

Abstract
During infection, the host response develops effector mechanisms to combat the parasite. However, this response can become uncontrolled or regulated by mechanisms that modulate the inflammatory reaction. The number of parasites that infects the host, such as trypomastigotes in Chagas disease, may also influence immune activation and disease pathology. We evaluated the inflammation and immune regulation that follows Trypanosoma cruzi infection with low (300), intermediate (3000) or high (30 000) parasite loads. Our results showed that the load of parasite inoculum influenced disease outcome: the higher the number of parasites in the inoculum, the lower were the survival rates. There was a strong association between parasitism and inflammatory infiltrate in the heart and the parasite inoculum determined cytokine interplay in this tissue, as shown by increased interferon-γ, tumour necrosis factor-α, interleukin-17 (IL-17) and IL-23 in the 300 and 30 000 inoculum groups, higher IL-4 and IL-10 in the intermediate-inoculum mice, and elevated IL-6 production in the heart of mice in the 3000 and 30 000 groups. The number of T cells and antigen-presenting cells was augmented in the infected groups, especially for the splenic CD4+ CD25+ regulatory T cells expressing CD45RBlow, GITR, PD-1 and FoxP3 in the group with the highest inoculum. Interestingly, these mice also presented an apparent decrease in CD4+ CD25+ FoxP3+ cells in the cardiac infiltrate, in contrast to the intermediate inoculum group, which showed elevated numbers of these regulatory leucocytes in the heart. Finally, our results demonstrated that parasite load during T. cruzi infection is linked to the response pattern that will result in parasite/inflammation control or tissue damage.
Keywords: immune response, inflammation, parasite inocula, regulatory T cells, Trypanosoma cruzi
Introduction
During an infection, the effector host response aimed primarily at parasite elimination may lead to uncontrolled inflammation, which can later be modulated by regulatory mechanisms to avoid tissue damage.1 On the other hand, pathogens have evolved diverse mechanisms to survive for a prolonged period and produce persistent or latent infection, mainly by interacting with these regulatory mechanisms of the host immune response.2 In this way, parasites may directly induce the production of regulatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) or indirectly act through the generation of regulatory T (Treg) cells in experimental infections.3–5
In Chagas disease, caused by the protozoan Trypanosoma cruzi, a strong inflammatory and preferentially T helper type 1 (Th1) effector response is essential to more efficient killing of the parasite.6 Along with the production of Th1 cytokines, the Th17 responses have also been linked to the pathogenesis of Chagas disease, because IL-17 is produced during the acute phase of T. cruzi infection and is thought to control cardiac inflammation by modulating the Th1 response.7 Conversely, in T. cruzi infection the production of regulatory cytokines may contribute to the persistence of the parasite inside host cells, causing inhibition of the production of nitric oxide (NO) and cytokines such as IL-12 and interferon-γ (IFN-γ), which are essential to parasite elimination.8,9 Moreover, it is known that in mice infected with T. cruzi, Treg cells migrate to the heart and treatment with anti-CD25 or anti-GITR monoclonal antibodies results in increased mortality of the animals.10 It is feasible that the extent of this regulatory process plays a key role in the outcome of the disease, by inhibiting the prejudicial inflammatory response that could cause tissue damage and by allowing parasite persistence. Therefore, it is possible that the modulation of the immune response against T. cruzi may determine the development of the different symptomatic forms of the disease,11,12 such as the acute myocarditis, which is characterized by a diffuse inflammatory reaction with or without T. cruzi nests, necrosis of cardiomyocytes and interstitial oedema.13
Another important feature that may be related to the pathogenesis of Chagas disease during the acute phase and its evolution to chronic forms is the number of trypomastigotes that naturally infect the host organism. Recent studies reported that oral infections may occur as a result of the ingestion of food or drinks contaminated with the metacyclic form of the parasite.14 In these cases the amount of inoculum and pathogenicity of the strain can determine the severity of the disease.15 The parasite load during T. cruzi infection may also influence the activation of the immune response and disease pathology in the chronic phase of Chagas disease.16 In addition, patients presenting specific clinical forms of Chagas disease, such as the cardiac form, may have parasite persistence that could maintain the immunological activation, which would be responsible for enhanced tissue damage in susceptible hosts.17
Even after several decades of research, the pathogenesis of Chagas disease remains unclear. In this context, unravelling the immune response that may follow infection and its correlation to the progression of the disease and parasite persistence is an important goal for the prognosis and treatment of infected individuals, especially regarding the low or high parasite loads to which patients may be exposed during infection. Accordingly, as we believe that the degree of the inflammatory response and the tissue injury that follows infection may be dependent on the amount of parasite in the first contact between the host and T. cruzi, we evaluated the inflammatory response outcome, tissue damage and immune regulation mechanisms that occur after T. cruzi infection with low, intermediate or high parasite loads.
Materials and methods
Animals
Male C57BL/6 mice (8–10 weeks old) were obtained and housed in our animal facility at Disciplina de Biologia Celular, UFTM, Uberaba, Brazil. Mice were given water and food ad libitum during the experimental period and all procedures were approved by the local ethical committee for animal research (CEUA – protocol number 96).
Parasites and infection
The ‘Colombian’ strain of T. cruzi was maintained by weekly passage in Swiss mice. Initially, for each experiment, one Swiss mouse was heparinized intraperitoneally, and then killed in a CO2 chamber for blood collection from the ophthalmic venous plexus. Five microlitres of blood was placed on slides covered with a coverslip size 22 × 22 mm to allow the microscopic parasites to be counted at 40 × magnification, in 50 microscope fields, according to Brener.18 After determining the concentration of parasites/ml in blood, these parasites were used for infection of the C57BL/6 mice. For the inoculum, the blood was diluted in saline (NaCl 0·9%) and inoculated subcutaneously at a volume of 100 μl per mouse. Natural infection with T. cruzi seems usually to occur with low parasite loads, so it was decided to first evaluate the effects of an initial inoculum considered low (300 parasites) to mimic a natural infection.19 The other inocula (3000 and 30 000) were based on preliminary studies developed by our research group to determine the inoculum necessary for differentiating low, medium and high infections in mice, besides the literature data regarding accidental infections with elevated numbers of parasites.14 Then, for experimental infection, C57BL/6 mice were divided into four groups according to the inoculum used: five control animals inoculated with saline, five mice inoculated with 300 bloodstream forms of T. cruzi, five mice inoculated with 3000 T. cruzi and five animals inoculated with 30 000 blood-stage trypomastigotes, injected subcutaneously, per experiment. Parasitaemia was evaluated in 5 μl blood drawn from the tail18 every 3 days, and mortality was monitored daily in infected mice. Before necropsy, mice were killed in a CO2 chamber.
Histology
Fragments of heart were fixed in Methacarn solution and embedded in paraffin. Sections, 5 μm thick, were stained with haematoxylin & eosin for standard histological procedures. The images for inflammatory infiltrate analysis were captured using the 40 × microscope objective through a digital video camera (Evolution MP 5.0–colour–Media Cybernetic, Silver Spring, MD) coupled to a light microscope (Nikon – Eclipse 50i, Melville, NY) that sends images to a computer. Analysis of the inflammatory infiltrate was performed on heart tissues from infected mice. The damaged areas were defined as the areas with breakdown of muscle fibres and accumulation of inflammatory cells, according to ‘Dallas’ criteria.20 We analysed 156 acquired images in left and right ventricles of the heart and the septum, totalling an area of 0·58 mm2, that was considered a statistically reliable quantified area, according to Hally.21 Then, we carried out a morphometric analysis to obtain the relationship between the delineated damaged area covered by inflammatory infiltrate and the total area of tissue visualized in the acquired image. The result was expressed as a percentage, using the following formula: P = (Da/Ta)*100, where P represents the percentage of inflammatory infiltrate, Da is the damaged area covered by inflammatory infiltrate and Ta is the total area of the acquired image.
Tissue extract preparation for cytokine measurements
The heart tissues were first weighed and then immersed in equal volumes of PBS (500 μl per tissue) containing Complete Protease Inhibitor Cocktail Tablets (Roche Applied Sciences, Indianapolis, IN). The protease inhibitor solution was prepared by adding one tablet to 50 ml PBS, according to the manufacturer's instructions. Extracts were obtained by homogenizing tissues with an electrical tissue homogenizer in the protease inhibitor buffer followed by centrifugation at 300 g for 15 min, after which the supernatants were collected and stored at −70° until use. Cytokines [IFN-γ, tumour necrosis factor-α (TNF-α), IL-4, IL-10, TGF-β, IL-6, IL-23 and IL-17] were measured according to the manufacturer's instructions, using commercially available ELISA kits (R&D Systems, Minneapolis, MN). The cytokine concentrations were normalized taking into account the weight of each tissue and the results were expressed as picograms per milligram of tissue.
Immunohistochemistry
The paraffin-embedded slides were processed and immunostained for detection of T. cruzi nests with anti-Trypanosoma cruzi sera. This serum was produced in rabbits inoculated with T. cruzi obtained from the blood of previously infected Swiss mice. After a period of 3 months, rabbit blood was collected, centrifuged and plasma containing polyclonal anti-T. cruzi antibodies was obtained. The antibody was used at a concentration of 1 : 200 diluted in 1% BSA in PBS.
Quantification of T. cruzi nests stained by immunohistochemistry was performed using imagej software (Wayne Rasband National Institutes of Health, Bethesda, MD) and the results were expressed in nests/cm2. The criteria for determining the number of nests were defined by the researchers, who considered nest regions to be those containing at least three grouped amastigotes per stained region. The software only reported the total number of nests previously delineated by the researcher.
Flow cytometry
For immunophenotyping spleen cells, the whole organs were minced with tweezers, the suspension was filtered and washed in PBS, and cells were layered on a tube containing Ficoll-Paque (GE Healthcare Bio-Sciences Corp, Pittsburgh, PA). Then, cells were centrifuged for 30 min at room temperature (400 g). Leucocytes were recovered from the interface, washed with PBS and the viability was evaluated by trypan blue exclusion in a Neubauer chamber. Cells were then counted and used for immunolabelling assays. Briefly, two-colour or three-colour cytometry was performed by incubating leucocytes with FITC-conjugated anti-CD8a, phycoerythrin Cychrome 5-conjugated anti-CD4 (anti-CD4-PE-Cy5) and anti-CD3e-PE; anti-CD25-FITC, anti-CD4-PE-Cy5 and one of the Treg markers (anti-FoxP3-PE, anti-GITR-PE, anti-CD45RB-PE or anti-PD-1-PE), besides monocytes staining with anti-CD11c-PE with anti-CD11b-FITC. All antibodies and their respective isotype controls were from BD Biosciences (San Jose, CA).
For flow cytometry analysis of the heart Treg cells, the organs collected from 10 mice at day 22 post-infection were pooled, washed, minced and incubated for 1 h at 37° with RPMI-1640 (Sigma-Aldrich®, St Louis, MO) and 0·05 mg/ml of liberase TL (Roche, Indianapolis, IN). After tissue digestion and maceration, we performed Ficoll-Paque (GE Healthcare Bio-Sciences Corp) gradient centrifugation for 30 min at room temperature (400 g) to obtain mononuclear cells. Leucocytes were recovered from the interface, washed with PBS and their viability was evaluated by trypan blue exclusion. Cells were then counted and used for an immunolabelling assay with anti-CD4-PE-Cy5, anti-CD25-FITC and anti-FoxP3-PE (BD Biosciences), as described above. Fluorocytometric acquisition of spleen and heart leucocytes was performed in a FACscalibur apparatus (Becton Dickson, San Jose, CA) and analysis was performed using flowjo software (Tree Star, Ashland, OR).
Statistical analysis
In all variables the normal distribution and homogeneous variance were tested. When the distribution was normal and there was homogeneous variance we used parametric tests like the analysis of variance with Tukey's multiple comparison, when three or more groups were compared. When the data did not reach a Gaussian distribution we used the non-parametric tests Kruskal–Wallis, followed by Dunn's multiple comparison. For correlations, we used the Spearman correlation test. The Kaplan–Meier method was used to compare survival rates. Differences were considered statistically significant when P < 0·05. All analyses were performed using the graphpad prism 5.0 software (San Diego, CA).
Results
The load of parasite inoculum influences infection outcome
First, to verify whether parasite loads were related to the infection outcome, C57BL/6 mice were infected with 300, 3000 or 30 000 bloodstream forms, the trypomastigotes, of the ‘Colombian’ strain of T. cruzi.
Results demonstrated that the higher the number of parasites in the inoculum, the lower were the survival rates observed during the experimental infection, pointing to the elevated mortality of mice infected with 30 000 trypomastigotes (Fig. 1a). This group also presented a shorter pre-patent period and augmented parasitaemia throughout the periods evaluated (Fig. 1b). Notably, the group with the lower inoculum showed a continuous increase in parasitaemia, compared with the other groups, which presented a decrease after day 24 post-infection. However, despite this later decrease, the mice given the higher inoculum maintained the most elevated levels of parasitaemia until day 30, when we observed elevated mortality in this group (Fig. 1a,b). In addition, mice exposed to the intermediate (3000) or low (300) inocula showed intermediate or lower parasitaemia levels and elevated survival compared with the high-inoculum group (Fig. 1a,b).
Figure 1.
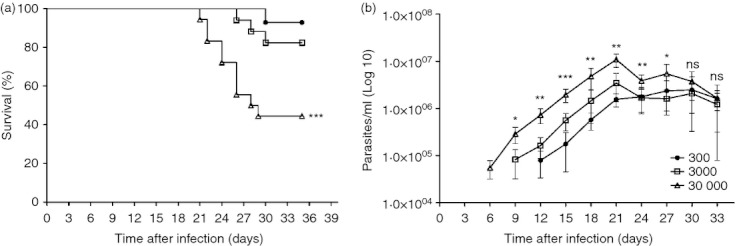
Mortality rate and parasitaemia of C57BL/6 mice infected with the ‘Colombian’ strain of Trypanosoma cruzi. (a) Percentages of mice that succumbed to infection. Mice were injected subcutaneously with 300, 3000 and 30 000 bloodstream forms of T. cruzi, n = 5 mice per group. (b) Parasitaemia levels were evaluated by counting numbers of parasites in 5 μl blood drawn from the tail vein, n = 5 mice per group. Data are representative from two independent experiments. *Statistical significance (P < 0·0001) between 30 000 inoculum and 3000 inoculum. **Statistical significance (P < 0·0001) between 30 000 inoculum and other groups. ***Statistical significance (P < 0·0001) between the three groups. ns, not significant.
Parasitism and inflammatory infiltrate in heart tissues are associated with the parasite load during infection
Because of the elevated parasitaemia and increased mortality rate observed in the higher inoculum groups, we asked whether parasites spread to the heart causing tissue damage and death. Histopathological analyses showed a greater inflammatory infiltrate in the group that received the highest inoculum (Fig. 2a), together with elevated tissue parasitism, which could be generating the inflammatory process (Fig. 2b), because we observed a strong association between tissue parasitism and inflammatory infiltrate in heart tissue (Fig. 2c).
Figure 2.
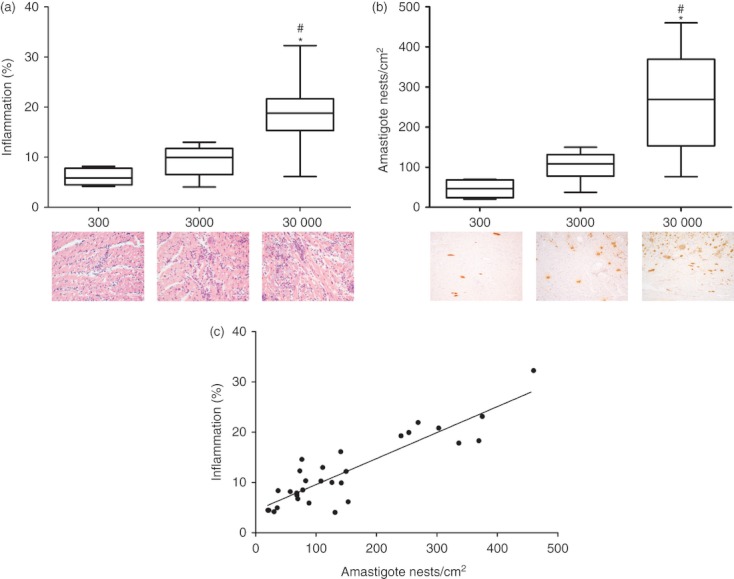
Morphometric analyses of inflammation, amastigote loads in hearts of infected mice and correlation between inflammation and amastigote nests. (a) Inflammatory infiltrate in hearts of mice after haematoxylin and eosin staining, n = 5 mice per group. (b) Immunohistochemical demonstration of antigen for Ttypanosoma cruzi (amastigote), n = 5 mice per group. (c) Correlation between inflammation and amastigote nests, calculated by Spearman correlation (P < 0·0001 and r = 0·7780), n = 5 mice per group. Data are representative from two independent experiments. *Statistical significance of P < 0·0001 between 30 000 inoculum group and 300 inoculum group. #Statistical significance of P < 0·0001 between 30 000 inoculum group and 3000 inoculum group.
The parasite load during infection determines cytokine interplay in the cardiac tissue
Trypanosoma cruzi is able to cause an intense immune response and focal areas of inflammation in the cardiac tissue,22 we next aimed to investigate if the inflammation and parasite accumulation in the heart was also associated with a local immune response that could influence the differential infection outcome observed previously. At 22 days post-infection, concentrations of IFN-γ and TNF-α (Fig. 3a,b) were increased in the groups given 300 and 30 000 trypomastigotes compared with the uninfected mice, whereas the mice infected with 30 000 trypomastigotes showed the highest levels of these cytokines in the heart, especially when compared with the group given 3000 trypomastigotes (Fig. 3a,b).
Figure 3.
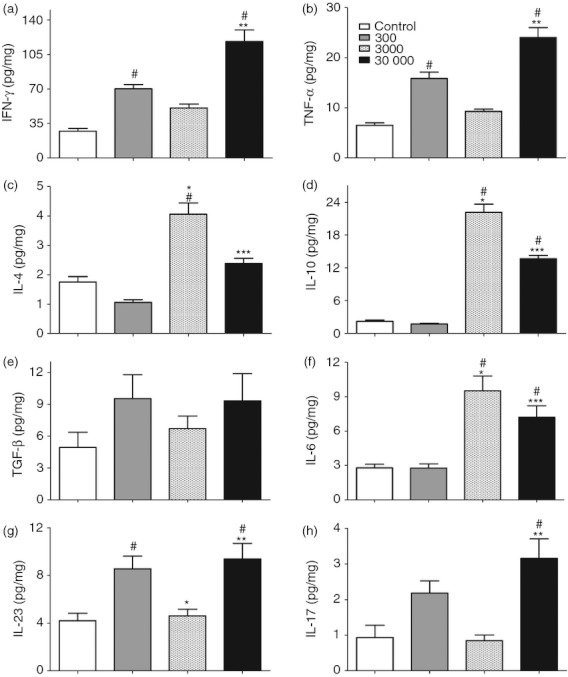
Cytokine production in heart tissue of mice infected with the ‘Colombian’ strain of Trypanosoma cruzi. Cytokine concentrations were determined by capture ELISA with specific antibodies to interferon-γ (IFN-γ) (a), tumour necrosis factor-α (TNF-α) (b), interleukin-4 (IL-4) (c), IL-10 (d), transforming growth factor-β (TGF-β) (e), IL-6 (f), IL-23 (g), and IL-17 (h) in the supernatants of heart tissue homogenates, n = 5 mice per group. Data are representative from two independent experiments. #Statistical significance of P < 0·05 compared with the control group. *Statistical significance of P < 0·05 compared between 300 and 3000 inocula groups. **Statistical significance of P < 0·05 compared with the 3000 inoculum group. ***Statistical significance of P < 0·05 compared with the 300 inoculum group.
Regarding counter-regulatory cytokines, levels of the anti-inflammatory cytokines IL-4 and IL-10 were increased in the groups of mice given 3000 and 30 000 trypomastigotes when compared with uninfected mice (Fig. 3c,d). The higher levels of IL-4 and IL-10 were found in the intermediate inoculum group (3000 forms), which also presented the lower levels of IFN-γ and TNF-α in the heart tissues, considering the infected mice (Fig. 3a–d).
In addition, we showed that TGF-β production, which can inhibit the activation of macrophages and their ability to produce pro-inflammatory cytokines,23 presented a similar profile to the IFN-γ and TNF-α levels, although no significant differences were observed among the groups (Fig. 3e). On the other hand, this cytokine may be involved in the commitment of Th17 cells and may generate tissue inflammation in T. cruzi infection.7 Levels of IL-6, also important in Th17 differentiation, were augmented in the intermediate and high inoculum groups (Fig. 3f), whereas the Th17-maitaining and signature cytokines, IL-23 and IL-17, respectively, were observed to have kinetics very similar to the other inflammatory cytokines described above, with emphasis on the increase in the groups given 300 and 30 000 trypomastigotes (Fig. 3g,h, respectively). In addition, we correlated the levels of parasitaemia or the tissue parasite burden with the immune response developed by the host and demonstrated that the production of IFN-γ, TNF-α and IL-17 in the hearts of infected mice was directly and strongly related to the parasitaemia and tissue parasite load in these animals (Fig. 4a–g). Then, as IL-17 may play an important role in regulating the production of IFN-γ, we plotted the levels of IL-17 against IFN-γ. The results showed a positive correlation between them, suggesting that in this case the production of IL-17 does not seem to be sufficient to control IFN-γ (Fig. 4d), probably because of the high tissue parasite burden (Fig. 4g). Altogether, these data suggested that cytokine interplay in the heart of infected mice was also influenced by the parasite load during T. cruzi infection and may be related to a differential outcome of the disease.
Figure 4.
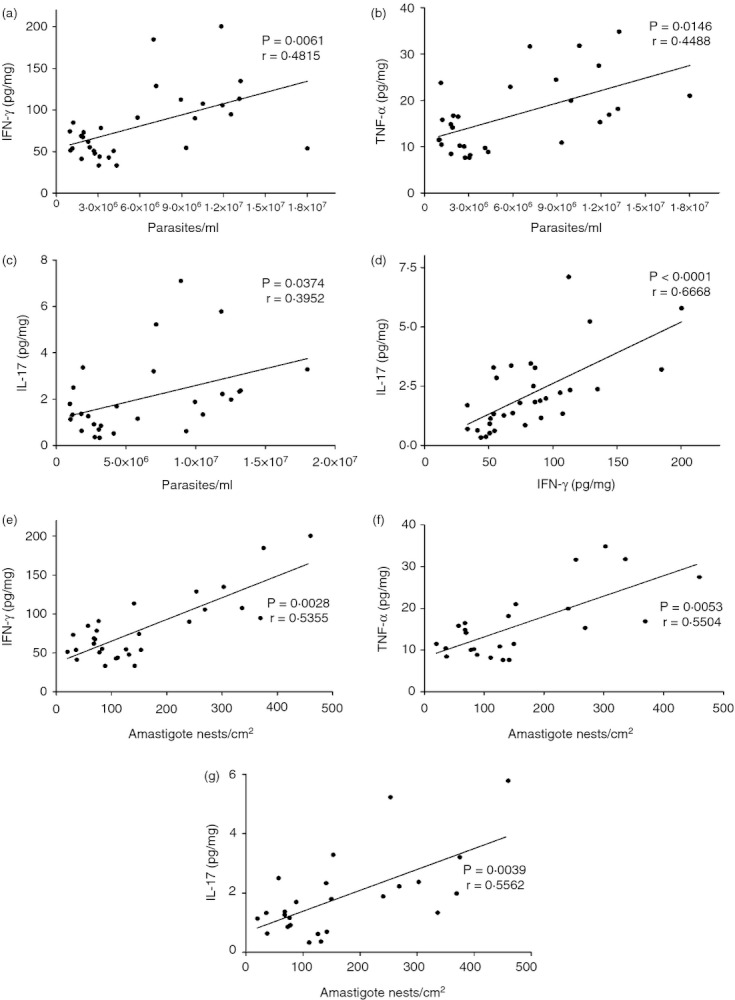
Correlation between parasitaemia, inflammatory cytokines and tissue parasite burden. (a) Relationship between parasitaemia and interferon-γ (IFN-γ). (b) Relationship between parasitaemia and tumour necrosis factor-α (TNF-α). (c) Relationship between parasitaemia and IL-17. (d) Relationship between IFN-γ and interleukin-17 (IL-17). (e) Relationship between tissue parasite burden and IFN-γ. (f) Relationship between tissue parasite burden and TNF-α. (g) Relationship between tissue parasite burden and IL-17. n = 5 mice per group. Data are representative from two independent experiments. Spearman Correlation.
Systemic immune response is also affected by different parasite loads during T. cruzi infection
Local cardiac alterations were observed that could be related to infection progression and later host survival, so we next asked if elevated mortality and disease worsening could also influence or be influenced by systemic responses induced by the different parasite loads. Infected mice showed an increase in spleen weight and in the absolute number of T cells compared with uninfected mice (Fig. 5a–c), with an apparent augmentation of CD4 and CD8 T cells in the highest inoculum group. Regarding the profile of antigen-presenting cells (CD11b+ CD11c+ and CD11b+ cells) in the spleen, we found a significant augmentation in the absolute number in groups of infected mice compared with uninfected controls, especially regarding CD11b+ CD11c+ dendritic cells (Fig. 5d) in the higher (30 000) and lower (300) inocula, besides an elevated number of CD11b+ cells (possibly macrophages) in the intermediate group (3000), in comparison to controls (Fig. 5e). These results suggested that antigen presentation may also be influenced by the number of parasites faced by the host during infection.
Figure 5.
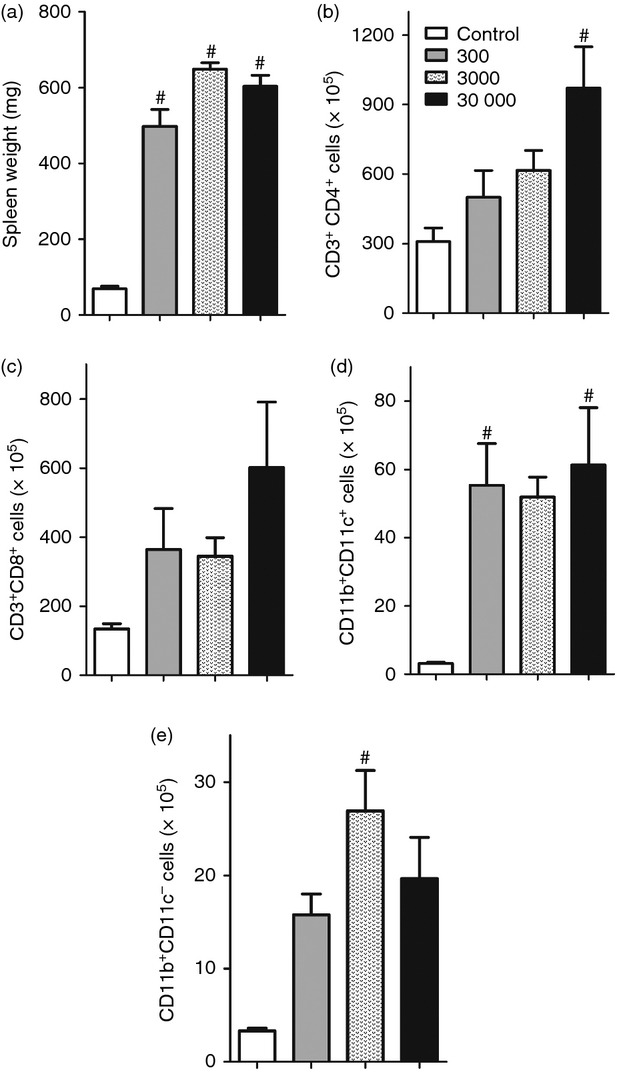
Systemic inflammatory alterations in spleen cells induced by T. cruzi infection. (a) Spleen weight expressed in mg. (b) The absolute number of CD4+ cells within CD3+ lymphocytes. (c) The absolute number of CD8+ cells within CD3+ lymphocytes. (d) The absolute number of CD11b+ within CD11c+ cells. (e) The absolute number of CD11b+ within CD11c− cells. n = 5 mice per group. Data are representative from two independent experiments. #Statistical significance of P < 0·05 compared with the control group.
Higher T. cruzi inocula up-regulate the number of splenic cells but not local Treg cells in the heart
As inflammation seemed to be up-regulated in the higher inocula groups, as well as parasitaemia and mortality, we next aimed to investigate if these mice failed to balance the systemic and local immune responses by a failure in number of Treg cells.
Our data showed a significant increase in the absolute number of spleen CD4+ CD25+ cells in the 3000 and 30 000 trypomastigote-infected mice compared with the control group and with the low inoculum mice (Fig. 6a). Therefore, as natural Treg-cell characterization requires the analysis of other phenotypic markers, we next aimed to evaluate if the difference in the number of CD4+ CD25+ cells was also accompanied by an augmentation in Treg cell molecules that could be related to the cell function (Fig. 6b). In fact, we observed a predominant increase of all markers in infected animals compared with controls. Regarding the CD4+ CD25+ population, we found an increase in the number of cells expressing the Treg markers analysed (CD45RBlow, GITR, PD-1 and FoxP3) in the spleen of animals in the high inoculum group (Fig. 6b). It is of note that, in general, the most inflamed group (30 000) was the one which presented the higher cell numbers in the spleen with regulatory markers (Fig. 6b), although it was not enough to avoid mortality. Furthermore, by analysing the CD4+ CD25− T cells expressing the same markers, we observed that the lowest and highest inocula groups had the greater number of cells labelled with GITR and CD45RBlow, although a statistically significant difference was found only when the 30 000 inoculum and control groups were compared. In addition, mice inoculated with the low and intermediate numbers of parasites showed higher counts of cells expressing PD-1 (Fig. 6c) whereas the number of CD4+ CD25− FoxP3+ cells was apparently elevated in the high inoculum group.
Figure 6.
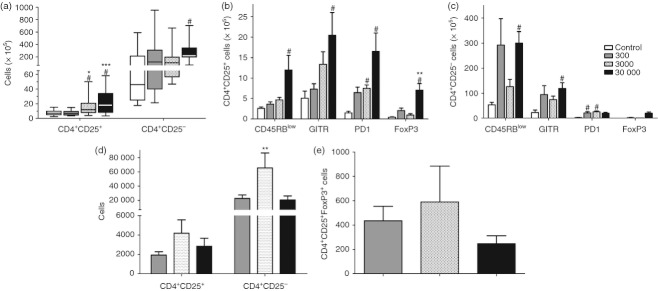
Quantification of regulatory T cells in the spleen and heart of mice infected with different Trypanosoma cruzi inocula. (a) The absolute number of CD4+ CD25+ and CD4+ CD25− spleen cells. (b) Number of CD4+ CD25+ spleen cells expressing several regulatory T-cell markers. (c) Number of CD4+ CD25− spleen cells expressing regulatory T-cell markers. (d) The absolute number of CD4+ CD25+ and CD4+ CD25− cells in the hearts of the different infected groups. (e) Absolute number of CD4+ CD25+ Foxp3+ cells in the hearts of the infected mice. From (a–c) n = 5 mice per group. In (d and e) 10 mice were pooled per analysis, which consisted of three pooled samples per infected group. Data are representative from one (d–e) or two (a–c) independent experiments. #Statistical significance of P < 0·05 compared with the control group. *Statistical significance of P < 0·05 compared between 300 and 3000 inocula groups. **Statistical significance of P < 0·05 compared between 3000 and 30 000 -inocula groups. ***Statistical significance of P < 0·05 compared with the 300 inoculum group.
In contrast to the spleen data, we found an increase in the number of CD4+ CD25+ and CD4+ CD25− cells in the hearts of the intermediate inoculum group, as well as an apparent augmentation of FoxP3+ cells in the CD4+ CD25+ population (Fig. 6d–e). Notably, the high inoculum group had the lowest number of Treg and CD4+ CD25− cells in the heart, accompanied by a tendency to reduced amounts of FoxP3+ cells (Fig. 6d–e). It is of note that, as expected, the control non-infected mice had no inflammatory infiltrate in the heart and we did not find CD4+ CD25− FoxP3+ cells in the hearts of most infected mice (data not shown).
Discussion
In the present study we demonstrated modulations in the immune response raised against different parasite loads in experimental T. cruzi infection. Our data showed that the increase in the inoculum used correlated with the higher parasite persistence and pro-inflammatory immune response in the heart. Conversely, the lower inocula led to a more restrained immune response and absence of extensive cardiac damage or mortality.
Immunopathogenesis of experimental infection with T. cruzi involves many mechanisms related to disease outcome during acute and chronic phases. In agreement with our results, the Colombian strain, which was used in the present study, is characterized by a tissue tropism for skeletal muscle and myocardium, high pathogenicity, virulence and the capacity to generate large lesions with necrosis and inflammation in the myocardium and skeletal muscle.24
Our results demonstrated that variations in the inoculum influenced the outcome of experimental Chagas disease, resulting in increased cardiac parasitism and inflammatory infiltration, especially when higher parasite loads were used during infection. In parallel, a study using mice infected with different parasitic loads of T. cruzi Y strain showed that the higher parasite load presented by mice infected with a high inoculum in the acute phase of infection influenced the development of the chronic phase, in which elevated tissue parasitism, injury and activation of the immune response were observed.16 Interestingly, in our study the group receiving an intermediate inoculum (3000) presented mortality, inflammatory infiltration and cardiac parasitism similar to the lower inoculum group, suggesting that a minimum number of parasites is required to induce a host response to T. cruzi, which can later become uncontrolled and cause tissue damage.
Initially, the immune response against infection with T. cruzi is carried out by the innate immune system through the action of antigen-presenting cells like dendritic cells. They recognize pathogen molecules and activate a sequence of signals with antigen presentation, co-stimulation and cytokine secretion.25,26 In this way, mouse dendritic cells increase IL-12 production after injection with a soluble tachyzoite extract of Toxoplasma gondii.27 In addition, the acute phase of T. cruzi infection induces an increase in dendritic cells, accompanied by a rise in parasitaemia levels.28 Our results showed a high absolute number of dendritic cells in infected groups, suggesting that the augmentation in this population occurs simultaneously with the immune response, which should be raised to counteract the infection. In parallel, macrophages seemed to play a relevant role in the intermediate inoculum infected group, because elevated numbers of CD11b+ cells (possibly macrophages) were found in the spleens of these mice. Macrophages also have an important role in the innate immune response; they are thought to be a major cell population involved in parasite uptake. Moreover, these cells can be activated by IFN-γ and produce several intracellular killing mediators, such as NO.29 Additionally, a huge increase in macrophage precursors (monocytes) has been found in the peripheral blood during acute T. cruzi infection.30 On the other hand, our results seem to partially agree with those of Souza et al.,31 who demonstrated that the regulatory cytokine profile of monocytes from Chagas disease patients correlated with the indeterminate form whereas those patients who presented with elevated TNF-α production by their monocytes had the cardiac clinical presentation of the disease. Furthermore, the IL-10 production by monocytes could be related to the development of M2 anti-inflammatory macrophages, because this group does not seem to die as a result of excess inflammation.32 However, although our results pointed to a role for CD11b+ cells along with elevated IL-10 in the control of the exacerbated inflammation induced by the intermediate inoculum of T. cruzi, further experiments are still necessary to test this hypothesis, which will be assessed in our next studies.
In our study the highest T. cruzi inoculum during experimental infection induced the strongest IFN-γ production. In the contrast, high levels of pro-inflammatory cytokines (IFN-γ and TNF-α) were also detected in the lower inoculum group. These results demonstrated that a small parasite load is sufficient to trigger an inflammatory response able to control the infection but not enough to cause extensive tissue damage. Indeed, it is largely known that Chagas disease is characterized by a predominantly Th1 response, which is necessary for the protection against T. cruzi. Macrophages are stimulated by this parasite to produce IL-12, and this cytokine is responsible for inducing IFN-γ production by natural killer and T cells, necessary for the subsequent control of T. cruzi proliferation in vivo.33
Another mechanism used by the immune system to control infections is by generating the differentiation of inflammatory Th17 cells, which requires IL-1β, IL-6 and TGF-β.34 Interleukin-17 can also be produced by CD8+ T cells, γδ T cells, neutrophils, monocytes and natural killer cells.35 Although usually related to inflammation rather than regulation, in T. cruzi infection there is production of IL-17, which controls IFN-γ production as well as the development of heart lesions during the course of infection.7 In contrast, our data demonstrated that increased levels of IL-17 in the groups with lower and higher inocula did not lead to a reduction in the concentration of IFN-γ, because levels of both cytokines remained elevated in these groups. Conversely, other studies showed that Th1 and Th17 cells can coexist under pathological conditions such as psoriasis,36 or infection with Mycobacterium37 or even T. cruzi, in which CD4 T cells from T-bet-deficient mice stimulated in vitro with anti-CD3 monoclonal antiboies were able to secrete both IFN-γ and IL-17.38 In addition, the cytokine IL-23 is responsible for stimulating the production of IL-17 by Th17 cells,39 but can also act on CD8 and γδ T cells, which also present high IL-23 receptor expression, which is necessary for IL-17 production.40,41 Our results showed the same profile of production of both cytokines in the infected groups, so corroborating previous data that demonstrated the dependence on IL-23 for Th17 maintenance.
The immune response against T. cruzi can also develop from CD8+ T cells through cytokine production and cytotoxic activity, being crucial for control of the infection.42,43 The CD8 T cells have little importance in the control of parasitaemia in the initial period of infection regardless of the inoculum used. However, after the peak of parasitaemia, cytotoxic T cells are critical for parasite control in the blood, directly influencing the mortality of animals.44 Tzelepis et al.44 also demonstrated that the intensity of cytotoxic response by CD8 T cells was directly correlated with the parasite inoculum, when different inocula were evaluated. In our results, consistent with this study, we observed an apparent increase in absolute number of cytotoxic T cells in the higher inoculum group (30 000). Hence we suggested that the amount of parasite inoculum was correlated with the acceleration of parasitaemia and the consequent increase in cytotoxic CD8 T cells in an attempt to control excess parasite load. In addition, CD4 as well as CD8 T cells are of great importance to the control of T. cruzi infection by the production of pro-inflammatory cytokines such as IFN-γ.45 Here we observed that the inoculum used influenced the degree of inflammatory response, as shown by the higher absolute numbers of CD4 and CD8 T cells and consequently elevated IFN-γ production in the 30 000 inoculum group.
On the other hand, the inflammatory response may be down-regulated by the anti-inflammatory cytokines IL-10 and TGF-β23,46 and by regulatory cells,10 in an attempt to balance this effector response.47 These suppressive mechanisms include a wide variety of cells with a unique capacity to inhibit the effector T-cell response,48 like inducible or natural Treg cells, that express a variety of phenotypic markers.49–52 However, as most of these markers can also be expressed by activated T cells, none of them are specific to natural Treg cells and the expression of the transcription factor FoxP3 has become the most expressive characteristic of natural Treg cells.53
Here we demonstrated that the variation of the inoculum used in the different groups was an outstanding factor in setting the level of regulation presented by each group. Trypanosoma cruzi induces non-specific immunosuppression during the acute phase of the infection,54as occurs in infections by Bordetella pertussis and Schistosoma mansoni, which induce IL-10 production by dendritic cells.55,56 In addition, the immune system itself tends to regulate the inflammatory response by producing regulatory cytokines,57,58 and apoptosis induction.59 Hence, we propose that the group given the low inoculum (300) showed a low absolute number of regulatory T cells (CD4+ CD25+) because the parasite burden in this group failed to induce a huge inflammatory response which, if it exists, should be regulated to avoid cardiac injury. Hence, T. cruzi induces the expression of PD-1 molecules by CD8 and CD4 T cells, regulating the immune response through decreasing inflammatory cytokine levels, inhibiting the proliferation of T cells, and inducing apoptosis.59 In parallel, the GITR molecule exerts a potent co-stimulatory role in both effector and regulatory T cells and this molecule may regulate diverse biological functions in T xcells, such as proliferation, activation, differentiation and cell survival.50 In addition, study using mice infected with the Y strain of T. cruzi treated with anti-GITR monoclonal antibody showed an exacerbated inflammatory reaction, probably as the result of the inhibition of the suppressor activity of Treg cells, and these cells are effectively involved in the pathogenesis of T. cruzi infection.10 Conversely, other studies showed that Treg cells have a limited role in the course of infection by T. cruzi and that Treg cells do not seem to suppress the CD8 T-cell cytolytic activity.47,60 The CD4+ CD45RBlow cells stimulate the production of IL-10 and IL-4 cytokines,52 and furthermore, protect, through IL-10, against the development of other immune-mediated diseases like colitis.61 Our data demonstrated that the CD4+ CD25+ CD45RBlow cells were present in higher absolute numbers in the spleens of the 30 000 inoculum group, suggesting that an attempt to regulate the immune response may be inducing the production of IL-10 and IL-4.
In addition, we demonstrated that the group receiving the intermediate inoculum (3000) also showed elevated numbers of CD4+ CD25+cells in the spleen, indicating that the host immune system may be regulating the pro-inflammatory response, or the parasite is inducing this regulatory activity; however, in this case, mediated by PD-1 and not by FoxP3, the most important and definitive Treg cell marker.53 On the contrary, we found elevated numbers of CD4+ CD25+ FOXP3+ cells in the heart of this intermediate inoculum group, indicating that such Treg cells may be regulating cardiac inflammation and controlling exacerbated damage, in accordance with the regulatory cytokine profile observed in these mice. Furthermore, we suggested that even with evident regulatory mechanisms, the T. cruzi multiplication and increased tissue parasite burden did not significantly occur in the intermediate inoculum group, because there was still a remaining basal inflammatory response that could have prevented this multiplication. On the other hand, although the 30 000 inoculum group had showed the highest number of regulatory cells in the spleen, it is possible that this systemic regulation was not sufficient to control the exaggerated inflammation caused by the high number of parasite antigenic molecules in the host and exacerbated the inflammatory cardiac response, as evidenced by a failure to induce local Treg cells in the heart. Therefore, any attempt to control the inflammatory response, either by the immune system or the induction of regulation by the parasite was not successful.
In the present work, the cytokine profile observed in the heart of infected mice corroborated with this effector/regulatory hypothesis, once our data showed that the low inoculum group developed a pro-inflammatory, Th1 and Th17 response that was able to control parasite growth while avoiding extensive tissue damage, with a small adjustment through the production of TGF-β. Moreover, the pro-inflammatory response raised against infection in the intermediate group was probably not avoided by the elevated production of regulatory cytokines and so was sufficient to provide satisfactory control of the parasite; however, this effector response was also not strong enough to induce host damage. On the other hand, the high inoculum group had an exacerbated and uncontrolled pro-inflammatory response, which was not adequate to successfully eliminate the elevated number of parasites nor were they satisfactorily controlled by regulatory mechanisms to avoid tissue injury. Indeed, the production of anti-inflammatory cytokines, IL-4, IL-10 and TGF-β and the elevated number of spleen, but not cardiac, Treg cells were not able to reduce the inflammatory response, leading to a high inflammatory infiltrate and mortality of these animals. However, it should still be clarified whether the main cause of death was the high number of parasites, the excess response triggered by them or both. These data suggested that a strict control of the immune response was necessary to eliminate parasite while avoiding tissue damage in T. cruzi infection and this control may be dependent on the parasite load to which the host is exposed during infection.
In summary, our results showed that the parasite load during T. cruzi infection is a primary factor in determining the pattern of the host immune response that will result in parasite/inflammation control or tissue damage. Therefore, it may also be essential to define the patient's prognosis, especially where there is elevated presence of parasites at the time of infection, such as in recent cases of oral infections with T. cruzi. However, we emphasize that Chagas disease pathogenesis remains unclear and further studies are still needed to clarify the relation between tissue damage by the host immune response versus damage caused by the parasite in the context of the uncontrolled immune response.
Acknowledgments
This work was supported by Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), grant APQ-01712-08, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). The authors thank João Batista Pereira for technical assistance with the histology procedures and Viviani Nardini for helping with figure preparation.
Disclosures
We declare no competing interests.
References
- 1.Perez AR, Silva-Barbosa SD, Roggero E, et al. Immunoendocrinology of the thymus in Chagas disease. NeuroImmunoModulation. 2011;18:328–38. doi: 10.1159/000329494. [DOI] [PubMed] [Google Scholar]
- 2.Mendez S, Reckling SK, Piccirillo CA, Sacks D, Belkaid Y. Role for CD4+ CD25+ regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J Exp Med. 2004;200:201–10. doi: 10.1084/jem.20040298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+ CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–7. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 4.Santello FH, Frare EO, dos Santos CD, Caetano LC, Alonso Toldo MP, do Prado JC., Jr Suppressive action of melatonin on the TH-2 immune response in rats infected with Trypanosoma cruzi. J Pineal Res. 2008;45:291–6. doi: 10.1111/j.1600-079X.2008.00589.x. [DOI] [PubMed] [Google Scholar]
- 5.Taylor MD, LeGoff L, Harris A, Malone E, Allen JE, Maizels RM. Removal of regulatory T cell activity reverses hyporesponsiveness and leads to filarial parasite clearance in vivo. J Immunol. 2005;174:4924–33. doi: 10.4049/jimmunol.174.8.4924. [DOI] [PubMed] [Google Scholar]
- 6.Hoft DF, Schnapp AR, Eickhoff CS, Roodman ST. Involvement of CD4+ Th1 cells in systemic immunity protective against primary and secondary challenges with Trypanosoma cruzi. Infect Immun. 2000;68:197–204. doi: 10.1128/iai.68.1.197-204.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.da Matta Guedes PM, Gutierrez FR, Maia FL, Milanezi CM, Silva GK, Pavanelli WR, Silva JS. IL-17 produced during Trypanosoma cruzi infection plays a central role in regulating parasite-induced myocarditis. PLoS Negl Trop Dis. 2010;4:e604. doi: 10.1371/journal.pntd.0000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michailowsky V, Silva NM, Rocha CD, Vieira LQ, Lannes-Vieira J, Gazzinelli RT. Pivotal role of interleukin-12 and interferon-γ axis in controlling tissue parasitism and inflammation in the heart and central nervous system during Trypanosoma cruzi infection. Am J Pathol. 2001;159:1723–33. doi: 10.1016/s0002-9440(10)63019-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva JS, Vespa GN, Cardoso MA, Aliberti JC, Cunha FQ. Tumor necrosis factor α mediates resistance to Trypanosoma cruzi infection in mice by inducing nitric oxide production in infected γ interferon-activated macrophages. Infect Immun. 1995;63:4862–7. doi: 10.1128/iai.63.12.4862-4867.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mariano FS, Gutierrez FR, Pavanelli WR, et al. The involvement of CD4+ CD25+ T cells in the acute phase of Trypanosoma cruzi infection. Microbes Infect. 2008;10:825–33. doi: 10.1016/j.micinf.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 11.da Silveira AB, de Araujo FF, Freitas MA, et al. Characterization of the presence and distribution of Foxp3+ cells in chagasic patients with and without megacolon. Hum Immunol. 2009;70:65–7. doi: 10.1016/j.humimm.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 12.Vitelli-Avelar DM, Sathler-Avelar R, Massara RL, et al. Are increased frequency of macrophage-like and natural killer (NK) cells, together with high levels of NKT and CD4+ CD25high T cells balancing activated CD8+ T cells, the key to control Chagas’ disease morbidity? Clin Exp Immunol. 2006;145:81–92. doi: 10.1111/j.1365-2249.2006.03123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higuchi ML, De Morais CF, Pereira Barreto AC, Lopes EA, Stolf N, Bellotti G, Pileggi F. The role of active myocarditis in the development of heart failure in chronic Chagas’ disease: a study based on endomyocardial biopsies. Clin Cardiol. 1987;10:665–70. doi: 10.1002/clc.4960101113. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida N. Trypanosoma cruzi infection by oral route: how the interplay between parasite and host components modulates infectivity. Parasitol Int. 2008;57:105–9. doi: 10.1016/j.parint.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Monteiro WM, Barbosa MG, Toledo MJ, Fe FA, Fe NF. [Series of acute Chagas’ disease cases attended at a tertiary-level clinic in Manaus, State of Amazonas, from 1980 to 2006] Rev Soc Bras Med Trop. 2010;43:207–10. doi: 10.1590/s0037-86822010000200021. [DOI] [PubMed] [Google Scholar]
- 16.Marinho CR, D'Imperio Lima MR, Grisotto MG, Alvarez JM. Influence of acute-phase parasite load on pathology, parasitism, and activation of the immune system at the late chronic phase of Chagas’ disease. Infect Immun. 1999;67:308–18. doi: 10.1128/iai.67.1.308-318.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monteon-Padilla V, Hernandez-Becerril N, Ballinas-Verdugo MA, Aranda-Fraustro A, Reyes PA. Persistence of Trypanosoma cruzi in chronic chagasic cardiopathy patients. Arch Med Res. 2001;32:39–43. doi: 10.1016/s0188-4409(00)00261-7. [DOI] [PubMed] [Google Scholar]
- 18.Brener Z. Therapeutic activity and criterion of cure on mice experimentally infected with Trypanosoma cruzi. Rev Inst Med Trop Sao Paulo. 1962;4:389–96. [PubMed] [Google Scholar]
- 19.Coura JR. Chagas disease: what is known and what is needed – a background article. Mem Inst Oswaldo Cruz. 2007;102(Suppl 1):113–22. doi: 10.1590/s0074-02762007000900018. [DOI] [PubMed] [Google Scholar]
- 20.Aretz HT. Myocarditis: the Dallas criteria. Hum Pathol. 1987;18:619–24. doi: 10.1016/s0046-8177(87)80363-5. [DOI] [PubMed] [Google Scholar]
- 21.Hally AD. A counting method for measuring the volumes of tissue components in microscopical sections. Q J Microsc Sci. 1964;s3-105:503–17. [Google Scholar]
- 22.Talvani A, Ribeiro CS, Aliberti JC, et al. Kinetics of cytokine gene expression in experimental chagasic cardiomyopathy: tissue parasitism and endogenous IFN-γ as important determinants of chemokine mRNA expression during infection with Trypanosoma cruzi. Microbes Infect. 2000;2:851–66. doi: 10.1016/s1286-4579(00)00388-9. [DOI] [PubMed] [Google Scholar]
- 23.Silva JS, Twardzik DR, Reed SG. Regulation of Trypanosoma cruzi infections in vitro and in vivo by transforming growth factor β (TGF-β) J Exp Med. 1991;174:539–45. doi: 10.1084/jem.174.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Camandaroba EL, Campos RF, Magalhaes JB, Andrade SG. Clonal structure of Trypanosoma cruzi Colombian strain (biodeme Type III): biological, isoenzymic and histopathological analysis of seven isolated clones. Rev Soc Bras Med Trop. 2001;34:151–7. doi: 10.1590/s0037-86822001000200001. [DOI] [PubMed] [Google Scholar]
- 25.Boonstra A, Asselin-Paturel C, Gilliet M, Crain C, Trinchieri G, Liu YJ, O'Garra A. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J Exp Med. 2003;197:101–9. doi: 10.1084/jem.20021908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol. 2002;20:29–53. doi: 10.1146/annurev.immunol.20.091101.091806. [DOI] [PubMed] [Google Scholar]
- 27.Reis e Sousa C, Yap G, Schulz O, Rogers N, Schito M, Aliberti J, Hieny S, Sher A. Paralysis of dendritic cell IL-12 production by microbial products prevents infection-induced immunopathology. Immunity. 1999;11:637–47. doi: 10.1016/s1074-7613(00)80138-7. [DOI] [PubMed] [Google Scholar]
- 28.Chaussabel D, Pajak B, Vercruysse V, et al. Alteration of migration and maturation of dendritic cells and T-cell depletion in the course of experimental Trypanosoma cruzi infection. Lab Invest. 2003;83:1373–82. doi: 10.1097/01.lab.0000087587.93781.6f. [DOI] [PubMed] [Google Scholar]
- 29.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 30.Melo RC, Machado CR. Trypanosoma cruzi: peripheral blood monocytes and heart macrophages in the resistance to acute experimental infection in rats. Exp Parasitol. 2001;97:15–23. doi: 10.1006/expr.2000.4576. [DOI] [PubMed] [Google Scholar]
- 31.Souza PE, Rocha MO, Rocha-Vieira E, Menezes CA, Chaves AC, Gollob KJ, Dutra WO. Monocytes from patients with indeterminate and cardiac forms of Chagas’ disease display distinct phenotypic and functional characteristics associated with morbidity. Infect Immun. 2004;72:5283–91. doi: 10.1128/IAI.72.9.5283-5291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez-Castejon G, Baroja-Mazo A, Pelegrin P. Novel macrophage polarization model: from gene expression to identification of new anti-inflammatory molecules. Cell Mol Life Sci. 2011;68:3095–107. doi: 10.1007/s00018-010-0609-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aliberti JC, Cardoso MA, Martins GA, Gazzinelli RT, Vieira LQ, Silva JS. Interleukin-12 mediates resistance to Trypanosoma cruzi in mice and is produced by murine macrophages in response to live trypomastigotes. Infect Immun. 1996;64:1961–7. doi: 10.1128/iai.64.6.1961-1967.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–48. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 35.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 36.Kryczek I, Bruce AT, Gudjonsson JE, et al. Induction of IL-17 + T cell trafficking and development by IFN-γ: mechanism and pathological relevance in psoriasis. J Immunol. 2008;181:4733–41. doi: 10.4049/jimmunol.181.7.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cruz A, Khader SA, Torrado E, Fraga A, Pearl JE, Pedrosa J, Cooper AM, Castro AG. Cutting edge: IFN-γ regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. J Immunol. 2006;177:1416–20. doi: 10.4049/jimmunol.177.3.1416. [DOI] [PubMed] [Google Scholar]
- 38.Guo S, Cobb D, Smeltz RB. T-bet inhibits the in vivo differentiation of parasite-specific CD4+ Th17 cells in a T cell-intrinsic manner. J Immunol. 2009;182:6179–86. doi: 10.4049/jimmunol.0803821. [DOI] [PubMed] [Google Scholar]
- 39.Vanden Eijnden S, Goriely S, De Wit D, Goldman M, Willems F. Preferential production of the IL-12(p40)/IL-23(p19) heterodimer by dendritic cells from human newborns. Eur J Immunol. 2006;36:21–6. doi: 10.1002/eji.200535467. [DOI] [PubMed] [Google Scholar]
- 40.Basile JI, Geffner LJ, Romero MM, et al. Outbreaks of Mycobacterium tuberculosis MDR strains induce high IL-17 T-cell response in patients with MDR tuberculosis that is closely associated with high antigen load. J Infect Dis. 2011;204:1054–64. doi: 10.1093/infdis/jir460. [DOI] [PubMed] [Google Scholar]
- 41.Kenna TJ, Davidson SI, Duan R, et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive γ/δ T cells in patients with active ankylosing spondylitis. Arthritis Rheum. 2011;64:1420–9. doi: 10.1002/art.33507. [DOI] [PubMed] [Google Scholar]
- 42.Martin D, Tarleton R. Generation, specificity, and function of CD8+ T cells in Trypanosoma cruzi infection. Immunol Rev. 2004;201:304–17. doi: 10.1111/j.0105-2896.2004.00183.x. [DOI] [PubMed] [Google Scholar]
- 43.Tarleton RL, Koller BH, Latour A, Postan M. Susceptibility of β2-microglobulin-deficient mice to Trypanosoma cruzi infection. Nature. 1992;356:338–40. doi: 10.1038/356338a0. [DOI] [PubMed] [Google Scholar]
- 44.Tzelepis F, Persechini PM, Rodrigues MM. Modulation of CD4+ T cell-dependent specific cytotoxic CD8+ T cells differentiation and proliferation by the timing of increase in the pathogen load. PLoS ONE. 2007;2:e393. doi: 10.1371/journal.pone.0000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Padilla A, Xu D, Martin D, Tarleton R. Limited role for CD4+ T-cell help in the initial priming of Trypanosoma cruzi-specific CD8+ T cells. Infect Immun. 2007;75:231–5. doi: 10.1128/IAI.01245-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silva JS, Morrissey PJ, Grabstein KH, Mohler KM, Anderson D, Reed SG. Interleukin 10 and interferon gamma regulation of experimental Trypanosoma cruzi infection. J Exp Med. 1992;175:169–74. doi: 10.1084/jem.175.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kotner J, Tarleton R. Endogenous CD4+ CD25+ regulatory T cells have a limited role in the control of Trypanosoma cruzi infection in mice. Infect Immun. 2007;75:861–9. doi: 10.1128/IAI.01500-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levings MK, Gregori S, Tresoldi E, Cazzaniga S, Bonini C, Roncarolo MG. Differentiation of Tr1 cells by immature dendritic cells requires IL-10 but not CD25+ CD4+ Tr cells. Blood. 2005;105:1162–9. doi: 10.1182/blood-2004-03-1211. [DOI] [PubMed] [Google Scholar]
- 49.Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–65. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanamaru F, Youngnak P, Hashiguchi M, Nishioka T, Takahashi T, Sakaguchi S, Ishikawa I, Azuma M. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J Immunol. 2004;172:7306–14. doi: 10.4049/jimmunol.172.12.7306. [DOI] [PubMed] [Google Scholar]
- 51.Shinohara T, Taniwaki M, Ishida Y, Kawaichi M, Honjo T. Structure and chromosomal localization of the human PD-1 gene (PDCD1) Genomics. 1994;23:704–6. doi: 10.1006/geno.1994.1562. [DOI] [PubMed] [Google Scholar]
- 52.Ten Hove T, The Olle F, Berkhout M, Bruggeman JP, Vyth-Dreese FA, Slors JF, Van Deventer SJ, Te Velde AA. Expression of CD45RB functionally distinguishes intestinal T lymphocytes in inflammatory bowel disease. J Leukoc Biol. 2004;75:1010–5. doi: 10.1189/jlb.0803400. [DOI] [PubMed] [Google Scholar]
- 53.Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60–73. doi: 10.1111/j.0105-2896.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 54.Sztein MB, Kierszenbaum F. Mechanisms of development of immunosuppression during Trypanosoma infections. Parasitol Today. 1993;9:424–8. doi: 10.1016/0169-4758(93)90053-i. [DOI] [PubMed] [Google Scholar]
- 55.McGuirk P, McCann C, Mills KH. Pathogen-specific T regulatory 1 cells induced in the respiratory tract by a bacterial molecule that stimulates interleukin 10 production by dendritic cells: a novel strategy for evasion of protective T helper type 1 responses by Bordetella pertussis. J Exp Med. 2002;195:221–31. doi: 10.1084/jem.20011288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van der Kleij D, Van Remoortere A, Schuitemaker JH, Kapsenberg ML, Deelder AM, Tielens AG, Hokke CH, Yazdanbakhsh M. Triggering of innate immune responses by schistosome egg glycolipids and their carbohydrate epitope GalNAc β1-4(Fuc α1-2Fuc α1-3)GlcNAc. J Infect Dis. 2002;185:531–9. doi: 10.1086/338574. [DOI] [PubMed] [Google Scholar]
- 57.Golgher D, Gazzinelli RT. Innate and acquired immunity in the pathogenesis of Chagas disease. Autoimmunity. 2004;37:399–409. doi: 10.1080/08916930410001713115. [DOI] [PubMed] [Google Scholar]
- 58.Savino W, Villa-Verde DM, Mendes-da-Cruz DA, et al. Cytokines and cell adhesion receptors in the regulation of immunity to Trypanosoma cruzi. Cytokine Growth Factor Rev. 2007;18:107–24. doi: 10.1016/j.cytogfr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 59.Gutierrez FR, Mariano FS, Oliveira CJ, et al. Regulation of Trypanosoma cruzi-induced myocarditis by programmed death cell receptor 1. Infect Immun. 2011;79:1873–81. doi: 10.1128/IAI.01047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sales PA, Jr, Golgher D, Oliveira RV, Vieira V, Arantes RM, Lannes-Vieira J, Gazzinelli RT. The regulatory CD4+ CD25+ T cells have a limited role on pathogenesis of infection with Trypanosoma cruzi. Microbes Infect. 2008;10:680–8. doi: 10.1016/j.micinf.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 61.Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]