

Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disorder affecting ∼1 % of people over the age of 65. Neuropathological hallmarks of PD are prominent loss of dopaminergic (DA) neurons in the substantia nigra and formation of intraneuronal protein inclusions termed Lewy bodies, composed mainly of α-synuclein (αSyn). Missense mutations in αSyn gene giving rise to production of degradation-resistant mutant proteins or multiplication of wild-type αSyn gene allele can cause rare inherited forms of PD. Therefore, the existence of abnormally high amount of αSyn protein is considered responsible for the DA neuronal death in PD. Normally, αSyn protein localizes to presynaptic terminals of neuronal cells, regulating the neurotransmitter release through the modulation of assembly of soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex. On the other hand, of note, pathological examinations on the recipient patients of fetal nigral transplants provided a prion-like cell-to-cell transmission hypothesis for abnormal αSyn. The extracellular αSyn fibrils can internalize to the cells and enhance intracellular formation of protein inclusions, thereby reducing cell viability. These findings suggest that effective removal of abnormal species of αSyn in the extracellular space as well as intracellular compartments can be of therapeutic relevance. In this review, we will focus on αSyn-triggered neuronal cell death and provide possible disease-modifying therapies targeting abnormally accumulating αSyn.
Keywords: α-Synuclein, Apoptosis, Dopaminergic neuron, Neuroprotection, Parkinson’s disease, Substantia nigra
Introduction
Parkinson’s disease (PD) is an age-related and the second most common neurodegenerative disorder beyond Alzheimer’s disease [1]. Clinical manifestation of PD is typical movement abnormalities that include resting tremor, rigidity, bradykinesia/akinesia, and postural instability. Neuropathological hallmarks in PD brains are (1) a prominent loss of dopaminergic (DA) neurons in the substantia nigra (SN) pars compacta (SNpc) projecting into the caudate/putamen (collectively called as striatum), and (2) formation of protein inclusions termed Lewy bodies and Lewy neurites that can be found in neuronal somas and processes, respectively. These aggregates are composed mainly of α-synuclein (αSyn) protein [2, 3]. Severe deprivation of striatal dopamine in PD can most effectively be treated with oral administration of dopamine precursor levodopa, whereas a long-term and pulsatile treatment with levodopa gradually induces adverse involuntary movements such as motor fluctuations and dyskinesias [4]. On the other hand, neurosurgical procedures including deep brain stimulation can partially normalize neuronal activities that have been agitated by the loss of the nigrostriatal DA pathway [5]. However, there have been no therapeutic options available that can reverse or even retard the progression of the disease, and such treatments are urgently required. To date, numerous efforts have been concentrated to elucidate the molecular mechanisms underlying the DA cell death in PD. In this article, we will review the relationship between abnormal αSyn and neuronal cell death. Several key molecules that can modulate the αSyn-induced neuronal death have hitherto been identified and investigated in αSyn-related animal models. We will also discuss such neuroprotective remedies for potential clinical interventions in PD (summarized in Fig. 1).
Fig. 1.
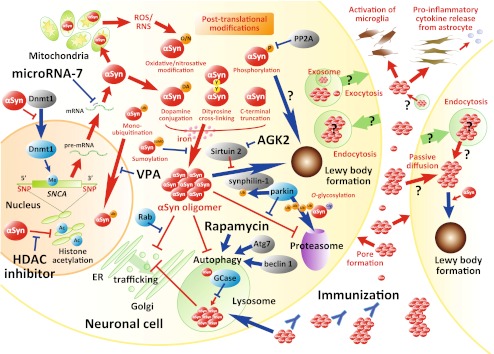
Schematic representation of molecular events and potential therapeutic targets associated with abnormal αSyn in PD. The molecular events that are reduced in PD and/or potentially neuroprotective, or considered to be neurotoxic, are shown in blue, or red arrows and inhibitory lines, respectively. Accumulation of αSyn oligomer, which can be modulated by several post-translational modification(s) of αSyn, leads to reduced neuronal cell viability by inhibiting ER-Golgi trafficking, autophagy, and/or proteasome. Mitochondrial translocation of αSyn induces production of ROS and RNS, further enhancing oxidative/nitrosative modification of αSyn. Oligomerized αSyn species can also be secreted into extracellular space, which might induce inflammatory glial reactions, pore formation on plasma membrane, or transmission to the neighboring neuronal cells to promote Lewy formation and/or cell death. These neurotoxic events can be ameliorated by several ways as indicated (also see the main text)
Neuronal Cell Death in PD Brains: Apoptotic or Non-apoptotic?
The way in which DA neurons die is the principal enigma in the field of PD research. In neurodegenerative environments, neurons die through distinct fashions that are distinguished by morphological features: (1) apoptosis (known as type 1 cell death) [6–16], (2) autophagy (type 2 cell death) [9–11], and (3) necrosis (type 3 cell death) or “necroptosis” [12–16]. Apoptosis is evolutionally conserved cell-suicide mechanism indispensable for fundamental biological processes such as normal development, elimination of malignant neoplasms, and establishment of neuronal circuitry [6]. The morphologic features of apoptosis include nuclear and cytoplasmic condensation, internucleosomal DNA cleavage, and packaging of the dying cell into apoptotic bodies that are engulfed by phagocytes, preventing release of intracellular components [7]. Pathogenic apoptosis cascade can be induced by (1) mitochondrial damage that involves B cell lymphoma 2 (Bcl-2) family proteins, apoptotic protease-activating factor 1 (apaf-1), and the cysteine proteases caspases (referred to as intrinsic pathway); and (2) agonistic ligands of death receptors such as tumor necrosis factor α (TNFα), Fas ligand (FasL), and TNF-related apoptosis-inducing ligand (TRAIL), which promote activation of caspase-8 inside the cell (extrinsic pathway) [10]. The involvement of apoptotic cascade in DA neuronal death has been controversial in PD [17–25]. We and other groups have previously reported the positive staining of DA neurons in PD for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and chromatin condensation, which is the typical process seen in apoptotic cell death [17, 21, 22]. However, other groups found no signs of apoptosis in the nigral DA neurons, regardless of disease duration, severity, drug treatment, and age of the patient [19, 20]. Using electron microscopy, Anglade et al. [18] showed the presence of condensed chromatin in the nucleus of neuromelanin-containing neurons and engulfment of apoptotic bodies in glial cells. Importantly, they also observed cells displaying the features of autophagic degeneration, implying that apoptosis may not be the sole form involved in DA neuronal death [18].
Autophagy is an evolutionally conserved mechanism for a bulk degradation of cellular components, including proteins and organelles, and serves as a cell survival mechanism during nutrient deprivation [9]. There exists a complex crosstalk between apoptosis and autophagy [10]. Common upstream signals sometimes result in combined autophagy and apoptosis at the single cell level. In other instances, the cell dictates autophagy or apoptosis in a mutually exclusive manner. Under certain circumstances, autophagy allows cells to adapt to stress, thereby avoiding apoptotic cell death, e.g., a harmful αSyn can be degraded by autophagic pathway (see below). By contrast, massive autophagy induces alternative cell death pathway that is called autophagic cell death (ACD) [9, 10]. ACD is characterized by the presence of autophagic vacuoles (autophagosomes), which can be identified as double-membraned vesicles, and autophagolysosomes, which arise from the fusion of autophagosomes and lysosomes and are defined by a single membrane, in dying cells [9, 10]. On the other hand, Kroemer and Levine [11] indicated that the term ACD may be a misnomer because that is, in many cases, cell death “with” autophagy rather than cell death “by” autophagy. They emphasized that the autophagic process is not the executioner of cell death, or rather, cytoprotective response under pro-apoptotic condition [11].
Energy depletion is a potent trigger of necrosis [13]. Morphologically, necrosis is characterized by extensive vacuolation of the cytoplasm, mitochondrial swelling, dilatation of the endoplasmic reticulum (ER) and nuclear membrane, condensation of chromatin into small, irregular, and circumscribed patches, and plasma membrane rupture. Necrotic cells are lysed and do not fragment into discrete corpses as their apoptotic counterparts do. As a consequence, cellular contents are liberated into the extracellular space, which might precipitate damage to neighboring cells and evoke inflammatory responses [13, 15]. Necrosis has traditionally been considered merely as an accidental, uncontrolled form of cell death that only occurs in pathological conditions. Also, apoptosis has long been believed the sole form of programmed cell death (PCD). However, accumulating evidence uncovered another route of PCD, a programmed necrosis termed necroptosis [reviewed in 14, 15]. While several articles have suggested the occurrence of the “non-apoptotic PCD” during neurodegenerative processes [12, 14, 15, 26, 27], there have been a limited number of reports documenting the necrotic cell death in PD brains. This might in part be attributed to a methodological difficulty to dissect necrotic cell explosion in the postmortem brain tissues. It is known that necroptosis is triggered by ligation of death receptors with TNFα, FasL, and TRAIL, the same ligands that activate apoptosis [14, 15]. A death domain-containing kinase receptor-interacting protein 1 (RIP1) and RIP3 are required to dictate necroptotic pathway. Caspase-8 inactivates RIP1 and RIP3 by proteolytic cleavage and initiates the pro-apoptotic caspase activation cascade [15]. By contrast, inhibition of caspase-8 results in execution of the programmed necrosis in primary DA cultures [16]. A small molecule inhibitor of necroptosis, necrostatin-1, attenuated RIP1 kinase activity [28] and prevented glutamate-induced hippocampal neuronal cell death [29]. It needs further explorations to determine the involvement of necroptosis in DA neuronal degeneration in PD.
Physiological Functions of αSyn
αSyn is a neuronal protein of 140 amino acids and normally localized to presynaptic terminals. The exact physiological function of αSyn remains yet defined, but several works have implicated its role in dopamine biosynthesis, synaptic plasticity, and vesicle dynamics [1, 30–32]. Indeed, αSyn directly binds to vesicle-associated membrane protein 2 (VAMP2; also called as synaptobrevin-2) and promotes assembly of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex through a nonclassical chaperone activity [33]. Orchestration of assembly/disassembly of SNARE complex is essential for the regulation of neurotransmission. Recent studies have implicated presynaptic dysfunction to be an initial event of neurodegeneration [34]. A presynaptic protein cysteine-string protein-α (CSPα) also promotes SNARE complex assembly through the formation of chaperone complex with heat shock cognate 70 (Hsc70) and the small glutamine-rich protein SGT [35, 36]. The CSPα-Hsc70-SGT complex binds directly to synaptosomal-associated protein of 25 kDa (SNAP-25), whereby promoting SNARE complex formation [36]. Depletion of CSPα in mice represents decreased level of SNAP-25 and corresponding reduced assembly of SNARE complex [36]. Intriguingly, the CSPα-knockout mice show a rapidly progressive neurodegeneration and premature death, both of phenotype counteracted by transgenic expression of αSyn [37]. On the other hand, increased expression of αSyn in the absence of overt cell toxicity markedly inhibited neurotransmitter release, which was attributed to a perturbed synaptic vesicle density at the active zone, due to a defective reclustering of synaptic vesicles after endocytosis [38]. In another study, overexpressed αSyn indirectly inhibited SNARE-mediated exocytosis by sequestering arachidonic acid, which upregulates syntaxin and enhances its engagement with SNARE complex [39]. The opposing actions of αSyn implicate that a tight regulation of subcellular level and distribution of αSyn is indispensable for the intrinsic functions of neuronal cells.
Pathogenic Roles of αSyn in PD
αSyn is one of the most extensively studied proteins in PD research [30–32, 40] (Fig. 1). The gene encoding αSyn (SNCA) is mutated in rare inherited forms of PD, resulting in amino acid substitutions (A53T [41], A30P [42], or E46K [43]; classified as PARK1), or multiplication of its allele (PARK4) [44, 45]. Moreover, αSyn is a major component of Lewy bodies and Lewy neurites found in sporadic cases [2, 3]. Therefore, the presence of abnormally high levels of αSyn protein due to unbalanced production and/or degradation is thought to trigger DA neuronal death in both familial and sporadic cases of PD (Fig. 1). Single nucleotide polymorphisms in the 5′-promoter and 3′-flanking regions of SNCA gene that influence αSyn protein level are associated with susceptibility to idiopathic PD [46–48]. Furthermore, genome-wide association studies identified SNCA as a common risk factor for PD [49, 50]. Recent two studies uncovered epigenetic regulation of SNCA gene expression. Reduced methylation in CpG islands at intron 1 of SNCA that leads to increased protein production was evident in the SN of sporadic patients with PD [51, 52]. Desplats et al. [53] showed reduction of nuclear level of DNA methyltransferase 1 (Dnmt1) and DNA methylation in human postmortem brains affected with PD and dementia with Lewy bodies (DLB). Physical association of αSyn with Dnmt1 might mediate the retention of Dnmt1 in the cytoplasm, which results in hypomethylation of DNA [53]. However, overexpressed αSyn protein sometimes functions as a neuroprotective molecule in cell types other than DA neurons [37, 54–56]. Also, a recent report indicated protective function of physiological level of αSyn in DA cells. In that study, αSyn was found to reduce p300/CBP level and its histone acetyltransferase activity, whereby suppressing the NFκB-mediated transcriptional expression of pro-apoptotic protein kinase Cδ [57]. Oxidative modification of αSyn by dopamine metabolites is considered responsible for the selective vulnerability to DA neurons [55, 58]. Dopamine-modified αSyn tends to form protofibrillar intermediates but not large fibrils [58]. Such “oligomeric” αSyn is supposed the real criminal in DA neuronal toxicity [59–66]. On the other hand, a recent important finding indicated that endogenous normal αSyn forms a helically folded tetrameric structure of 58 kDa in neuronal and non-neuronal cell lines, brain tissue, and human red blood cells [67]. The tetrameric αSyn had high lipid-binding capacity and little or no propensity for amyloid-like aggregation. They proposed that destabilization of the tetramer precedes the misfolding and aggregation of αSyn in pathogenic conditions with PD and other α-synucleinopathies [67]. Another group also indicated that bacterially produced αSyn forms a stable tetramer [68] (To avoid misconceptions, hereafter, the nomenclature “oligomer” will be applied for the toxic species of αSyn formed in the diseased situations).
A 22-kDa O-glycosylated form of αSyn (αSp22) is destined for proteasomal degradation by receiving polyubiquitin moieties through the action of E3 ligase parkin, which is linked to a recessively inherited young-onset PD, PARK2 [69, 70]. Overexpression of wild-type or familial PD-linked mutant of αSyn in cell culture impairs proteasome activity and induces apoptosis or ACD, depending on the experimental conditions [71–75]. Soluble oligomeric αSyn impaired proteasome activity and likely impeded access of other proteasomal substrates [76, 77]. αSyn is degraded not only via ubiquitin-proteasome system but also autophagy [78, 79]. Both macroautophagy and chaperone-mediated autophagy (CMA) are involved in the clearance of accumulating αSyn [80]. Overexpressed wild-type αSyn compromised macroautophagy by inhibiting Rab1a [81], and pathogenic mutant and dopamine-modified αSyn prevented their own degradation and that of other substrates in CMA [82, 83]. As a result, DA cells harboring abnormal αSyn are sensitized to degenerative stimuli.
The majority of cellular source of energy is produced in mitochondria in the form of ATP. Because of the electrons being transported along the respiratory chain to potentiate mitochondrial intermembranous proton gradient, the prerequisite for oxidative phosphorylation, this organelle can intrinsically be a primary source of reactive oxygen species (ROS). A number of studies have demonstrated mitochondrial dysfunction and oxidative (and nitrosative) stresses linked to neuronal cell degeneration in PD [reviewed in 84]. This is well illustrated in an animal model of PD generated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which inhibits complex I in the electron transport system [85, 86]. αSyn protein has a non-canonical mitochondrial targeting sequence at its N-terminus and is indeed translocated to mitochondria in human fetal DA neuronal culture and postmortem normal brain tissues [87]. The mitochondrial αSyn accumulation is enhanced in PD brains. αSyn interacts with complex I and interferes with its function, promoting the production of ROS [87]. Particularly, superoxide radical rapidly reacts with nitric oxide to yield highly reactive peroxynitrite anion and ensuing reactive nitrogen species (RNS) [84]. ROS/RNS covalently modify lipids, nucleic acids, and proteins. αSyn can be modified with these compounds, augmenting the formation of toxic oligomeric αSyn (see below) [88].
Previous studies have implicated an increased iron level in the SN of postmortem brains of idiopathic PD and parkin-deficient PARK2 patients [89–91]. In MPTP-treated hemiparkinsonian monkeys, we and another group reported that DA cell death preceded iron accumulation [92, 93], suggesting that the elevation of iron may be a secondary event in nigral degeneration. On the contrary, several recent studies indicate that intraneuronal iron overload can be a primary cause of DA cell death in part through enhancing the formation of toxic radicals by Fenton reaction [94–96]. An iron transporter, divalent metal transporter 1 (DMT1), is upregulated and contributes to nigral DA neuronal death in MPTP and 6-hydroxydopamine rodent models of PD [94, 95]. Importantly, parkin regulates uptake of iron via degradation of DMT1 in ubiquitin-proteasome system [96]. These results suggest that DMT1-mediated iron overload can cause DA cell loss in parkinsonian brains. Iron promotes aggregation of αSyn protein [97, 98], and formation of pore-forming toxic oligomer species [99]. Moreover, DMT1-mediated cell death was aggravated in the presence of mutant αSyn as a result of excessive autophagic activity [100].
Recent studies revealed the association of Gaucher disease, the lysosomal storage disorder, with αSyn pathology [101–105]. Gaucher disease is caused by mutations in the gene encoding lysosomal protein glucocerebrosidase (GCase) that also increase the risk for PD and DLB [reviewed in 105]. A direct physical interaction between GCase and αSyn that prefers lysosomal acidic condition has been demonstrated [102]. In another study, importantly, glucosylceramide, which is the substrate of GCase and accumulated in Gaucher disease brains, directly influenced amyloid formation of αSyn by stabilizing soluble oligomeric intermediates [103]. The oligomeric αSyn in turn inhibited intracellular trafficking of GCase and decreased lysosomal GCase function. Such bidirectional effects of αSyn and GCase form a positive feedback loop that may lead to a self-propagating disease [103]. Genetic mouse model of Gaucher disease exhibited αSyn accumulation in the SN, cortex, or hippocampus [103, 104], and adeno-associated viral (AAV) vector-mediated delivery of GCase ameliorated pathological and behavioral aberrations in the Gaucher mice [104].
Prion-Like Cell-to-Cell Transmission of αSyn
In the last 20 years, more than 300 patients with PD have received striatal transplantation of midbrain tissues that were isolated from aborted fetuses [106, 107]. Many of these patients experienced a transient improvement of motor symptoms [106, 107], while severe off-phase dyskinesia remains a major concern [108–110]. On the other hand, intriguingly, more than a decade after the fetal transplantation, Lewy-like inclusions were depicted to be present in the surviving DA cells in the grafts [111, 112]. These findings led to the current provocative hypothesis that αSyn protein itself might transmit from neuron to neuron like as prion proteins, whereby spreading the pathologies in the brains of PD and other α-synucleinopathies [113–115].
Indeed, αSyn and its oligomeric forms are localized in the lumen of vesicles in differentiated neuronal cells and rat synaptosomal preparations, and secreted via non-classical ER/Golgi-independent exocytosis like as a part of the normal life cycle of this protein [116]. The intravesicular αSyn was found more prone to aggregation compared with cytosolic αSyn [116]. Another group showed that soluble monomeric and oligomeric αSyn were externalized via the vesicles that have characteristic hallmarks of exosomes in a calcium-dependent manner, and significantly reduced cell viability [117]. Danzer et al. [63] showed that different species of extracellular αSyn oligomers can exert distinct effects on cells; some oligomeric αSyn induced cell death by presumably pore-forming mechanism, and the other form of oligomer directly entered the cell and enhanced aggregation of αSyn. They proposed that heterogeneous populations of oligomeric forms coexist in equilibrium [63]. A solution structure of the pore-forming αSyn oligomer has been determined by small angle X-ray scattering [65]. On the other hand, cationic liposome-mediated forced transduction of exogenously produced fibrils of αSyn could seed the intracellular formation of Lewy-like inclusion in cultured cells [118, 119]. Furthermore, several groups reported that the extracellular αSyn can be uptaken by cells through endocytotic mechanism, and the internalized αSyn enhanced aggregation of (endogenous or overexpressed) αSyn and neuronal cell death [120–122]. Importantly, Mougenot et al. [123] demonstrated prion-like propagation of αSyn pathology in αSyn-transgenic mice. Brain homogenates from old αSyn-transgenic mice, which display motor clinical signs and contain insoluble Ser129-phosphorylated αSyn, were intracerebrally inoculated to young αSyn-transgenic mice. This triggered an early onset of characteristic motor signs and a prominent formation of inclusions that contain Ser129-phosphorylated αSyn, compared with uninoculated αSyn-transgenic mice or mice inoculated with brain homogenate from young healthy αSyn-transgenic mouse [123]. In that experiment, αSyn-null mice showed no abnormalities when inoculated with the brain homogenate of old disease-state αSyn-transgenic mice, indicating the crucial role for the presence of (pre-abnormal) αSyn in the host brain [123]. Extracellular αSyn is also capable of inducing microglial activation [124] and pro-inflammatory cytokine release from astrocytes [125] that may enhance neuronal toxicity. Accordingly, removal of extracellular αSyn species may be relevant to disease modification. Vaccination or passive immunization targeting the overloaded αSyn has successfully cured mice from neuronal degeneration (see below) [126, 127].
αSyn-Transgenic Animals
αSyn-transgenic models have been generated in mice [reviewed in 31, 128–130] and other organisms including nematode Caenorhabditis elegans [131–133] and fruit fly Drosophila melanogaster [134, 135]. Nematode models of αSyn overexpression exhibited neuronal or dendritic loss of DA cells and corresponding behavioral deficits [131–133]. Drosophila models of αSyn overexpression show adult-onset loss of DA neurons and locomotor dysfunction [134, 135]. These invertebrate models well recapitulate several key features of human PD and are relevant for comprehensive genetic analyses and drug screening towards elucidating the molecular pathogenesis and developing therapies for α-synucleinopathies [131–135].
On the other hand, a single transgenic expression of wild-type or familial PD-associated αSyn mutant in mice hardly represents a progressive loss of DA cells in the SNpc [31, 128–130]. Masliah et al. [136] reported the decrease of the striatal DA terminals and corresponding motor impairment induced by the overexpression of wild-type αSyn under the regulatory control of human platelet-derived growth factor-β (PDGF-β) promoter. Thereafter, several lines of αSyn-transgenic mice were generated and displayed severe movement disorders, loss of neuronal cells other than DA ones, and/or synaptic dysfunction before overt neuronal loss. In the transgenic mice of αSyn A53T mutant driven by the mouse prion promoter, which were originally reported by Lee et al. [137], Martin et al. [138] found that neocortical, brainstem, and motor neurons developed Lewy-like intraneuronal inclusions, axonal degeneration, and mitochondrial damage, as well as p53- and caspase-3-mediated apoptotic death. This report provided a mechanistic insight into the severe movement disorder of the αSyn A53T-transgenic mice. Sotiriou et al. [140] recently showed that the mouse prion promoter-αSyn A53T-transgenic mice, originally reported by Giasson et al. [139], had selective vulnerability for noradrenergic systems in the spinal cord, olfactory bulb (OB), and striatum in an age-dependent manner, while DA cells in the SN and noradrenergic cells in the locus coeruleus were not affected [140]. Lim et al. [141] generated inducible line of αSyn-transgenic mice with a tet-off system and the calcium/calmodulin-dependent protein kinase IIα (CaMKIIα) promoter, in which A53T mutant can be conditionally expressed in neuronal cells mainly in the cortex and hippocampus, to model human DLB. αSyn pathology and age-dependent neuronal cell loss was observed in cortical and hippocampal areas that resulted in memory impairment. Drug-induced suppression of αSyn transgene partially cleared pre-existing αSyn pathology and reverted defects in presynaptic proteins including synaptophysin, CSPα, synaptotagmin, SNAP-25, and syntaxin, and corresponding memory functions [141]. These results emphasize that targeted removal of αSyn pathology can reverse cognitive decline in DLB.
On the other hand, oxidation and nitration of αSyn induces the formation of stable dimers and oligomers through intermolecular dityrosine cross-linking [88]. αSyn possesses four tyrosine residues at positions 39, 125, 133, and 136 and lacks cysteine. When cysteine was substituted for tyrosine 39 and 125, these mutants increased intracellular inclusions and induced apoptosis in a rat DA cell line [142]. They indicated that cross-linking at critical positions in αSyn molecule can increase dimer formation, and accelerate protein aggregation and cellular toxicity of αSyn [142]. αSyn-transgenic mice carrying Y39C substitution under the murine Thy1 promoter were then generated and analyzed [143]. The mice showed age-dependent formation of αSyn oligomer and aggregate, progressive apoptotic cell loss in the cortex, and motor and cognitive deficits similar to DLB. Midbrain DA neurons and spontaneous locomotor activity were not affected in the αSyn Y39C-transgenic mice [143].
The murine prion promoter-αSyn-transgenic mice carrying E46K mutation, initially reported to cause PD and DLB [43], displayed detrimental age-dependent motor impairment, although DA neurons in the SN did not produce αSyn E46K protein [144]. These animals accumulated intracytoplasmic neuronal inclusions of αSyn in the cerebellum and pons that more closely resemble nigral Lewy bodies in PD than the previously described transgenic mice of human A53T αSyn. Intriguingly, phosphorylated tau-positive inclusions were found in the motor cortex and pons of the αSyn E46K-transgenic mice [144].
αSyn can be processed by C-terminal truncation in normal and PD brains [145, 146], and this modification promotes aggregation of αSyn [145–149]. The transgenic mice, that express C-terminally truncated form of αSyn [αSyn(1–120)] under the control of rat tyrosine hydroxylase (TH) promoter on a mouse αSyn-null background, exhibited the formation of pathological αSyn-positive inclusions in the SN and OB, reduction of the striatal dopamine levels, and a progressive reduction in spontaneous locomotion, in the absence of DA cell death [150]. In the following study, they investigated the presynaptic SNARE proteins in the striatum of the αSyn(1–120)-transgenic mice [151]. Synaptic accumulation of αSyn was accompanied by age-dependent redistribution of SNAP-25, syntaxin-1, and synaptobrevin-2, as well as reduced exocytosis of dopamine. A similar redistribution of the SNARE proteins was detected in PD brains [151]. Of note, Wakamatsu et al. [152] reported a selective loss of DA neurons in the SNpc of the transgenic mice carrying human αSyn(1–130). This truncated form of αSyn further caused reduction of the striatal DA axon terminals and dopamine level with corresponding reduction of locomotor activity, which can be reversed by administration of levodopa. However, the loss of nigral DA neurons was not progressive and seemed to occur during embryogenesis along with the onset of transgene expression [152].
Mutations in leucine-rich repeat kinase 2 (LRRK2) gene have been linked not only to a dominantly inherited PARK8 [153, 154] but also to sporadic form of PD [155]. The gene product LRRK2 possesses multiple functional domains including GTPase and kinase domains [156, 157]. A commonly found mutation, G2019S, increased its kinase activity, suggesting a gain-of-function mechanism for the pathogenesis of LRRK2-linked PD [157]. Intriguingly, LRRK2 accelerated the progression of neuropathology of αSyn [158]. Lin et al. [158] produced inducible line of LRRK2- or αSyn A53T-transgenic mice with a tet-off system and CaMKIIα promoter, in which the PD-related transgene can be expressed at high-level (LRRK2: about 8- to 16-fold; and αSyn A53T: about 30-fold) in neuronal cells in the striatum and cortex. While LRRK2 alone did not cause neurodegeneration, the presence of excess LRRK2 G2019S exacerbated abnormal accumulation and aggregation of αSyn A53T, which likely stemmed from the impairment of microtubule dynamics, Golgi organization, and the ubiquitin-proteasome pathway. Morphological abnormality of mitochondria and superoxide production was also promoted in the presence of high amount of LRRK2. In their αSyn A53T mice, genetic ablation of LRRK2 preserved the Golgi structure and suppressed the accumulation/aggregation of αSyn, and then delayed the progression of neuropathology [158]. This study elegantly demonstrated that suppression of LRRK2 can be a potential therapeutic target to ameliorate αSyn-induced neurodegeneration. In another report, by contrast, a single LRRK2-knockout mouse, which has a normal nigrostriatal DA system, developed accumulation and aggregation of αSyn and ubiquitinated proteins in the kidneys during aging [159]. This was possibly due to impairment of autophagy-lysosomal pathway. Furthermore, the ablation of LRRK2 gene dramatically increased apoptotic cell death, inflammatory responses, and oxidative damage in the kidney. These mice implicated that loss-of-function mutations of LRRK2 may cause cell death via impairment of protein degradation pathways, which lead to αSyn accumulation and aggregation [159].
Viral Vector-Mediated αSyn Overexpression Models
PD models have also been generated by viral vector-mediated overexpression of αSyn in rodents and nonhuman primates [160–166]. The AAV and lentiviral vectors have efficient tropism for DA neurons when injected into the SN and the ability of long-term stable gene expression with low accompanying cytotoxicity [167, 168]. In rodents, the viral vector-mediated overexpression of wild-type and familial PD-associated αSyn mutants can cause a progressive loss of DA cell bodies with neuritic pathology [160–163]. Representative images are shown in Fig. 2. The DA cell death was accompanied with phosphorylation of αSyn at Ser129 residue and apoptotic cascade with activation of caspase-9 in our examination [163]. We have also reported that, in the presence of the PARK5-linked ubiquitin carboxy-terminal hydrolase-L1 (UCH-L1) I93M mutant in mice, the AAV-αSyn-induced accumulation of αSyn and apoptotic DA cell death was enhanced, but not influenced in the absence of wild-type UCH-L1, indicating that PARK5-linked PD might be caused by gain-of-function mutation in UCH-L1 [166]. Importantly, Chung et al. [169] found that disturbance of the proteins relevant to synaptic transmission and axonal transport preceded the AAV-αSyn A53T-induced DA neuronal loss. It is known that a majority of the virally αSyn-challenged rodents lacks significant behavioral abnormalities, although they finally exhibit a profound DA neurodegeneration [160, 161, 163].
Fig. 2.

AAV vector-mediated expression of foreign gene in mouse brain. a The AAV vector can be injected stereotaxically into the SN of mice. b–e Representative images for the AAV vector-mediated overexpression of human αSyn (hαSyn) or GFP in DA cells. Nigral sections of the AAV-GFP- (b, d) or AAV-hαSyn-injected mice (c, e) were immunostained for GFP (GFP; b, d; shown in green) or hαSyn (c, e; green) and tyrosine hydroxylase (TH; b–e; red; merged with anti-GFP or hαSyn, yellow). Images for DAPI are also merged (d, e; blue). Note that the overexpression of hαSyn caused a profound loss of DA cell bodies with neuritic pathology. The overexpressed hαSyn was localized to nucleus and cytoplasm in a heterogeneous pattern in the remaining DA cells, while GFP distributed uniformly. Scale bars: (b, c) 20 μm and (d, e) 10 μm
By contrast, adult common marmosets (Callithrix jacchus) injected with αSyn-encoding AAV exhibited a severe neuronal pathology with a significant motor impairment such as head position bias in a short-term (16-weeks) study [164]. In a long-term examination for 1 year, the αSyn-treated monkey displayed behavioral impairments including full body rotation, head turn bias, and slowed and decreased use of contralateral hand [165]. These motor abnormalities were most pronounced in αSyn A53T-transduced group compared with wild-type αSyn and control GFP groups. About half of the αSyn A53T monkeys analyzed further developed slips of contralateral limbs (hand and foot) and persistent head tilts down on the contralesional side in the later phase [165]. Pathologically, wild-type αSyn-transduced monkeys exhibited a notably lower density of fibers immunopositive for αSyn in the caudate and putamen than for GFP in the GFP-transduced monkeys. The αSyn-containing aggregates were also found in the striatal fibers. This finding was even more pronounced in the αSyn A53T group, where only a sparse network of αSyn-positive fibers was seen in the caudate/putamen. In the αSyn A53T group, the ectopic αSyn protein appeared to have cleared from the SN, and there were fewer surviving αSyn-positive cell bodies compared with wild-type αSyn and GFP groups. When the Ser129-phosphorylated αSyn was examined by immunostaining, some of the neurons in the SN appeared normal while other cells were atrophic with shrunken cell bodies or had dystrophic dendrites, some with beaded aggregations. Interestingly, in several cases, the Ser129-phosphorylated αSyn-positive staining was localized to the nucleus (see below) [165]. In the wild-type and A53T αSyn groups, a substantial loss of TH-positive DA axon terminals and numerous pathological TH-positive accumulations were found in the striatum, suggesting that some of the affected but surviving cells were nonetheless dysfunctional. In the SN, the αSyn A53T-transduced monkeys showed a clear and consistent DA neurodegeneration in the injected side, which was significantly different when compared with GFP and wild-type αSyn groups [165]. The nonhuman primate model of α-synucleinopathy will be greatly useful for preclinical researches potentially preventing or retarding the behavioral and pathological progressions of the disease.
Phosphorylation and Neurotoxicity of αSyn
As described above, αSyn receives several post-translational modifications in diseased brains. In particular, Fujiwara et al. [170] found that about 90 % of αSyn proteins deposited in the brains of α-synucleinopathy are phosphorylated at Ser129 residue. Thereafter, the relationship between phosphorylation and neuronal toxicity of αSyn has been investigated. In dopamine-producing cells, rotenone treatment induced Ser129 phosphorylation of αSyn and formation of Lewy-like aggregates, with increased apoptotic cell death through the unfolded protein response [171]. In another study, increased oxidative stress or proteasomal inhibition caused significant elevation of soluble and non-aggregated form of Ser129-phosphorylated αSyn with increased DA cell death [172]. These in vitro studies suggested that Ser129 phosphorylation of αSyn is toxic to DA cells. Chen and Feany [173] reported that phosphorylation at Ser129 is essential for αSyn to have neuronal toxicity in a Drosophila model of PD. The toxicity was abolished by amino acid substitution S129A that is no longer phosphorylated, and reproduced by S129D that carries a negative charge mimicking phosphate on serine residue [173]. On the other hand, phosphoprotein phosphatase 2A (PP2A) dephosphorylates αSyn at Ser129, and this activity is enhanced by carboxyl methylation of the catalytic C subunit of PP2A [174]. αSyn-transgenic mice raised on a diet supplemented with eicosanoyl-5-hydroxytryptamide, an agent that enhances PP2A methylation, dramatically reduced both Ser129 phosphorylation and aggregation of αSyn in the brain [174]. These mice displayed enhanced neuronal activity, increased dendritic arborizations, and reduced astroglial and microglial activation, as well as improved motor performance [174].
There exist opposing reports as to the neurotoxicity of the Ser129-phosphorylated αSyn in the viral vector-mediated rodent model of αSyn overexpression. Alteration of Ser129 to nonphosphorylated Ala resulted in enhanced [175, 176] or unchanged toxicity of αSyn [177], and alteration of Ser129 to a phospho-mimetic Asp resulted in eliminated [175, 176] or unchanged toxicity of αSyn [177]. These studies suggest that the Ser129 phosphorylation of αSyn has, if any, protective effect on DA neurons. We recently reported that viral vector-mediated delivery of parkin prevented DA neuronal loss induced by a chronic MPTP in mice [178]. The osmotic minipump-mediated MPTP infusion caused accumulation of the Ser129-phosphorylated αSyn in DA cells, which was enhanced by overexpression of parkin, suggesting that the phosphorylation resulted in reduced toxicity of αSyn [178]. This result is in line with the report by Lo Bianco et al. [179] who demonstrated that lentiviral-parkin attenuated αSyn-induced DA cell loss by increasing the number of the Ser129-phosphorylated αSyn-positive inclusions in rats. The discrepancy in the neurotoxic consequence of the αSyn Ser129 phosphorylation makes difficulties in developing disease-modifying therapies. More elaborate time-series examinations in primates might be required to target this post-translational modification.
Prevention of αSyn-Induced Neuronal Cell Death/Dysfunction
αSyn-induced neuronal cell death and dysfunction can be targeted by several strategies. Masliah’s group has reported effective treatment of αSyn-transgenic mice with active and passive immunization protocols, which enabled clearance of toxic αSyn in multiple neuronal populations simultaneously [126, 127]. Passive immunization with a monoclonal antibody directed against C-terminus of αSyn (epitope: 118–126 amino acids of αSyn) that crossed into the central nervous system ameliorated behavioral deficits and synaptic abnormalities in αSyn-transgenic mice [127]. Moreover, the monoclonal antibody reduced the accumulation of calpain-cleaved and oligomerized αSyn aggregates in neuronal cells via lysosomal-degradation pathway [127]. They further indicated that lentiviral vector-mediated transduction of beclin 1, a regulator of autophagic pathway, ameliorated the synaptic and dendritic pathology in αSyn-transgenic mice [79]. The reduced accumulation of αSyn induced by the beclin 1 transduction was accompanied by enhanced lysosomal activation. These studies demonstrated that beclin 1-mediated autophagy pathway plays an important role in the intracellular degradation of αSyn and may present a novel therapeutic target for DLB and PD [79].
A number of studies demonstrated that parkin, PARK2-associated ubiquitin E3 ligase, protects against αSyn-induced cell death in vitro. Petrucelli et al. [72] showed that αSyn A53T-mediated toxicity in primary neuronal culture, which could be mimicked by the application of proteasome inhibitor, was reduced by E3 ligase activity of parkin. This study implicated that parkin and αSyn are linked in a common pathway associated with selective DA neuronal cell death. Another group reported that parkin could restore the reduced cell viability induced by wild-type αSyn via activation of calpain [180]. The calpain-mediated cleavage of accumulated αSyn occurred independently of proteasomal degradation [180]. In Drosophila model of PD, parkin suppressed DA neuronal death induced by overproduction of αSyn as well as parkin-associated endothelin receptor-like receptor (Pael-R) [181]. In rats, we have shown that AAV vector-mediated parkin delivery ameliorated DA cell loss induced by overexpression of wild-type αSyn [182]. On the other hand, parkin is known to interact with and ubiquitinate synphilin-1 [183], which was isolated as αSyn-interacting protein by yeast two-hybrid screen [184], through nonclassical K63-linked fashion [185]. Co-expression of αSyn and synphilin-1 resulted in the formation of Lewy body-like ubiquitin-positive cytosolic inclusions [183–185], which were found to be cytoprotective under proapoptotic stimuli [186]. A recent study indicated that transgenic expression of synphilin-1 attenuated αSyn-induced cell death in mice [187]. A double-transgenic mouse for αSyn A53T-mutant and synphilin-1 exhibited longer lifespan, improved motor performance, and reduced neuronal degeneration in the brainstem as compared to their single αSyn A53T-transgenic counterparts. Increased expression of beclin 1 and enhanced formation of aggresome-like structures were observed in the double αSyn A53T/synphilin-1-transgenic mice [187]. On the other hand, αSyn can directly be modified with small ubiquitin-related modifier (SUMO) at the positions of lysine 96 and 102 residues [188, 189]. The sumoylated αSyn showed increased solubility, whereas unmodified αSyn formed fibrils. Simultaneous substitution of K96 and K102 to arginine residues, which significantly impaired the sumoylation but did not affect the ubiquitination status of αSyn, was manifested by increased aggregation propensity and neuronal toxicity in vitro and in vivo [189]. Regulation of αSyn sumoylation may thus have a therapeutic potential.
We have shown previously that downregulation of SNCA transcripts by the AAV-mediated transduction of ribozymes provided rat DA neurons with a resistance to neurotoxin-induced αSyn accumulation and cell death [190]. Recently, Junn et al. [191] reported that downregulation of αSyn expression via microRNA-7 was effective for protection of αSyn A53T-expressing cells against oxidative stress. MicroRNA-7 was abundantly expressed in neurons in the SN, striatum, and OB in mice, the most affected areas in PD. Intoxication of mice with MPTP caused 50 % decrease of microRNA-7 in ventral midbrain, raising the possibility that the reduction of microRNA-7 in PD may cause degeneration of the nigrostriatal system, likely through upregulating αSyn production [191].
Sirtuin family of the class III NAD+-dependent histone deacetylases (HDACs) is involved in a variety of biological processes and several age-associated diseases [192, 193]. One of the family members, sirtuin 1, the mammalian ortholog of yeast Sir2, is upregulated under the conditions of caloric restriction and resveratrol treatment, and has a critical role in cell survival [192, 193]. On the other hand, sirtuin 2 induces neuronal cell death through its protein deacetylase activity [194]. The opposing mode of function is called as yin and yang of sirtuins [195]. A potent inhibitor of the deacetylase activity of sirtuin 2, AGK2, alleviated αSyn-induced DA neuronal cell death in primary cell culture and Drosophila models of PD [194]. In αSyn-aggregation experiment, where αSyn and synphilin-1 were co-introduced, AGK2 decreased the number and increased the size of αSyn aggregates, suggesting that the formation of large aggregates of αSyn might affect neuronal survival [194]. On the other hand, αSyn was found localized to the nucleus of DA neurons in mice that were exposed to neurotoxic herbicide paraquat, and associated with histones in vitro [196]. Kontopoulos et al. [197] have shown in DA cell line and Drosophila models that wild-type αSyn, C-terminally tagged with nuclear localization sequence or nuclear export sequence, enhanced or attenuated the neuronal toxicity, respectively. The inherited PD-linked A53T or A30P mutation promoted the nuclear localization of αSyn. Intranuclear αSyn inhibited histone acetylation and administration of HDAC inhibitors, sodium butyrate or suberoylanilide hydroxamic acid (SAHA), protected against the αSyn-induced DA cell loss [197]. Valproic acid (VPA), another HDAC inhibitor selective for class I and IIa HDACs, has been clinically used for the treatment of bipolar mood disorder, schizophrenia, and convulsive seizures [198, 199]. Leng and Chuang [56] reported that VPA induced upregulation of αSyn through hyperacetylation of histone H3 in the SNCA promoter region in rat cerebellar granule cells and cortical neurons. The increased αSyn protein participated in neuroprotection against glutamate-induced excitotoxicity. By contrast, recent study indicated that VPA showed neuroprotective effect in rotenone-induced PD model rats [200]. In the minipump-mediated rotenone rats, monoubiquitinated αSyn increased its localization into the nuclei, suggesting that the monoubiquitinated αSyn functions in the nucleus to promote DA neuronal cell death. The intranuclear translocation of αSyn and subsequent DA cell death was attenuated by VPA treatment [200].
On the other hand, abnormally accumulating αSyn was found to induce ER stress via blocking the vesicular trafficking from ER to Golgi network [201]. In genome-wide screen, Rab guanosine triphosphatase YPT1 (a member of Rab subfamily that belongs to Ras superfamily) was identified to modify the cell toxicity of abnormal αSyn and associate with cytoplasmic αSyn inclusions in yeast cells. Transgenic expression of Rab1 (the murine YPT1 ortholog) rescued the loss of DA neuronal cells in Drosophila and C. elegans models of αSyn overexpression [201]. This research group indicated in the following experiments that αSyn disrupts localization of several Rab proteins [202]. Transduction of RAB8A, the human homolog of yeast Sec4p and a close paralog to Rab1, and another neuron-specific RAB3A that functions at the synapse were also able to provide substantial rescue against αSyn-induced DA neurodegeneration in C. elegans and primary midbrain culture [202].
Rapamycin is an allosteric inhibitor of mammalian target of rapamycin (mTOR), an intracellular serine/threonine protein kinase involved in various cellular processes including cell growth and proliferation, protein synthesis, and autophagy [reviewed in 203]. Rapamycin has been clinically used as immunosuppressant drug to prevent the graft rejection and is now extensively studied as promising anticancer agents because of its anti-proliferative properties [203]. Recent two reports indicated that systemic treatment with rapamycin protected DA neurons from death in MPTP mouse model of PD [204, 205]. The neuroprotective molecular cascade upregulated by systemic rapamycin was distinct in these two reports. One indicated that rapamycin blocked mTOR complex 1-induced upregulation of pro-cell death RTP801 protein, which inactivate mTOR complex 2-mediated phosphorylation of pro-survival Akt kinase [203, 204]. In the other report, mitochondria-derived ROS induced permeabilization of lysosomal membranes that resulted in accumulation of altered mitochondria and undegraded autophagosomes [203, 205]. The lysosomal membrane permeabilization also induced ectopic release of lysosomal proteases cathepsin B and D to the cytosol, which can cause the digestion of vital proteins or the activation of additional hydrolases, including caspases. All of these pathogenic events, including apoptotic cell death, can be attenuated by rapamycin treatment. In particular, rapamycin was shown to restore impairment of lysosome-mediated clearance of autophagosome, by boosting lysosomal biogenesis and promoting autophagolysosome formation [203, 205]. αSyn-transgenic mouse model of DLB and PD [136], which displayed elevation of mTOR, reduction of autophagy-related protein 7 (Atg7) levels, and the presence of abundant and abnormal autophagosomes, was also healed with rapamycin [206]. Intracerebral infusion of rapamycin into the lateral ventricle of αSyn-transgenic mice enhanced clearance of αSyn protein accumulating in neuronal cell bodies and synapses and redistribution to the axons, through upregulation of autophagy pathway. This study further indicated lentiviral vector-mediated Atg7 expression resulted in reduced accumulation of αSyn and amelioration of associated neurodegenerative alterations [206].
Neurotrophic factors including glial cell line-derived neurotrophic factor (GDNF) and its closely related family protein Neurturin have provided promising therapeutic effects in various animal neurotoxin models and phase I clinical trials for PD [reviewed in 207]. However, surprisingly, AAV or lentiviral vector-mediated GDNF delivery did not prevent DA neuronal cell loss induced by the virally overexpressed αSyn of wild-type or A30P mutant [208, 209]. The difference in neuroprotective efficacy of GDNF raises important issues pertinent to the relevance for the therapeutic use of GDNF and Neurturin in the patients with PD.
Conclusion
αSyn has a central role in the pathogenesis of PD and other α-synucleinopathies, and a proper regulation of production, distribution, modification, and degradation of αSyn is crucial for neuronal functions and viability. Correction of the impairments in these multiple aspects of αSyn protein in its life cycle should provide disease modification remedies for the patients suffering from the devastating neurological disorders.
Acknowledgments
This work was supported by grants from Japan Science and Technology Agency (JST), Core Research for Evolutional Science and Technology (CREST); Grants-in-Aid from the Research Committee of CNS Degenerative Diseases, the Ministry of Health, Labour and Welfare of Japan; the Research Grant for Longevity Sciences from the Ministry of Health, Labour and Welfare of Japan; and grants (#S0801035) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan.
Conflicts of interest
The authors declare there is no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Abbreviations
- AAV
Adeno-associated virus
- ACD
Autophagic cell death
- αSyn
α-Synuclein
- CaMKIIα
Calcium/calmodulin-dependent protein kinase IIα
- CMA
Chaperone-mediated autophagy
- CSPα
Cysteine-string protein-α
- DA
Dopaminergic
- DLB
Dementia with Lewy bodies
- DMT1
Divalent metal transporter 1
- Dnmt1
DNA methyltransferase 1
- ER
Endoplasmic reticulum
- GCase
Glucocerebrosidase
- GDNF
Glial cell line-derived neurotrophic factor
- HDAC
Histone deacetylase
- Hsc70
Heat shock cognate 70
- LRRK2
Leucine-rich repeat kinase 2
- MPTP
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- mTOR
Mammalian target of rapamycin
- OB
Olfactory bulb
- PCD
Programmed cell death
- PD
Parkinson’s disease
- PP2A
Phosphoprotein phosphatase A2
- RIP
Receptor-interacting protein
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- SN
Substantia nigra
- SNAP-25
Synaptosomal-associated protein of 25 K
- SNARE
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- SNpc
Substantia nigra pars compacta
- TH
Tyrosine hydroxylase
- UCH-L1
Ubiquitin carboxy-terminal hydrolase-L1
- VPA
Valproic acid
References
- 1.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Shults CW. Lewy bodies. Proc Natl Acad Sci U S A. 2006;103:1661–1668. doi: 10.1073/pnas.0509567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- 5.Deep-Brain Stimulation for Parkinson’s Disease Study Group Deep-brain stimulation of the subthalamic nucleus or the pars interna of the globus pallidus in Parkinson’s disease. N Engl J Med. 2001;345:956–963. doi: 10.1056/NEJMoa000827. [DOI] [PubMed] [Google Scholar]
- 6.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hengartner MO. Apoptosis: corralling the corpses. Cell. 2001;104:325–328. doi: 10.1016/s0092-8674(01)00219-7. [DOI] [PubMed] [Google Scholar]
- 8.Yuan J, Lipinski M, Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40:401–413. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]
- 9.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 10.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 11.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2:589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- 13.Artal-Sanz M, Tavernarakis N. Proteolytic mechanisms in necrotic cell death and neurodegeneration. FEBS Lett. 2005;579:3287–3296. doi: 10.1016/j.febslet.2005.03.052. [DOI] [PubMed] [Google Scholar]
- 14.Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vandenabeele P, Galluzzi L, Vanden BT, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 16.Hartmann A, Troadec JD, Hunot S, Kikly K, Faucheux BA, Mouatt-Prigent A, Ruberg M, Agid Y, Hirsch EC. Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson’s disease, but pathway inhibition results in neuronal necrosis. J Neurosci. 2001;21:2247–2255. doi: 10.1523/JNEUROSCI.21-07-02247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mochizuki H, Goto K, Mori H, Mizuno Y. Histochemical detection of apoptosis in Parkinson’s disease. J Neurol Sci. 1996;137:120–123. doi: 10.1016/0022-510x(95)00336-z. [DOI] [PubMed] [Google Scholar]
- 18.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 19.Kosel S, Egensperger R, von Eitzen U, Mehraein P, Graeber MB. On the question of apoptosis in the parkinsonian substantia nigra. Acta Neuropathol. 1997;93:105–108. doi: 10.1007/s004010050590. [DOI] [PubMed] [Google Scholar]
- 20.Banati RB, Daniel SE, Blunt SB. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov Disord. 1998;13:221–227. doi: 10.1002/mds.870130205. [DOI] [PubMed] [Google Scholar]
- 21.Tatton NA, Maclean-Fraser A, Tatton WG, Perl DP, Olanow CW. A fluorescent double-labeling method to detect and confirm apoptotic nuclei in Parkinson’s disease. Ann Neurol. 1998;44:S142–S148. doi: 10.1002/ana.410440721. [DOI] [PubMed] [Google Scholar]
- 22.Hirsch EC, Hunot S, Faucheux B, Agid Y, Mizuno Y, Mochizuki H, Tatton WG, Tatton N, Olanow WC. Dopaminergic neurons degenerate by apoptosis in Parkinson’s disease. Mov Disord. 1999;14:383–385. doi: 10.1002/1531-8257(199903)14:2<383::aid-mds1037>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 23.Barzilai A, Melamed E. Molecular mechanisms of selective dopaminergic neuronal death in Parkinson’s disease. Trends Mol Med. 2003;9:126–132. doi: 10.1016/s1471-4914(03)00020-0. [DOI] [PubMed] [Google Scholar]
- 24.Vila M, Przedborski S. Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci. 2003;4:365–375. doi: 10.1038/nrn1100. [DOI] [PubMed] [Google Scholar]
- 25.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 26.Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci U S A. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyllie AH, Golstein P. More than one way to go. Proc Natl Acad Sci U S A. 2001;98:11–13. doi: 10.1073/pnas.98.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu X, Chua CC, Kong J, Kostrzewa RM, Kumaraguru U, Hamdy RC, Chua BH. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem. 2007;103:2004–2014. doi: 10.1111/j.1471-4159.2007.04884.x. [DOI] [PubMed] [Google Scholar]
- 30.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 31.Lotharius J, Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- 32.Auluck PK, Caraveo G, Lindquist S. Alpha-synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu Rev Cell Dev Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- 33.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burgoyne RD, Morgan A. Chaperoning the SNAREs: a role in preventing neurodegeneration? Nat Cell Biol. 2011;13:8–9. doi: 10.1038/ncb0111-8. [DOI] [PubMed] [Google Scholar]
- 35.Tobaben S, Thakur P, Fernandez-Chacon R, Sudhof TC, Rettig J, Stahl B. A trimeric protein complex functions as a synaptic chaperone machine. Neuron. 2001;31:987–999. doi: 10.1016/s0896-6273(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 36.Sharma M, Burre J, Sudhof TC. CSP-alpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat Cell Biol. 2011;13:30–39. doi: 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- 37.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 38.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darios F, Ruiperez V, Lopez I, Villanueva J, Gutierrez LM, Davletov B. Alpha-synuclein sequesters arachidonic acid to modulate SNARE-mediated exocytosis. EMBO Rep. 2010;11:528–533. doi: 10.1038/embor.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yasuda T, Mochizuki H. The regulatory role of alpha-synuclein and parkin in neuronal cell apoptosis; possible implications for the pathogenesis of Parkinson’s disease. Apoptosis. 2010;15:1312–1321. doi: 10.1007/s10495-010-0486-8. [DOI] [PubMed] [Google Scholar]
- 41.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 42.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 43.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez TE, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 44.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 45.Nishioka K, Hayashi S, Farrer MJ, Singleton AB, Yoshino H, Imai H, Kitami T, Sato K, Kuroda R, Tomiyama H, Mizoguchi K, Murata M, Toda T, Imoto I, Inazawa J, Mizuno Y, Hattori N. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson’s disease. Ann Neurol. 2006;59:298–309. doi: 10.1002/ana.20753. [DOI] [PubMed] [Google Scholar]
- 46.Winkler S, Hagenah J, Lincoln S, Heckman M, Haugarvoll K, Lohmann-Hedrich K, Kostic V, Farrer M, Klein C. Alpha-synuclein and Parkinson disease susceptibility. Neurology. 2007;69:1745–1750. doi: 10.1212/01.wnl.0000275524.15125.f4. [DOI] [PubMed] [Google Scholar]
- 47.Fuchs J, Tichopad A, Golub Y, Munz M, Schweitzer KJ, Wolf B, Berg D, Mueller JC, Gasser T. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB J. 2008;22:1327–1334. doi: 10.1096/fj.07-9348com. [DOI] [PubMed] [Google Scholar]
- 48.Mata IF, Shi M, Agarwal P, Chung KA, Edwards KL, Factor SA, Galasko DR, Ginghina C, Griffith A, Higgins DS, Kay DM, Kim H, Leverenz JB, Quinn JF, Roberts JW, Samii A, Snapinn KW, Tsuang DW, Yearout D, Zhang J, Payami H, Zabetian CP. SNCA variant associated with Parkinson disease and plasma alpha-synuclein level. Arch Neurol. 2010;67:1350–1356. doi: 10.1001/archneurol.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 50.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jowaed A, Schmitt I, Kaut O, Wullner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci. 2010;30:6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsumoto L, Takuma H, Tamaoka A, Kurisaki H, Date H, Tsuji S, Iwata A. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One. 2010;5:e15522. doi: 10.1371/journal.pone.0015522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, Adame A, Rockenstein E, Masliah E. Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem. 2011;286:9031–9037. doi: 10.1074/jbc.C110.212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.da Costa CA, Ancolio K, Checler F. Wild-type but not Parkinson’s disease-related ala-53 – > Thr mutant alpha-synuclein protects neuronal cells from apoptotic stimuli. J Biol Chem. 2000;275:24065–24069. doi: 10.1074/jbc.M002413200. [DOI] [PubMed] [Google Scholar]
- 55.Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA. Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- 56.Leng Y, Chuang DM. Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci. 2006;26:7502–7512. doi: 10.1523/JNEUROSCI.0096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. Alpha-synuclein negatively regulates protein kinase C delta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci. 2011;31:2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 59.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldberg MS, Lansbury PT., Jr Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson’s disease? Nat Cell Biol. 2000;2:E115–E119. doi: 10.1038/35017124. [DOI] [PubMed] [Google Scholar]
- 61.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 62.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 63.Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, Spencer B, Rockenstein E, Trejo M, Platoshyn O, Yuan JX, Masliah E. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS One. 2008;3:e3135. doi: 10.1371/journal.pone.0003135. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Giehm L, Svergun DI, Otzen DE, Vestergaard B. Low-resolution structure of a vesicle disrupting alpha-synuclein oligomer that accumulates during fibrillation. Proc Natl Acad Sci U S A. 2011;108:3246–3251. doi: 10.1073/pnas.1013225108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bartels T, Choi JG, Selkoe DJ. Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LTT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ (2011) A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A 108:17797–17802 [DOI] [PMC free article] [PubMed]
- 69.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- 70.Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, Hirsch EC, Farrer M, Schapira AH, Halliday G. Missing pieces in the Parkinson’s disease puzzle. Nat Med. 2010;16:653–661. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- 71.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- 73.Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, Dawson VL, Dawson TM, Ross CA. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- 74.Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–11759. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- 75.Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- 76.Lindersson E, Beedholm R, Hojrup P, Moos T, Gai W, Hendil KB, Jensen PH. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem. 2004;279:12924–12934. doi: 10.1074/jbc.M306390200. [DOI] [PubMed] [Google Scholar]
- 77.Emmanouilidou E, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging. 2010;31:953–968. doi: 10.1016/j.neurobiolaging.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 78.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 79.Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O’Kane CJ, Rubinsztein DC. Alpha-synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 83.Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsang AH, Chung KK. Oxidative and nitrosative stress in Parkinson’s disease. Biochim Biophys Acta. 2009;1792:643–650. doi: 10.1016/j.bbadis.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 85.Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503–2508. doi: 10.1016/0024-3205(85)90146-8. [DOI] [PubMed] [Google Scholar]
- 86.Nicklas WJ, Youngster SK, Kindt MV, Heikkila RE. MPTP, MPP+ and mitochondrial function. Life Sci. 1987;40:721–729. doi: 10.1016/0024-3205(87)90299-2. [DOI] [PubMed] [Google Scholar]
- 87.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- 89.Dexter DT, Wells FR, Agid F, Agid Y, Lees AJ, Jenner P, Marsden CD. Increased nigral iron content in postmortem parkinsonian brain. Lancet. 1987;2:1219–1220. doi: 10.1016/s0140-6736(87)91361-4. [DOI] [PubMed] [Google Scholar]
- 90.Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, Marsden CD. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem. 1989;52:1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x. [DOI] [PubMed] [Google Scholar]
- 91.Takanashi M, Mochizuki H, Yokomizo K, Hattori N, Mori H, Yamamura Y, Mizuno Y. Iron accumulation in the substantia nigra of autosomal recessive juvenile parkinsonism (ARJP) Parkinsonism Relat Disord. 2001;7:311–314. doi: 10.1016/s1353-8020(00)00050-x. [DOI] [PubMed] [Google Scholar]
- 92.Mochizuki H, Imai H, Endo K, Yokomizo K, Murata Y, Hattori N, Mizuno Y. Iron accumulation in the substantia nigra of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced hemiparkinsonian monkeys. Neurosci Lett. 1994;168:251–253. doi: 10.1016/0304-3940(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 93.He Y, Thong PS, Lee T, Leong SK, Mao BY, Dong F, Watt F. Dopaminergic cell death precedes iron elevation in MPTP-injected monkeys. Free Radic Biol Med. 2003;35:540–547. doi: 10.1016/s0891-5849(03)00385-x. [DOI] [PubMed] [Google Scholar]
- 94.Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nunez MT, Garrick MD, Raisman-Vozari R, Hirsch EC. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc Natl Acad Sci U S A. 2008;105:18578–18583. doi: 10.1073/pnas.0804373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hirsch EC. Iron transport in Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:S209–S211. doi: 10.1016/S1353-8020(09)70816-8. [DOI] [PubMed] [Google Scholar]
- 96.Roth JA, Singleton S, Feng J, Garrick M, Paradkar PN. Parkin regulates metal transport via proteasomal degradation of the 1B isoforms of divalent metal transporter 1. J Neurochem. 2010;113:454–464. doi: 10.1111/j.1471-4159.2010.06607.x. [DOI] [PubMed] [Google Scholar]
- 97.Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci. 2000;20:6048–6054. doi: 10.1523/JNEUROSCI.20-16-06048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Golts N, Snyder H, Frasier M, Theisler C, Choi P, Wolozin B. Magnesium inhibits spontaneous and iron-induced aggregation of alpha-synuclein. J Biol Chem. 2002;277:16116–16123. doi: 10.1074/jbc.M107866200. [DOI] [PubMed] [Google Scholar]
- 99.Kostka M, Hogen T, Danzer KM, Levin J, Habeck M, Wirth A, Wagner R, Glabe CG, Finger S, Heinzelmann U, Garidel P, Duan W, Ross CA, Kretzschmar H, Giese A. Single particle characterization of iron-induced pore-forming alpha-synuclein oligomers. J Biol Chem. 2008;283:10992–11003. doi: 10.1074/jbc.M709634200. [DOI] [PubMed] [Google Scholar]
- 100.Chew KC, Ang ET, Tai YK, Tsang F, Lo SQ, Ong E, Ong WY, Shen HM, Lim KL, Dawson VL, Dawson TM, Soong TW. Enhanced autophagy from chronic toxicity of iron and mutant A53T alpha-synuclein: implications for neuronal cell death in Parkinson disease. J Biol Chem. 2011;286:33380–33389. doi: 10.1074/jbc.M111.268409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, Morrison A, Lwin A, Colegial C, Allman JM, Schiffmann R. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 2004;82:192–207. doi: 10.1016/j.ymgme.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 102.Yap TL, Gruschus JM, Velayati A, Westbroek W, Goldin E, Moaven N, Sidransky E, Lee JC. Alpha-synuclein interacts with glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J Biol Chem. 2011;286:28080–28088. doi: 10.1074/jbc.M111.237859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, Passini MA, Grabowski GA, Schlossmacher MG, Sidman RL, Cheng SH, Shihabuddin LS. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A. 2011;108:12101–12106. doi: 10.1073/pnas.1108197108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Velayati A, Yu WH, Sidransky E. The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr Neurol Neurosci Rep. 2010;10:190–198. doi: 10.1007/s11910-010-0102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lindvall O, Brundin P, Widner H, Rehncrona S, Gustavii B, Frackowiak R, Leenders KL, Sawle G, Rothwell JC, Marsden CD, et al. Grafts of fetal dopamine neurons survive and improve motor function in Parkinson’s disease. Science. 1990;247:574–577. doi: 10.1126/science.2105529. [DOI] [PubMed] [Google Scholar]
- 107.Freed CR, Greene PE, Breeze RE, Tsai WY, DuMouchel W, Kao R, Dillon S, Winfield H, Culver S, Trojanowski JQ, Eidelberg D, Fahn S. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med. 2001;344:710–719. doi: 10.1056/NEJM200103083441002. [DOI] [PubMed] [Google Scholar]
- 108.Hagell P, Piccini P, Bjorklund A, Brundin P, Rehncrona S, Widner H, Crabb L, Pavese N, Oertel WH, Quinn N, Brooks DJ, Lindvall O. Dyskinesias following neural transplantation in Parkinson’s disease. Nat Neurosci. 2002;5:627–628. doi: 10.1038/nn863. [DOI] [PubMed] [Google Scholar]
- 109.Olanow CW, Goetz CG, Kordower JH, Stoessl AJ, Sossi V, Brin MF, Shannon KM, Nauert GM, Perl DP, Godbold J, Freeman TB. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol. 2003;54:403–414. doi: 10.1002/ana.10720. [DOI] [PubMed] [Google Scholar]
- 110.Olanow CW, Gracies JM, Goetz CG, Stoessl AJ, Freeman T, Kordower JH, Godbold J, Obeso JA. Clinical pattern and risk factors for dyskinesias following fetal nigral transplantation in Parkinson’s disease: a double blind video-based analysis. Mov Disord. 2009;24:336–343. doi: 10.1002/mds.22208. [DOI] [PubMed] [Google Scholar]
- 111.Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 112.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 113.Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 2010;11:155–159. doi: 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010;33:317–325. doi: 10.1016/j.tins.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 115.Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Are synucleinopathies prion-like disorders? Lancet Neurol. 2010;9:1128–1138. doi: 10.1016/S1474-4422(10)70213-1. [DOI] [PubMed] [Google Scholar]
- 116.Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of alpha-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem. 2010;285:34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P. Alpha-synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121:715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchere J, Lakhdar L, Legastelois S, Baron T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging. 2012;33:2225–2228. doi: 10.1016/j.neurobiolaging.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 124.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 125.Klegeris A, Giasson BI, Zhang H, Maguire J, Pelech S, McGeer PL. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 2006;20:2000–2008. doi: 10.1096/fj.06-6183com. [DOI] [PubMed] [Google Scholar]
- 126.Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 127.Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, Mueller-Steiner S, Seubert P, Barbour R, McConlogue L, Buttini M, Games D, Schenk D. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011;6:e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 128.Fernagut PO, Chesselet MF. Alpha-synuclein and transgenic mouse models. Neurobiol Dis. 2004;17:123–130. doi: 10.1016/j.nbd.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 129.Kahle PJ. Alpha-synucleinopathy models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:87–95. doi: 10.1007/s00401-007-0302-x. [DOI] [PubMed] [Google Scholar]
- 130.Crabtree DM, Zhang J. Genetically engineered mouse models of Parkinson’s disease. Brain Res Bull. 2012;88:13–32. doi: 10.1016/j.brainresbull.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lakso M, Vartiainen S, Moilanen AM, Sirvio J, Thomas JH, Nass R, Blakely RD, Wong G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem. 2003;86:165–172. doi: 10.1046/j.1471-4159.2003.01809.x. [DOI] [PubMed] [Google Scholar]
- 132.Kuwahara T, Koyama A, Gengyo-Ando K, Masuda M, Kowa H, Tsunoda M, Mitani S, Iwatsubo T. Familial Parkinson mutant alpha-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J Biol Chem. 2006;281:334–340. doi: 10.1074/jbc.M504860200. [DOI] [PubMed] [Google Scholar]
- 133.Cao P, Yuan Y, Pehek EA, Moise AR, Huang Y, Palczewski K, Feng Z. Alpha-synuclein disrupted dopamine homeostasis leads to dopaminergic neuron degeneration in Caenorhabditis elegans. PLoS One. 2010;5:e9312. doi: 10.1371/journal.pone.0009312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 135.Mizuno H, Fujikake N, Wada K, Nagai Y. Alpha-synuclein transgenic Drosophila as a model of Parkinson’s disease and related synucleinopathies. Parkinsons Dis. 2010;2011:212706. doi: 10.4061/2011/212706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 137.Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 – > Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 140.Sotiriou E, Vassilatis DK, Vila M, Stefanis L. Selective noradrenergic vulnerability in alpha-synuclein transgenic mice. Neurobiol Aging. 2010;31:2103–2114. doi: 10.1016/j.neurobiolaging.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 141.Lim Y, Kehm VM, Lee EB, Soper JH, Li C, Trojanowski JQ, Lee VM. Alpha-syn suppression reverses synaptic and memory defects in a mouse model of dementia with Lewy bodies. J Neurosci. 2011;31:10076–10087. doi: 10.1523/JNEUROSCI.0618-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou W, Freed CR. Tyrosine-to-cysteine modification of human alpha-synuclein enhances protein aggregation and cellular toxicity. J Biol Chem. 2004;279:10128–10135. doi: 10.1074/jbc.M307563200. [DOI] [PubMed] [Google Scholar]
- 143.Zhou W, Milder JB, Freed CR. Transgenic mice overexpressing tyrosine-to-cysteine mutant human alpha-synuclein: a progressive neurodegenerative model of diffuse Lewy body disease. J Biol Chem. 2008;283:9863–9870. doi: 10.1074/jbc.M710232200. [DOI] [PubMed] [Google Scholar]
- 144.Emmer KL, Waxman EA, Covy JP, Giasson BI. E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairments. J Biol Chem. 2011;286:35104–35118. doi: 10.1074/jbc.M111.247965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu CW, Giasson BI, Lewis KA, Lee VM, Demartino GN, Thomas PJ. A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J Biol Chem. 2005;280:22670–22678. doi: 10.1074/jbc.M501508200. [DOI] [PubMed] [Google Scholar]
- 146.Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jakala P, Hartmann T, Price DL, Lee MK. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci U S A. 2005;102:2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 1998;436:309–312. doi: 10.1016/s0014-5793(98)01146-6. [DOI] [PubMed] [Google Scholar]
- 148.Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci. 2007;27:3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tofaris GK, Garcia RP, Humby T, Lambourne SL, O’Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, Spillantini MG. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J Neurosci. 2006;26:3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Garcia-Reitbock P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, Ghetti B, Della CL, Spano P, Tofaris GK, Goedert M, Spillantini MG. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain. 2010;133:2032–2044. doi: 10.1093/brain/awq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wakamatsu M, Ishii A, Iwata S, Sakagami J, Ukai Y, Ono M, Kanbe D, Muramatsu S, Kobayashi K, Iwatsubo T, Yoshimoto M. Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha-synuclein in mice. Neurobiol Aging. 2008;29:574–585. doi: 10.1016/j.neurobiolaging.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 153.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 154.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 155.Zabetian CP, Samii A, Mosley AD, Roberts JW, Leis BC, Yearout D, Raskind WH, Griffith A. A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology. 2005;65:741–744. doi: 10.1212/01.wnl.0000172630.22804.73. [DOI] [PubMed] [Google Scholar]
- 156.Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ, 3rd, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107:9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Klein RL, King MA, Hamby ME, Meyer EM. Dopaminergic cell loss induced by human A30P alpha-synuclein gene transfer to the rat substantia nigra. Hum Gene Ther. 2002;13:605–612. doi: 10.1089/10430340252837206. [DOI] [PubMed] [Google Scholar]
- 161.Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Bjorklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lo BC, Ridet JL, Schneider BL, Deglon N, Aebischer P. Alpha-synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2002;99:10813–10818. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yamada M, Iwatsubo T, Mizuno Y, Mochizuki H. Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson’s disease. J Neurochem. 2004;91:451–461. doi: 10.1111/j.1471-4159.2004.02728.x. [DOI] [PubMed] [Google Scholar]
- 164.Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Bjorklund A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2003;100:2884–2889. doi: 10.1073/pnas.0536383100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Eslamboli A, Romero-Ramos M, Burger C, Bjorklund T, Muzyczka N, Mandel RJ, Baker H, Ridley RM, Kirik D. Long-term consequences of human alpha-synuclein overexpression in the primate ventral midbrain. Brain. 2007;130:799–815. doi: 10.1093/brain/awl382. [DOI] [PubMed] [Google Scholar]
- 166.Yasuda T, Nihira T, Ren YR, Cao XQ, Wada K, Setsuie R, Kabuta T, Wada K, Hattori N, Mizuno Y, Mochizuki H. Effects of UCH-L1 on alpha-synuclein over-expression mouse model of Parkinson’s disease. J Neurochem. 2009;108:932–944. doi: 10.1111/j.1471-4159.2008.05827.x. [DOI] [PubMed] [Google Scholar]
- 167.Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ, Muzyczka N. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther. 2004;10:302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 168.Mandel RJ, Manfredsson FP, Foust KD, Rising A, Reimsnider S, Nash K, Burger C. Recombinant adeno-associated viral vectors as therapeutic agents to treat neurological disorders. Mol Ther. 2006;13:463–483. doi: 10.1016/j.ymthe.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 169.Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci. 2009;29:3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 171.Sugeno N, Takeda A, Hasegawa T, Kobayashi M, Kikuchi A, Mori F, Wakabayashi K, Itoyama Y. Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J Biol Chem. 2008;283:23179–23188. doi: 10.1074/jbc.M802223200. [DOI] [PubMed] [Google Scholar]
- 172.Chau KY, Ching HL, Schapira AH, Cooper JM. Relationship between alpha synuclein phosphorylation, proteasomal inhibition and cell death: relevance to Parkinson’s disease pathogenesis. J Neurochem. 2009;110:1005–1013. doi: 10.1111/j.1471-4159.2009.06191.x. [DOI] [PubMed] [Google Scholar]
- 173.Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- 174.Lee KW, Chen W, Junn E, Im JY, Grosso H, Sonsalla PK, Feng X, Ray N, Fernandez JR, Chao Y, Masliah E, Voronkov M, Braithwaite SP, Stock JB, Mouradian MM. Enhanced phosphatase activity attenuates alpha-synucleinopathy in a mouse model. J Neurosci. 2011;31:6963–6971. doi: 10.1523/JNEUROSCI.6513-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, Mandel RJ, Muzyczka N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci U S A. 2008;105:763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Azeredo da Silveira S, Schneider BL, Cifuentes-Diaz C, Sage D, Abbas-Terki T, Iwatsubo T, Unser M, Aebischer P. Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum Mol Genet. 2009;18:872–887. doi: 10.1093/hmg/ddn417. [DOI] [PubMed] [Google Scholar]
- 177.McFarland NR, Fan Z, Xu K, Schwarzschild MA, Feany MB, Hyman BT, McLean PJ. Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J Neuropathol Exp Neurol. 2009;68:515–524. doi: 10.1097/NEN.0b013e3181a24b53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yasuda T, Hayakawa H, Nihira T, Ren YR, Nakata Y, Nagai M, Hattori N, Miyake K, Takada M, Shimada T, Mizuno Y, Mochizuki H. Parkin-mediated protection of dopaminergic neurons in a chronic MPTP-minipump mouse model of Parkinson disease. J Neuropathol Exp Neurol. 2011;70:686–697. doi: 10.1097/NEN.0b013e3182269ecd. [DOI] [PubMed] [Google Scholar]
- 179.Lo BC, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, Aebischer P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:17510–17515. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kim SJ, Sung JY, Um JW, Hattori N, Mizuno Y, Tanaka K, Paik SR, Kim J, Chung KC. Parkin cleaves intracellular alpha-synuclein inclusions via the activation of calpain. J Biol Chem. 2003;278:41890–41899. doi: 10.1074/jbc.M306017200. [DOI] [PubMed] [Google Scholar]
- 181.Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron. 2003;37:911–924. doi: 10.1016/s0896-6273(03)00143-0. [DOI] [PubMed] [Google Scholar]
- 182.Yamada M, Mizuno Y, Mochizuki H. Parkin gene therapy for alpha-synucleinopathy: a rat model of Parkinson’s disease. Hum Gene Ther. 2005;16:262–270. doi: 10.1089/hum.2005.16.262. [DOI] [PubMed] [Google Scholar]
- 183.Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- 184.Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, Margolis RL, Troncoso JC, Lanahan AA, Worley PF, Dawson VL, Dawson TM, Ross CA. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet. 1999;22:110–114. doi: 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- 185.Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci. 2005;25:2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004;279:4625–4631. doi: 10.1074/jbc.M310994200. [DOI] [PubMed] [Google Scholar]
- 187.Smith WW, Liu Z, Liang Y, Masuda N, Swing DA, Jenkins NA, Copeland NG, Troncoso JC, Pletnikov M, Dawson TM, Martin LJ, Moran TH, Lee MK, Borchelt DR, Ross CA. Synphilin-1 attenuates neuronal degeneration in the A53T alpha-synuclein transgenic mouse model. Hum Mol Genet. 2010;19:2087–2098. doi: 10.1093/hmg/ddq086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dorval V, Fraser PE. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. 2006;281:9919–9924. doi: 10.1074/jbc.M510127200. [DOI] [PubMed] [Google Scholar]
- 189.Krumova P, Meulmeester E, Garrido M, Tirard M, Hsiao HH, Bossis G, Urlaub H, Zweckstetter M, Kugler S, Melchior F, Bahr M, Weishaupt JH. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J Cell Biol. 2011;194:49–60. doi: 10.1083/jcb.201010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hayashita-Kinoh H, Yamada M, Yokota T, Mizuno Y, Mochizuki H. Down-regulation of alpha-synuclein expression can rescue dopaminergic cells from cell death in the substantia nigra of Parkinson’s disease rat model. Biochem Biophys Res Commun. 2006;341:1088–1095. doi: 10.1016/j.bbrc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 191.Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106:13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Gan L, Mucke L. Paths of convergence: sirtuins in aging and neurodegeneration. Neuron. 2008;58:10–14. doi: 10.1016/j.neuron.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins–novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841–853. doi: 10.1038/nrd2665. [DOI] [PubMed] [Google Scholar]
- 194.Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 195.Dillin A, Kelly JW. The yin-yang of sirtuins. Science. 2007;317:461–462. doi: 10.1126/science.1146585. [DOI] [PubMed] [Google Scholar]
- 196.Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA, Uversky VN, Fink AL. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry. 2003;42:8465–8471. doi: 10.1021/bi0341152. [DOI] [PubMed] [Google Scholar]
- 197.Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- 198.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 199.Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and therapeutic potential and toxicity of valproic acid. J Biomed Biotechnol. 2010;2010:479364. doi: 10.1155/2010/479364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Monti B, Gatta V, Piretti F, Raffaelli SS, Virgili M, Contestabile A. Valproic acid is neuroprotective in the rotenone rat model of Parkinson’s disease: involvement of alpha-synuclein. Neurotox Res. 2010;17:130–141. doi: 10.1007/s12640-009-9090-5. [DOI] [PubMed] [Google Scholar]
- 201.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, Caldwell KA, Caldwell GA, Rochet JC, McCaffery JM, Barlowe C, Lindquist S. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A. 2008;105:145–150. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12:437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- 204.Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J Neurosci. 2010;30:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 207.Yasuda T, Mochizuki H. Use of growth factors for the treatment of Parkinson’s disease. Expert Rev Neurother. 2010;10:915–924. doi: 10.1586/ern.10.55. [DOI] [PubMed] [Google Scholar]
- 208.Lo BC, Deglon N, Pralong W, Aebischer P. Lentiviral nigral delivery of GDNF does not prevent neurodegeneration in a genetic rat model of Parkinson’s disease. Neurobiol Dis. 2004;17:283–289. doi: 10.1016/j.nbd.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 209.Decressac M, Ulusoy A, Mattsson B, Georgievska B, Romero-Ramos M, Kirik D, Bjorklund A. GDNF fails to exert neuroprotection in a rat alpha-synuclein model of Parkinson’s disease. Brain. 2011;134:2302–2311. doi: 10.1093/brain/awr149. [DOI] [PubMed] [Google Scholar]