

Abstract
Familial hypercholesterolemia (FH) is a genetic disorder characterized by extremely high levels of plasma low-density lipoprotein (LDL), due to defective LDL receptor-apolipoprotein B (APOB) binding. Current therapies such as statins or LDL apheresis for homozygous FH are insufficiently efficacious at lowering LDL cholesterol or are expensive. Treatments that target APOB100, the structural protein of LDL particles, are potential therapies for FH. We have developed a series of APOB-directed splice-switching oligonucleotides (SSOs) that cause the expression of APOB87, a truncated isoform of APOB100. APOB87, like similarly truncated isoforms expressed in patients with a different condition, familial hypobetalipoproteinemia, lowers LDL cholesterol by inhibiting very low–density lipoprotein (VLDL) assembly and increasing LDL clearance. We demonstrate that these “APO-skip ” SSOs induce high levels of exon skipping and expression of the APOB87 isoform, but do not substantially inhibit APOB48 expression in cell lines. A single injection of an optimized APO-skip SSO into mice transgenic for human APOB resulted in abundant exon skipping that persists for >6 days. Weekly treatments generated a sustained reduction in LDL cholesterol levels of 34–51% in these mice, superior to pravastatin in a head-to-head comparison. These results validate APO-skip SSOs as a candidate therapy for FH.
Introduction
Familial hypercholesterolemia (FH) is a condition characterized by extremely high concentrations of low-density lipoprotein (LDL) cholesterol, most commonly due to genetic defects in the hepatic LDL receptor. Patients with the most severe form, homozygous FH, develop life-threatening cardiovascular disease (CVD) in early adulthood.1 Lowering LDL cholesterol is known to reduce mortality and morbidity, delaying the onset of CVD. However, current drug treatments for FH such as statins are insufficiently efficacious and are unable to reduce LDL cholesterol and the risks of CVD to baseline. Another therapy in use is LDL apheresis which is highly effective at reducing cholesterol levels, but is expensive and requires patients to attend specialist centers for treatment every 1–2 weeks.2 Alternative therapies to lower LDL cholesterol are needed.
Apolipoprotein B (APOB) has emerged as a key target for LDL cholesterol-lowering therapies. Two natural protein isoforms are generated from the APOB gene, APOB100 and APOB48. The principal structural apolipoprotein in LDL particles is the full-length APOB100 isoform. APOB100 is expressed in the liver and assembled into very low–density lipoprotein (VLDL) particles. These are catabolized to form LDL particles, which are normally recycled via the liver when APOB100 binds to the LDL receptor. Disruption of this interaction, e.g., due to mutations in LDL receptor or APOB100, cause accumulation of LDL particles in the circulation and hypercholesterolemia, ultimately leading to atherosclerosis and CVD. APOB48 is generated by cell-specific RNA editing of a CAA codon to a premature UAA termination codon in intestinal enterocytes and is identical to the N-terminal 48% of APOB100 (Figure 1). APOB48 is not assembled into VLDL particles but instead into chylomicrons, whose function is to transport dietary fat and fat-soluble vitamins from the intestine. Disruption of intestinal APOB48 expression and chylomicron assembly is characterized by fat intolerance and malabsorption of fat and fat-soluble vitamins such as vitamins A, D, E, and K.3,4 Ideally, any treatment aimed at reducing LDL cholesterol by interfering with APOB should be selective for APOB100 and should have no effect on APOB48.
Figure 1.

APOB gene structure with normal and exon-skipped protein isoforms. Exons 26–29 at the 3′ end of the APOB gene are shown with the respective introns represented by connecting lines. The constitutively expressed APOB100 isoform is translated from the APOB100 mRNA, which includes the 115 nt exon 27 (top). The shorter APOB48 isoform found in the intestine is translated from the APOB100 mRNA after RNA editing of a CAA to a UAA stop codon (marker in exon 26). Exclusion of exon 27 by exon skipping leads to the APOB87 mRNA with frame-shifted exon 28 and 29 sequences and a premature stop codon (marker in exon 28). Thus, a truncated APOB protein, APOB87, is translated. Exon 27 skipping does not interfere with the exon 26 sequence, so APOB48 RNA editing and expression can proceed as normal whether or not exon 27 is skipped.
In contrast to FH, the antithetical phenotype of familial hypobetalipoproteinemia results from expression of mutant, C-terminally truncated APOB isoforms and is characterized by VLDL cholesterol, protection from CVD, and increased longevity. These shortened APOB100 isoforms reduce VLDL assembly and secretion and increase LDL clearance from the bloodstream, leading to profoundly low LDL cholesterol levels typically <1.8 mmol/l, protection from atherosclerosis and a longer lifespan.5,6 In order to mimic hypobetalipoproteinemia for therapy, we have developed “APO-skip ” splice-switching oligonucleotides (SSOs) that induce the skipping of APOB exon 27 to generate a truncated APOB isoform, APOB87.7
Here, we show that sequence optimization of our previously published SSOs7 is able to increase the in vitro exon-skipping efficiency of APO-skip SSOs by up to 15-fold. We confirm that treatment induced expression of the truncated isoform APOB87 and that this did not affect APOB48 production. In mice transgenic for human APOB, intravenous injection of APO-skip SSOs resulted in exon 27 skipping which persisted for at least 6 days after a single dose. Weekly administration resulted in reduced LDL cholesterol compared to controls.
Results
In vitro optimization of “APO-skip ” SSOs increases their efficiency 15-fold
In the treatment of Duchenne muscular dystrophy with SSOs, targeting sequences within the exon is more effective than targeting splice sites.8 However, this strategy was not notably successful for inducing skipping of exon 27 in the APOB gene (Supplementary Figure S1). As previously demonstrated, SSOs can induce significantly more skipping of APOB exon 27 when directed against branch point binding sites, the polypyrimidine tract and the 3́ splice site of APOB exon 27.7
Therefore, starting with the most effective previously published oligonucleotide skip27-3B, herein designated SSO 1, we systematically optimized the SSO sequence and backbone modifications to increase efficiency. All SSOs were 2′-O-methyl RNA oligonucleotides unless otherwise stated.
For accurate quantification of the full-length (APOB100) and skipped (APOB87) mRNAs, we developed two reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR) assays specific for the APOB26–27 and APOB26–28 exon–exon junctions. As recommended by Vandenbroucke et al.,9 each assay used a single standard curve generated from a plasmid containing both the APOB26–27 and APOB26–28 PCR products. The plasmid was serially diluted resulting in a seven point standard curve of 107–101 copies per reaction under the assay conditions. The use of a single common standard curve in both assays ensures direct proportionality of the RT-qPCR copy number values and allows calculation of skipping efficiency as a ratio within each sample without the need for reference genes for normalization.
We selected the human hepatocellular carcinoma cell line HepG2 as our model system because these cells express APOB100 and have been shown to support exon skipping and production of the APOB87 isoform.7 Cells were transfected with the various SSOs as described in the methods, and exon-skipping efficiency was analyzed 48 hours later using the RT-qPCR assays described above. As shown in Figure 2, the prototype SSO 1 targets the 3′ splice site (splice acceptor) and two potential branch point sites in intron 26–27. The newly developed RT-qPCR assay demonstrated that SSO 1 has a relatively low skipping efficiency of 4.14% in comparison to its scrambled control, SSO sc1, or mock-transfected control (Figure 2). Attempts to reduce the SSO length or further illuminate the essential target sequences resulted in severe reductions in skipping efficiency in all cases (Figure 2—SSOs 3–13, 17–21). Addition of the intron 27–28 5′ splice site (splice donor) to the target sequence led to a significant increase in efficiency (SSO 14: 14.17%), while modification of the 5′ end with fluorescein (SSO 1-F) or biotin (SSO 1-B) also increased the skipping efficiency compared with the parental SSO 1 (Figure 2). Similarly, the SSO 14 sequence modified with 5′ fluorescein (SSO 14-F) and biotin (SSO 14-B) showed increased skipping efficiency in comparison to SSO 14. However, the largest rise in skipping efficiency occurred when the backbone of SSO 14 was modified with five phosphorothioate (PS) modifications on the 5′ and 3′ ends (SSO 14-53P: 58.73%). In comparison to the respective non-PS modified SSOs, full PS modification of SSO 14 (SSO 14-P) resulted in reduced efficiency, while full PS modification of SSO 1 (SSO 1-P) resulted in increased efficiency. Additional 5′ fluorescein modification on fully PS modified SSOs did not lead to further improvement in skipping levels for either the SSO 1 (SSO 1-PF) or the SSO 14 (SSO 14-PF) sequence.
Figure 2.
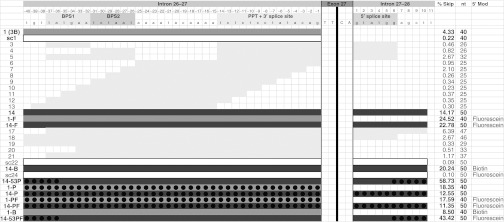
SSO design and in vivo skipping efficiency. The introns and splice elements surrounding exon 27 are shown in detail on the top row (exon 27 itself is shortened). The intronic splice elements necessary for RNA splicing are indicated: BPS, branchpoint sequence; PPT+3′ splice, polypyrimidine tract plus 3′ splice site; 5′ splice, 5′ splice site. The light gray and dark gray bars represent the targets for each indicated SSO. SSO 1 is the prototype targeting the BPS and the 3′ splice site. SSO 14 targets the BPS, 3′ splice site, and 5′ splice site. The black circles indicate the nucleotides that were modified with phosphorothioate, where used in SSOs 14-53P, 14-P, 1-PF, 14-PF, 14-53PF. For the determination of skipping efficiencies, total RNA was extracted from HepG2 cells 48 hours after transfection with 250 nmol/l of SSO. RT-qPCR was performed on the total RNA samples to determine the copy numbers of APOB26–28 and APOB26–27, and skipping efficiencies were calculated as % skipping (% Skip—3rd column from right) = APOB26–28/(APOB26–28 + APOB26–27) × 100%. Mean skipping efficiencies are presented in the figure and were as follows (mean ± SD, with n numbers referring to independent experiments): SSO 1 (4.33 ± 2.39%; n = 15), sc1 (0.22 ± 0.21%, n = 10), 14 (14.17 ± 8.15%, n = 15), 1-F (24.52 ± 5.73%, n = 8), 14-F (22.78 ± 4.87%, n = 8), 14-B (20.24 ± 10.46%, n = 4), 14-53P (58.73 ± 5.26%, n = 3), 1-P (18.35%, n = 2), 14-P (12.55 ± 5.19%, n = 3), 1-PF (17.59%, n = 2), 14-PF (11.35 ± 4.40%, n = 3), 1-B (8.50%, n = 2) and mock-transfected control (0.18 ± 0.15%; n = 17). The nucleotide length of each SSO is indicated in the 2nd column from right. The right-most column indicates the 5′ modification, where used in SSOs 1-F, 14-F, 14-B, sc24, 1-PF, 14-PF, 1-B, 14-53PF. RT-qPCR, reverse transcriptase-quantitative polymerase chain reaction; SSO, splice-switching oligonucleotides.
SSOs result in expression of the APOB87 protein isoform in HepG2 and Caco-2 cells
As previously reported, the exon 27-skipped APOB mRNA isoform escapes nonsense mediated decay and is translated into a secreted protein, APOB87, in HepG2 cells.7 Here, we confirm this finding in cultured Caco-2 cells, a human epithelial colorectal adenocarcinoma line, which after reaching confluence differentiate to resemble enterocytes of the small intestine.10 These cells have been used as a model for lipoprotein synthesis in the intestine and express both the APOB100 and APOB48 protein isoforms once cell confluence and differentiation is achieved.11,12 As with HepG2 cells, secretion of apolipoproteins can be stimulated by incubation with oleic acid.13 However, achieving efficient transfection on differentiated, fully confluent cells proved impossible, so we reverse transfected near-confluent Caco-2 cells, plated at high density, and incubated for 72 hours before collection of supernatant for western blotting and extraction of cellular RNA. Differential toxic effects of transfection resulted in different cell densities, in turn leading to inconsistent differentiation and consequent variation in the levels of APOB expression. Nevertheless, as shown in Figure 3a, transfection with each functional SSO (14, 1-F, 14-F, 14-53P and 14-53PF) clearly resulted in expression of the intended APOB87 isoform, while transfection with control scrambled SSOs sc14-53P and sc14-53PF did not. Mock-transfected cells also did not express the APOB87 isoform.
Figure 3.

Western blotting confirms expression of the exon-skipped APOB87 isoform. (a) Caco-2 and (b) HepG2 cells were transfected with 250 nmol/l of the stated SSOs, active or control scrambled as indicated, and one mock-transfected (–SSO) control was included. For western blot analysis of secreted APOB isoforms 200 µg in a and 250 µg in b of total protein was loaded per lane. APOB protein isoforms were detected using a polyclonal anti-APOB antibody which recognizes all APOB isoforms. The respective skipping efficiencies measured by RT-qPCR are shown below the blots. Diff, differentiated; RT-qPCR, reverse transcriptase-quantitative polymerase chain reaction; SSO, splice-switching oligonucleotides. Undiff, Undifferentiated.
The APOB48 isoform is clearly visible in the blots of cells treated with SSO 14, 1-F and 14-F showing that transfection with these oligonucleotides, and the resulting exon skipping, does not substantially inhibit APOB48 expression. The greater nonspecific toxicity resulting from Lipofectamine 2000 transfection of the longer PS modified SSOs resulted in a lower cell density and lower level expression of both APOB100 and APOB48 by Caco-2 cells treated with SSOs 14-53P and 14-53PF, sc14-53P and sc14-53PF (Figure 3a). Despite this, the data show that APOB48 expression was not substantially diminished, relative to APOB100, in the cells treated with functional SSOs 14-53P and 14-53PF when compared with their respective negative controls, sc14-53P and sc14-53PF.
Similar western blotting and skipping efficiency data are shown for HepG2 cells (Figure 3b). As expected, APOB48 was not expressed in these cells, but treatment with the functional SSOs 1, 14, and 14-F also resulted in the expression of the APOB87 isoform, whereas nonfunctional, scrambled control SSO sc1 and mock-transfected cells showed only the APOB100 isoform. The respective skipping efficiencies are given below each blot and clearly demonstrate that as little as 3% skipping results in detectable APOB87, while skipping levels of >10% lead to substantial expression and secretion of this isoform (Figure 3a,b).
SSOs induce exon skipping in vivo
Having shown that our best SSO candidates cause up to 60% skipping in HepG2 and Caco-2 cells, we proceeded to evaluate the skipping efficiency of selected SSOs in transgenic mice expressing human APOB.14 We chose to use SSO 14-53PF for these studies because it is a fluorescently labeled version of SSO 14-53P and thus allowed tracking of SSO delivery to the liver using fluorescence microscopy.
A standard IV injection of naked SSO 14-53PF (25 mg/kg) via the tail vein resulted in minimal delivery and skipping efficiencies not significantly above background, with little improvement seen with hydrodynamic injection (Supplementary Figure S2). Examination of tissues and urine with fluorescence microscopy showed that SSO 14-53PF was cleared rapidly in the urine (data not shown). This is likely to be due to the length of the SSO and the relatively small number of PS modifications used, leading to insufficient serum protein binding. In order to achieve efficient hepatic uptake, a cationic complexing reagent, Invivofectamine 2.0 (IVF2.0), was used which allowed efficient delivery to all hepatocytes at both 12.5 and 25 mg/kg doses (Supplementary Figure S2).
Measuring skipping efficiencies after IVF2.0 delivery (Figure 4a), the lower dose of 12.5 mg/kg resulted in relatively low efficiency 24 hours after injection (~6%). Doubling the dose to 25 mg/kg increased skipping efficiency by nearly fivefold (~29%), although the highest dose of 50 mg/kg did not provide further increases. As expected, control mice injected with vehicle only showed background level skipping. To investigate the hepatotoxicity of the treatment, we measured aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels. The vehicle only controls (IVF2.0 without SSO) showed a massive increase of AST (>3,500 U/l) and ALT (>2,800 U/l) levels over baseline (95 ± 23 U/l, n = 11 and 62 ± 17 U/l, n = 11; respectively). Similarly, large increases of AST and ALT levels were observed with the 50 mg/kg dose, but encouragingly for the 25 mg/kg group the increases were smaller (AST 959 ± 433 U/l, ALT 511 ± 316 U/l, n = 5), and for the 12.5 mg/kg group the increases were close to baseline values (AST 134 ± 35 U/l, ALT 60 ± 13 U/l, n = 3).
Figure 4.

Exon skipping in transgenic human APOB mice. Mice were injected intravenously via the tail vein with IVF2.0-formulated SSO at the indicated doses and killed at the indicated times for collection of tissue samples from liver. RNA was extracted and skipping efficiencies were determined by RT-qPCR. (a) Dose escalation study with SSO 14-53PF. Samples taken 24 hours after injection: 12.5 mg/kg (5.80 ± 3.65%), 25 mg/kg (29.07 ± 8.30%), 50 mg/kg (20.40 ± 11.80%) and vehicle only control consisting of IVF2.0 without complexed SSO (0.39 ± 0.13%). (b) Time course after single injection with 25 mg/kg of SSO 14-53PF: 24 hour (29.07 ± 8.30%, n = 5), 48 hour (22.04%, n = 2), 72 hour (20.57%, n = 2), and 6 days (7.55 ± 3.13%, n = 5). (c) SSO comparison, samples taken at 6 days after injection, n = 3 each group: 14-53P (5.95 ± 1.13%), 14-53PF (5.39 ± 0.35%), sc14-53P (scrambled control; 0.5 ± 0.05%), 1-F (0.72 ± 0.21%), and 14-F (0.72 ± 0.26%). (d) Quantification of APOB26–27 (27 inc), APOB26–28 (skip), APOB25–26 (total), and mouse Apob (ms) in mice with successful treatment (SSO 14-53P and 14-53PF) relative to control/unsuccessful treatment (SSO sc14-53PF/1-F and 14-F), presented as whisker plots, where the line within the box marks the median value, the box denotes the interquartile range (25th–75th centile) and the whiskers show the maximum and minimum observations. IVF2.0, Invivofectamine 2.0; RT-qPCR, reverse transcriptase-quantitative polymerase chain reaction; SSO, splice-switching oligonucleotides.
Based on these results, we selected the 25 mg/kg dose as the best combination of high efficiency and acceptable liver toxicity. We then investigated the persistence of the effects of SSO treatment. After injection of a 25 mg/kg dose, exon-skipping levels were the highest after 24 hours (29%), dropped by only a third within 72 hours, and after 144 hours (6 days), exon-skipping levels still remained above 5% (Figure 4b).
To allow for the possibility that in vivo exon-skipping efficiencies are only partly predictive of the results of in vitro experiments, we compared several SSOs from our in vitro screen by measuring exon skipping 6 days after a single injection with SSO. As shown in Figure 4c, only SSOs 14-53P and 14-53PF demonstrated efficacy at this time point with no significant difference in efficiency between them. We performed relative quantification of splice variant expression levels between successful treatment (SSO 14-53P and 14-53PF) and control/unsuccessful treatment (SSO sc14-53PF/1-F and 14-F). Murine B2M and ACTB were used as reference genes after geNORM analysis established these as the two most stable (V2/3 = 0.103).15 Calculation with the REST software16 demonstrated significant tenfold upregulation of the exon 27 skipped isoform (APOB26–28 assay; P = 0.001), whereas the expression levels of the exon 27 included isoform (huAPOB26–27), total human APOB (APOB25–26), and murine Apob did not change (Figure 4d).
Skipping of exon 27 results in reduction of LDL
Having determined that exon skipping persists for at least 6 days after a single dose, we investigated whether 8 weekly treatments would change the plasma lipid profile. To reduce toxicity, we elected to decrease the dose slightly from 25 mg/kg to 20 mg/kg. This proved successful, as no significant differences in AST or ALT levels were found when comparing treatment groups (SSO 14-53P, SSO sc14-53P, and pravastatin) with the untreated control group. With the functional SSO 14-53P this regimen resulted in a modest 6.76% skipping efficiency when sampled 72 hours after the last injection (Figure 5a). As anticipated, skipping levels after treatment with control SSO sc14-53P or 0.25 mg/ml pravastatin in drinking water did not differ from levels in the untreated control group.
Figure 5.

Exon skipping induces LDL reduction in transgenic human APOB mice. Transgenic human APOB mice were injected with weekly doses of 20 mg/kg SSOs as indicated over 8 weeks, and blood samples were taken 2 weeks before the first injection and 3 days after the 3rd, 6th, and 8th injections. Animals were killed 3 days after the last injection, and liver tissue samples were taken for RNA extraction and subsequent RT-qPCR. (a) Skipping efficiencies: SSO 14-53P (6.76 ± 3.55%; n = 9), SSO sc14-53P (scrambled control; 0.34 ± 0.08%; n = 6), untreated control group (UT; 0.25 ± 0.03%; n = 6), and 0.25 mg/ml pravastatin in water ad libitum (statin; 0.25 ± 0.02%; n = 6). (b) Quantification of APOB26–27 (27 inc), APOB26–28 (skip), APOB25–26 (total), and mouse Apob (ms) in SSO 14-53P treated mice relative to untreated and SSO 14-53P controls, presented as whisker plots as in Figure 4d. (c) Baseline-corrected reduction in ΔLDL values presented. The percentage change in LDL cholesterol was calculated for each mouse at each timepoint using the following formula: 100% × (LDLtimepoint − LDLbaseline)/LDLbaseline. The mean of these percentage changes for the placebo groups (SSO sc14-53P serving as control for 14-53P; untreated group serving as control for the pravastatin group) was calculated as the expected baseline change for LDL cholesterol. This baseline change was then subtracted from the percentage changes observed for the experimental groups (SSO 14-53P and pravastatin): week 3 SSO 14-53P (black; −51.08 ± 19.37%) and pravastatin (gray; −13.02 ± 13.88%), week 6 SSO 14-53P (−33.50 ± 23.11%) and pravastatin (−4.35 ± 5.50%), and week 8 SSO 14-53P (−34.44 ± 11.81%) and pravastatin (−2.86 ± 9.38%). LDL, low-density lipoprotein; RT-qPCR, reverse transcriptase-quantitative polymerase chain reaction; SSO, splice-switching oligonucleotides.
Relative quantification of treatment with SSO 14-53P versus the control SSO sc14-53P using murine GAPDH, HPRT, SDHA, B2M, and ACTB as reference genes (geNORM V4/5 = 0.007) showed statistically significant 18-fold upregulation of the exon 27 skipped isoform (P = 0.001). The expression levels of the exon 27 included isoform, total human APOB and murine Apob did not differ between SSO 14-53P and control treatments (Figure 5b).
To assess the effects of SSO treatments on lipid concentrations, we measured LDL cholesterol in serum before commencing treatment and after 3, 6, and 8 weekly doses of SSO/IVF2.0 complex. The lipid analysis was complicated by a confounding nonspecific effect of IVF2.0, which results in an increase in measured LDL cholesterol (Supplementary Figure S3). Nevertheless, by baseline-correcting the ΔLDL values achieved during treatment (SSO 14-53P and pravastatin) with their respective control values (ΔLDL of SSO sc14-53P and no treatment, respectively), we show that APO-skip treatment with SSO 14-53P resulted in a 51% relative reduction after three injections and a sustained 34% relative reduction over the rest of the test period (Figure 5c). In contrast, treatment with pravastatin decreased LDL cholesterol by 8–12% over time.
Discussion
The potential therapeutic use of SSOs to modify splicing was first described in 1993, to correct aberrant processing of mutant β-globin pre-mRNAs,17 but has been developed furthest for treatment of Duchenne muscular dystrophy where promising data from a Phase 2 study have been published recently.18 For exon skipping in Duchenne muscular dystrophy, SSOs targeting exonic sequences such as exonic splice enhancers (ESEs) have been more efficient than SSOs targeting donor and acceptor sites in the flanking introns.8 In the case of APOB exon 27, the converse appears to be true, as to date a number of SSOs targeting exon sequences, some of them designed according to guidelines published by Aartsma-Rus,19 have shown little activity (Khoo et al.7 and Supplementary Figure S1). This may reflect the lack of alternative splicing of APOB and/or the extraordinary length of APOB exon 26; notably, the first half of exon 27 contains curiously few matches for ESEs and many for splicing silencer motifs according to Human Splicing Finder.20
RNA structure modeling of the exon 27 sequence plus 50 nucleotides from the flanking introns with mfold21 and RNA structure22 suggests that the 3′ end of intron 26–27 remains single stranded, allowing effective binding of 3′ spliceosome components. The beginning of exon 27 shows a high probability of a short stem-loop structure with the actual splice site situated at the apex of the loop followed by a 10 nucleotide unstructured region and a more complex stem-loop presenting a SRp55 ESE motif. The 3′ half of exon 27 is largely predicted to form double-stranded structures with the beginning of intron 27–28, but again displaying the 5′ splice site in a short loop. Bimolecular RNA folding with various SSOs suggests that the exon-internal SSOs do not change the single-stranded nature of the intron 26–27 3′ end, the 3′ splice site loop or the double-stranded exon 27/intron 27–28 region. Although the SRp55 ESE motif can be masked with exonic SSOs, this is clearly not sufficient for effective exon skipping. In contrast, SSO 1 is predicted to sequester the 3′ end of intron 26–27 into a double strand, forcing the 3′ splice site loop to move along into the exon 27 sequence closer to the SRp55 ESE. Thus, the actual 3′ splice site is hidden while SRp55 binding may be inhibited by the proximity of the 3′ loop. Still, the double-stranded nature of exon 27/intron 27–28 border region remains unchanged. Only when this border region is modified to a more open single-stranded formation and the 5′ splice site is blocked in addition, as is predicted to be the case with SSO 14-53P, could we achieve significantly higher exon-skipping efficiencies.
We found that two types of modification appear to increase SSO exon-skipping efficiency. First, partial PS modification increased exon skipping, whereas full PS modification seemed to reduce this somewhat. PS modification increases oligonucleotide stability to nucleases23 but reduces binding affinity to RNA. This can be overcome by using a “mixed-backbone ” strategy as applied here with the partial PS modification to the 5′ and 3′ ends of our SSOs.24 Second, SSOs bearing 5′ fluorescein or biotin compared to nonmodified SSOs of the same sequence were more efficient at exon skipping. This observation has not been reported before for other exons. A possible explanation for this phenomenon might be that the 5′ fluorescein group is more hydrophobic and enables charged SSO translocation across the membrane, although this cannot explain the effectiveness of 5′ biotinylation, as biotin is a relatively polar moiety. A more unifying explanation for our observations is that the presence of these groups near the 5′ splice site of exon 27 interferes with recognition of this splice site, consistent with our structural view of APOB exon 27 skipping. Although early attempts to reduce the length of the resulting 50-mer SSO led to severe efficiency losses, our more recent experiments which took RNA structure modeling into account suggest that exon-skipping efficiencies comparable to 14-53P can be achieved with much shorter SSOs (P. Disterer et al., unpublished data). These shorter SSOs, together with more extensive PS modification, should also lead to more efficient hepatocyte delivery when given “naked ” without the requirement for a delivery reagent. However, the extent of PS modifications must be balanced against the propensity of these polyanionic modifications to trigger nonspecific effects by binding to other plasma proteins.24
However achieved, the downregulation of APOB100 expression is a validated means of reducing LDL cholesterol. This is particularly important in homozygous FH patients who present with extremely high LDL cholesterol levels, resulting in premature CVD morbidity and mortality in late childhood or early adulthood.25 Unfortunately, aggressive treatment with potent statins achieves only around 25% decline in LDL in this population since statins mainly work through upregulation of LDL receptor in the liver, which is nonfunctional in FH patients. Nevertheless, even small LDL cholesterol reductions that do not come close to achieving normal levels are directly related to significant survival benefits, and further decreases are likely to accrue even more benefits.25 Our in vivo data demonstrate that APO-skip SSOs resulted in a sustained 34–51% drop in LDL levels in human transgenic APOB mice, compared to control SSOs. This large reduction was associated with a relatively low exon-skipping efficiency of 6.8%. This seemingly paradoxical finding may be explained by the similarity of APOB87 to truncated human APOB100 isoforms such as APOB89 and APOB87-Padova. In subjects expressing these truncated isoforms, cholesterol levels are well below normal, typically <1.7 mmol/l,26,27,28 due to greater catabolic rates of particles containing the shortened APOB isoforms.29 Aguilar-Salinas et al.30 also report that production rates of VLDL-APOB100 and LDL-APOB100 particles in truncated APOB heterozygotes were ~30% of normal, suggesting a dominant negative effect. Thus, even low levels of exon skipping will likely be enough for significant therapeutic effects, suggesting that lower doses of APO-skip treatment will achieve clinically relevant effects compared to other oligonucleotide-based therapies in development.
Current approaches to the reduction of APOB100 expression include antisense gap-mer oligonucleotides such as mipomersen,31 locked nucleic acid oligonucleotides developed by Santaris,32 and RNA interference variants such as an AAV-delivered shRNA33 or liposome-packed siRNA.34 None of these approaches is selective for APOB100, and each could potentially cause downregulation of all APOB expression, including the APOB48 isoform necessary for fat and fat-soluble vitamin absorption. It is claimed that this is not an issue due to tissue tropism33 or the natural distribution to liver when SSO is injected subcutaneously.35 However, a study in mice, using the mouse equivalent of mipomersen, showed a 54% downregulation of intestinal APOB100 mRNA, although curiously APOB48 protein levels appeared unchanged.36
A conceivable future oral formulation of SSO or siRNA to replace subcutaneous injections would certainly bring the SSO or siRNA in contact with intestinal cells. Downregulation of APOB48 in the intestine would thus expose patients to the side-effect of fat and fat-soluble vitamin malabsorption. In contrast, our approach does not interfere with APOB48 expression, thus avoiding these potential side effects. This is consistent with the fact that normal APOB48 expression is found in subjects with familial hypobetalipoproteinemia expressing a truncated APOB89 isoform.37
In summary, APO-skip SSOs are able to reduce LDL cholesterol significantly even at relatively low exon-skipping efficiencies and offer advantages over other therapies in current development. At the present early stage, the SSO designs presented here are not practical therapies due to the length of the SSO and requirement for vehicle. To translate the APO-skip concept to the bedside, we anticipate that shortening the SSOs with sequence optimization and consideration of other oligonucleotide chemistries will eventually enable hepatic delivery without vehicle and with reduced manufacturing costs; our designs offer a basis from which to develop these newer generation APO-skip SSOs.
Materials and Methods
Materials. HepG2 (85011430) and Caco-2 (86010202) cells were acquired from the European Collection of Cell Cultures (Salisbury, UK) and used for no more than 25 passages. Cell culture media, FBS, GlutaMAX, nonessential amino acids, Lipofectamine 2000, Opti-MEM, pCR2.1, Invivofectamine 2.0, and all western blotting reagents were obtained from Life Technologies (Paisley, UK); phosphate-buffered saline from the Royal Free Hospital Pharmacy (London, UK); insulin (Actrapid) from Novo Nordisk (Crawley, UK); and all other reagents from Sigma Aldrich (Gillingham, UK) except where stated. All reagents, machines, and consumables for RNA extraction and RT-qPCR were from Qiagen (Crawley, UK) unless otherwise noted. SSOs and DNA primers were purchased from Eurogentec (Fawley, UK) or Integrated DNA Technologies (Coralville, IA) and their sequences can be found in Figure 2, Supplementary Figure S1 and Supplementary Table S1. Human APOB transgenic mice14 (B6.SJL-Tg(APOB)1102Sgy N20+?) were obtained from Taconic (Germantown, NY) and given standard chow and water ad libitum. All studies were approved by the UK Home Office under the Animals Scientific Procedures Act 1986. Mice were intravenously injected via the tail vein and killed by cervical dislocation.
Cell culture and transfection, RNA extractions, RNA quality control, reverse transcription, qPCR and standard curve creation. These methods have been described in detail in Disterer and Khoo,38 but may also be found in the Supplementary Materials and Methods. MIQE guideline39 compliant data are available in Supplementary Tables S1,S2 and Supplementary Figure S4.
Western blot analysis of APOB isoforms. Caco-2 cells were seeded at 1 × 107 cells in 10 cm plates and transfected as described above. For protein extraction, plates were washed three times with phosphate-buffered saline containing Complete Protease Inhibitor Cocktail tablets (Roche, Burgess Hill, UK) and then incubated with 5 ml Opti-MEM I containing 100 µg per ml oleic acid/bovine serum albumin for 4 hours to encourage APOB synthesis and secretion. The supernatant was collected and stored at −80 °C. The supernatants were concentrated tenfold using Vivaspin 2 columns with polyethersulfone membranes and a molecular weight cutoff of 30,000 Da (Sartorius Stedim, Binbrook, UK). Protein concentration was determined using a Novagen BCA kit from Merck (Feltham, UK) as per the provided instructions. The appropriate amount of total protein (20–25 µg) in water containing 1x NuPAGE LDS sample buffer and reducing agent was denatured at 70 °C for 10 minutes and then loaded onto a NuPAGE Novex 3–8% Tris-Acetate mini gel together with HiMark prestained protein standard and human APOB protein (ab77903; Abcam, Cambridge, UK). Gels were run in NuPAGE Tris-Acetate SDS running buffer containing 1x NuPAGE Antioxidant at a constant 150 V for 90–120 minutes. Proteins were transferred from the gel to a Immobilon-P PVDF membrane (Millipore, Watford, UK) in ice-cold CAPS buffer [pH 11.0] containing 10% ethanol, 0.01% SDS, and 1x NuPAGE Antioxidant with an electric potential of 55 V for 10 minutes and 30 V for 1 hour. Membranes were blocked overnight in 5% nonfat milk and 0.1% Tween-20 and then incubated with a polyclonal goat antihuman APOB antibody (ab742, Millipore) at a 1:3,000 dilution for 1 hour at RT. The membrane was then stained with antigoat IgG-HRP (A5420, Sigma Aldrich) secondary antibody at a 1:10,000 dilution for 1 h at RT. Protein bands were visualized with the ECL western blot detection kit and ECL Hyperfilm (GE Healthcare, Chalfont St Giles, UK).
IVF2.0 oligonucleotide complexation. SSOs were complexed with IVF2.0 for liver-specific delivery in vivo. SSOs at a concentration of 3 mg/ml were diluted with complexation buffer supplied with IVF2.0 to achieve 1.5 mg/ml. This was then added directly to an equal volume of IVF2.0 in 50 ml tubes to obtain a 1 ml IVF2.0 to 1.5 mg SSO ratio and immediately vortexed. This solution was then incubated at 50 °C under constant rotation for 30 minutes. After incubation 14–17 volumes of phosphate-buffered saline were added and 12 ml of this was transferred into a prewashed Amicon Ultra-15 Centrifugal Devices with Ultracel-50 membrane (Millipore). The Amicon filter was centrifuged at 1,600g until the volume was reduced to <1 ml and another 12 ml of the diluted IVF2.0/SSO complexation solution were added. The process was repeated until the whole volume was concentrated to 2–4 mg/ml, as appropriate to achieve the required dose in 150–200 µl of injection volume.
Lipoprotein profiles and AST/ALT levels. For cholesterol profiles, mice were fasted for 2 hours before blood collection. Blood was collected in microcentrifuge tubes and allowed to clot at room temperature for at least 2 hours or at 4 °C overnight (consistent within experiment). Samples were spun at 9,500g at 4 °C for 10 minutes and then the supernatant was carefully transferred to fresh tubes to avoid carryover of erythrocytes. This step was repeated with the supernatant samples. Sera were aliquoted and stored at −80 °C. For analysis, aliquots were sent to the MRC Mary Lyon Centre (Harwell, UK). Samples were diluted 1:3 and analyzed on Olympus AU400 Clinical Chemistry (Olympus, Nyon, Switzerland) platform assays.
Data analysis. Data analysis was performed using GraphPad Prism 5 (GraphPad, San Diego, CA). Values in text and figures reflect the mean ± SD unless otherwise stated. Statistical significance was determined with one-tailed unpaired t-test when comparing two groups only or else with analysis of variance using the Bonferroni post-hoc test.
SUPPLEMENTARY MATERIAL Figure S1. SSO designs targeting APOB exon 27 sequences. Figure S2. Delivery of SSO with standard IV injection, hydrodynamic (HD) injection, and Invivofectamine (IVF) 2.0. Figure S3. Invivofectamine 2.0 treatment increases LDL cholesterol. Figure S4. Validation of RT-qPCR assays APOB26–27 and APOB26–28. Table S1. RT-qPCR assays. Table S2. PCR products of the APOB26–27 and APOB26–28 RT-qPCR assays. Materials and Methods.
Acknowledgments
The authors thank Xavier de Mollerat du Jeu (Life Technologies) for technical advice with IVF2.0 and Tertius Hough (MRC Mary Lyon Centre, Harwell, UK) for performing the serum analyses. This work was supported by the MRC Developmental Pathway Funding Scheme (G0802469) and by UCL Business. B.K., J.S.O., J.P.S., and P.D. are inventors of an associated patent. The authors declared no conflict of interest.
Supplementary Material
References
- Marks D, Thorogood M, Neil HA., and, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis. 2003;168:1–14. doi: 10.1016/s0021-9150(02)00330-1. [DOI] [PubMed] [Google Scholar]
- Thompson GR, HEART-UK LDL Apheresis Working Group Recommendations for the use of LDL apheresis. Atherosclerosis. 2008;198:247–255. doi: 10.1016/j.atherosclerosis.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Tarugi P, Averna M, Di Leo E, Cefalù AB, Noto D, Magnolo L.et al. (2007Molecular diagnosis of hypobetalipoproteinemia: an ENID review Atherosclerosis 195e19–e27. [DOI] [PubMed] [Google Scholar]
- Bouma ME, Beucler I, Aggerbeck LP, Infante R., and, Schmitz J. Hypobetalipoproteinemia with accumulation of an apoprotein B-like protein in intestinal cells. Immunoenzymatic and biochemical characterization of seven cases of Anderson's disease. J Clin Invest. 1986;78:398–410. doi: 10.1172/JCI112590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glueck CJ, Gartside PS, Mellies MJ., and, Steiner PM. Familial hypobeta-lipoproteinemia: studies in 13 kindreds. Trans Assoc Am Physicians. 1977;90:184–203. [PubMed] [Google Scholar]
- Schonfeld G. Familial hypobetalipoproteinemia: a review. J Lipid Res. 2003;44:878–883. doi: 10.1194/jlr.R300002-JLR200. [DOI] [PubMed] [Google Scholar]
- Khoo B, Roca X, Chew SL., and, Krainer AR. Antisense oligonucleotide-induced alternative splicing of the APOB mRNA generates a novel isoform of APOB. BMC Mol Biol. 2007;8:3. doi: 10.1186/1471-2199-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A, Houlleberghs H, van Deutekom JC, van Ommen GJ., and, 't Hoen PA. Exonic sequences provide better targets for antisense oligonucleotides than splice site sequences in the modulation of Duchenne muscular dystrophy splicing. Oligonucleotides. 2010;20:69–77. doi: 10.1089/oli.2009.0215. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke II, Vandesompele J, Paepe AD., and, Messiaen L. Quantification of splice variants using real-time PCR. Nucleic Acids Res. 2001;29:E68–E68. doi: 10.1093/nar/29.13.e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy E, Mehran M., and, Seidman E. Caco-2 cells as a model for intestinal lipoprotein synthesis and secretion. FASEB J. 1995;9:626–635. doi: 10.1096/fasebj.9.8.7768354. [DOI] [PubMed] [Google Scholar]
- Reisher SR, Hughes TE, Ordovas JM, Schaefer EJ., and, Feinstein SI. Increased expression of apolipoprotein genes accompanies differentiation in the intestinal cell line Caco-2. Proc Natl Acad Sci USA. 1993;90:5757–5761. doi: 10.1073/pnas.90.12.5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner RD, Krul ES, Moberly JB, Alpers DH., and, Schonfeld G. Apolipoprotein expression and cellular differentiation in Caco-2 intestinal cells. Am J Physiol. 1992;263 2 Pt 1:E374–E382. doi: 10.1152/ajpendo.1992.263.2.E374. [DOI] [PubMed] [Google Scholar]
- Moberly JB, Cole TG, Alpers DH., and, Schonfeld G. Oleic acid stimulation of apolipoprotein B secretion from HepG2 and Caco-2 cells occurs post-transcriptionally. Biochim Biophys Acta. 1990;1042:70–80. doi: 10.1016/0005-2760(90)90058-6. [DOI] [PubMed] [Google Scholar]
- Linton MF, Farese RV, Jr, Chiesa G, Grass DS, Chin P, Hammer RE.et al. (1993Transgenic mice expressing high plasma concentrations of human apolipoprotein B100 and lipoprotein(a) J Clin Invest 923029–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A.et al. (2002Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes Genome Biol 3RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW., and, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominski Z., and, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc Natl Acad Sci USA. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K.et al. (2011Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study Lancet 378595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A. Overview on AON design. Methods Mol Biol. 2012;867:117–129. doi: 10.1007/978-1-61779-767-5_8. [DOI] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M., and, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JS., and, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke ST. Progress in antisense technology. Annu Rev Med. 2004;55:61–95. doi: 10.1146/annurev.med.55.091902.104408. [DOI] [PubMed] [Google Scholar]
- Agrawal S, Jiang Z, Zhao Q, Shaw D, Cai Q, Roskey A.et al. (1997Mixed-backbone oligonucleotides as second generation antisense oligonucleotides: in vitro and in vivo studies Proc Natl Acad Sci USA 942620–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raal FJ, Pilcher GJ, Panz VR, van Deventer HE, Brice BC, Blom DJ.et al. (2011Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy Circulation 1242202–2207. [DOI] [PubMed] [Google Scholar]
- Krul ES, Kinoshita M, Talmud P, Humphries SE, Turner S, Goldberg AC.et al. (1989Two distinct truncated apolipoprotein B species in a kindred with hypobetalipoproteinemia Arteriosclerosis 9856–868. [DOI] [PubMed] [Google Scholar]
- Talmud P, King-Underwood L, Krul E, Schonfeld G., and, Humphries S. The molecular basis of truncated forms of apolipoprotein B in a kindred with compound heterozygous hypobetalipoproteinemia. J Lipid Res. 1989;30:1773–1779. [PubMed] [Google Scholar]
- Gabelli C, Bilato C, Martini S, Tennyson GE, Zech LA, Corsini A.et al. (1996Homozygous familial hypobetalipoproteinemia. Increased LDL catabolism in hypobetalipoproteinemia due to a truncated apolipoprotein B species, apo B-87Padova Arterioscler Thromb Vasc Biol 161189–1196. [DOI] [PubMed] [Google Scholar]
- Parhofer KG, Barrett PH, Aguilar-Salinas CA., and, Schonfeld G. Positive linear correlation between the length of truncated apolipoprotein B and its secretion rate: in vivo studies in human apoB-89, apoB-75, apoB-54.8, and apoB-31 heterozygotes. J Lipid Res. 1996;37:844–852. [PubMed] [Google Scholar]
- Aguilar-Salinas CA, Barrett PH, Parhofer KG, Young SG, Tessereau D, Bateman J.et al. (1995Apoprotein B-100 production is decreased in subjects heterozygous for truncations of apoprotein B Arterioscler Thromb Vasc Biol 1571–80. [DOI] [PubMed] [Google Scholar]
- Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC.et al. (2010Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial Lancet 375998–1006. [DOI] [PubMed] [Google Scholar]
- Straarup EM, Fisker N, Hedtjärn M, Lindholm MW, Rosenbohm C, Aarup V.et al. (2010Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates Nucleic Acids Res 387100–7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koornneef A, Maczuga P, van Logtenstein R, Borel F, Blits B, Ritsema T.et al. (2011Apolipoprotein B knockdown by AAV-delivered shRNA lowers plasma cholesterol in mice Mol Ther 19731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN.et al. (2006RNAi-mediated gene silencing in non-human primates Nature 441111–114. [DOI] [PubMed] [Google Scholar]
- Yu RZ, Lemonidis KM, Graham MJ, Matson JE, Crooke RM, Tribble DL.et al. (2009Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100 Biochem Pharmacol 77910–919. [DOI] [PubMed] [Google Scholar]
- Crooke RM, Graham MJ, Lemonidis KM, Whipple CP, Koo S., and, Perera RJ. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J Lipid Res. 2005;46:872–884. doi: 10.1194/jlr.M400492-JLR200. [DOI] [PubMed] [Google Scholar]
- Krul ES, Tang J, Kettler TS, Clouse RE., and, Schonfeld G. Lengths of truncated forms of apolipoprotein B (apoB) determine their intestinal production. Biochem Biophys Res Commun. 1992;189:1069–1076. doi: 10.1016/0006-291x(92)92313-m. [DOI] [PubMed] [Google Scholar]
- Disterer P., and, Khoo B. Antisense-mediated exon-skipping to induce gene knockdown. Methods Mol Biol. 2012;867:289–305. doi: 10.1007/978-1-61779-767-5_19. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M.et al. (2009The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments Clin Chem 55611–622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.