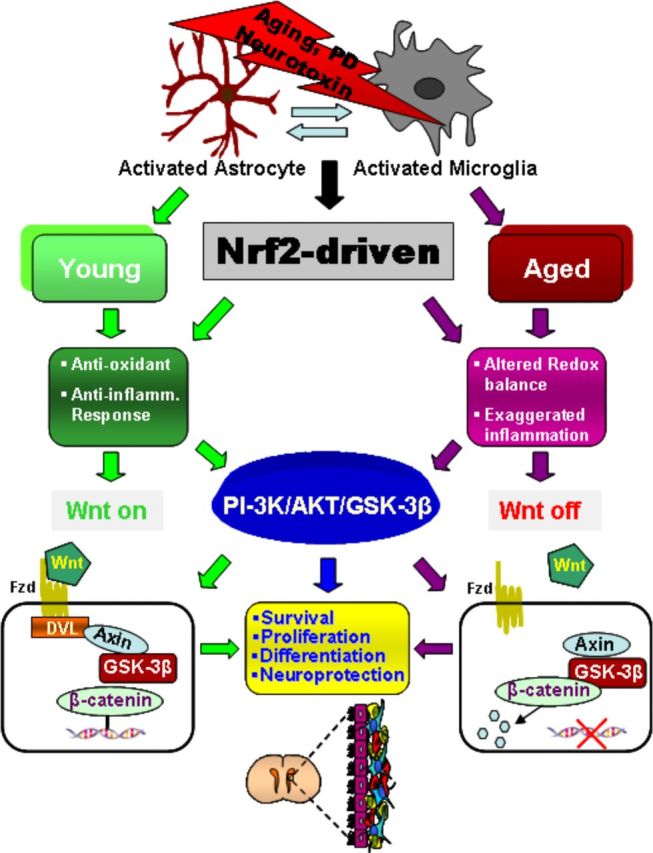
Simplified scheme summarizing the effect of aging and MPTP on Nrf2-ARE axis dysfunction-mediated Wnt/β-catenin signaling dysregulation in NPCs associated with neurogenic impairment. In young mice, a regulatory circuit linking microglial activation and proinflammatory cytokine to Nrf2-ARE protective pathway in SVZ, provides an efficient self-adaptive mechanism against inflammatory/neurotoxin-induced oxidative stress. In addition to governing the redox balance within the SVZ niche, the Nrf2-induced Hmox target gene may simultaneously protect astrocytes, thereby upregulating the expression of vital Wnt signaling elements that switch on key components required for maintaining SVZ cells in a proliferative state, for promoting differentiation, and/or for exerting neuroprotective effects. Cross talk between two pivotal pathways, the PI3-K/Akt/GSK-3β and Wnt/β-catenin signaling cascades, appears to finely control the transcriptional activator, β-catenin, which in turn represents a point of convergence to direct proliferation/differentiation/survival in the SVZ stem niche. Importantly, SVZ “rejuvenation” may have beneficial consequences for DAergic neuroprotection, and vice versa. Astrocytes (blue), neuroblasts (red), transit-amplifying cells (yellow), and ependymal cells (purple) in SVZ niche are schematically illustrated.
Discussion
The critical role of microglia in adult neurogenesis and the potential for anti-inflammatory drug treatment to modulate this system have been emphasized in both early and more recent studies (Ekdahl et al., 2003; Monje et al., 2003;Butovsky et al., 2006; Jakubs et al., 2008; Pluchino et al., 2008; Ehninger et al., 2011; Ekdahl, 2012; L'Episcopo et al., 2012). Additionally, not only local CNS inflammation, but also age-related molecular changes in the systemic milieu correlated to age-related decline in adult neurogenesis (Villeda et al., 2011). However, within the aging SVZ niche, the cell–cell interactions and signaling mechanisms impairing NPC homeostasis are not completely clarified. Here, we reveal that a major risk factor for PD, namely aging, drives a long-lasting SVZ impairment at least in part via reduced Nrf2-mediated SVZ tolerance to inflammation and oxidative stress associated with dysfunctional astrocyte–microglial dialogue, in turn interrupting key molecular signaling mechanisms finely regulating SVZ cell homeostasis. In particular, when “primed” microglia of aged mice become hyperactivated upon a second hit, MPTP exposure, the generation of highly toxic mediators in the face of impaired antioxidant self-protective NPC response dramatically inhibits neurogenesis, suggesting that glial age is of critical importance in directing promotion versus inhibition of neurogenesis. Of special interest, with age, the exaggerated microglial activation can impair an astrocyte's ability to express critical antioxidant, anti-inflammatory, and neurogenic factors within the niche, including Hmox and Wnt1, thus predisposing aged SVZ cells to reduced Wnt's sensitivity also via Fzd-1 downregulation, thereby resulting in an overall reduction of glial proneurogenic capacities. Interestingly, these processes may disrupt the cross talk between two pivotal pathways in SVZ, namely the PI3-K/Akt and the Wnt/Fzd/β-catenin signaling cascades (Fig. 12). That the differential and timely coordination of these transduction signaling mechanisms may be linked to SVZ cell survival, proliferation, and/or differentiation, appears documented by pharmacological agonist/antagonist studies showing the potential for direct modulation of aged SVZ cells in vitro. The significance of this circuitry is suggested in vivo by HCT1026-induced switch of aged microglial exacerbated phenotype resulting in SVZ cell “rejuvenation,” at least in part via Nrf2/PI3-K/Akt–Wnt/Fzd-1/β-catenin cooperation. Interestingly, the manipulation of these age-related SVZ-Nrf2 pathways at middle age is associated with significant DAergic neuroprotection upon MPTP challenge. Together, these results can advance our understanding of the role and function of signaling pathways in regulating neural stem cells and generation/integration of newborn neurons in the aged inflamed brain, and may illuminate novel targets to develop CNS pharmacologic approaches for neurodegenerative diseases including PD (Cusimano et al., 2012; Höglinger et al., 2012; Rueger et al., 2012; Sakata et al., 2012; Vukovic et al., 2012; Wallenquist et al., 2012).
Failure to adapt to an imbalanced redox/proinflammatory milieu predisposes the aged SVZ niche to long-lasting impairment upon MPTP exposure
Aged mice exhibit a unique gene-expression profile in the brain, particularly when the immune system is activated. Aged microglia appear dysfunctional and adopt a potent neurotoxic phenotype (Streit et al., 2009; Njie et al., 2012). The transcription factor Nrf2 and its target gene products elicit an antioxidant/anti-inflammatory response. In particular, Nrf2 governs basal and inducible expression of Hmox, an inducible phase II enzyme endowed with cytoprotective and anti-inflammatory properties (Lee et al., 2003; Gennuso et al., 2004; Surh et al., 2009), but little is known on the putative role of Nrf2-Hmox axis within the adult/aged SVZ niche. Here, the identification that Nrf2-Hmox pathway is active in young SVZ of MPTP-challenged mice, as opposed to aging mice, appears of specific interest. Hence, two most potent pro-oxidant and proinflammatory gene transcripts, gp91Phox and Nos2, exhibited life-long upregulation in response to MPTP, thereby indicating aging-induced reduced SVZ resistance (the first hit) as a potential predisposing factor to further NPC impairment upon a second hit (e.g., gene mutations and exposure to neurotoxicants) Then, aging-induced Nrf2-ARE disruption in the SVZ likely contributes to reduced SVZ plasticity, with potential consequences for DAergic neuronal death exacerbation and/or failure to recover (Frank-Cannon et al., 2008; Gao et al., 2008; Marchetti et al., 2011; Lastres-Becker et al., 2012; Fig. 12).
Glial age directs activation/inhibition of SVZ neurogenesis: involvement of Nrf2-PI3K/Akt axis
According to the activation stage, microglia can determine the fate of differentiating adult NPCs (Butovsky et al., 2006; Thored et al., 2009; Ekdahl, 2012). Here, we found that glial age is important for directing promotion versus inhibition of SVZ neurogenesis, in line with very recent findings on hippocampal microglia differentially influencing NPC activity in situ as a function of aging and exercise (Vukovic et al., 2012). Additionally, by reducing the exaggerated microglia activation with the NO-donating derivative of flurbiprofen, HCT1026, in vitro and in vivo, we found upregulated Nrf2-Hmox axis in SVZ, thus reverting aged microglia to a beneficial “younger” proneurogenic phenotype. In fact, ROS production is a critical component of cellular signaling, and increased ROS production by switching on Nrf2 promotes its translocation into the nucleus, the binding to ARE leading to a coordinated activation of gene expression contributing to self-adaptation (Smith et al., 2000). However, ROS overproduction may interrupt key signaling pathways regulating cell homeostasis (Smith et al., 2000; Kim and Wong, 2009). Hence, we observed that an exacerbated microglia phenotype can inhibit pathways associated with enhanced cell proliferation and survival, such as the PI3K/Akt pathway (Ojeda et al., 2011), and we defined the PI-3K signaling cascade as a necessary upstream pathway in HCT1026-mediated effects.
Active GSK-3β upregulation is linked to oxidative stress-induced cell-death mechanisms. Because active GSK-β may phosphorylate Nrf2, leading to Nrf2 degradation (Rada et al., 2011; Rojo et al., 2012). Then, the observation that aged microglia-activated oxidative stress-dependent GSK-3β signaling could be reverted by GSK-3β-specific antagonism with AR, favoring NPC rescue, strongly supported the notion that young microglial cell activation is associated with PI-3K/Akt signaling and active GSK-3β downregulation that promotes the switching on of Nrf2. By contrast, aging is associated with Akt deactivation and active GSK-3β upregulation, with consequent switching off of Nrf2, resulting in NPC impairment, with potential consequences for DAergic neuron death aggravation upon injury (Williamson et al., 2012). Moreover, the beneficial effects of HCT1026 acting upstream of GSK-3β in SVZ cells identified these players as potential targets for pharmacological modulation of DAergic neurorescue (Figs. 11, 12).
Nrf2-Hmox axis and PI-3K/Akt cooperate to inhibit GSK-3β activation and converge in canonical Wnt1/β-catenin signaling activation-induced SVZ and DAergic neurorescue
One way for GSK-3β to regulate many intracellular signaling pathways is by phosphorylating substrates, such as β-catenin (Gordon and Nusse, 2006). In fact, in the absence of Wnt activity, GSK-3β is known to phosphorylate β-catenin at serine or threonine residues of the N-terminal region to predispose degradation of β-catenin through ubiquination (Gordon and Nusse, 2006). Activation of Wnt/β-catenin signaling in type-B and type-C cells of the adult SVZ is sufficient to increase the percentage of dividing C cells that give rise to new neurons in the SVZ (Adachi et al., 2007). In stark contrast, MPTP challenge sharply inhibits β-catenin expression and signaling in SVZ cells, a process mimicked by intracerebroventricular infusion of the Wnt/β-catenin antagonist, Dkk1 (L'Episcopo et al., 2012). In keeping with these findings, activation of Wnt/β-catenin signaling by either intracerebroventricular infusion or systemic injections of the GSK-3β antagonist, AR, can rescue NPC proliferation in the SVZ of young MPTP-exposed mice, further underscoring SVZ plasticity to neurotoxin-induced SVZ injury (L'Episcopo et al., 2012). Here, we further reveal that the imbalance in redox/proinflammatory milieu of aging SVZ can, on the one hand, inhibit PI-3K/Akt leading to active GSK-3β upregulation and loss of the transcriptional activator β-catenin, and on the other hand reduce the sensitivity of aged NPCs to Wnts, resulting in further inhibition of Wnt/β-catenin-induced SVZ homeostatic regulation. Hence, Nrf2-ARE dysfunction-induced dysregulated Wnt signaling may represent a common final pathway to SVZ impairment (Fig. 12).
Activated astrocytes promote neurogenesis from adult NPCs (Lim and Alvarez-Buylla, 1999; Jiao and Chen, 2008) also via the activation of the Wnt/β-catenin pathway (Lie et al., 2005; Kuwabara et al., 2009; L'Episcopo et al., 2011b, 2012). In addition, astrocyte-derived Wnt1 was defined as a critical component of astrocyte-induced DAergic neuroprotection against different neurotoxic stimuli in vitro and as a prosurvival factor in vivo, thereby linking astrocytes and the activation of Wnt/β-catenin pathway to DAergic survival, self-defense, and neurorepair (L'Episcopo et al., 2011b,c). It should be recalled that the Wnt/β-catenin pathway plays a central role in the generation of midbrain DAergic neurons (Prakash et al., 2006; Inestrosa and Arenas, 2010). Importantly, emerging evidence has recently linked PD gene mutations (i.e., PARK8-LRRK2) to the impairment of canonical Wnt signaling activation (Berwick and Harvey, 2012), which is associated with dysfunctional microglial responses (Gillardon et al., 2012; Kim et al., 2012). Here, dysfunctional microglia of aged mice was further linked to (1) reduced expression of Wnt1 in aged astrocytes, (2) reduced astrocyte ability to promote neurogenesis via Wnt/β-catenin activation, and (3) failure to rescue aged NPCs, suggesting that a derangement in the cross talk between inflammatory and Wnt/β-catenin signaling mechanism(s) may represent an early event of aging-induced reduced SVZ plasticity, thus inhibiting DAergic self-repair and increasing vulnerability (L'Episcopo et al., 2011a,b, 2012; Marchetti and Pluchino, 2013). Hence, aged SVZ cell impairment is mimicked by aged microglia exposure, but efficiently reversed by pharmacological modulation of inflammation with HCT1026, both in vitro and in vivo, supporting the role of Wnt/β-catenin in regulating aged neurogenesis as a function of the inflammatory microenvironment (Marchetti and Pluchino, 2013).
Together, these findings provide significant insights into the signaling mechanism underlying inflammation-dependent neurogenic impairment of aging SVZ and hold promise for the development of targeted therapies aimed at modulating endogenous adult neurogenesis to enhance neuronal outcome in age-dependent diseases, such as PD.
Footnotes
We thank the Italian Ministry of Health (Contract 82; Ps-CARDIO ex 56 and PS-NEURO ex 56 to B.M.; Young Investigator Award GR08-7 to S.P.); the Italian Ministry of Research (Curr. Res. Progr. 2008-2012 to B.M.); the Italian Multiple Sclerosis Foundation (Grants 2004/R/15 to S.P.); the National Multiple Sclerosis Society (partial Grants RG-4001-A1 to S.P.); the Italian Ministry of Education, Universities and Research (to B.M.); Wings for Life (Grant XBAG/163 to S.P.); Banca Agricola Popolare di Ragusa (unrestricted grant to S.P.); the Oasi Maria SS. Istituto di Ricovero e Cura a Caraterre Scientifico Institution for Research and Care on Mental Retardation and Brain Aging, Troina (to B.M.); and the European Research Council (ERC) under the ERC-2010-StG Grant agreement no. 260511-SEM_SEM. S.P. holds a John and Lucille van Geest University Lecturership in Brain Repair at the Cambridge Centre for Brain Repair, University of Cambridge, United Kingdom.
The authors declare no competing financial interests.
References
- Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi K, Mirzadeh Z, Sakaguchi M, Yamashita T, Nikolcheva T, Gotoh Y, Peltz G, Gong L, Kawase T, Alvarez-Buylla A, Okano H, Sawamoto K. β-Catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells. 2007;25:2827–2836. doi: 10.1634/stemcells.2007-0177. [DOI] [PubMed] [Google Scholar]
- Ahlenius H, Visan V, Kokaia M, Lindvall O, Kokaia Z. Neural stem and progenitor cells retain their potential for proliferation and differentiation into functional neurons despite lower number in aged brain. J Neurosci. 2009;29:4408–4419. doi: 10.1523/JNEUROSCI.6003-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Buylla A, García-Verdugo JM, Tramontin AD. A unified hypothesis on the lineage of neural stem cells. Nat Rev Neurosci. 2001;2:287–293. doi: 10.1038/35067582. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Ajmone-Cat MA, Gasparini L, Ongini E, Minghetti L. Nuclear receptor peroxisome proliferator-activated receptor-gamma is activated in rat microglial cells by the anti-inflammatory drug HCT1026, a derivative of flurbiprofen. J Neurochem. 2005;92:895–903. doi: 10.1111/j.1471-4159.2004.02932.x. [DOI] [PubMed] [Google Scholar]
- Berwick DC, Harvey K. LRRK2 functions as a Wnt signaling scaffold, bridging cytosolic proteins and membrane-localized LRP6. Hum Mol Genet. 2012;21:4966–4979. doi: 10.1093/hmg/dds342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitar MS, Al-Mulla F. A defect in Nrf2 signaling constitutes a mechanism for cellular stress hypersensitivity in a genetic rat model of type 2 diabetes. Am J Physiol Endocrinol Metab. 2011;301:E1119–E1129. doi: 10.1152/ajpendo.00047.2011. [DOI] [PubMed] [Google Scholar]
- Borta A, Höglinger GU. Dopamine and adult neurogenesis. J Neurochem. 2007;100:587–595. doi: 10.1111/j.1471-4159.2006.04241.x. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31:149–160. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, Ascherio A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease. Ann Neurol. 2005;58:963–967. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, Johnson JA. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: critical role for the astrocyte. Proc Natl Acad Sci. 2009;106:2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TJ, Lipton J, Daley BF, Palfi S, Chu Y, Sortwell C, Bakay RA, Sladek JR, Jr, Kordower JH. Aging-related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates: diminished compensatory mechanisms as a prelude to parkinsonism. Neurobiol Dis. 2007;26:56–65. doi: 10.1016/j.nbd.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusimano M, Biziato D, Brambilla E, Donegà M, Alfaro-Cervello C, Snider S, Salani G, Pucci F, Comi G, Garcia-Verdugo JM, De Palma M, Martino G, Pluchino S. Transplanted neural stem/precursor cells instruct phagocytes and reduce secondary tissue damage in the injured spinal cord. Brain. 2012;135:447–460. doi: 10.1093/brain/awr339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, García-Verdugo JM, Alvarez-Buylla A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci. 1997;17:5046–5061. doi: 10.1523/JNEUROSCI.17-13-05046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002;36:1021–1034. doi: 10.1016/s0896-6273(02)01133-9. [DOI] [PubMed] [Google Scholar]
- Duka T, Duka V, Joyce JN, Sidhu A. α-Synuclein contributes to GSK-3β-catalyzed Tau phosphorylation in Parkinson's disease models. FASEB J. 2009;23:2820–2830. doi: 10.1096/fj.08-120410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Wang LP, Klempin F, Römer B, Kettenmann H, Kempermann G. Enriched environment and physical activity reduce microglia and influence the fate of NG2 cells in the amygdala of adult mice. Cell Tissue Res. 2011;345:69–86. doi: 10.1007/s00441-011-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT. Microglial activation: tuning and pruning adult neurogenesis. Front Pharmacol. 2012;3:41. doi: 10.3389/fphar.2012.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in the adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience. 2009;158:1021–1029. doi: 10.1016/j.neuroscience.2008.06.052. [DOI] [PubMed] [Google Scholar]
- Enwere E, Shingo T, Gregg C, Fujikawa H, Ohta S, Weiss S. Aging results in reduced epidermal growth factor receptor signaling, diminished olfactory neurogenesis, and deficits in fine olfactory discrimination. J Neurosci. 2004;24:8354–8365. doi: 10.1523/JNEUROSCI.2751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada C, Murillo-Carretero M. Nitric oxide and adult neurogenesis in health and disease. Neuroscientist. 2005;11:294–307. doi: 10.1177/1073858404273850. [DOI] [PubMed] [Google Scholar]
- Fernandez-Gonzalez A, Pérez-Otaño I, Morgan JI. MPTP selectively induces haem oxygenase-1 expression in striatal astrocytes. Eur J Neurosci. 2000;12:1573–1583. doi: 10.1046/j.1460-9568.2000.00044.x. [DOI] [PubMed] [Google Scholar]
- Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, Hartley M, Treviño I, O'Brien DE, Casey B, Goldberg MS, Tansey MG. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008;28:10825–10834. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Furlan R, Kurne A, Bergami A, Brambilla E, Maucci R, Gasparini L, Butti E, Comi G, Ongini E, Martino G. A nitric oxide releasing derivative of flurbiprofen inhibits experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;150:10–19. doi: 10.1016/j.jneuroim.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Gao HM, Hong JS. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29:357–365. doi: 10.1016/j.it.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS. Critical role of microglia NADPH-oxidase-derived free radicals in the in vitro MPTP model of Parkinson's disease. FASEB J. 2003;17:1954–1966. doi: 10.1096/fj.03-0109fje. [DOI] [PubMed] [Google Scholar]
- Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of α-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28:7687–7698. doi: 10.1523/JNEUROSCI.0143-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Verdugo JM, Doetsch F, Wichterle H, Lim DA, Alvarez-Buylla A. Architecture and cell types of the adult subventricular zone: in search of the stem cells. J Neurobiol. 1998;36:234–248. doi: 10.1002/(sici)1097-4695(199808)36:2<234::aid-neu10>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Gennuso F, Fernetti C, Tirolo C, Testa N, L'Episcopo F, Caniglia S, Morale MC, Ostrow JD, Pascolo L, Tiribelli C, Marchetti B. Bilirubin protects astrocytes from its own toxicity inducing up-regulation and translocation of multigrug resistance-associated protein 1 (Mrp 1) Proc Natl Acad Sci U S A. 2004;101:2470–2475. doi: 10.1073/pnas.0308452100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillardon F, Schmid R, Draheim H. Parkinson's disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience. 2012;208:41–48. doi: 10.1016/j.neuroscience.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Gordon MD, Nusse R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281:22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- Gritti A, Bonfanti L, Doetsch F, Caille I, Alvarez-Buylla A, Lim DA, Galli R, Verdugo JM, Herrera DG, Vescovi AL. Multipotent neural stem cells reside into the rostral extension and olfactory bulb of adult rodents. J Neurosci. 2002;22:437–445. doi: 10.1523/JNEUROSCI.22-02-00437.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindle JV. Ageing, neurodegeneration and Parkinson's disease. Age Ageing. 2010;39:156–161. doi: 10.1093/ageing/afp223. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- Ho A, Blum M. Induction of interleukin-1 associated with compensatory dopaminergic sprouting in the denervated striatum of young mice: model of aging and neurodegenerative disease. J Neurosci. 1998;18:5614–5629. doi: 10.1523/JNEUROSCI.18-15-05614.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglinger GU, Rizk P, Muriel MP, Duyckaerts C, Oertel WH, Caille I, Hirsch EC. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat Neurosci. 2004;7:726–735. doi: 10.1038/nn1265. [DOI] [PubMed] [Google Scholar]
- Höglinger GU, Barker RA, Hagg T, Arias-Carrión O, Caldwell MA, Hirsch EC. Quantitative evaluation of the human subventricular zone. Brain. 2012;135:e221, 1–4. doi: 10.1093/brain/aws087. [DOI] [PubMed] [Google Scholar]
- Idris AI, Ralston SH, van't Hof RJ. The nitrosylated flurbiprofen derivative HCT1026 inhibits cytokine-induced signalling through a novel mechanism of action. Eur J Pharmacol. 2009;602:215–222. doi: 10.1016/j.ejphar.2008.11.023. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Arenas E. Emerging role of Wnts in the adult nervous system. Nat Rev Neurosci. 2010;11:77–86. doi: 10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Przedborski S. Protocol for the MPTP model of Parkinson's disease. Nat Protocols. 2007;2:141–151. doi: 10.1038/nprot.2006.342. [DOI] [PubMed] [Google Scholar]
- Jakubs K, Bonde S, Iosif RE, Ekdahl CT, Kokaia Z, Kokaia M, Lindvall O. Inflammation regulates functional integration of neurons born in adult brain. J Neurosci. 2008;28:12477–12488. doi: 10.1523/JNEUROSCI.3240-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, Chen DF. Induction of neurogenesis in nonconventional neurogenic regions of the adult central nervous system by niche astrocyte-produced signals. Stem Cells. 2008;26:1221–1230. doi: 10.1634/stemcells.2007-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalani MY, Cheshier SH, Cord BJ, Bababeygy SR, Vogel H, Weissman IL, Palmer TD, Nusse R. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc Natl Acad Sci U S A. 2008;105:16970–16975. doi: 10.1073/pnas.0808616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanis I. The subependymal zone neurogenic niche: a beating heart in the centre of the brain: how plastic is adult neurogenesis? Opportunities for therapy and questions to be addressed. Brain. 2009;132:2909–2921. doi: 10.1093/brain/awp237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeble JE, Moore PK. Pharmacology and potential therapeutic application of nitric oxide-releasing nonsteroidal anti-inflammatory and related nitric oxide-donating drugs. Br J Pharmacol. 2002;137:295–310. doi: 10.1038/sj.bjp.0704876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Yang MS, Choi D, Kim JH, Kim HS, Seol W, Choi S, Jou I, Kim EY, Joe EH. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One. 2012;7:e34693. doi: 10.1371/journal.pone.0034693. (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wong PK. Loss of ATM impairs proliferation of neural stem cells through oxidative stress-mediated p38 MAPK signaling. Stem Cells. 2009;27:1987–1998. doi: 10.1002/stem.125. [DOI] [PubMed] [Google Scholar]
- Kim WY, Snider WD. Functions of GSK-3 signaling in development of the nervous system. Front Mol Neurosci. 2011;4:44. doi: 10.3389/fnmol.2011.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara T, Hsieh J, Muotri A, Yeo G, Warashina M, Lie DC, Moore L, Nakashima K, Asashima M, Gage FH. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat Neurosci. 2009;12:1097–1105. doi: 10.1038/nn.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Ulusoy A, Innamorato NG, Sahin G, Rábano A, Kirik D, Cuadrado A. α-Synuclein expression and Nrf2-deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson's disease. Hum Mol Genet. 2012;21:3173–3192. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Marchetti B. Glia as a turning point in the therapeutic strategy of Parkinson's disease. CNS Neurol Disord Drug Targets. 2010a;9:349–372. doi: 10.2174/187152710791292639. [DOI] [PubMed] [Google Scholar]
- L'Episcopo F, Tirolo C, Caniglia S, Testa N, Serra PA, Impagnatiello F, Morale MC, Marchetti B. Combining nitric oxide release with anti-inflammatory activity preserves nigrostriatal dopaminergic innervation and prevents motor impairment in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neuroinflammation. 2010b;7:83. doi: 10.1186/1742-2094-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Cossetti C, D'Adamo P, Zardini E, Andreoni L, Ihekwaba AE, Serra PA, Franciotta D, Martino G, Pluchino S, Marchetti B. Reactive astrocytes and Wnt/β-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Neurobiol Dis. 2011a;41:508–527. doi: 10.1016/j.nbd.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L'Episcopo F, Serapide MF, Tirolo C, Testa N, Caniglia S, Morale MC, Pluchino S, Marchetti B. A Wnt1 regulated Frizzled-1/β-catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: therapeutical relevance for neuron survival and neuroprotection. Mol Neurodegener. 2011b;13:6–49. doi: 10.1186/1750-1326-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Impagnatiello F, Marchetti B. Switching the microglial harmful phenotype promotes lifelong restoration of subtantia nigra dopaminergic neurons from inflammatory neurodegeneration in aged mice. Rejuvenation Res. 2011c;14:411–424. doi: 10.1089/rej.2010.1134. [DOI] [PubMed] [Google Scholar]
- L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Deleidi M, Serapide MF, Pluchino S, Marchetti B. Plasticity of subventricular zone neuroprogenitors in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model of Parkinson's disease involves crosstalk between inflammatory and Wnt/β-catenin signaling pathways: functional consequences for neuroprotection and repair. J Neurosci. 2012;32:2062–2085. doi: 10.1523/JNEUROSCI.5259-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie DC, Colamarino SA, Song HJ, Désiré L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signaling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Lim DA, Alvarez-Buylla A. Interaction between astrocytes and adult subventricular zone precursors stimulates neurogenesis. Proc Natl Acad Sci U S A. 1999;96:7526–7531. doi: 10.1073/pnas.96.13.7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Daniels SB, Lennington JB, Notti RQ, Conover JC. The aging neurogenic subventricular zone. Aging Cell. 2006;5:139–152. doi: 10.1111/j.1474-9726.2006.00197.x. [DOI] [PubMed] [Google Scholar]
- Marchetti B, Abbracchio MP. To be or not to be (inflammed) is that the question in anti-inflammatory drug therapy of neurodegenerative diseases? Trends Pharmacol Sci. 2005;26:517–525. doi: 10.1016/j.tips.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Marchetti B, Pluchino S. Wnt your brain be inflamed? Yes, it Wnt! Trends Mol Med. 2013 doi: 10.1016/j.molmed.2012.12.001. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti B, Morale MC, Brouwer J, Tirolo C, Testa N, Caniglia S, Barden N, Amor S, Smith PA, Dijkstra CD. Exposure to a dysfunctional glucocorticoid receptor from early embryonic life programs the resistance to experimental autoimmune encephalomyelitis via nitric oxide-induced immunosuppression. J Immunol. 2002;168:5848–5859. doi: 10.4049/jimmunol.168.11.5848. [DOI] [PubMed] [Google Scholar]
- Marchetti B, Kettenmann H, Streit WJ, editors. Glia-neuron crosstalk in neuroinflammation, neurodegeneration and neuroprotection. Brain Res Rev Special. 2005a;48:129–408. [Google Scholar]
- Marchetti B, Serra PA, Tirolo C, L'Episcopo F, Caniglia S, Gennuso F, Testa N, Miele E, Desole S, Barden N, Morale MC. Glucocorticoid receptor-nitric oxide crosstalk and vulnerability to experimental Parkinsonism: pivotal role for glia-neuron interactions. Brain Res Rev. 2005b;48:302–321. doi: 10.1016/j.brainresrev.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Marchetti B, L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC. Vulnerability to Parkinson's disease: towards an unifying theory of disease etiology. In: Nriagu JO, editor. Encyclopedia of environmental health. Vol. 5. Burlington, VT: Elsevier; 2011. pp. 690–704. [Google Scholar]
- Martino G, Pluchino S, Bonfanti L, Schwartz M. Brain regeneration in physiology and pathology: the immune signature driving therapeutic plasticity of neural stem cells. Physiol Rev. 2011;91:1281–1304. doi: 10.1152/physrev.00032.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov AY, Barone TA, Plunkett RJ, Pruitt SC. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J Neurosci. 2004;24:1726–1733. doi: 10.1523/JNEUROSCI.4608-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Glial reactions in Parkinson‘s disease. Mov Disord. 2008;23:474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Morale MC, Serra PA, Delogu MR, Migheli R, Rocchitta G, Tirolo C, Caniglia S, Testa N, L'Episcopo F, Gennuso F, Scoto GM, Barden N, Miele E, Desole MS, Marchetti B. Glucocorticoid receptor deficiency increases vulnerability of the nigrostriatal dopaminergic system: critical role of glial nitric oxide. FASEB J. 2004;18:164–166. doi: 10.1096/fj.03-0501fje. [DOI] [PubMed] [Google Scholar]
- Morale MC, L'Episcopo F, Tirolo C, Giaquinta G, Caniglia S, Testa N, Arcieri P, Serra PA, Lupo G, Alberghina M, Harada N, Honda S, Panzica GC, Marchetti B. Loss of aromatase cytochrome P450 function as a risk factor for Parkinson's disease? Brain Res Rev. 2008;57:431–443. doi: 10.1016/j.brainresrev.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Munji RN, Choe Y, Li G, Siegenthaler JA, Pleasure SJ. Wnt signalling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci. 2011;31:1676–1687. doi: 10.1523/JNEUROSCI.5404-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njie MG, Boelenb E, Stassenb FR, Steinbuschc HWM, Borchelta DR, Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging. 2012;33:195. doi: 10.1016/j.neurobiolaging.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda L, Gao J, Hooten KG, Wang E, Thonhoff JR, Dunn TJ, Gao T, Wu P. Critical role of Pl3K/Akt/GSK3β in motoneuron specification from human neural stem cells in response to FGF2 and EGF. Plos One. 2011;6:e23414. doi: 10.1371/journal.pone.0023414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keeffe GC, Tyers P, Aarsland D, Dalley JW, Barker RA, Caldwell MA. Dopamine-induced proliferation of adult neural precursor cells in the mammalian subventricular zone is mediated through EGF. Proc Natl Acad Sci U S A. 2009;106:8754–8759. doi: 10.1073/pnas.0803955106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Schapira AHV, Agid Y. Neurodegeneration and prospects for neuroprotection and rescue in Parkinson's disease. Annal Neurol. 2003;53(suppl 3):S1–S2. doi: 10.1002/ana.10566. [DOI] [PubMed] [Google Scholar]
- Osakada F, Ooto S, Akagi T, Mandai M, Akaike A, Takahashi M. Wnt signalling promotes regeneration in the retina of adult mammals. J Neurosci. 2007;27:4210–4219. doi: 10.1523/JNEUROSCI.4193-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 3rd edition. San Diego: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- Petit-Paitel A, Brau F, Cazareth J, Chabry J. Involment of cytosolic and mitochondrial GSK-3beta in mitochondrial dysfunction and neuronal cell death of MPTP/Mpp+-treated neurons. Plos One. 2009;4:e5491. doi: 10.1371/journal.pone.0005491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccin D, Morshead CM. Wnt signaling regulates symmetry of division of neural stem cells in the adult brain and in response to injury. Stem Cells. 2011;29:528–538. doi: 10.1002/stem.589. [DOI] [PubMed] [Google Scholar]
- Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, Galli R, Del Carro U, Amadio S, Bergami A, Furlan R, Comi G, Vescovi AL, Martino G. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:688–694. doi: 10.1038/nature01552. [DOI] [PubMed] [Google Scholar]
- Pluchino S, Zanotti L, Rossi B, Brambilla E, Ottoboni L, Salani G, Martinello M, Cattalini A, Bergami A, Furlan R, Comi G, Constantin G, Martino G. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature. 2005;436:266–271. doi: 10.1038/nature03889. [DOI] [PubMed] [Google Scholar]
- Pluchino S, Muzio L, Imitola J, Deleidi M, Alfaro-Cervello C, Salani G, Porcheri C, Brambilla E, Cavasinni F, Bergamaschi A, Garcia-Verdugo JM, Comi G, Khoury SJ, Martino G. Persistent inflammation alters the function of the endogenous brain stem cell compartment. Brain. 2008;131:2564–2578. doi: 10.1093/brain/awn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash N, Brodski C, Naserke T, Puelles E, Gogoi R, Hall A, Panhuysen M, Echevarria D, Sussel L, Weisenhorn DM, Martinez S, Arenas E, Simeone A, Wurst W. A Wnt1-regulated genetic network controls theidentity and fate of midbrain-dopaminergic progenitors in vivo. Development. 2006;133:89–98. doi: 10.1242/dev.02181. [DOI] [PubMed] [Google Scholar]
- Przedborski S. Inflammation and Parkinson's disease pathogenesis. Mov Disord. 2010;25:S55–S57. doi: 10.1002/mds.22638. [DOI] [PubMed] [Google Scholar]
- Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31:1121–1133. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricaurte GA, DeLanney LE, Irwin I, Langston JW. Older dopaminergic neurons do not recover from the effects of MPTP. Neuropharmacology. 1987a;26:97–99. doi: 10.1016/0028-3908(87)90051-7. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Irwin I, Forno LS, DeLanney LE, Langston E, Langston JW. Aging and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced degeneration of dopaminergic neurons in the substantia nigra. Brain Res. 1987b;403:43–51. doi: 10.1016/0006-8993(87)90120-x. [DOI] [PubMed] [Google Scholar]
- Rojo AI, Medina-Campos ON, Rada P, Zúñiga-Toalá A, López-Gazcón A, Espada S, Pedraza-Chaverri J, Cuadrado A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: Role of glycogen synthase kinase-3. Free Radic Biol Med. 2012;52:473–487. doi: 10.1016/j.freeradbiomed.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Rueger MA, Muesken S, Walberer M, Jantzen SU, Schnakenburg K, Backes H, Graf R, Neumaier B, Hoehn M, Fink GR, Schroeter M. Effects of minocycline on endogenous neural stem cells after experimental stroke. Neuroscience. 2012;215:174–183. doi: 10.1016/j.neuroscience.2012.04.036. [DOI] [PubMed] [Google Scholar]
- Sakata H, Niizuma K, Yoshioka H, Kim GS, Jung JE, Katsu M, Narasimhan P, Maier CM, Nishiyama Y, Chan PH. Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J Neurosci. 2012;32:3462–3473. doi: 10.1523/JNEUROSCI.5686-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih PH, Yen GC. Differential expressions of antioxidant status in aging rats: the role of transcriptional factor Nrf2 and MAPK signaling pathway. Biogerontology. 2007;8:71–80. doi: 10.1007/s10522-006-9033-y. [DOI] [PubMed] [Google Scholar]
- Smith J, Ladi E, Mayer-Proschel M, Noble M. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc Natl Acad Sci U S A. 2000;97:10032–10037. doi: 10.1073/pnas.170209797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Stevens CF, Gage FH. Astroglia induce neurogenesis from adult neural stem cells. Nature. 2002;417:39–44. doi: 10.1038/417039a. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh YJ, Kundu JK, Li MH, Na HK, Cha YN. Role of Nrf2-mediated heme oxygenase-1 upregulation in adaptive survival response to nitrosative stress. Arch Pharm Res. 2009;32:1163–1176. doi: 10.1007/s12272-009-1807-8. [DOI] [PubMed] [Google Scholar]
- Tepavčević V, Lazarini F, Alfaro-Cervello C, Kerninon C, Yoshikawa K, Garcia-Verdugo JM, Lledo PM, Nait-Oumesmar B, Baron-Van Evercooren A. Inflammation-induced subventricular zone dysfunction leads to olfactory deficits in a targeted mouse model of multiple sclerosis. J Clin Invest. 2011;121:4722–4734. doi: 10.1172/JCI59145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thored P, Heldmann U, Gomes-Leal W, Gisler R, Darsalia V, Taneera J, Nygren JM, Jacobsen SE, Ekdahl CT, Kokaia Z, Lindvall O. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia. 2009;57:835–849. doi: 10.1002/glia.20810. [DOI] [PubMed] [Google Scholar]
- Tropepe V, Craig CG, Morshead CM, van der Kooy D. Transforming growth factor-α null and senescent mice show decreased neural progenitor cell proliferation in the forebrain subependyma. J Neurosci. 1997;17:7850–7859. doi: 10.1523/JNEUROSCI.17-20-07850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard-Després S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–94. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukovic J, Colditz MJ, Blackmore DG, Ruitenberg MJ, Bartlett PF. Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J Neurosci. 2012;32:6435–6443. doi: 10.1523/JNEUROSCI.5925-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallenquist U, Holmqvist K, Hånell A, Marklund N, Hillered L, Forsberg-Nilsson K. Ibuprofen attenuates the inflammatory response and allows formation of migratory neuroblasts from grafted stem cells after traumatic brain injury. Restor Neurol Neurosci. 2012;30:9–19. doi: 10.3233/RNN-2011-0606. [DOI] [PubMed] [Google Scholar]
- Warner TT, Schapira AH. Genetic and environmental factors in the cause of Parkinson's disease. Ann Neurol. 2003;53:S16–S25. doi: 10.1002/ana.10487. [DOI] [PubMed] [Google Scholar]
- Williamson TP, Johnson DA, Johnson JA. Activation of the Nrf2-ARE pathway by siRNA knockdown of Keap1 reduces oxidative stress and provides partial protection from MPTP-mediated neurotoxicity. Neurotoxicology. 2012;33:272–279. doi: 10.1016/j.neuro.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SZ, Taylor MM, Bordey A. Neurotransmitters couple brain activity to subventricular zone neurogenesis. Eur J Neurosci. 2011;33:1123–1132. doi: 10.1111/j.1460-9568.2011.07611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yang X, Yang S, Zhang J. The Wnt/β-catenin signaling pathway in the adult neurogenesis. Eur J Neurosci. 2011;33:1–8. doi: 10.1111/j.1460-9568.2010.7483.x. [DOI] [PubMed] [Google Scholar]
- Zhang W, Dallas S, Zhang D, Guo JP, Pang H, Wilson B, Miller DS, Chen B, Zhang W, McGeer PL, Hong JS, Zhang J. Microglial PHOX and Mac-1 essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant Alpha-Synuclein. Glia. 2007;55:1178–1188. doi: 10.1002/glia.20532. [DOI] [PubMed] [Google Scholar]
- Ziv Y, Schwartz M. Orchestrating brain-cell renewal: the role of immune cells in adult neurogenesis in health and disease. Trends Mol Med. 2008;14:471–478. doi: 10.1016/j.molmed.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]