

Abstract
The formation of supramolecular activation clusters within the immunological synapse, crucial for sustained signaling and T lymphocyte activation, requires costimulation-dependent reorganization of the actin cytoskeleton. Here we have identified the actin-remodeling protein cofilin as a key player in this process. Cell-permeable peptides that block costimulation-induced cofilin/F-actin interactions in untransformed human T lymphocytes impair receptor capping and immunological synapse formation at the interface between T cells and antigen-presenting cells. As a consequence, T cell activation, as measured by cytokine production and proliferation, is inhibited.
Once T cells encounter their cognate antigen on an antigen-presenting cell (APC), initial adhesive contacts are strengthened and a zone of close contact with the APC is built, leading to formation of the immunological synapse (IS). The latter consists of highly organized clusters of surface receptors and signaling molecules (supramolecular activation clusters or SMACs) (1, 2). The IS is thought to be crucial for T cell activation, and its formation and maintenance represents an active actin cytoskeleton guided process (3–5). T cell receptor (TCR) engagement (signal 1) alone is not sufficient for T cell activation; it rather leads to T cell anergy or apoptosis (6–8). Thus, T cells require costimulatory signals (signal 2) mediated via accessory receptors, e.g., CD28 (6, 7) or CD2 (9, 10), to proliferate and to secrete IL-2. Costimulation initiates active directional transport of receptors (11–13) and lipid microdomains (rafts) (14) to the T cell/APC contact site. In addition, to allow sustained signaling required for full T cell activation, costimulation-dependent reorganization of the actin cytoskeleton is required to stabilize the IS (5, 15, 16). As yet, little information is available on molecules that link accessory receptor engagement to such a reorganization of the actin cytoskeleton.
We have identified the actin-binding protein cofilin as a component of a costimulatory signaling pathway (17, 18). Cofilin binds to globular- and filamentous (F)-actin and induces F-actin depolymerization as well as F-actin severing (cleavage), thus playing a key role for actin cytoskeleton remodeling processes (5, 19). In resting peripheral blood T lymphocytes (PBT), cofilin is mainly phosphorylated at serine 3 and inactive. After costimulation via accessory receptors (e.g., CD2 or CD28) but not on TCR engagement alone, cofilin is activated through dephosphorylation (17, 20, 21). Activated cofilin transiently associates with the actin cytoskeleton (18).
Here, we have developed cell permeable peptides homologous to the actin binding sites within the human cofilin sequence [cofilin peptide homologs (CPH)] to investigate the functional role of cofilin in T cell activation. Introduction of these peptides into human PBT prevents the interaction of cofilin with the actin cytoskeleton in vivo and results in profound effects on polyclonal and antigen-induced T cell activation.
Methods
Cells and Antibodies. Human PBT were obtained from healthy volunteers as described (18). The human B lymphoma cell line Raji was grown in culture medium (RPMI medium 1640 with 10% FCS). mAbs against human CD2 (M1 and M2) and rabbit antisera against cofilin were raised in our laboratory. The rabbit actin antiserum and the FLAG mAb were obtained from Sigma. CD3, CD4, CD25, CD28, CD49d, CD69, and IFN-γ mAbs were from BD Biosciences, and secondary Abs were from Dianova. The CD2 mAb 3PT2H9 was kindly provided by S. F. Schlossman (Dana–Farber Cancer Institute, Boston).
Peptides. For solid phase synthesis of peptides M (CDYKDDDDKMASGVAVSDGVIK), W (CDYKDDDDKWAPESAPLKSKM), and Q (CDYKDDDDKWAPESAPLQSQM) we used the fluorenylmethoxycarbonyl (Fmoc) strategy. Cofilin peptides were coupled to activated Penetratin (QBiogene, Heidelberg, Germany) via disulfide bond by incubation of equimolar amounts for 2 h in ddH2O. Penetratin/peptide conjugates were purified by HPLC, and peptide masses were confirmed by laser desorption mass spectrometry. Peptides were solubilized in ddH2O at 1 mg/ml.
Stimulation of PBT by Means of CD2 and Peptide Treatment. PBT were incubated at 37°C with a mitogenic mixture of 0.5 μg/ml each of mAbs directed against three different epitopes of CD2 (M1, M2, and 3PT2H9) unless otherwise indicated. For peptide inhibition, cells were preincubated for 2 h with peptides M plus W or Q. During stimulation, the total peptide concentration was 2.5 μM unless stated otherwise.
Preparation of Cytoskeletal Fractions and Immunoblotting. After 2 h of CD2 stimulation (5 μg/ml M1, M2, and 3PT2H9), actin cytoskeletal fractions were analyzed as described (18).
Proliferation Assay and Cytokine ELISA. Proliferation was analyzed by [3H]thymidine incorporation as described (21). Cell culture supernatants were harvested 48 h after stimulation and subjected to IL-2, IL-10, and IFN-γ ELISAs (Beckman Coulter).
Mixed Lymphocyte Reaction. Peripheral blood mononuclear cells (PBMC) were separated into sheep erythrocyte (SRBC) rosetting-positive cells (responder cells/T cells) and rosetting-negative cells (stimulator cells/monocytes and B cells). Responder cells were preincubated without or with a total concentration of 2 μM peptides M/W. Responder cells (1 × 105) were cocultured with irradiated (30 Gy) stimulator cells (1 × 105) of an unrelated donor. After 48 h, proliferation was analyzed.
Quantitative RT-PCR. Total RNA from 2 × 106 PBT was purified with the High Pure RNA Isolation Kit. Employing the 1st Strand cDNA Synthesis Kit for RT-PCR, templates for PCR were created. Target sequences were amplified by using LightCycler Primer Sets (Search-LC, Heidelberg) with the LightCycler Fast-Start DNA Sybr Green I Kit (all kits were from Roche Diagnostics). RNA input was normalized by average expression of the two housekeeping genes β-Actin and Cyclophilin-B. The data of two independent analyses for each sample and parameter were averaged (for details, see ref. 22).
Stimulation of PBMC with Cytomegalovirus (CMV) Antigen or Superantigen. SRBC rosetting-positive cells (T cells) and rosetting-negative cells (APCs) were isolated as described. T cells were preincubated without or with 2.5 μM peptides M/W and washed once with culture medium. Stimulations were performed essentially as described (23, 24) with 80% T cells plus 20% autologous APCs. As stimulus, lysate from CMV-infected fibroblasts (complement fixation reagent; Virion, Rüschlikon, Switzerland) was used. As negative controls, cells were stimulated with lysate from uninfected fibroblasts (Virion). Alternative stimulations were carried out by using 2.5 μg/ml Staphylococcus aureus enterotoxin B (SEB) (Sigma) or water as a solvent control.
Flow Cytometry. Cellular viability was assessed by staining with 5 μg/ml propidium iodide (Sigma). For detection of surface expression of CD25, PBT were stained for 30 min with directly labeled CD25 mAbs. For analysis of antigen-specific stimulation, fixed PBMC were permeabilized for 10 min with 0.1% saponin (Sigma) in PBS/5% filtered FCS/0.5% BSA/0.07% NaN3 and stained for 30 min by using directly labeled antibodies against CD69, IFN-γ, and CD4 [because CD4+ T cells dominate the CMV-specific immune response during secondary antigen exposure (25)]. At least 30,000 CD4-positive lymphocytes per sample were evaluated on a FACSCalibur (with cellquest software, BD Biosciences).
Receptor Cap Formation and Immunofluorescence Labeling. PBT were pretreated without or with 8 μM peptides M/W, washed in RPMI medium 1640, and allowed to adhere for 15 min onto adhesion slides (Marienfeld, Lauda-Königshofen, Germany). After washing with PBS and blocking with PBS/1% BSA (20 min) cells were incubated at room temperature with 15 μg/ml 3PT2H9 (15 min) in culture medium. Primary antibodies were crosslinked with 1.5 μg/ml Cy3-labeled anti-mouse mAb (45 min). After two washes with PBS/0.2% sodium azide, cells were fixed in 4% paraformaldehyde (30 min) and quenched in 50 mM NH4Cl. After permeabilization (0.3% Triton X-100, 7 min), cells were counterstained with cofilin antiserum (30 min) and Cy2-coupled anti-rabbit Ab (30 min).
T Cell/B Cell Conjugate Formation. PBT were preincubated without or with 5 μM of peptides M/W and washed once with RPMI medium 1640. Raji cells were loaded for 15 min with 2.5 μg/ml each of SEB and S. aureus enterotoxin F (SEF) (Sigma) and washed with RPMI medium 1640. PBT and Raji cells were mixed at a ratio of 1.5:1 and pipetted onto adhesion slides. After 30 min at 37°C, wells were washed with PBS/0.2% sodium azide. Fixation, permeabilization, and staining were performed as for receptor capping. For blocking, slides were incubated in PBS/0.2% fish skin gelatin (Sigma) for 15 min.
Confocal Laser Scanning Microscopy. After immunofluorescence staining, digitized confocal images were generated by using a confocal laser scanning microscope (Leica DMRBE microscope with tcs nt software). Single XY optical sections were acquired. Images for each fluorochrome were overlayed electronically.
Results
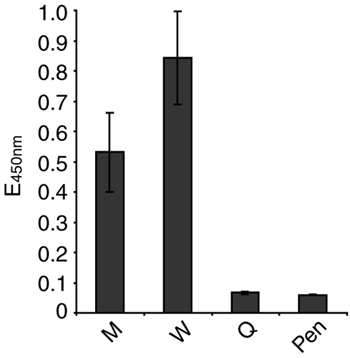
Penetratin-Coupled CPH Inhibit Cofilin/F-Actin Interactions in PBT. With the aim of blocking the interaction of cofilin with the actin cytoskeleton in PBT in vivo, we synthesized peptides corresponding to potential actin binding sites in the human cofilin sequence: residues 1–13 (peptide M) (26) and residues 104–115 (peptide W) (26, 27) and a control peptide with lysines 112 and 114 substituted by glutamines (peptide Q), which should not bind to actin (28). A FLAG epitope was added at the N terminus of each peptide to allow their detection. To enable entry into cells, peptides were coupled to the carrier peptide Penetratin (29). As determined by ELISA, Penetratin-coupled peptides M and W bound to F-actin, whereas the control peptide Q did not, as expected (Fig. 8, which is published as supporting information on the PNAS web site).
We next analyzed whether the CPH block the activation induced association of cofilin with the actin cytoskeleton in freshly isolated human PBT in vivo. PBT comprise a multitude of different antigen specificities. Therefore, polyclonal stimulation via lectins (e.g., PHA) or mitogenic monoclonal antibodies directed against nonvariable membrane receptors expressed by all cells represents the most efficient means to experimentally assess T cell activation. Here, we used a mitogenic combination of three different CD2 mAbs providing signals 1 and 2 simultaneously (alternative pathway of T cell activation) (9, 17, 30). This well established experimental protocol allowed the initial discovery of cofilin dephosphorylation on T cell activation (17). Meanwhile, we have shown that the same effects occur after costimulation through CD3 plus CD28 mAbs (18, 21). As reported before (18), cofilin does not bind to F-actin in unstimulated PBT. After CD2 stimulation, however, cofilin associates with the actin cytoskeleton. Fig. 1 demonstrates that pretreatment of cells with peptides M/W inhibited this association in a dose-dependent manner. Importantly, this finding is not due to interference with cofilin dephosphorylation (data not shown). As expected, control peptide Q did not reduce the amounts of cofilin in the actin cytoskeletal fractions of CD2-activated T cells. Peptides M or W alone also inhibited the association of cofilin with F-actin (data not shown). To ensure maximal effects, we used both peptides together in all further experiments.
Fig. 1.

CPH inhibit the CD2-induced interaction of cofilin with the actin cytoskeleton in vivo. PBT were incubated without (None) or with 0.5, 2, or 5 μM peptides M plus W (M/W) or peptide Q (Q) and stimulated via CD2. Actin and cofilin in actin cytoskeletal fractions were detected by immunoblotting. This experiment was performed three times with comparable results.
CPH Block CD2- and Alloantigen-Induced Proliferation of PBT. In the next set of experiments, we addressed the question of whether this treatment would influence T cell activation. As shown in Fig. 2A, CD2-induced T cell proliferation was clearly reduced by peptides M/W, whereas peptide Q had no inhibitory effect. Parallel determination of cellular viability ruled out peptide toxicity (Fig. 2B).
Fig. 2.

CPH inhibit CD2-induced T cell proliferation without affecting cell viability. (A) PBT were incubated with peptides M/W or Q or without peptides (None) and stimulated via CD2 (CD2) or left unstimulated (RT). After 48 h, cell proliferation was quantified by [3H]thymidine incorporation. Proliferation of CD2-stimulated cells without CPH was taken as 1 (means ± SE of four independent experiments). (B) PBT were pretreated and stimulated as in A. After 72 h, cell viability was assessed through propidium iodide exclusion (means ± SE of three independent experiments).
Polyclonal T cell proliferation is to a substantial degree driven by the IL-2/IL-2R system (31). The observed inhibition of T cell proliferation could, therefore, be due to inhibition of IL-2 secretion, IL-2R (CD25) expression, or both. To investigate this point, secretion of IL-2 and expression of CD25 were measured after CD2 stimulation. Preincubation with M/W caused an inhibition of IL-2 secretion by 89% and reduced CD25 expression by 68% (Fig. 9, which is published as supporting information on the PNAS web site).
To assess the effects of the peptides on antigen-induced T cell proliferation, we tested their influence on mixed lymphocyte reactions (Fig. 3). As expected, allogenic stimulator cells induced proliferation of responder PBT. When responder T cells were, however, preincubated with peptides M/W, alloresponsive T cell proliferation was dramatically reduced.
Fig. 3.

CPH inhibit alloantigen-induced T cell proliferation. Responder T cells (R1) were preincubated with peptides M/W or without peptides (None). R1 were mixed with irradiated stimulator cells of a second donor (R1+*S2) or as control with autologous stimulator cells (R1+*S1). Irradiated stimulator cells of both donors incubated alone (*S1 and *S2) served as further controls. Proliferation was determined after 48 h by 3H incorporation. Two independent experiments are shown (means ± SE of triplicate wells).
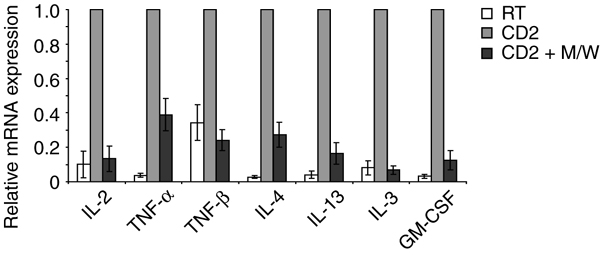
TH1- and TH2-Type Cytokine Responses Are Inhibited by CPH. According to their lymphokine pattern, two major subsets of T helper (TH) cells have been proposed: TH1 cells mainly produce IL-2, IFN-γ, and tumor necrosis factor (TNF) β, whereas TH2 cells express IL-4, -5, -6, -10, and -13 (32). We measured the secretion of IFN-γ as a representative TH1 type cytokine and IL-10 as a major TH2 type cytokine in cell culture supernatants of CD2-stimulated PBT. Fig. 4A demonstrates that the secretion of both cytokines was inhibited by M/W. To investigate whether the peptides interfere with proximal steps in the events leading to cytokine production, the expression levels of IFN-γ and IL-10 mRNA were quantified by means of RT-PCR analysis. Peptides M/W indeed reduced IFN-γ and IL-10 mRNA levels (Fig. 4B). These results suggest that TH1- and TH2-type responses are equally susceptible to inhibition by CPH. To support this conclusion, additional TH1 cytokines (IL-2, TNF-α, and TNF-β), TH2 cytokines (IL-4 and -13), and cytokines produced by both groups [IL-3 and granulocyte/macrophage colony-stimulating factor (GM-CSF)] were evaluated. CPH prevented induction of all cytokines already at the mRNA level (Fig. 10, which is published as supporting information on the PNAS web site).
Fig. 4.

CPH inhibit production of TH1 and TH2 cytokines. PBT were incubated with peptides M/W (black bars) or without peptides (white and gray bars) and then stimulated via CD2 (gray and black bars) for 48 h (A) or 4 h (B) or left unstimulated (white bars). (A) Secretion of IFN-γ and IL-10 into culture supernatants. Data are means ± SE of four (IFN-γ) and three (IL-10) independent experiments with cytokine amounts produced by stimulated cells in the absence of CPH taken as 1. (B) mRNA expression (quantitative RT-PCR) of IFN-γ and IL-10. Normalized mRNA levels of stimulated cells without CPH were taken as 1 (means ± SE of four blood donors). Effects were similar after 2 h of CD2 stimulation (not shown).
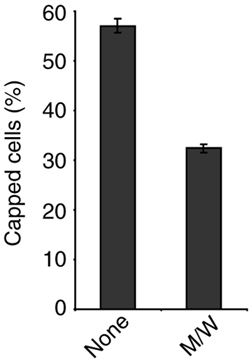
CPH Interfere with Receptor Capping. Receptor clustering after ligand binding, an early event that requires actin cytoskeletal rearrangements (3, 4), plays a crucial role for the recruitment of signaling molecules and the induction of cell proliferation (33). It was, therefore, tempting to speculate that peptides M/W act through interference with receptor capping. As shown in Fig. 5, in the absence of CD2 crosslinking, cofilin is present in the cytoplasm of PBT. Crosslinking of CD2 receptors by mAbs induced CD2 receptor caps to which cofilin clearly colocalized. Pretreatment with peptides M/W markedly reduced CD2 receptor cap formation to small patches of receptor clusters. Quantitative evaluation of four independent experiments yielded a statistically significant inhibition of CD2 receptor cap formation from 57% to 32% capped cells (P < 0.01, two-tailed paired t test; Fig. 11, which is published as supporting information on the PNAS web site). This result implies that the interaction of cofilin with the actin cytoskeleton is crucial for receptor polarization on the T cell surface.
Fig. 5.

CPH inhibit CD2 receptor capping. PBT were preincubated without (Top and Middle) or with (Bottom) peptides M/W. Caps were induced with CD2 mAb and anti-mouse-Cy3 mAb (CD2 crosslinking) or cells were only incubated with CD2 mAb (no crosslinking). After fixation and permeabilization, cells were counterstained for cofilin (green, Left). For samples without crosslinking, anti-mouse Cy3-mAb was included to visualize CD2 distribution (red, Center). Colocalization of red and green fluorescence appears yellow (Right).
Cofilin in T Cell/APC Interactions. Naturally, receptor polarization occurs on encounter of T cells with APCs, leading to formation of the IS. To determine the localization of cofilin with respect to the organization of the IS (1), T cell/APC conjugation experiments were performed. Given the high variety of individual antigen specificities in untransformed human PBT, induction of a detectable number of antigen-elicited synapses is only feasible by using superantigens. Human Raji B lymphoma cells were chosen as APCs (34). In the presence of the superantigens SEB and SEF, T cells displayed a marked polarization of CD2, CD3, and cofilin toward the contact interface (Fig. 6 A and B). As expected, CD2 distributed broadly along the interface including the peripheral SMAC (pSMAC) (2, 35) (Fig. 6A), whereas CD3 concentrated in the center of the IS (cSMAC) (1, 2) (Fig. 6B). In T cells, cofilin was consistently observed at the periphery of the IS (Fig. 6 A and B). Within APCs, in contrast, cofilin was clearly not recruited to the contact interfaces. This latter finding is in agreement with a report that the B cell cytoskeleton is dispensable for IS formation (4).
Fig. 6.

CPH inhibit T cell/B cell conjugate formation and cofilin and CD2 colocalization to the T cell/B cell contact zone. (A and B) PBT (labeled T) were preincubated without (Top and Middle) or with (Bottom) peptides M/W and conjugated with superantigen-loaded (SEB/SEF) or unloaded (no Ag) Raji cells (labeled B). After fixation and permeabilization, cells were stained for cofilin (A, green, Left) and CD2 (A, red, Center) or for cofilin (B, green, Left) and CD3 (B, red, Center). Colocalization of red and green fluorescence appears yellow (Right). (C) Quantification of PBT/Raji conjugation in the presence (M/W) or absence (None) of CPH. Shown are mean percentages of T cells engaged in T cell/B cell contacts ± SE from three independent experiments performed as in A. In each experiment, at least 500 (No Ag) or 1,000 (SEB/SEF) cells per sample were analyzed. Peptide inhibition of contact formation was significant (two-tailed paired t test, P < 0.05). (D) Quantification of the accumulation of cofilin, CD2, or CD3 at the contact interfaces between PBT and superantigen-loaded Raji cells in the presence (M/W) or absence (None) of CPH. Shown are mean percentages of conjugates with accumulation of cofilin, CD2, or CD3 ± SE from three independent experiments performed as in A. In each experiment, at least 75 conjugates per sample were evaluated. Inhibition of cofilin and CD2 accumulation was significant (two-tailed paired t test, P < 0.05). Changes in CD3 accumulation were not significant.
We next determined whether CPH interfere with T cell/APC contact and IS formation. Contact frequencies were quantified in a flow cytometry-based conjugation assay (data not shown), as well as by confocal laser scanning microscopy (Fig. 6C). Pretreatment of PBT with peptides M/W reduced SEB/SEF-inducible conjugate formation significantly (P < 0.05). Yet, still a substantial number of T cell/APC conjugates was formed. Therefore, we examined the composition of the contact interfaces of these remaining T cell/APC pairs (Fig. 6 A, B, and D). Accumulation of cofilin and CD2 within these contact zones was significantly inhibited by pretreatment of PBT with CPH (Fig. 6 A, B, and D). Interestingly, TCR/CD3 recruitment remained unaffected (Fig. 6 B and D). Taken together, CPH decreased T cell/APC conjugate formation and altered the molecular composition of the remaining conjugates.
CPH Affect Antigen-Induced Cytokine Production. The functional consequences of the effects of the CPH on antigen-elicited IS formation were investigated in a flow cytometric assay developed to characterize antigen-specific T cell activation on the basis of intracellular cytokine production (23). PBT were stimulated for 6 h with APCs loaded with either the superantigen SEB or with CMV antigen (Fig. 7). As markers of activation, CD69 and intracellular IFN-γ were determined. All stimuli and control antigen promoted CD69 expression. This response was, however, not susceptible to inhibition by CPH. In contrast, IFN-γ production, as induced by SEB and CMV antigen, was dramatically reduced when T cells had been pretreated with CPH. This finding indicates that CPH efficiently inhibit antigen-triggered cytokine production.
Fig. 7.

CPH inhibit antigen-induced intracellular IFN-γ production. T cells were preincubated without (None) or with peptides M/W. Stimulations were performed with sheep erythrocyte rosetting-negative peripheral blood mononuclear cells as APCs loaded with SEB, control antigen (Control Ag), or CMV antigen (CMV Ag) or without antigen (No Ag). The frequency of antigen-reactive CD4+ T cells is given as the percentage of IFN-γ+ cells [including mean fluorescence index (MFI) of IFN-γ+ cells]. This experiment was performed three times with comparable results.
Discussion
We have investigated the function of the actin remodeling protein cofilin for activation of untransformed human PBT. Productive T cell activation requires costimulation-dependent rearrangements in the T cell actin cytoskeleton that enable ordered and stable formation of cell/cell contacts (i.e., IS formation) (5, 11, 15, 16). After costimulation through accessory receptors (e.g., CD2 or CD28) but not after TCR/CD3 stimulation alone, cofilin is dephosphorylated and associates with the actin cytoskeleton (17, 18, 21), enabling its F-actin severing and depolymerizing activities, which are crucial for enhanced polymerization and the remodeling of the actin cytoskeleton (5, 19). Because cofilin is an essential protein (36–38), cofilin-deficient T cells are not available for functional investigations. Therefore, we synthesized nontoxic cell-permeable Penetratin-coupled peptides homologous with two actin binding motifs of the human cofilin sequence [peptides M (amino acids 1–13) and W (amino acids 104–115)] that compete with cofilin for its association with the actin cytoskeleton. Note that these peptides do not prevent cofilin dephosphorylation. For control, we used the peptide Q (amino acids 104–115 with amino acids 112/114 substituted by Q), which does not inhibit F-actin/cofilin interactions in vivo.
In PBT, cofilin is recruited to CD2 receptor caps and superantigen-induced T cell/B cell interfaces. Inhibition of the cofilin interaction with the actin cytoskeleton through CPH impairs the formation of receptor caps, as well as T cell/B cell conjugate and IS formation. Moreover, T cell proliferation, as well as TH1- and TH2-type cytokine production, are blocked. Note that the effects of the individual peptides M and W were qualitatively identical to the effects of the combined peptides M and W (data not shown).
Importantly, CPH do not influence all T cell activation events. After stimulation with CMV antigen (lysate of infected cells) or control lysate, expression of the T cell activation marker CD69 was even enhanced by CPH. The large number of CD69-positive cells suggests that undefined components in the cellular lysates induce CD69, likely via TCR-independent mechanisms. A resistance of CD69 expression toward therapeutic immunosuppression as compared to other T cell activation markers has been reported by others as well (39).
Although CPH competitively prevent cofilin from its association with the actin cytoskeleton, we cannot formally exclude similar effects of CPH on other F-actin binding proteins. Note, however, that F-actin binding of a second costimulation-related actin-binding protein, L-Plastin (30), is not reduced by CPH (data not shown). Moreover, the effects of CPH clearly differ from those of cytochalasins, well accepted inhibitors of actin polymerization (40, 41). Thus, although in parallel experiments cytochalasins inhibited T cell proliferation, they did not reduce IL-2 production (data not shown).
Using naive human PBT and superantigen-loaded Raji cells as APCs, cofilin is recruited to the T cell/APC contact site. There, it does not colocalize with TCR/CD3 in the cSMAC but accumulates at the periphery of the IS. The CPH reduce superantigen-induced T cell/APC contact formation, implying that cofilin/F-actin interaction is important for this process. Moreover, synapse organization of the remaining T cell/APC couples is profoundly disturbed, showing a significant reduction of cofilin and CD2 recruitment. In contrast, CD3 redistribution to the remaining contact zones is not inhibited by the presence of CPH, a finding that underlines the particular relationship of cofilin activity with accessory receptor signaling/costimulation.
Our data are in accordance with several studies showing that TCR accumulation in the contact zone and T cell activation represent independent events. For example, in the absence of CD28/CD80 interactions, lipid bilayer-supported synapses with central TCR accumulation are formed yet are not sufficient to induce T cell proliferation (42). Another study showed that although in vivo CD28 costimulation is necessary for IL-2 production, TCRs already redistribute toward APCs in the absence of CD28 (43). Finally, inhibition of the CD2/CD58 interaction prevents IFN-γ production but not TCR-triggered tyrosine phosphorylation at the T cell/B cell interface (12). Other groups observed changes in the IS structure affecting the extent and stability of central TCR accumulation after antibody-mediated blockade of costimulation (13, 15). Whether CPH also elicit such subtle changes in TCR accumulation remains to be examined.
A recent study suggested that costimulation contributes decisively to sustained actin dynamics at the T cell/APC contact zone. Prolonged cytoskeletal dynamics in turn is essential for the formation of a complete IS and T cell proliferation (16). Because cofilin is a central regulator of actin rearrangements (5, 19), activation of cofilin through costimulation (17, 18) may provide a molecular link between these processes. Thus, through its actin severing activity dephosphorylated cofilin enables enhanced actin polymerization. At the same time, the depolymerizing activity of cofilin allows removal of actin filaments at sites where they are no longer required. Therefore, activation of cofilin through costimulation of accessory receptors may explain both the actin-dependent recruitment of accessory receptors to the T cell/APC contact zone as well as the actin-dependent organization and stabilization of the IS.
In conclusion, the actin-remodeling protein cofilin appears to represent a central integrator of T cell costimulation, at least in part through its involvement in IS formation. Importantly, because cofilin activation is not influenced by common immunosuppressive drugs (e.g., Cyclosporin A/FK 506, rapamycin, dexamethasone, leflunomide, or mycophenolic acid) (21), this signaling pathway may contain novel target structures for potent immunomodulatory therapeutics.
Supplementary Material
Acknowledgments
We thank M. Klemke, M. Konstandin, and M. Sester for critical reading of the manuscript. This work was supported by Deutsche Forschungsgemeinschaft Grant SFB 405/A4.
Abbreviations: APC, antigen-presenting cell; CMV, cytomegalovirus; CPH, cofilin peptide homologs; IS, immunological synapse; PBT, peripheral blood T cells; SMAC, supramolecular activation cluster; SEB, S. aureus enterotoxin B; SEF, S. aureus enterotoxin F; TCR, T cell receptor; TH, T helper.
References
- 1.Monks, C. R., Freiberg, B. A., Kupfer, H., Sciaky, N. & Kupfer, A. (1998) Nature 395, 82–86. [DOI] [PubMed] [Google Scholar]
- 2.Grakoui, A., Bromley, S. K., Sumen, C., Davis, M. M., Shaw, A. S., Allen, P. M. & Dustin, M. L. (1999) Science 285, 221–227. [DOI] [PubMed] [Google Scholar]
- 3.Valitutti, S., Dessing, M., Aktories, K., Gallati, H. & Lanzavecchia, A. (1995) J. Exp. Med. 181, 577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wülfing, C., Sjaastad, M. D. & Davis, M. M. (1998) Proc. Natl. Acad. Sci. USA 95, 6302–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samstag, Y., Eibert, S. M., Klemke, M. & Wabnitz, G. H. (2003) J. Leukocyte Biol. 73, 30–48. [DOI] [PubMed] [Google Scholar]
- 6.Harding, F. A., McArthur, J. G., Gross, J. A., Raulet, D. H. & Allison, J. P. (1992) Nature 356, 607–609. [DOI] [PubMed] [Google Scholar]
- 7.Radvanyi, L. G., Shi, Y., Vaziri, H., Sharma, A., Dhala, R., Mills, G. B. & Miller, R. G. (1996) J. Immunol. 156, 1788–1798. [PubMed] [Google Scholar]
- 8.Gimmi, C. D., Freeman, G. J., Gribben, J. G., Gray, G. & Nadler, L. M. (1993) Proc. Natl. Acad. Sci. USA 90, 6586–6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meuer, S. C., Hussey, R. E., Fabbi, M., Fox, D., Acuto, O., Fitzgerald, K. A., Hodgdon, J. C., Protentis, J. P., Schlossman, S. F. & Reinherz, E. L. (1984) Cell 36, 897–906. [DOI] [PubMed] [Google Scholar]
- 10.Bierer, B. E., Peterson, A., Gorga, J. C., Herrmann, S. H. & Burakoff, S. J. (1988) J. Exp. Med. 168, 1145–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wülfing, C. & Davis, M. M. (1998) Science 282, 2266–2269. [DOI] [PubMed] [Google Scholar]
- 12.Zaru, R., Cameron, T. O., Stern, L. J., Muller, S. & Valitutti, S. (2002) J. Immunol. 168, 4287–4291. [DOI] [PubMed] [Google Scholar]
- 13.Wülfing, C., Sumen, C., Sjaastad, M. D., Wu, L. C., Dustin, M. L. & Davis, M. M. (2002) Nat. Immunol. 3, 42–47. [DOI] [PubMed] [Google Scholar]
- 14.Viola, A., Schroeder, S., Sakakibara, Y. & Lanzavecchia, A. (1999) Science 283, 680–682. [DOI] [PubMed] [Google Scholar]
- 15.Wetzel, S. A., McKeithan, T. W. & Parker, D. C. (2002) J. Immunol. 169, 6092–6101. [DOI] [PubMed] [Google Scholar]
- 16.Tskvitaria-Fuller, I., Rozelle, A. L., Yin, H. L. & Wülfing, C. (2003) J. Immunol. 171, 2287–2295. [DOI] [PubMed] [Google Scholar]
- 17.Samstag, Y., Eckerskorn, C., Wesselborg, S., Henning, S., Wallich, R. & Meuer, S. C. (1994) Proc. Natl. Acad. Sci. USA 91, 4494–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee, K. H., Meuer, S. C. & Samstag, Y. (2000) Eur. J. Immunol. 30, 892–899. [DOI] [PubMed] [Google Scholar]
- 19.Condeelis, J. (2001) Trends Cell Biol. 11, 288–293. [DOI] [PubMed] [Google Scholar]
- 20.Samstag, Y., Bader, A. & Meuer, S. C. (1991) J. Immunol. 147, 788–794. [PubMed] [Google Scholar]
- 21.Ambach, A., Saunus, J., Konstandin, M., Wesselborg, S., Meuer, S. C. & Samstag, Y. (2000) Eur. J. Immunol. 30, 3422–3431. [DOI] [PubMed] [Google Scholar]
- 22.Autschbach, F., Giese, T., Gassler, N., Sido, B., Heuschen, G., Heuschen, U., Zuna, I., Schulz, P., Weckauf, H., Berger, I., et al. (2002) Virchows Arch. 441, 500–513. [DOI] [PubMed] [Google Scholar]
- 23.Waldrop, S. L., Pitcher, C. J., Peterson, D. M., Maino, V. C. & Picker, L. J. (1997) J. Clin. Invest. 99, 1739–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sester, M., Sester, U., Gartner, B., Kubuschok, B., Girndt, M., Meyerhans, A. & Kohler, H. (2002) J. Virol. 76, 3748–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sester, M., Sester, U., Gartner, B. C., Girndt, M., Meyerhans, A. & Kohler, H. (2002) J. Am. Soc. Nephrol. 13, 2577–2584. [DOI] [PubMed] [Google Scholar]
- 26.Lappalainen, P., Fedorov, E. V., Fedorov, A. A., Almo, S. C. & Drubin, D. G. (1997) EMBO J. 16, 5520–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yonezawa, N., Homma, Y., Yahara, I., Sakai, H. & Nishida, E. (1991) J. Biol. Chem. 266, 17218–17221. [PubMed] [Google Scholar]
- 28.Moriyama, K., Yonezawa, N., Sakai, H., Yahara, I. & Nishida, E. (1992) J. Biol. Chem. 267, 7240–7244. [PubMed] [Google Scholar]
- 29.Derossi, D., Chassaing, G. & Prochiantz, A. (1998) Trends Cell Biol. 8, 84–87. [PubMed] [Google Scholar]
- 30.Henning, S. W., Meuer, S. C. & Samstag, Y. (1994) J. Immunol. 152, 4808–4815. [PubMed] [Google Scholar]
- 31.Meuer, S. C., Hussey, R. E., Cantrell, D. A., Hodgdon, J. C., Schlossman, S. F., Smith, K. A. & Reinherz, E. L. (1984) Proc. Natl. Acad. Sci. USA 81, 1509–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mosmann, T. R., Cherwinski, H., Bond, M. W., Giedlin, M. A. & Coffman, R. L. (1986) J. Immunol. 136, 2348–2357. [PubMed] [Google Scholar]
- 33.Holsinger, L. J., Graef, I. A., Swat, W., Chi, T., Bautista, D. M., Davidson, L., Lewis, R. S., Alt, F. W. & Crabtree, G. R. (1998) Curr. Biol. 8, 563–572. [DOI] [PubMed] [Google Scholar]
- 34.Roumier, A., Olivo-Marin, J. C., Arpin, M., Michel, F., Martin, M., Mangeat, P., Acuto, O., Dautry-Varsat, A. & Alcover, A. (2001) Immunity 15, 715–728. [DOI] [PubMed] [Google Scholar]
- 35.van der Merwe, P. A., Davis, S. J., Shaw, A. S. & Dustin, M. L. (2000) Semin. Immunol. 12, 5–21. [DOI] [PubMed] [Google Scholar]
- 36.Samstag, Y., Dreizler, E. M., Ambach, A., Sczakiel, G. & Meuer, S. C. (1996) J. Immunol. 156, 4167–4173. [PubMed] [Google Scholar]
- 37.Iida, K., Moriyama, K., Matsumoto, S., Kawasaki, H., Nishida, E. & Yahara, I. (1993) Gene 124, 115–120. [DOI] [PubMed] [Google Scholar]
- 38.Moon, A. L., Janmey, P. A., Louie, K. A. & Drubin, D. G. (1993) J. Cell Biol. 120, 421–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hutchinson, P., Divola, L. A. & Holdsworth, S. R. (1999) Cytometry 38, 244–249. [DOI] [PubMed] [Google Scholar]
- 40.Freed, B. M., Lempert, N. & Lawrence, D. A. (1989) Int. J. Immunopharmacol. 11, 459–465. [DOI] [PubMed] [Google Scholar]
- 41.Chiaffarino, F., Biffi, M., Luciano, A., Gromo, G. & Leoni, F. (1994) Cell. Immunol. 153, 39–51. [DOI] [PubMed] [Google Scholar]
- 42.Bromley, S. K., Iaboni, A., Davis, S. J., Whitty, A., Green, J. M., Shaw, A. S., Weiss, A. & Dustin, M. L. (2001) Nat. Immunol. 2, 1159–1166. [DOI] [PubMed] [Google Scholar]
- 43.Reichert, P., Reinhardt, R. L., Ingulli, E. & Jenkins, M. K. (2001) J. Immunol. 166, 4278–4281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}