

Abstract
Puumala virus (PUUV) is a causative agent of hemorrhagic fever with renal syndrome (HFRS). Although PUUV-associated HFRS does not result in high case-fatality rates, the social and economic impact is considerable. There is no licensed vaccine or specific therapeutic to prevent or treat HFRS. Here we report the synthesis of a codon-optimized, full-length M segment open reading frame and its cloning into a DNA vaccine vector to produce the plasmid pWRG/PUU-M(s2). pWRG/PUU-M(s2) delivered by gene gun produced high-titer neutralizing antibodies in hamsters and nonhuman primates. Vaccination with pWRG/PUU-M(s2) protected hamsters against infection with PUUV but not against infection by related HFRS-associated hantaviruses. Unexpectedly, vaccination protected hamsters in a lethal disease model of Andes virus (ANDV) in the absence of ANDV cross-neutralizing antibodies. This is the first evidence that an experimental DNA vaccine for HFRS can provide protection in a hantavirus lethal disease model.
INTRODUCTION
Puumala virus (PUUV), a virus of the genus Hantavirus in the Bunyaviridae family, is responsible for the vast majority of cases of hemorrhagic fever with renal syndrome (HFRS) in Scandinavia, Europe, and Western Russia (1). Other notable hantaviruses that cause disease in humans include Hantaan virus (HTNV) and Seoul virus (SEOV), which cause disease predominantly in Asia; Dobrava virus (DOBV), which causes disease predominantly in the Balkans; and Sin Nombre virus (SNV) and Andes virus (ANDV), which cause disease predominantly in the southwestern United States and southern South America, respectively. Like all members of the Bunyaviridae family, PUUVs are enveloped viruses with trisegmented (S, M, and L), negative-sense RNA genomes. The S gene segment encodes the nucleoprotein (N), which interacts with the genomic RNA to form nucleocapsids. The M gene segment encodes the Gn and Gc surface glycoproteins, which are the targets of neutralizing antibodies found in infected animals (2). The L genome segment encodes the RNA-dependent RNA polymerase (3).
PUUV is carried by persistently infected bank voles (Myodes glareolus). Human disease occurs when persons are exposed to infected rodent excreta or secreta, by either inhalation, ingestion, or a bite (4, 5). PUUV does not have the high lethality of other hantaviruses, such as HTNV (15% case-fatality rate) or the viruses that cause hantavirus pulmonary syndrome (HPS), SNV and ANDV (35% case-fatality rate); however, there is a high level of morbidity (3). There are no vaccines to prevent HFRS caused by PUUV, and there are no specific drugs to treat this disease (6). Patients receive supportive care including dialysis (7).
Attempts to produce vaccines against PUUV have included killed virus vaccines (8), virus-like particles (9), and subunit vaccines. Most of the subunit vaccines have involved the N protein produced in Escherichia coli, yeasts, mammalian cells, and chimeric hepatitis B virus (HBV) core particles (10–13). Hitherto, attempts to produce vaccines that elicit high-titer neutralizing antibodies against PUUV have been unsuccessful. Our approach to developing hantavirus vaccines has been to construct DNA vaccines containing the full-length hantavirus M gene. We used this approach to produce SEOV, HTNV, and ANDV M gene-based DNA vaccines (14–16). We also constructed a plasmid containing both the HTNV and ANDV M gene open reading frames (ORFs), controlled by separate promoters (17). These constructs elicited high-titer neutralizing antibodies in rodents or rabbits and nonhuman primates (NHPs). None of these vaccines elicited high levels of PUUV cross-neutralizing antibodies, although the HTNV-ANDV combination plasmid elicited detectable PUUV cross-neutralizing antibodies after a long-range boost (17). Importantly, none of these vaccines cross-protected against PUUV infection in a hamster infection model. Recently, we developed a synthetic PUUV DNA vaccine, pWRG/PUU-M(s2), that was shown to be immunogenic in hamsters and humans (18, 19). Details of the construction of the vaccine and protection data have not yet been reported. Here we report the experiments that led to the development of pWRG/PUU-M(s2) and describe protection experiments demonstrating that this experimental vaccine is capable of protecting hamsters not only from infection with PUUV but also against lethal disease caused by Andes virus.
MATERIALS AND METHODS
Viruses, cells, and medium.
PUUV strain K27 (20), PUUV strain Sotkamo (21), HTNV strain 76-118 (22), ANDV strain Chile-9717869 (23), SEOV strain SR11 (24), and DOBV strain Dobrava (25) were propagated in Vero E6 cells (Vero C1008; ATCC CRL 1586). Transient-expression experiments were performed with COS cells (COS-7; ATCC CRL 1651). Both cell types were maintained in Eagle's minimum essential medium with Earle's salts (EMEM) containing 10% fetal bovine serum (FBS), 10 mM HEPES, pH 7.4, and antibiotics (penicillin [100 U/ml], streptomycin [100 μg/ml], and gentamicin sulfate [50 μg/ml]) at 37°C in a 5% CO2 incubator.
Construction of hantavirus M gene vaccine plasmids.
Construction of the HTNV M gene DNA vaccine plasmid pWRG/HTN-M(x) was described previously (15). pWRG/PUU-M(x22) was constructed by reverse transcription of viral RNA, followed by PCR amplification of cDNA, and standard cloning techniques. RNA was removed by digestion with RNase H at 37°C for 20 min. Forward and reverse primers were based on the published primers used to clone the Hantaan virus M gene into pWRG/HTN-M(x) and on PUUV sequences, respectively. The forward primer was HTNMX (5′-GGCCGCGGCCGCGGATCTGCAGGAATTCGGCACGAGAGTAGTAGACTCCGCAAGAAACAGCA), and the reverse primer was PUUM-R (5′-GCGCGGATCCTAGTAGTATGCTCCGCAGGAAC). The forward primer included a NotI restriction site (underlined) and 24 nucleotides upstream of the M genome segment noncoding region that are important for expressing G1 in pWRG/HTN-M(x) (15). The reverse primer included a BamHI restriction site (underlined). cDNA was purified by use of a PCR purification kit (Qiagen) and used as a template in a PCR. Primers HTNMX and PUUM-R were included in the PCR mix, which also included Platinum Taq High Fidelity DNA polymerase (Invitrogen); the PCR conditions were one 3-min cycle at 94°C followed by 30 cycles of 94°C for 30 s and 68°C for 8 min. The PCR product was cut with NotI and BamHI and then ligated into NotI-BglII-cut pWRG7077 vector to produce pWRG/PUU-M(x22). Note that the BamHI and BglII sites are compatible for ligation, but the resulting sequence cannot be digested by either enzyme.
A plasmid expressing PUUV Gn alone (pWRG/PUU-Gn) was made using pWRG/PUU-M(x22) as the template. The Gn sequence was amplified by PCR and subcloned into the NotI and BglII sites of pWRG7077. The forward primer was HTNMX (see above), and the reverse primer was PUUG1R (5′-GCGCAGATCTAGCACTGGCAGCCCATACAATTAACTCTAGGACCAATAACACGCACC). The underlined G represents a nucleotide change to knock out the XbaI site. A plasmid expressing PUUV Gc alone (pWRG/PUU-Gc) was also made using pWRG/PUU-M(x22) as the template. The Gc sequence was amplified by PCR and subcloned into the NotI and BamHI sites of pWRG7077. The forward primer was PUUG2F (5′-GGCCGCGGCCGCCACCATGTGGTGCGTGTTATTGGTCCTAGAGTTAATTGTATGGGCTGCC), and the reverse primer was PUUMXNEF (5′-GCGCGGATCCGGGGTAATTTAATTAGTAAAG). The forward primer included a NotI restriction site (underlined), and the single underlined C represents a nucleotide change to knock out the XbaI site.
To produce optimized plasmids, codon-optimized genes were synthesized by GeneArt (26) and subcloned into the NotI-BglII sites of the DNA vaccine vector pWRG7077. The plasmids containing synthetic gene 1 (s1) and synthetic gene 2 (s2) were denoted pWRG/PUU-M(s1) and pWRG/PUU-M(s2), respectively. Note that the sequences from the NotI site to the M gene start codon and from the M gene stop codon to the BamHI site in all three PUUV plasmids [i.e., pWRG/PUU-M(x22), pWRG/PUU-M(s1), and pWRG/PUU-M(s2)] are identical.
PMED vaccinations.
Vaccinations using an XR1 particle-mediated epidermal delivery (PMED) device (gene gun) (Powderject-XR1 delivery device; Powderject Vaccines, Inc.) have been described previously (15, 16). Gene gun cartridges consisting of ∼0.75 μg of plasmid DNA coating 0.5 mg of gold were prepared and stored at 4°C, desiccated, until use. Anesthetized outbred Syrian hamsters were vaccinated with four administrations per vaccination, at nonoverlapping sites on the shaved abdominal epidermis, using 400 lb/in2 of helium pressure. Female macaques were vaccinated with similar cartridges, and the same gene gun conditions were used to vaccinate the hamsters; however, the monkeys received eight administrations (four on the abdomen and four over the inguinal lymph nodes) per vaccination. Hamsters and monkeys were anesthetized during the nonpainful gene gun procedure. The only visible effect was mild erythema at the site of vaccination.
ND10 vaccinations.
Hamsters were anesthetized using isoflurane. Fur was removed from the vaccination site (i.e., abdomen) by use of clippers. Vaccines composed of DNA-coated gold beads were delivered to the ventral abdominal epidermis by use of a handheld disposable gene gun designated an ND10 device. The ND10 device utilizes compressed helium (500 lb/in2) to deliver 1.0 mg of ∼2-μm gold beads (coated with 2 μg of plasmid DNA) through the intact stratum corneum into the underlying ventral abdominal stratum epidermis in a skin target area that is ∼1.5 cm in diameter. A stand allowed the ND10 device to be discharged without applying pressure to the abdomen of the hamster. In the clinic, the ND10 device is pressed on the vaccination site to release a safety mechanism allowing discharge of the helium. In addition, this stand allows ∼4-mm spacing between the barrel of the ND10 device and the skin surface.
Challenge with hantaviruses.
Anesthetized Syrian hamsters were exposed to infectious virus by either intramuscular (i.m.) injection to the caudal thigh or intranasal (i.n.) administration. Hantaviruses administered i.m. (2,000 PFU of PUUV or ANDV) were diluted in sterile phosphate-buffered saline (PBS) (pH 7.4) and administered in a volume of 0.2 ml. Hantaviruses administered i.n. (2,000 PFU of DOBV or 4,000 PFU of ANDV) were administered in a volume of 50 μl, at approximately 25 μl per naris, with a micropipette.
Immunoprecipitation.
Radioimmunoprecipitation assays (RIPAs) using COS cell lysates labeled with Promix ([35S]methionine and [35S]cysteine; Amersham) were performed exactly as described previously (15). The plasmid pWRG/PUU-M(x22) or the empty vector pWRG7077 was transfected into COS cells. Cells were starved for 30 min in methionine- and cysteine-free medium and then radiolabeled for 17 h in medium containing 600 μCi of Promix (Amersham) per T-75 flask. Lysates were made using 4% Zwittergent 3-14 (Calbiochem-Bering) RIPA lysis buffer as described previously (27). Anti-PUUV polyclonal rabbit serum, anti-PUUV Gn monospecific serum, or anti-PUUV Gc serum was used to immunoprecipitate the viral glycoproteins. Reduced samples were run in 4 to 12% Bis-Tris SDS-PAGE gradient gels with morpholinepropanesulfonic acid (MOPS) running buffer (Invitrogen) at 200 V.
N-specific ELISA.
The enzyme-linked immunosorbent assay (ELISA) used to detect N-specific antibodies (N-ELISA) was described previously (15, 16, 28). The antigen consisted of either truncated SEOV N (amino acids 1 to 117) or truncated PUUV N (amino acids 1 to 117), expressed as a histidine-tagged fusion protein in Escherichia coli BL21(DE3) (Novagen, Inc.) by use of the pRSET plasmid (Invitrogen) and purified by affinity chromatography on Ni-nitrilotriacetic acid (Ni-NTA) columns (Qiagen). The endpoint titer was determined as the highest dilution that had an optical density (OD) greater than the mean OD for serum samples from negative-control wells plus 3 standard deviations. The SEOV N antigen was used to detect HTNV and DOBV N-specific antibodies, and the PUUV N antigen was used to detect PUUV and ANDV N-specific antibodies.
PRNT.
Plaque-reduction neutralization tests (PRNT) were performed using Vero E6 cells as previously described (15). HTNV, DOBV, and ANDV plaques were stained with 5% neutral red (Gibco) after 1 week. PUUV plaques were fed with an additional overlay containing Earle's basal minimal essential medium, 10 mM HEPES, 0.6% agarose, 8 mM l-glutamine, 10% FBS, 1× nonessential amino acids, penicillin (100 U/ml), streptomycin (100 μg/ml), and gentamicin (50 μg/ml) on day 7 and then stained with neutral red on day 10. Plates were incubated for 2 to 3 days at 37°C after staining to allow resolution of visible plaques. The 50% PRNT titer (PRNT50 titer) was the highest serum dilution reducing the number of plaques by 50% relative to the average number of plaques in control wells that received medium alone.
Ethics.
This research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to the principles stated in the Guide for the Care and Use of Laboratory Animals (29). The facilities where this research was conducted are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All animal experiments were approved by USAMRIID's Institutional Animal Care and Use Committee.
Statistical analysis.
Protection from challenge with PUUV neutralizing antibodies was determined using logistic regression. Estimated probabilities were calculated using logistic regression by Probit. Comparison of neutralization titers was done using Student's t test. The effect of vaccine on survival outcome was assessed using a logistic regression model. Analyses were conducted using Prism 5 (GraphPad) and SAS, version 8.2 (SAS Institute Inc.).
RESULTS
The first-generation PUUV DNA vaccine plasmid, pWRG/PUU-M(x22), elicits Gn and Gc antibodies in nonhuman primates, but not in hamsters, and does not protect against infection.
We previously demonstrated that SEOV and HTNV M gene-based DNA vaccines failed to cross-protect against PUUV in a hamster infection model (15). Thus, to develop a candidate vaccine to protect against PUUV, we used reverse transcription and PCR to amplify the PUUV M gene from RNA purified from PUUV strain K27. This cDNA was cloned into our DNA vaccine vector, pWRG7077, to produce pWRG/PUU-M(x22). The M gene open reading frame sequence contained no insertions or deletions; however, there were 17 changes in the amino acid sequence compared to the published sequences for strain K27 and other closely related strains (Table 1). pWRG/PUU-M(x22) was evaluated for in vitro expression of Gn and Gc, and both proteins were detected by immunoprecipitation (see Fig. S1A in the supplemental material). pWRG/PUU-M(x22) was tested for immunogenicity in hamsters, and the results indicated that there was no antibody response, even after five vaccinations delivered by gene gun (data not shown). In addition, hamsters were not protected from PUUV infection as determined by ELISA 1 month after a 20,000-PFU intramuscular challenge (∼100 50% infective doses [ID50]) (data not shown). In an earlier study, it was determined that the ANDV M gene-based DNA vaccine failed to elicit a detectable immune response or protection in hamsters but nevertheless elicited high-titer neutralizing antibodies in nonhuman primates (17). Therefore, we tested the pWRG/PUU-M(x22) plasmid for immunogenicity in nonhuman primates. Three cynomolgus macaques were vaccinated with pWRG/PUU-M(x22). One of the three (animal 6064) produced antibodies against the Gn and Gc proteins and weak neutralizing antibodies (PRNT50 titer = 40) (data not shown). In a second NHP experiment, five rhesus macaques were vaccinated with pWRG/PUU-M(x22) five times by use of a gene gun. Four of the five animals developed antibodies against Gn or Gc (see Fig. S1B). Only one animal (animal CH20) produced detectable levels of neutralizing antibodies (PRNT50 titer = 20). These data were encouraging, because it was the first time that any PUUV DNA vaccine had produced antibodies to the PUUV glycoproteins. However, the data indicated that improvements in the quality of the immune response, i.e., the neutralizing antibody response, were needed.
Table 1.
Comparison of differences in deduced M segment open reading frame amino acids for four strains of PUUV and three DNA vaccine constructsf
| Amino acid positiona | PUUV M segment amino acid |
DNA vaccine amino acid |
|||||
|---|---|---|---|---|---|---|---|
| DTK/Ufa-97b | K27c | P360d | Hallnas B1e | PUU-M(x22) | PUU-M(s1) | PUU-M(s2) | |
| 10 | Y | Y | C | Y | Y | Y | Y |
| 38 | V | I | I | I | I | I | I |
| 187 | Q | Q | Q | D | Q | Q | Q |
| 207 | H | H | H | H | Y | Y | H |
| 312 | A | A | A | V | A | A | A |
| 416 | L | S | L | L | L | L | L |
| 448 | M | M | M | M | T | T | M |
| 492 | W | W | W | W | R | R | W |
| 501 | F | L | F | F | F | F | F |
| 604 | T | T | T | R | T | T | T |
| 649 | L | L | L | H | L | L | L |
| 650 | E | E | E | H | E | E | E |
| 675 | G | G | G | R | G | G | G |
| 690 | P | P | P | Q | P | P | P |
| 972 | Q | Q | Q | Q | R | R | Q |
| 1077 | G | G | G | P | G | G | G |
| 1097 | S | S | S | S | L | L | S |
M gene segment open reading frame amino acid position.
DTK/Ufa-97, PUUV strain DTK/Ufa (GenBank accession no. BAF49040).
K27, PUUV strain K27 (GenBank accession no. P41265).
P360, PUUV strain P360 (GenBank accession no. P41266).
Hallnas B1, PUUV strain Hallnas B1 (GenBank accession no. P21400).
Amino acids shown in bold are differences with respect to the PUUM(s2) sequence.
Synthetic, mammalian codon-optimized Puumala virus M gene-based DNA vaccine pWRG/PUU-M(s2) elicits neutralizing antibodies in nonhuman primates.
As noted above, the amino acid sequence of GnGc encoded by pWRG/PUU-M(x22) was not identical to any published PUUV GnGc sequences (Table 1). It was possible that one or more of these amino acid differences could disrupt the conformation of the protein, including the conformations of epitopes bound by neutralizing antibodies. To detect possible deviations in the amino acid sequence, an alignment of several published PUUV M gene ORFs was made. Five possible nonconsensus amino acid residues were detected (Table 1). To test whether optimizing the gene or changing to consensus amino acids could improve the neutralizing antibody response, two genes were synthesized and cloned into pWRG7077. The first synthetic gene (s1) was a mammalian codon-optimized version of the ORF in pWRG/PUU(x22). This new plasmid was called pWRG/PUU-M(s1). The second synthetic gene was identical to that in pWRG/PUU-M(s1); however, the five amino acids that were unique to pWRG/PUU-M(x22) were changed to the consensus amino acids identified in Table 1. The new plasmid containing this gene was called pWRG/PUU-M(s2).
To determine if nucleic acid changes in pWRG/PUU-M(s1) or nucleic acid and amino acid changes in pWRG/PUU-M(s2) could affect the immune response elicited by these DNA vaccines, an experiment was performed in which groups of three rhesus macaques were vaccinated with pWRG/PUU-M(x22), pWRG/PUU-M(s1), or pWRG/PUU-M(s2). Sera collected at the indicated times were tested for the capacity to neutralize PUUV strain K27. PRNT data indicated that one or more of the five amino acid changes made in the synthetic gene in the pWRG/PUU-M(s2) DNA vaccine resulted in a dramatic increase in the neutralizing antibody response (Fig. 1A). Titers were all higher than 100 after the second vaccination, increased 2-fold to 4-fold after the third vaccination, and then declined to just detectable levels by week 18. Upon booster vaccination on week 19, the titers in all animals increased 4- to 64-fold. Neither the pWRG/PUU-M(s1) nor pWRG/PUU-M(x22) vaccine elicited neutralizing antibodies with a titer of >20, indicating that gene optimization alone was insufficient to correct the deficiencies in the original pWRG/PUU-M(x22) plasmid.
Fig 1.
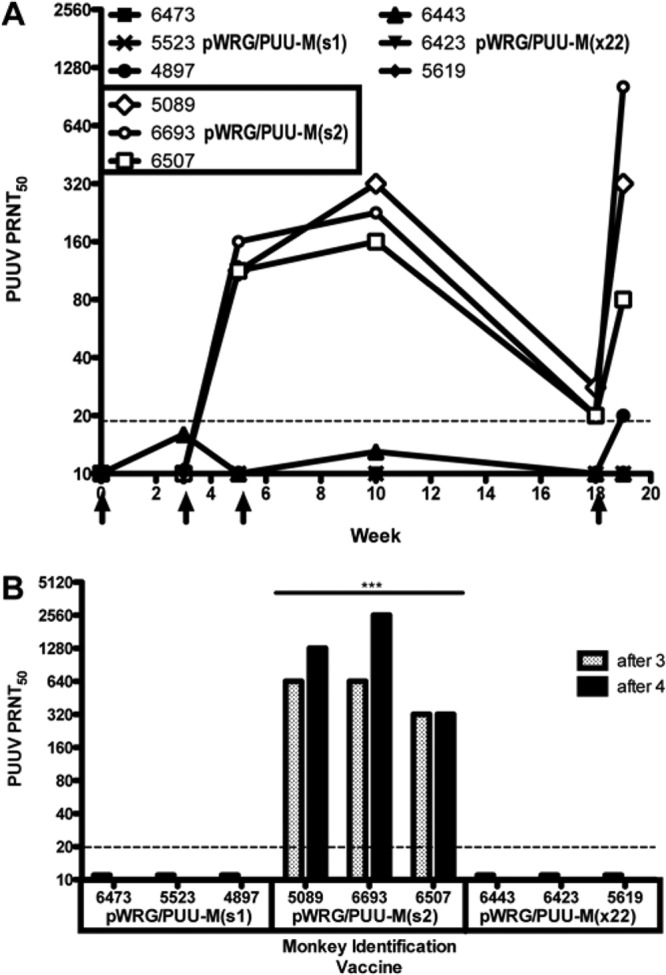
PUUV DNA vaccine testing in nonhuman primates. (A) Neutralization of PUUV strain K27. Groups of three rhesus macaques were vaccinated (indicated by arrows) with either pWRG/PUU-M(s1), pWRG/PUU-M(s2), or pWRG/PUU-M(x22). Sera collected on the indicated weeks were tested for the capacity to neutralize PUUV strain K27. Each symbol represents the GMT for 2 or 3 PRNT50 assays. (B) Cross-neutralization of PUUV strain Sotkamo. Sera collected after 3 (week 5) or 4 (week 19) vaccinations were tested by a PRNT using PUUV strain Sotkamo. The PRNT limit of detection was a titer of 20 (dashed line).
Having determined that pWRG/PUU-M(s2) was capable of eliciting neutralizing antibodies against PUUV strain K27, we tested the sera from week 10 (after 3 vaccinations) or week 18 (after 4 vaccinations) for the capacity to neutralize the distantly related Sotkamo strain of PUUV. The Sotkamo strain of PUUV, isolated in Finland, has ∼63 amino acids that differ from the M segment sequence encoded by the PUUV M gene in pWRG/PUU-M(s2). Nevertheless, antibodies produced in the NHPs vaccinated with pWRG/PUU-M(s2) efficiently neutralized PUUV strain Sotkamo, and the differences in titers were statistically significant [P < 0.0001 for comparing each titer to its pWRG/PUU-M(s1) and pWRG/PUU-M(x22) counterparts, which did not elicit detectable neutralizing antibodies] (Fig. 1B). The PRNT50 titers were similar for both strain K27 and strain Sotkamo. These data suggested that the pWRG/PUU-M(s2) vaccine would cross-protect against divergent PUUV strains found in Northern Europe and Western Russia.
The week 10 and 18 sera were also tested for the capacity to cross-neutralize HTNV, SEOV, and DOBV. There was no cross-neutralizing antibody against SEOV or DOBV, and only one of three NHPs (animal 6693, week 19) exhibited weak (titer of 20) cross-neutralizing activity against HTNV (data not shown).
The pWRG/PUU-M(s2) DNA vaccine delivered using a handheld particle-mediated epidermal delivery device (i.e., ND10 device) elicits antibodies in hamsters.
We tested the pWRG/PUU-M(s2) PUUV DNA vaccine in hamsters by using a research gene gun and found that the vaccine efficiently elicited neutralizing antibodies (19). Thus, the changes in pWRG/PUU-M(s2) overcame the absence of immunity in hamsters observed for vaccination with pWRG/PUU-M(x22). This was an important finding because it allowed us to use a small-animal model for testing vaccine potency and protective efficacy.
Having successfully constructed immunogenic hantavirus M gene-based DNA vaccines against the HFRS hantaviruses, we initiated preclinical testing of these vaccines, delivered using a handheld particle-mediated epidermal delivery device (gene gun) designed for human use, known as an ND10 device. Two lots of ND10 devices loaded with pWRG/PUU-M(s2) and two lots of ND10 devices loaded with pWRG/HTN-M(x), hitherto referred to as the PUUV DNA vaccine and HTNV DNA vaccine, respectively, were manufactured according to good manufacturing practices (GMP). Product stability studies were performed on the four GMP lots of vaccine. In the first study, two lots of vaccine were evaluated after 0, 3, 6, 9, and 12 months of storage at 4°C. As part of the stability studies, the vaccines were tested for potency by measuring their capacity to elicit neutralizing antibodies in hamsters. At each of the aforementioned time points, groups of 8 to 10 Syrian hamsters were vaccinated by use of ND10 devices on a compressed schedule (weeks 0, 2, and 3), which was used instead of a more lengthy schedule to comply with necessary potency assay timelines. Sera collected on vaccination weeks 0 and 4 were tested for neutralizing antibodies by PRNT (Fig. 2A). The HTNV DNA vaccine and PUUV DNA vaccine resulted in 98% (44/45 animals) and 88% (44/50 animals) seroconversion, respectively. Over the course of 1 year, the neutralizing antibodies produced in individual potency test hamsters ranged from <20 to 5,120, and geometric mean titers (GMTs) for the groups at different time points ranged from 302 to 666 for the HTNV DNA vaccine. The neutralizing antibody titers for the PUUV DNA vaccine ranged from <20 to 20,480, and GMTs for groups at different time points ranged from 53 to 408. In the second stability study, two lots of vaccine were evaluated after 0, 3, 6, 9, 12, 18, or 24 months of storage at 4°C (Fig. 2B). The HTNV DNA vaccine and PUUV DNA vaccine resulted in 100% (67/67 animals) and 95% (63/66 animals) seroconversion, respectively. Over the course of 2 years, the neutralizing antibody titers ranged from 40 to 40,960, and GMTs ranged from 226 to 1,631 for the HTNV DNA vaccine. The neutralizing antibody titers for the PUUV DNA vaccine ranged from <20 to 40,960, and GMTs ranged from 113 to 3,883. These data clearly demonstrated that the candidate PUUV DNA vaccine and the HTNV DNA vaccine remained immunogenic for at least 2 years when stored at 4°C.
Fig 2.

In vivo stability testing of HTNV and PUUV DNA vaccines delivered using a handheld single-use gene gun (ND10). (A) Lot 1 of each vaccine was tested for immunogenicity over 1 year. (B) Lot 2 of each vaccine was tested for immunogenicity over 2 years. Symbols represent the neutralizing antibody PRNT50 titers for individual hamsters vaccinated with either pWRG/HTN-M(x) or pWRG/PUU-M(s2). Hamsters vaccinated with the HTNV DNA vaccine were evaluated for HTNV neutralizing antibodies, and hamsters vaccinated with the PUUV DNA vaccine were evaluated for PUUV strain K27 neutralizing antibodies. The PRNT limit of detection was a titer of 20 (dashed line).
PUUV DNA vaccine (delivered via ND10 device) protects hamsters against infection with PUUV.
Having found that the PUUV DNA vaccine elicited neutralizing antibodies, it was important to determine if the vaccine could confer protective immunity. The PUUV DNA vaccine was tested for the capacity to protect against infection with homotypic or heterotypic hantaviruses. In the first two experiments, groups of hamsters were vaccinated with either the PUUV DNA vaccine or the HTNV DNA vaccine and then challenged with PUUV by the i.m. route either 1 month (experiment 1) or 4 months (experiment 2) after vaccination. PUUV does not cause disease in hamsters; however, evidence of infection can be detected by measuring the presence or absence of antibodies to the viral nucleocapsid (N) protein after challenge (30). Pre- and postchallenge sera were run in N-ELISA. All prechallenge serum samples were negative by ELISA (data not shown). In both experiments, 80% and 90% (4 weeks and 10 weeks after vaccination, respectively) of the hamsters did not have detectable anti-N antibodies, indicating that these animals were protected against PUUV infection (Fig. 3A). These results were statistically significant (P < 0.0001 compared to unvaccinated hamsters). In contrast, the hamsters that were not vaccinated or were vaccinated with the HTNV DNA vaccine were not protected (i.e., anti-N antibodies were detected in all of the hamsters). The latter data confirmed our earlier finding that the HTNV DNA vaccine does not protect against PUUV infection (15).
Fig 3.
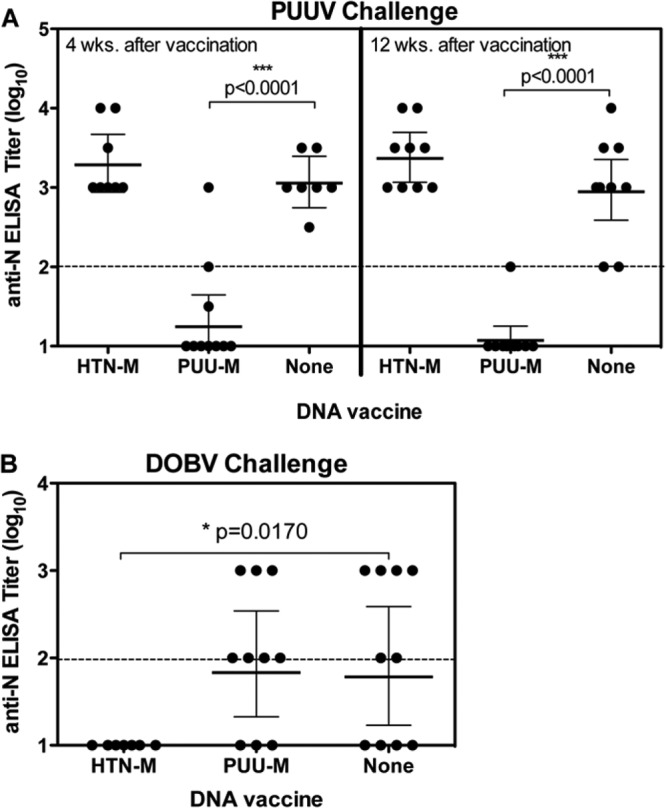
Evaluation of protection (PUUV) and cross-protection (DOBV) in hamster infection models. Groups of 6 to 8 hamsters were vaccinated (week 0, 2, or 3) with PUUV DNA vaccine, HTNV DNA vaccine, or no vaccine and then challenged with PUUV strain K27 or DOBV. (A) Hamsters were challenged either 4 weeks or 12 weeks after the last vaccination, using an intramuscular challenge of 2,000 PFU PUUV. (B) Hamsters were challenged 11 weeks after the last vaccination, using an intranasal challenge of 2,000 PFU DOBV. Sera were collected at 35 days postchallenge, and N-ELISA log10 titers were plotted. The N-ELISA limit of detection was a titer of 2 log10 (dashed line).
We next determined if the PUUV neutralizing antibody titer could predict protective immunity against PUUV. PRNT and ELISA titers for sera collected pre- and postchallenge for experiments 1 and 2 in Fig. 3A were evaluated to determine the relationship between evidence of a vaccine “take” (i.e., positive PRNT titer after vaccination) and protection (i.e., negative ELISA titer after challenge). In general, the presence of PUUV neutralizing antibodies in the prechallenge sera was predictive of protective immunity (Table 2). There were two exceptions that are discussed further in Discussion. Results of logistic regression indicated that the log10 titer was significantly associated with the protection outcome. Each unit log10 increase in the prechallenge PUUV PRNT50 titer was associated with an increase in the odds of protection against PUUV infection of 7.8 times (P < 0.0001).
Table 2.
PUUV neutralizing antibodies and protection against PUUV infection
| Vaccinea | Hamster ID | Prechallenge PUUV PRNT50 titerd | Postchallenge N-ELISA titer (log10)b,d | Protectionc |
|---|---|---|---|---|
| pWRG/PUU-M(s2) | 269 | 5,120 | 3 | N |
| 262 | 2,560 | 1 | Y | |
| 261, 263 | 1,280 | 1 | Y | |
| 243 | 640 | 1 | Y | |
| 248 | 320 | 2 | N | |
| 266 | 320 | 1 | Y | |
| 245, 264, 267 | 160 | 1 | Y | |
| 242, 244, 249, 270 | 80 | 1 | Y | |
| 241 | 40 | 1 | Y | |
| 246, 250, 268 | 20 | 1 | Y | |
| 265 | 1 | 2 | N | |
| None | 298 | 40 | 3 | N |
| 281, 284 | 20 | 3 | N | |
| 282 | 20 | 2 | N | |
| 283 | 1 | 2 | N | |
| 285, 286 | 1 | 3 | N | |
| 287 | 1 | 4 | N | |
| 288, 290, 978, 980 | 1 | 3 | N | |
| 291 | 1 | 2 | N | |
| 292, 293–295, 297 | 1 | 3 | N | |
| pWRG/HTN-M(x) | 251, 252, 254, 258–260, 232, 236, 239, 240 | 1 | 3 | N |
| 253, 256, 231, 237 | 1 | 4 | N |
Hamsters were vaccinated on weeks 0, 2, and 3 by use of ND10 delivery devices.
Log10 endpoint titers represent GMTs for two N-ELISAs.
A postchallenge N-ELISA titer of <2 indicates that the hamster was protected. N, not protected; Y, protected.
A value of 1 indicates that the titer was below the limit of detection. The limit of detection for the PRNT was 20, and the limit of detection for the ELISA was 2 (log10).
PUUV DNA vaccine (delivered via ND10 device) does not cross-protect hamsters against infection with DOBV.
We next determined if the PUUV DNA vaccine could cross-protect against infection with DOBV. To test this, groups of hamsters were vaccinated with the PUUV DNA vaccine as described above. A control group of hamsters was vaccinated with the HTNV DNA vaccine, which had previously been shown to protect hamsters against DOBV (15). Eleven weeks after the last vaccination, hamsters were challenged with DOBV by the i.n. route. The i.n. route of exposure was tested to gain additional information on the capacity of hantavirus DNA vaccines to confer protection against a mucosal challenge. Like PUUV, DOBV does not cause disease in hamsters but readily infects hamsters by the i.m. or i.n. route (15; unpublished data). Sera collected 35 days after i.n. DOBV challenge were evaluated by N-ELISA (Fig. 3B). The PUUV DNA vaccine did not protect against DOBV infection. In contrast, the HTNV DNA vaccine did protect against an i.n. challenge with DOBV (P = 0.0170). In this experiment, there were no DOBV cross-neutralizing antibodies detected in hamsters vaccinated with the PUUV or HTNV DNA vaccine (data not shown). Thus, the HTNV M gene-based DNA vaccine, but not the PUUV DNA vaccine, was protective against a mucosal challenge with DOBV, even in the absence of detectable levels of cross-neutralizing antibodies.
PUUV DNA vaccine cross-protects hamsters against a lethal challenge with ANDV.
The PUUV and DOBV infection models described above require sterilizing immunity (i.e., no evidence of an antibody response to the challenge virus nucleocapsid protein) for a positive readout. It is possible that a vaccine that fails to protect against infection might still be capable of preventing disease. There are no practical HFRS disease models for testing candidate hantavirus vaccines; however, there is a disease model for hantavirus pulmonary syndrome (HPS). When Syrian hamsters are exposed to ANDV, they develop a lethal disease that closely resembles HPS in humans (14, 23, 31–36). Recombinant adenovirus and vesicular stomatitis virus vectors encoding ANDV N or glycoproteins have been shown to protect against lethal HPS in the hamster model (34, 37). In an earlier study, we found that the HTNV M gene-based DNA vaccine conferred partial but not statistically significant protection against an i.m. challenge with ANDV (14). Nevertheless, we hypothesized that the PUUV DNA vaccine might be capable of protecting against lethal disease caused by ANDV, because PUUV and ANDV Gn and Gc are more closely related on the amino acid level than the HTNV and ANDV proteins (66.5% versus 54.9% identity). To test this, we first performed two experiments where groups of hamsters were vaccinated with either the PUUV DNA vaccine or the HTNV DNA vaccine or not vaccinated and then challenged (either 14 or 20 weeks later) with 4,000 PFU of ANDV by the i.n. route. The PUUV DNA vaccine, but not the HTNV DNA vaccine, conferred a significant level of protection against lethal HPS in both experiments (Fig. 4A and B), with P values of 0.0428 (14 weeks) and <0.0001 (20 weeks).
Fig 4.
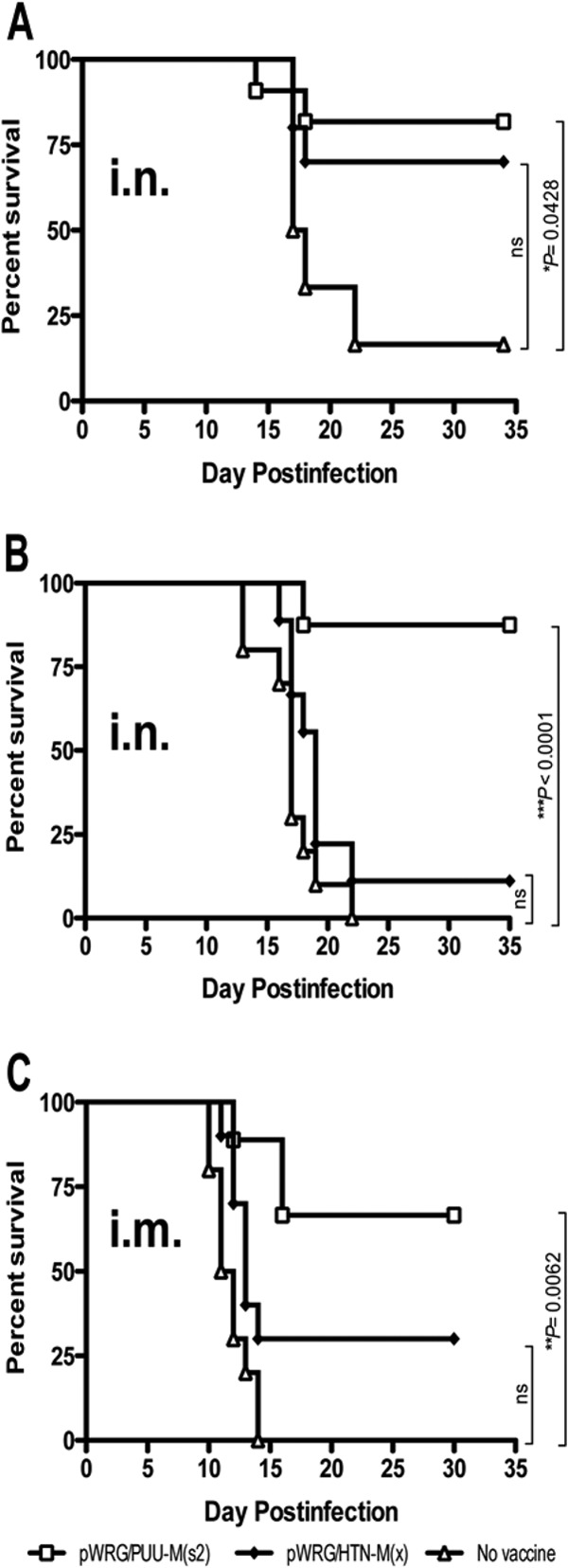
PUUV DNA vaccine protects against lethal hantavirus disease. Groups of 8 to 10 hamsters were vaccinated with the indicated vaccine or not vaccinated. Sixteen weeks (A) or 20 weeks (B) elapsed before an intranasal challenge of 4,000 PFU ANDV. (C) Groups of 9 or 10 hamsters were vaccinated with the indicated vaccine or not vaccinated before an intramuscular challenge of 2,000 PFU ANDV. Survival of all animals was monitored for 35 days postchallenge. ns, not significant.
Another experiment was conducted using the more rigorous i.m. route of exposure (the 50% lethal dose [LD50] by the i.n. route is 95 PFU, and the LD50 by the i.m. route is 8 PFU) (23, 33). Hamsters were vaccinated with the PUUV DNA, HTNV DNA, or no vaccine as described above and then challenged (7 weeks later) by the i.m. route with ANDV. The PUUV DNA vaccine elicited a significant level of protection against intramuscular challenge (Fig. 4C), with a P value of 0.0062. As reported previously, the HTNV DNA vaccine elicited partial but not significant protection. Interestingly, the PUUV DNA vaccine elicited only low-level (PRNT50 titer = 20) ANDV cross-neutralizing antibodies in prechallenge sera of half of the hamsters (data not shown). Thus, we concluded that neutralizing antibodies produced by the DNA vaccines, while sufficient to protect, are not necessary to confer cross-protection against lethal hantavirus disease.
DISCUSSION
Early attempts to clone full-length hantavirus M segments into expression plasmids resulted in clones with nonsense mutations in the ORFs that typically resulted in truncated Gn or Gc proteins, or other disruptive mutations. These included single nucleotide deletions/substitutions and large insertions (i.e., integration of insertional elements such as IS5). It is possible that the M gene nucleotide sequence is somehow toxic to E. coli and that this toxicity results in negative selection against full-length clones with the correct sequence. When we finally succeeded in cloning a full-length PUUV M gene with an intact ORF [plasmid pWRG/PUU-M(x22)], there were still a number of nucleotide changes that resulted in unique amino acid changes relative to published sequences. At least one of these changes presumably altered the Gn-Gc structure such that the pWRG/PUU-M(x22) DNA vaccine was only capable of eliciting nonneutralizing or low-titer neutralizing antibodies. By employing synthetic biology, it was possible not only to correct the amino acid changes but also to alter the entire ORF such that the sequence was no longer toxic in E. coli (via codon optimization). These changes resulted in a DNA vaccine, pWRG/PUU-M(s2), that was readily propagated in E. coli (data not shown) and capable of eliciting high-titer neutralizing antibodies. This was the first time that any molecular PUUV vaccine had elicited high-titer neutralizing antibodies in any species. Future research will employ the same strategy to develop candidate M gene-based DNA vaccines against other hantaviruses. This is a powerful approach because all that is required is the full-length M gene ORF sequence information.
Using synthetic biology, it would be possible to produce DNA vaccine plasmids against all of the known pathogenic strains of hantaviruses; however, from a vaccine development perspective, this would be expensive, impractical, and unnecessary. We and others have demonstrated that neutralizing antibodies are sufficient to protect against infection and/or disease (14, 15, 31, 37, 38). Thus, the production of cross-neutralizing antibodies is a reasonable predictor that the vaccine will be cross-protective. For example, pWRG/PUU-M(s2) is based on PUUV strain K27 from Western Russia, and we demonstrated that the neutralizing antibodies produced by this vaccine readily cross-neutralize PUUV strain Sotkamo, from Scandinavia. There is approximately 94.3% identity in the M segment amino acid sequences between these viruses. From this, we predict that pWRG/PUU-M(s2) will be protective against strains of PUUV found in Russia and Scandinavia. Additional cross-neutralization experiments utilizing other PUUV strains will be needed to determine the breadth of cross-reactivity.
To start defining the level of neutralizing antibodies that predicts protective immunity, we performed a statistical analysis comparing prechallenge neutralizing antibody titers with protection data. A PRNT50 titer of >22 gave an estimated probability of protective immunity of 60%, and a titer of >119 gave an estimated probability of protective immunity of 90%, with 95% certainty (Table 3). Thus, neutralizing antibodies produced by vaccination with hantavirus vaccines can be used to predict the probability that the vaccination will be protective. There were individual animals that were exceptions. For example, the hamster (animal 269) with the highest PUUV PRNT titer (5,120) was nevertheless infected with PUUV as measured by ELISA (Table 2). It is possible that the PRNT or ELISA results for animal 269 were produced in error (e.g., by tube mislabeling), or it is possible that an animal with a very potent neutralizing antibody response can still be infected. Because we cannot rule out the possibility of a technical error, and because the number of exceptions is low, we are basing our conclusions on the aggregate data set shown in Table 2 and the statistical analysis shown in Table 3. In that analysis, the presence of neutralizing antibodies is predictive of protection. As more neutralizing antibody/protection data are compiled, the precise relationship between neutralizing antibodies and protection will become evident, and a better estimate of what constitutes a protective titer will be generated. In addition, passive transfer experiments will be used to determine what neutralizing antibody titer, in the absence of a cell-mediated immune response, is sufficient to confer protection.
Table 3.
Calculation of estimated probabilities of protective immunity against PUUV infection based on the protection data from Table 2
| Estimated probability of protective immunitya | Prechallenge PUUV PRNT50 titer | Lower 95% fiducial limit | Upper 95% fiducial limit |
|---|---|---|---|
| 0.40 | 24 | 8 | 77 |
| 0.50 | 37 | 13 | 148 |
| 0.60 | 59 | 22 | 305 |
| 0.70 | 97 | 35 | 713 |
| 0.80 | 177 | 58 | 2,122 |
| 0.90 | 438 | 119 | 11,700 |
Determined by logistic regression by Probit. Protection status by titer was used to calculate estimated probabilities of protective immunity.
The HTNV DNA vaccine did not elicit DOBV cross-neutralizing antibodies in the ND10 experiment shown in Fig. 1, but nevertheless, it protected hamsters against DOBV infection. It is possible that low levels of neutralizing or otherwise protective antibodies were present but not detected by classical PRNT. In support of this, previous reports have demonstrated that the HTNV DNA vaccine delivered by gene gun can elicit anti-DOBV cross-neutralizing antibodies in hamsters and nonhuman primates (15). It is possible that the levels elicited by the ND10 experiment were below detection but nevertheless capable of preventing infection. Another possibility is that a cellular response (e.g., the CD8+ T cell response) elicited by the HTNV DNA vaccine, but not the PUUV DNA vaccine, is sufficient to protect hamsters from DOBV infection. Others have shown that recombinant adenovirus vectors containing ANDV Gn or Gc are capable of protecting hamsters against lethal HPS in the absence of neutralizing antibodies (34). Presently, we do not have assays to measure specific hamster T cell responses to hantavirus vaccines.
The PUUV DNA vaccine did not elicit ANDV cross-neutralizing antibodies, but it nevertheless protected hamsters against lethal infection with ANDV. This finding demonstrates that although neutralizing antibodies are sufficient to confer protection, they are not necessary. In an earlier study, the HTNV DNA vaccine partially protected hamsters from lethal HPS, but these results did not reach statistical significance (14). The fact that PUUV-vaccinated hamsters were protected from lethal HPS regardless of the route of ANDV challenge (either i.n. or i.m.) demonstrates that an HFRS vaccine might have broader protective efficacy than that predicted by cross-neutralization. If the mechanism of cross-protection in the absence of neutralizing antibodies is mediated by CD8+ T cells, then our results indicate that PUUV Gn and Gc, but not HTNV Gn and Gc, share a sufficient protective epitope(s) with ANDV Gn and Gc to confer protection. This corresponds to the overall amino acid identity of the glycoproteins: PUUV Gn and Gc are 66.5% identical to ANDV Gn and Gc, whereas HTNV Gn and Gc are only 54.9% identical to ANDV Gn and Gc. It is possible that the PUUV DNA vaccine cross-protects against disease caused by HTNV (the Gn-Gc identity of these viruses is 55.0%). At this time, an animal model of HFRS disease suitable for vaccine testing does not exist. Suckling mice injected intracerebrally with HTNV develop a lethal neurological disease (39); however, suckling mice are not amenable to active vaccine studies. Similarly, PUUV has been shown to cause some signs of disease in macaques; however, the observed tissue pathology and abnormalities in blood hematology and chemistry are not consistent in all exposed animals (40–43). Thus, large numbers of nonhuman primates would be needed to determine if a medical countermeasure was effective at preventing disease. A caveat to this would be if the medical countermeasure were effective at preventing infection. For such medical countermeasures, it would be possible to use either rodent (e.g., hamster) or nonhuman primate models of infection (40–42, 44, 45).
As mentioned above, the HTNV and PUUV DNA vaccines delivered by the ND10 gene gun were recently tested in a phase 1 clinical trial (18). In that trial, both hantavirus DNAs were shown to be safe and immunogenic (i.e., elicited neutralizing antibodies) in some but not all vaccines. Here we have demonstrated that these vaccines delivered by the ND10 device can protect hamsters not only against infection with PUUV and DOBV but also against lethal disease caused by ANDV. This is the first time that active vaccination with any DNA vaccine has been shown to protect against lethal hantavirus disease in an animal model. Future clinical trials will involve testing the immunogenicity of these vaccines delivered by alternative technologies, such as muscle and skin electroporation or needle-free jet injection. Although we know that protection can occur in the absence of neutralizing antibodies, a path to licensure of these vaccines will likely involve the use of neutralizing antibodies produced in vaccinated humans as surrogate markers of protective efficacy. Still, our finding that the PUUV DNA vaccine can provide protection in an animal model of lethal hantavirus disease (HPS caused by ANDV) might allow an alternative pathway to licensure involving the animal rule (rule 21 CFR 601.90 for biological products).
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Thompson and D. M. Custer for expert technical assistance in the cloning and characterization of the full-length PUUV M gene and D. Fisher for performing statistical analyses. We also thank F. Elgh for providing the pSEOSXdelta and pPUUSXdelta plasmids used to produce ELISA antigen and O. Vapalahti for providing anti-Gn and anti-Gc rabbit serum pools used in RIPA.
This work was funded by the Military Infectious Disease Research Program (MIDRP), Program Area T. R.L.B. and V.W.-J. performed this work as participants in the National Research Council Associate Program. V.W.-J. also performed part of this work as an employee of Tunnell Consulting, Inc., a subcontractor to Battelle Memorial Institute under its prime contract with NIAID, under contract HHSN272200200016I.
Opinions, interpretations, conclusions, and recommendations are ours and are not necessarily endorsed by the U.S. Army or the Department of Defense. The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services or of the institutions and companies affiliated with the authors.
Footnotes
Published ahead of print 12 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00546-12.
REFERENCES
- 1. Clement J, Maes P, Barrios M, Verstraeten WW, Amirpour Haredasht S, Docoffre G, Aerts J-M, Van Ranst M. 2011. Global warming and epidemic trends of an emerging viral disease in western-Europe: the nephropathia epidemica case, p 39–52 In Casalegno S. (ed), Global warming impacts—case studies on the economy, human health, and on urban and natural environments. InTech, Rijeka, Croatia [Google Scholar]
- 2. Lundkvist A, Niklasson B. 1992. Bank vole monoclonal antibodies against Puumala virus envelope glycoproteins: identification of epitopes involved in neutralization. Arch. Virol. 126:93–105 [DOI] [PubMed] [Google Scholar]
- 3. Schmaljohn C, Nichol S. 2006. Bunyaviridae, p 1741–1789 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4. Kallio ER, Klingstrom J, Gustafsson E, Manni T, Vaheri A, Henttonen H, Vapalahti O, Lundkvist A. 2006. Prolonged survival of Puumala hantavirus outside the host: evidence for indirect transmission via the environment. J. Gen. Virol. 87:2127–2134 [DOI] [PubMed] [Google Scholar]
- 5. Vapalahti O, Mustonen J, Lundkvist A, Henttonen H, Plyusnin A, Vaheri A. 2003. Hantavirus infections in Europe. Lancet Infect. Dis. 3:653–661 [DOI] [PubMed] [Google Scholar]
- 6. Schmaljohn C. 2009. Vaccines for hantaviruses. Vaccine 27(Suppl 4):D61–D64 [DOI] [PubMed] [Google Scholar]
- 7. Mustonen J, Brummer-Korvenkontio M, Hedman K, Pasternack A, Pietila K, Vaheri A. 1994. Nephropathia epidemica in Finland: a retrospective study of 126 cases. Scand. J. Infect. Dis. 26:7–13 [DOI] [PubMed] [Google Scholar]
- 8. Lee HW, Chu YK, Woo YO, An CN, Kim H, Tkachenko EA, Gligic A. 1999. Vaccines against hemorrhagic fever with renal syndrome, p 267–272 In Saluzzo JF, Dodet B. (ed), Factors in the emergence and control of rodent-borne viral diseases (hantaviral and arenal diseases). Elsevier, Paris, France [Google Scholar]
- 9. Ulrich R, Koletzki D, Lachmann S, Lundkvist A, Zankl A, Kazaks A, Kurth A, Gelderblom HR, Borisova G, Meisel H, Kruger DH. 1999. New chimaeric hepatitis B virus core particles carrying hantavirus (serotype Puumala) epitopes: immunogenicity and protection against virus challenge. J. Biotechnol. 73:141–153 [DOI] [PubMed] [Google Scholar]
- 10. Dargeviciute A, Brus Sjolander K, Sasnauskas K, Kruger DH, Meisel H, Ulrich R, Lundkvist A. 2002. Yeast-expressed Puumala hantavirus nucleocapsid protein induces protection in a bank vole model. Vaccine 20:3523–3531 [DOI] [PubMed] [Google Scholar]
- 11. de Carvalho Nicacio C, Gonzalez Della Valle M, Padula P, Bjorling E, Plyusnin A, Lundkvist A. 2002. Cross-protection against challenge with Puumala virus after immunization with nucleocapsid proteins from different hantaviruses. J. Virol. 76:6669–6677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kallio-Kokko H, Leveelahti R, Brummer-Korvenkontio M, Lundkvist A, Vaheri A, Vapalahti O. 2001. Human immune response to Puumala virus glycoproteins and nucleocapsid protein expressed in mammalian cells. J. Med. Virol. 65:605–613 [PubMed] [Google Scholar]
- 13. Ulrich R, Lundkvist A, Meisel H, Koletzki D, Sjolander KB, Gelderblom HR, Borisova G, Schnitzler P, Darai G, Kruger DH. 1998. Chimaeric HBV core particles carrying a defined segment of Puumala hantavirus nucleocapsid protein evoke protective immunity in an animal model. Vaccine 16:272–280 [DOI] [PubMed] [Google Scholar]
- 14. Custer DM, Thompson E, Schmaljohn CS, Ksiazek TG, Hooper JW. 2003. Active and passive vaccination against hantavirus pulmonary syndrome with Andes virus M genome segment-based DNA vaccine. J. Virol. 77:9894–9905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hooper JW, Custer DM, Thompson E, Schmaljohn CS. 2001. DNA vaccination with the Hantaan virus M gene protects hamsters against three of four HFRS hantaviruses and elicits a high-titer neutralizing antibody response in rhesus monkeys. J. Virol. 75:8469–8477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hooper JW, Kamrud KI, Elgh F, Custer D, Schmaljohn CS. 1999. DNA vaccination with hantavirus M segment elicits neutralizing antibodies and protects against Seoul virus infection. Virology 255:269–278 [DOI] [PubMed] [Google Scholar]
- 17. Hooper JW, Custer DM, Smith J, Wahl-Jensen V. 2006. Hantaan/Andes virus DNA vaccine elicits a broadly cross-reactive neutralizing antibody response in nonhuman primates. Virology 347:208–216 [DOI] [PubMed] [Google Scholar]
- 18. Boudreau EF, Josleyn M, Ullman D, Fisher D, Dalrymple L, Sellers-Myers K, Loudon P, Rusnak J, Rivard R, Schmaljohn C, Hooper JW. 2012. A phase 1 clinical trial of Hantaan virus and Puumala virus M-segment DNA vaccines for hemorrhagic fever with renal syndrome. Vaccine 30:1951–1958 [DOI] [PubMed] [Google Scholar]
- 19. Spik KW, Badger C, Mathiessen I, Tjelle T, Hooper JW, Schmaljohn C. 2008. Mixing of M segment DNA vaccines to Hantaan virus and Puumala virus reduces their immunogenicity in hamsters. Vaccine 26:5177–5181 [DOI] [PubMed] [Google Scholar]
- 20. Xiao SY, Spik KW, Li D, Schmaljohn CS. 1993. Nucleotide and deduced amino acid sequences of the M and S genome segments of two Puumala virus isolates from Russia. Virus Res. 30:97–103 [DOI] [PubMed] [Google Scholar]
- 21. Lee PW, Gibbs CJ, Jr, Gajdusek DC, Yanagihara R. 1985. Serotypic classification of hantaviruses by indirect immunofluorescent antibody and plaque reduction neutralization tests. J. Clin. Microbiol. 22:940–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee HW, Lee PW, Johnson KM. 1978. Isolation of the etiologic agent of Korean hemorrhagic fever. J. Infect. Dis. 137:298–308 [DOI] [PubMed] [Google Scholar]
- 23. Hooper JW, Larsen T, Custer DM, Schmaljohn CS. 2001. A lethal disease model for hantavirus pulmonary syndrome. Virology 289:6–14 [DOI] [PubMed] [Google Scholar]
- 24. Kitamura T, Morita C, Komatsu T, Sugiyama K, Arikawa J, Shiga S, Takeda H, Akao Y, Imaizumi K, Oya A, Hashimoto N, Urasawa S. 1983. Isolation of virus causing hemorrhagic fever with renal syndrome (HFRS) through a cell culture system. Jpn. J. Med. Sci. Biol. 36:17–25 [DOI] [PubMed] [Google Scholar]
- 25. Avsic-Zupanc T, Xiao SY, Stojanovic R, Gligic A, van der Groen G, LeDuc JW. 1992. Characterization of Dobrava virus: a hantavirus from Slovenia, Yugoslavia. J. Med. Virol. 38:132–137 [DOI] [PubMed] [Google Scholar]
- 26. Fath S, Bauer AP, Liss M, Spriestersbach A, Maertens B, Hahn P, Ludwig C, Schafer F, Graf M, Wagner R. 2011. Multiparameter RNA and codon optimization: a standardized tool to assess and enhance autologous mammalian gene expression. PLoS One 6:e17596 doi: 10.1371/journal.pone.0017596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmaljohn C, Vanderzanden L, Bray M, Custer D, Meyer B, Li D, Rossi C, Fuller D, Fuller J, Haynes J, Huggins J. 1997. Naked DNA vaccines expressing the prM and E genes of Russian spring summer encephalitis virus and Central European encephalitis virus protect mice from homologous and heterologous challenge. J. Virol. 71:9563–9569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elgh F, Lundkvist A, Alexeyev OA, Stenlund H, Avsic-Zupanc T, Hjelle B, Lee HW, Smith KJ, Vainionpaa R, Wiger D, Wadell G, Juto P. 1997. Serological diagnosis of hantavirus infections by an enzyme-linked immunosorbent assay based on detection of immunoglobulin G and M responses to recombinant nucleocapsid proteins of five viral serotypes. J. Clin. Microbiol. 35:1122–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.National Research Council 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC [Google Scholar]
- 30. Sjolander KB, Elgh F, Kallio-Kokko H, Vapalahti O, Hagglund M, Palmcrantz V, Juto P, Vaheri A, Niklasson B, Lundkvist A. 1997. Evaluation of serological methods for diagnosis of Puumala hantavirus infection (nephropathia epidemica). J. Clin. Microbiol. 35:3264–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brocato R, Josleyn M, Ballantyne J, Vial P, Hooper JW. 2012. DNA vaccine-generated duck polyclonal antibodies as a postexposure prophylactic to prevent hantavirus pulmonary syndrome (HPS). PLoS One 7:e35996 doi: 10.1371/journal.pone.0035996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hammerbeck CD, Hooper JW. 2011. T cells are not required for pathogenesis in the Syrian hamster model of hantavirus pulmonary syndrome. J. Virol. 85:9929–9944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hooper JW, Ferro AM, Wahl-Jensen V. 2008. Immune serum produced by DNA vaccination protects hamsters against lethal respiratory challenge with Andes virus. J. Virol. 82:1332–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Safronetz D, Hegde NR, Ebihara H, Denton M, Kobinger GP, St Jeor S, Feldmann H, Johnson DC. 2009. Adenovirus vectors expressing hantavirus proteins protect hamsters against lethal challenge with Andes virus. J. Virol. 83:7285–7295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wahl-Jensen V, Chapman J, Asher L, Fisher R, Zimmerman M, Larsen T, Hooper JW. 2007. Temporal analysis of Andes virus and Sin Nombre virus infections of Syrian hamsters. J. Virol. 81:7449–7462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Safronetz D, Zivcec M, Lacasse R, Feldmann F, Rosenke R, Long D, Haddock E, Brining D, Gardner D, Feldmann H, Ebihara H. 2011. Pathogenesis and host response in Syrian hamsters following intranasal infection with Andes virus. PLoS Pathog. 7:e1002426 doi: 10.1371/journal.ppat.1002426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brown KS, Safronetz D, Marzi A, Ebihara H, Feldmann H. 2011. Vesicular stomatitis virus-based vaccine protects hamsters against lethal challenge with Andes virus. J. Virol. 85:12781–12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuan ZG, Luo SJ, Xu HJ, Wang XH, Li J, Yuan LG, He LT, Zhang XX. 2010. Generation of E3-deleted canine adenovirus type 2 expressing the Gc glycoprotein of Seoul virus by gene insertion or deletion of related terminal region sequences. J. Gen. Virol. 91:1764–1771 [DOI] [PubMed] [Google Scholar]
- 39. Tsai TF, Bauer S, McCormick JB, Kurata T. 1982. Intracerebral inoculation of suckling mice with Hantaan virus. Lancet ii:503–504 [DOI] [PubMed] [Google Scholar]
- 40. Groen J, Gerding M, Koeman JP, Roholl PJ, van Amerongen G, Jordans HG, Niesters HG, Osterhaus AD. 1995. A macaque model for hantavirus infection. J. Infect. Dis. 172:38–44 [DOI] [PubMed] [Google Scholar]
- 41. Klingstrom J, Plyusnin A, Vaheri A, Lundkvist A. 2002. Wild-type Puumala hantavirus infection induces cytokines, C-reactive protein, creatinine, and nitric oxide in cynomolgus macaques. J. Virol. 76:444–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Klingstrom J, Stoltz M, Hardestam J, Ahlm C, Lundkvist A. 2008. Passive immunization protects cynomolgus macaques against Puumala hantavirus challenge. Antivir. Ther. 13:125–133 [PubMed] [Google Scholar]
- 43. Sironen T, Klingstrom J, Vaheri A, Andersson LC, Lundkvist A, Plyusnin A. 2008. Pathology of Puumala hantavirus infection in macaques. PLoS One 3:e3035 doi: 10.1371/journal.pone.0003035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McElroy AK, Bray M, Reed DS, Schmaljohn CS. 2002. Andes virus infection of cynomolgus macaques. J. Infect. Dis. 186:1706–1712 [DOI] [PubMed] [Google Scholar]
- 45. Yanagihara R, Amyx HL, Lee PW, Asher DM, Gibbs CJ, Jr, Gajdusek DC. 1988. Experimental hantavirus infection in nonhuman primates. Arch. Virol. 101:125–130 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.