

Abstract
There are multiple sources of reactive oxygen species (ROS) in the cell. As a major site of ROS production, mitochondria have drawn considerable interest because it was recently discovered that mitochondrial ROS (mtROS) directly stimulate the production of proinflammatory cytokines and pathological conditions as diverse as malignancies, autoimmune diseases, and cardiovascular diseases all share common phenotype of increased mtROS production above basal levels. Several excellent reviews on this topic have been published, but ever-changing new discoveries mandated a more up-to-date and comprehensive review on this topic. Therefore, we update recent understanding of how mitochondria generate and regulate the production of mtROS and the function of mtROS both in physiological and pathological conditions. In addition, we describe newly developed methods to probe or scavenge mtROS and compare these methods in detail. Thorough understanding of this topic and the application of mtROS-targeting drugs in the research is significant towards development of better therapies to combat inflammatory diseases and inflammatory malignancies.
Keywords: Mitochondria, ROS, Inflammatory diseases
Introduction
Free radicals and other ROS are generated in a wide range of normal physiological conditions. However, ROS also participate in many pathological conditions including cardiovascular diseases, malignancies, autoimmune diseases, and neurological degenerative diseases. Despite intensive investigations in this field, current anti-oxidant therapeutics are not clinically effective in combating these pathological conditions suggesting that our understanding of this field is limited, and there is a need to narrow the “knowledge gap” in order to develop more effective new therapies [1]. Although ROS are historically considered toxic by-products of cellular metabolism, recent studies have suggested that cells “have learned” to harness the power of ROS for cell signaling purposes. In analogous to phosphorylation modification of proteins, the term “redox signaling” is emerging in reference to events of oxidation modification of proteins by ROS. Indeed, there are multiple sources of ROS in the cell including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) [2], xanthine oxidase (XO), uncoupling of nitric oxide synthase (NOS), cytochrome P450, and mitochondrial electron transport chain (ETC). Among these potential sources, however, mtROS have drawn increasing attentions because it was recently discovered that mtROS directly contribute to inflammatory cytokine production and innate immune responses [3] by activation of newly characterized RIG-I-like receptors (RLRs) [4], inflammasomes [5], and mitogen-activated protein kinases (MAPK) [6].
Cardiovascular disease (CVD) is the leading cause of morbidity and mortality in the western world. Nearly 75% of the CVD-related death results from atherosclerosis which is found in 80-90% of Americans over the age of 30. Early atherosclerotic lesions can be detected in youths as young as 7 years of age [7,8]. As a form of chronic autoimmune inflammatory condition associated with specific CVD risk factors, development of atherosclerosis is fueled by aberrant response of the innate immune system and overproduction of proinflammatory cytokines [9,10]. A recent progress in characterizing mtROS has led to the generation of a new paradigm, in which blockade of mtROS production may serve as a promising therapy for inhibiting proinflammatory cytokine production and in turn atherosclerosis. Although there were several excellent reviews published 5 years ago in this topic [11,12], new recent discoveries have mandated a more up-to-date and comprehensive review [13-15]. Therefore, in this review we consider current understandings of several compelling questions: 1) how mitochondria generate and dispose of ROS; 2) how production of mtROS is regulated; and 3) what signaling pathways are targeted by mtROS. In addition, we describe the methods to probe mtROS and analyze the merits and flaws of these different methods. Furthermore, we demonstrate how mtROS regulate important vascular function in physiological conditions and activate inflammatory pathways in response to CVD risk factors. In-depth understanding of these processes is critical to developing novel therapeutic drugs against chronic inflammatory conditions such as atherosclerosis.
Production of mtROS
Mitochondria have a four-layer structure, including outer mitochondrial membrane, intermembrane space, inner mitochondrial membrane and matrix (Figure 1). Generation of mtROS mainly takes place at the ETC located on the inner mitochondrial membrane during the process of oxidative phosphorylation (OXPHOS). Oxidative phosphorylation is an essential cellular process that uses oxygen and simple sugars to create adenosine triphosphate (ATP), which is the cell's main energy source. Five big protein complexes are involved in this process (HUGO Gene Nomenclature Committee Website, Table 1). These ETC complexes are named complex I (NADH dehydrogenase (ubiquinone), 45 protein subunits), complex II (succinate dehydrogenase, 4 protein subunits), complex III (ubiquinol-cytochrome c reductase, 10 protein subunits), complex IV (cytochrome c oxidase, 19 protein subunits), and complex V (ATP synthase, 19 protein subunits). Electrons donated from nicotine adenine dinucleotide (NADH) at complex I and flavin adenine dinucleotide (FADH2) at complex II pass through ETC and ultimately reduce O2 to water at complex IV. Meanwhile, positively charged protons (H+) are actively being pumped from the mitochondrial matrix into the intermembrane space, resulting in the increased negative charges in the mitochondrial matrix and the upregulated positive charges in the intermembrane space, and thus creating a mitochondrial membrane potential (Δψm) across the inner mitochondrial membrane. This proton-motive force allows complex V - ATP synthase (ATP-ase) to generate ATP from adenosine diphosphate (ADP) and inorganic phosphate when protons re-enter the mitochondrial matrix through the complex V enzyme. However, either by accident or by design, the process of ETC is not perfect. Leakage of electrons at complex I and complex III leads to partial reduction of oxygen to form superoxide (O2 .-). It has been estimated that 0.2% to 2.0% of O2 consumed by mitochondria generates O2 .-[11]. There are three leak events: complex I leaks O2 .- towards the mitochondrial matrix, while complex III leaks O2 .- towards both the intermembrane space and mitochondrial matrix [11,16]. Subsequently, O2 .- is quickly dismutated to hydrogen peroxide (H2O2) by two dismutases including superoxide dismutase 2 (SOD2) in mitochondrial matrix and superoxide dismutase 1 (SOD1) in mitochondrial intermembrane space. Collectively, both O2 .- and H2O2 generated in this process are considered as mtROS. These two mtROS have different fates however. Given its electrophilic property and short half-life, O2 .- can hardly pass through mitochondrial outer membrane and unlikely to become the candidate of signaling transduction molecule in the cell. Instead, O2 .- can undergo radical-radical reaction with nitric oxide (NO) to form peroxynitrite (ONOO2 .-) within mitochondria, a detrimental oxidant capable of induction of DNA damage, disruption of mitochondrial integrity, and irreversible modification of proteins [11]. In contrast, H2O2 is electrophobic and more stable than O2 .-. Indeed, the concentrations of H2O2 in mitochondria are 100 times greater than that of O2 .-[17]. These properties render mitochondrial H2O2 an ideal signaling molecule in the cells.
Figure 1.
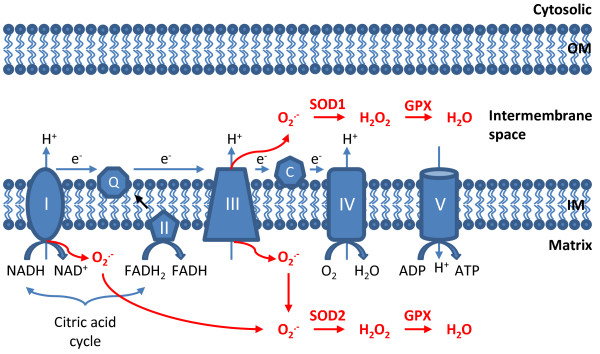
Production and disposal of mtROS. Electrons (e-) donated from NADH and FADH2 pass through the electron transport chain and ultimately reduce O2 to form H2O at complex IV. MtROS are produced from the leakage of e- to form superoxide (O2 .-) at complex I and complex III. O2 .- is produced within matrix at complex I, whereas at complex III O2 .- is released towards both the matrix and the intermembrane space. Once generated, O2 .- is dismutated to H2O2 by superoxide dismutase 1 (SOD1) in the intermembrane space and by SOD2 in the matrix. Afterwards, H2O2 is fully reduced to water by glutathione peroxidase (GPX). Both O2 .- and H2O2 produced in this process are considered as mtROS. OM: outer membrane; IM: inner membrane.
Table 1.
Mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) encoded subunits for the electron transport chain
| Complex | ||||||
|---|---|---|---|---|---|---|
| Genome |
I |
II |
III |
IV |
V |
Total |
| mtDNA |
7 |
0 |
1 |
3 |
2 |
13 |
| nDNA |
38 |
4 |
9 |
16 |
17 |
84 |
| Total | 45 | 4 | 10 | 19 | 19 | 97 |
Scavenging of mtROS
Owing to the high reactivity and toxicity of mtROS, mammalian cells have evolved a number of antioxidant enzyme systems to scavenge mtROS as soon as they are generated. As mentioned in the previous section, the SOD family of antioxidant enzymes catalyze the dismutation of O2 .- to H2O2. Subsequently, H2O2 is quickly reduced to water by two other enzymes, catalase and glutathione peroxidase (GPx) (Figure 1). It should be noted that all the mitochondrial antioxidant enzymes are encoded by the nuclear genome but not mitochondrial genome, and these enzymes are subsequently imported into the mitochondria after their protein translation.
SODs
Three isoforms of SOD have been identified, including SOD1/copper-zinc SOD (CuZn-SOD), SOD2/manganese SOD (Mn-SOD), and extracellular SOD3 (EC-SOD). SOD1 is widely distributed throughout the cell cytoplasm, nucleus, and intermembrane space of mitochondria [18]. SOD2 is expressed only in the mitochondrial matrix [18], and SOD3 is found in the extracellular space. The physiological importance of SOD2 is high-lighted by the finding that in contrast to other SOD isoforms, the deficiency of SOD2 causes early neonatal death in gene knockout mice [19] and endothelial dysfunction in carotid artery of proatherogenic apolipoprotein E (ApoE)-deficient mice [11,20].
Catalase
Catalase is a heme-containing tetramer of four polypeptide chains that reduces H2O2 to water. Although catalase is highly efficient at reducing hydrogen peroxide, it may not play a central role in scavenging ROS in the mitochondria since it is localized mainly in peroxisomes except that rat heart mitochondria does partially depend on catalase to scavenge ROS [21]. Nevertheless, overexpression of catalase in ApoE−/− mice results in the retardation of atherosclerosis [22]. In addition, overexpression of catalase in the mitochondria decreased oxidative damage, inhibited cardiac pathology, and extended the lifespan of mice [23]. These results suggest the importance of catalase in suppressing cardiovascular inflammation and damage and atherosclerosis.
GPx
GPx catalyzes the reductive inactivation of H2O2 using reduced glutathione (GSH) as a cofactor. GSH is a tripeptide containing of three amino acid residues including glutamate, cysteine, and glycine. During the process of reducing H2O2, GSH is oxidized to oxidized glutathione (GSSG). GSSG is then recycled back to GSH by the enzyme glutathione reductase (GR) using NADPH as a substrate [13]. Thus, the maintenance of GSH for optimal scavenging capacity is dependent on the bioavailability of NADPH stores. Deficiency of GPx results in acceleration of atherogenesis in ApoE−/− mice, highlighting the importance of glutathione peroxidase in suppressing vascular inflammation and atherosclerosis [24].
Peroxiredoxin
Peroxiredoxins are a family of antioxidant enzymes that regulate cytokine-induced peroxide levels and mediate signal pathways [25]. There are six peroxiredoxins in this family. Importantly, H2O2 has the highest affinity to peroxiredoxin 2 (100%), then to GSH (<0.01%), to Cdc25B (<0.0001%) and to protein tyrosine phosphatase 1B (<0.000001%), demonstrating the importance of peroxiredoxins [1]. Overexpression of mitochondrial matrix peroxiredoxin (peroxiredoxin-3) prevents left ventricular remodeling and failure after myocardial infarction in mice [26].
Thioredoxins
Thioredoxins are small proteins that play a variety of roles depending upon binding interactions and oxidoreductase activity. Mammalian thioredoxin-2 (Trx2) is a mitochondrial protein. Trx2 deficiency results in embryonic lethal at gestational day 10.5 and embryos show massive apoptosis. The timing coincides with the maturation of mitochondrial function. In addition, Trx2 protects against vascular pathology in the ApoE-knockout mouse model for CVD [27]. Furthermore, the accumulated data strongly support a role for Trx2 in protecting against oxidant-induced apoptosis via regulating mitochondrial permeability transition [28].
Synthetic mtROS scavengers
Although several natural antioxidants including vitamin E have been shown to decrease mtROS; however their effectiveness was limited as they did not accumulated within mitochondria nor could they cross the blood–brain barrier efficiently [29,30]. To address this issue, several synthetic mtROS scavengers have been developed. These compounds easily pass through all biological membranes, including the blood–brain barrier, into cells and tissues affected by mtROS [31]. The first targeted ROS scavenger is MitoVit-E in which vitamin E is covalently attached to a triphenylphosphonium cation [32]. MitoVit-E decreases ROS production and apoptosis in aortic endothelial cells induced by oxidative stress, but it is ineffective against hypoxic–ischemic striatal injury in neonatal rats [33]. The second ROS scavenger is MitoQ10 which consists of a lipophilic triphenylphosphonium cation covalently attached via an aliphatic linker to a ubiquinone derivative [34]. After detoxifying an oxidant species, MitoQ10 can be regenerated by the respiratory chain. MitoQ10 inhibits mitochondrial oxidative damage in rodent models of cardiac ischemia and reperfusion injury [33]. Moreover, the results of animal studies using MitoQ10 or an alternative compound termed SkQ1 are promising. When perfused through isolated heart preparations or fed to rats, SkQ1 is able to reduced ischemia-induced arrhythmia and infarct size; SkQ1 is used at a concentration that is an astounding one million fold lower than that of MitoQ. The results of ongoing clinical studies are awaited with great interest [33].
Regulation of mtROS
ROS production in mitochondria is determined by the rates of both mtROS production and disposal, and it is regulated by a number of factors, such as mitochondrial membrane potential, metabolic state of mitochondria, and O2 levels [35] (Figure 2).
Figure 2.
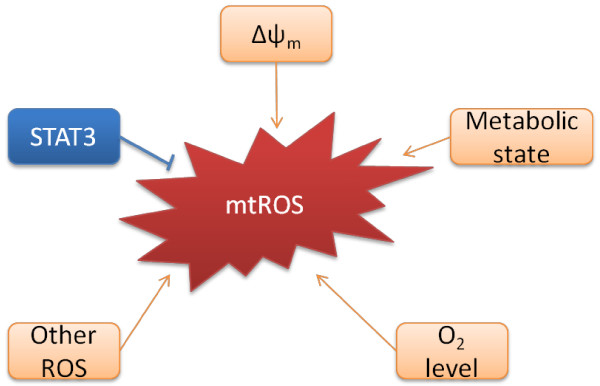
Regulation of mtROS production. A number of factors including mitochondrial membrane potential (Δψm), metabolic state of mitochondrial, O2 concentration regulate the production of mtROS. Non-mitochondrial generated ROS can also augment mtROS production, a process known as “ROS-induced ROS”. Meanwhile, transcription factor STAT3 has recently been found to suppress mtROS production independent of its nuclear factor activity.
Mitochondrial membrane potential (Δψm)
As described above, Δψm is created when protons are pumped from the mitochondrial matrix to the intermembrane space as electrons pass through the ETC. The concept that higher (more polarized) Δψm is associated with greater mtROS generation is widespread in literature, and this is thought to be due to the slowed electron transport [11,20]. This idea is supported by the observation of decreased ROS generation when Δψm is dissipated by either chemical uncouplers [36], such as carbonyl cyanide p-(tri-fluromethoxy)phenyl-hydrazone (FCCP) [37], or overexpression of mitochondrial uncoupling proteins (UCPs) [38]. However, it has also been shown that uncoupling of the mitochondrial ETC in cardiomyocytes using chemical uncouplers in fact increased ROS accumulation [15]. To reconcile this obvious discrepancy, a redox-optimized ROS balance hypothesis is proposed, stating that physiological ROS signaling occurs within an optimized mitochondrial membrane potential, and oxidative stress can happen at either the extreme of high Δψm or low Δψm[15]. This hypothesis is based on the fact that the redox couples involved in substrate oxidation (NADH) are closely linked to the redox couples involved in antioxidant defenses (NADPH). Hence, it is vital to balance an adequate level of Δψm to maintain matrix NADPH rather than NADP+, which is necessary for mitochondrial antioxidant enzyme systems. In other word, an increase in mitochondrial uncoupling of the ETC can increase ROS production primarily because the antioxidant system of the cell is compromised.
Metabolic state of mitochondria
Metabolic state of mitochondria is another important factor that modulates mtROS production. The concept of metabolic states of mitochondria was proposed by Britton Chance and G.R. Williams in 1955 [39]. State I is the first respiratory state observed when isolated mitochondria are added to mitochondrial respiration medium containing oxygen and inorganic phosphate, but no ADP and no reduced respiratory substrates [39]. In State I, leak respiration may be supported to some extent by undefined endogenous substrates, which are oxidized and slowly exhausted. State II is the substrate-limited state of residual oxygen consumption, after addition of ADP to isolated mitochondria suspended in mitochondrial respiration medium in the absence of reduced substrates [39]. State III respiration is the ADP-stimulated respiration of isolated coupled mitochondria in the presence of high ADP and phosphate concentrations, supported by a defined substrate or substrate combination at saturating oxygen levels [39]. State IIIu (u for uncoupled) has been used frequently in bioenergetics on the fundamental difference between OXPHOS capacity and non-coupled ETS capacity. State IV is the respiratory state obtained in isolated mitochondria after State III, when added ADP is phosphorylated completely to ATP driven by electron transfer from defined respiratory substrates to O2[39]. State V is the respiratory state obtained in a protocol with isolated mitochondria after a sequence of State I to State IV, when the concentration of O2 is depleted in the closed oxygraph chamber and zero oxygen (the anaerobic state) is reached [39]. The State V is defined in the original publication in two ways - State V may be obtained by antimycin A treatment and by anaerobiosis. Resting mitochondria (State IV) are characterized by low electron flow and ATP synthesis, low rates of O2 consumption, and high NADH/NAD+ ratio leading to high ROS production. When mitochondria are synthesizing ATP (State III), the opposite happens (high electron flow and ATP synthesis, high rates of O2 consumption, high NADH/NAD+) which results in lower ROS production [40].
Importantly, endogenous modulators such as NO and Ca2+ can regulate the production of mtROS by regulating the metabolic states of mitochondria. NO is a diffusible gas synthesized by three NOS enzymes including endothelial NOS (eNOS), inducible NOS (iNOS) and neuronal NOS (nNOS). These three enzymes share 50–60% homology at the amino acid sequence and have an N-terminal oxygenase domain with heme-, L-arginine-, tetrahydrobiopterin (BH4)-binding domains, a central calmodulin (CaM)-binding region, and a C-terminal reductase domain with NADPH, FAD, and FMN binding sites [41]. The identification of NOS in the mitochondria [42] and the fact that the ETC has several NO. reactive-redox metal centers [11] strongly argue NO’s role as an important modulator of mtROS production. NO can modulate mitochondrial respiration and oxygen consumption through reversible binding and inhibition at complex IV, leading to the accumulation of NADH and increases in ROS production [43]. Mitochondria also participate in Ca2+ homeostasis by serving as a high-capacity, low-affinity transient Ca2+ store. Unlike NO, it seems that a moderate increase in mitochondrial Ca2+ stimulates the rate of electron flow in the ETC and thus decreases mtROS generation [44]. However, it should be noted that mitochondrial Ca2+ overload increases mtROS production, which is independent of the metabolic states of mitochondria [45].
O2 concentration
MtROS production also depends on O2 concentration. As cellular O2 concentration increases, the rate of mtROS production increases linearly [46]. However, during hypoxia, a paradoxical increase in mtROS release was reported [47]. This mtROS release appears to come from complex III and functions as a regulator of hypoxia-inducible factor 1α (HIF-1α). Nevertheless, the precise molecular basis underlying the seemingly controversial relationship between ambient oxygen levels and mtROS production remains obscure. The redox-optimized ROS balance hypothesis mentioned previously can be used to account for this discrepancy. It is postulated that hypoxic cells would exhibit high Δψm and augmented mtROS production due to the low electron flow [15]. In this setting, the increased generation of mtROS could then be relieved by overexpressing mitochondrial UCPs [48].
Mitochondrial mass
MtROS may be additionally altered by mitochondrial mass. Theoretically, the level of mtROS generation should be positively correlated to the quantity of mitochondria in the cell. Nevertheless, it has been shown that the mitochondrial biogenesis factor peroxisome-proliferator-activated receptor-γ coactivator 1α (PGC1α) not only increases mitochondrial mass but also increases the expression of many antioxidant enzymes including GPx and SOD2 [49]. It therefore stands to reason that mitochondrial mass is not an important factor that regulates mtROS production.
Mitochondrial fusion
Mitochondria are dynamic organelles which frequently change their number, size, shape, and distribution in response to intra- and extracellular stimuli. After proliferated from pre-existing ones, fresh mitochondria enter constant cycles of fission and fusion that can be classified into two distinct states - individual state and network state. When compromised with various injuries, solitary mitochondria are subjected to organelle degradation, which relies on autophagy, a self-eating process that plays key roles in manifold cell activities. Recent reports reveal that defects in autophagic degradation selective for mitochondria (mitophagy) are associated with neurodegenerative diseases, highlighting the physiological relevance of mitophagy to cellular functions [50]. The fission and fusion processes are important for mitochondria to redistribute their proteins, protecting the cells from the harmful effects of mitochondrial DNA (mtDNA) mutations. These processes are regulated by N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-like proteins which include mitofusin-1 and −2 [51]. Whether mtROS regulate mitochondrial fusion remains unclear, however it has been reported that mtROS enhance mitochondrial fragmentation [52].
Transcription factors
Several nuclear transcription factors (TFs) with well-characterized functions in the nucleus are also present in the mitochondria and become mitochondrial TFs (mitoTFs). MitoTFs include those of the nuclear hormone receptor family as well as TFs such as p53, nuclear factor kappa B (NF-κB) and the signal transducer and activator of transcription (STATs) that are activated downstream of the binding of growth hormones and cytokines to cell-surface receptors [53]. These TFs have several different mechanisms in regulating mitochondrial function and ROS levels. P53 can bind to the Bcl-2 family members and induces apoptosis. Also, p53 can inhibit SOD2. In addition, interferon regulatory transcription factor (IRF) family 3 (IRF3) can interact with proapoptotic protein Bax. Moreover, TFs including cAMP-responsive transcription factor (CREB), NF-κB, myocyte enhancer factor-2D (MEF2D) and STAT3 all can regulate gene expression.
STAT3, initially identified as a transcription factor that regulates gene expression in response to cytokines such as interleukin (IL)-6 and IL-10, has recently been found to modulate mtROS through mechanisms independent of its nuclear factor activity, but dependent on its ability to directly modulate the activity of the ETC [13,54]. It has been shown that STAT3 is present in the mitochondrial matrix, and deficiency of STAT3 in murine hearts leads to decreased activities of complexes I and II while increasing mtROS at complex I [13]. However, the molecular mechanism by which STAT3 modulates ETC activity is not well understood and it remains to be determined whether STAT3 is unique among STAT proteins in localizing to mitochondrial matrix and regulating mtROS. Since STAT3 responds to cytokines of the IL-6 and IL-10 families, which themselves regulate cellular metabolism process, it is tempting to speculate that by modulating mitochondrial ETC activity and mtROS generation, STAT3 links cytokine signaling pathways to cellular metabolism.
HIF-1 mediates adaptive responses to chronic hypoxia with reduced oxygen availability by regulating gene expression. HIF-1 reduces mtROS production under hypoxic conditions by multiple mechanisms including: i) a subunit switch in cytochrome c oxidase from the cytochrome c oxidase subunit IV (COX4)-1 to COX4-2 regulatory subunit that increases the efficiency of mitochondrial complex IV; ii) induction of pyruvate dehydrogenase kinase 1, which shunts pyruvate away from the mitochondria; iii) induction of BCL2/adenovirus E1B 19 kDa protein-interacting protein 3, which triggers mitochondrial selective autophagy; and iv) induction of microRNA-210, which blocks assembly of Fe/S clusters that are required for oxidative phosphorylation [55].
Epigenetic regulatory enzyme protein deacetylases
Recent progress has demonstrated that the Class III (NAD + −dependent) deacetylases termed sirtuins play a critical role in suppressing inflammation. Specifically, the subcellular locations of these sirtuins including the nucleus (SIRT-1,-2,-6,-7), cytosol (SIRT-1,-2) and mitochondria (SIRT-3,-4,-5) [56] suggest the important functions of these Sirtuins in these locations [33,57]. Endothelial overexpression of SIRT1 has been shown to suppress atherosclerosis and maintain normal endothelial function in mice fed a high-fat diet [58] and can also protect against hyperglycemia-induced vascular cell senescence [59]. Recent progress has demonstrated that sirtuins’ complicated regulatory functions can be activated by resveratrol, an antioxidant polyphenol compound isolated from grape skin. Resveratrol has shown promising clinical benefits as anti-aging [60], anti-inflammatory [61], anti-diabetic [62], anti-viral [63], anti-neoplastic [64], and anti-CVD agents [65]. Several mechanisms underlying sirtuins functions have been found, for example, Sirt1 induces eNOS function and promotes NO generation. Sirt1 inhibits type 1 angiotensin receptor and enhances tissue inhibitor of metalloproteinase. Sirt1 also induce superoxide dismutase and other antioxidant genes and suppress cellular burden of ROS.
Cytokines
As early as a decade ago, mtROS were already found to be induced by tumor necrosis factor (TNF)-α which is mediated by ceramide-dependent signaling pathways [66]. However, mtROS do not appear to have a role in TNF-α triggered NF-κB activation and ICAM-1 expression in endothelial cells [67]. It is later unveiled that TNF-α induces a calcium-dependent increase in mtROS that causes the shedding of TNF-α receptor-1 and reduces the severity of microvascular inflammation [68]. Another proinflammatory cytokine, interferon-γ (IFN-γ) is capable of upregulating the expression of many nuclear genes encoding mitochondrial ETC and inducing mtROS by activation of estrogen-related receptor alpha (ERRα) and coactivator peroxisome proliferator-activated receptor gamma coactivator-1 beta (PGC1β).
Adipose tissue is not only an organ of energy storage but also an endocrine organ capable of producing a number of cytokines termed as adipokines [69]. Two members of the adipokine family, leptin and resistin, have been shown to increase mtROS production [70,71]. Leptin is a circulating adipokine involved in the control of body weight. Not surprisingly, leptin could “talk” with mitochondria and induces mtROS by increasing fatty acid oxidation via protein kinase A (PKA) activation [70]. Resistin is another adipocyte-derived cytokine that play an important role in insulin resistance, adipogenesis, and inflammation. By reducing mitochondrial Δψm and activities of antioxidants including catalase and SOD, resistin could induce eNOS downregulation through overproduction of mtROS in endothelial cells [71].
A recent report showed that CD8+ memory T cells, but not CD8+ T effector (Teff) cells, possess substantial mitochondrial spare respiratory capacity (SRC). SRC is an extra capacity available in cells to produce energy in response to increased stress or work, which is associated with cellular survival. Interleukin-15 (IL-15), a cytokine critical for CD8+ memory T cells, regulates SRC and oxidative metabolism by upregulating mitochondrial biogenesis and expression of carnitine palmitoyl transferase (CPT1a), a metabolic enzyme that controls the rate-limiting step to mitochondrial fatty acid oxidation (FAO). These results demonstrate how cytokines control the bioenergetic stability of memory T cells after infection by regulating mitochondrial metabolism [72]. In addition, IL-15 transgenic mice run twice as long as control littermates in a run-to-exhaustion trial and preferentially uses fat for energy metabolism. Skeletal muscles in IL-15 transgenic mice have high expression of intracellular mediators of oxidative metabolism that are induced by exercise, including sirtuin 1, peroxisome proliferator-activated receptor (PPAR)-δ, PPAR-γ coactivator-1α, and PPAR-γ coactivator-1β [73].
Non-mitochondrial ROS sources
Under certain conditions, non-mitochondrial generated ROS can augment mtROS production, a process known as “ROS-induced ROS”. It has been demonstrated that many other ROS-producing enzymes, including NADPH oxidase [74], xanthine oxidase [75], and uncoupled eNOS [76], can stimulate mtROS production. The “ROS-induced ROS” system downstream of angiotensin II (Ang II) signaling pathway is well characterized. Ang II is a well-known stimulator of NADPH-oxidase-derived ROS [77], but a role of mtROS downstream of Ang II-mediated cellular signaling has also emerged recently [74]. In fact, it has been suggested that by activating NADPH oxidase, Ang II induces mtROS, which in turn leads to further activation of NADPH oxidase. Moreover, scavenging of mtROS using mitochondria-targeted antioxidant can interrupt this vicious cycle and significantly decrease blood pressure after the onset of Ang II-induced hypertension [78].
The question of how other sources of ROS induce mtROS remains. However, the importance of p66shc in this process is highlighted by the fact that p66shc is localized within the mitochondrial intermembrane space and can directly transfer electrons from cytochrome c to O2 to generate mtROS [79]. Importantly, intracellular antioxidants such as GSH are thought to maintain the mitochondrial form of p66shc in an inactive state [80]. Thus, p66shc may serve as a thiol-based redox sensor that signals to mitochondria to induce mtROS when the ROS level in cytoplasm becomes high. As such, the atherosclerotic risk factor, oxidized LDL, activates p66shc through NADPH oxidase [81]. Furthermore, deficiency of p66shc gene renders mice resistant to complications of atherosclerosis [82].
MtROS levels and signaling of mtROS
Once thought as merely the by-products of cellular metabolism, mtROS are increasingly viewed as important signaling molecules [83]. At low levels, mtROS are considered to be important for metabolic adaptation as seen in hypoxia. Moderate levels of mtROS, stimulated by danger signals such as Toll-like receptor 4 ligand bacterial endotoxin lipopolysaccharide (LPS), are involved in regulating inflammatory response. Finally, high levels of mtROS activate apoptosis/autophagy pathways capable of inducing cell death [83] (Figure 3). But, how does mtROS signal in the cell? Much like the events of phosphorylation modification of proteins, mtROS promote cell signaling via the oxidation of certain reactive cysteine residues of proteins [84]. Cysteine residues can exist in a number of oxidative states, including sulfenic form (RSOH), sulfinic form (RSO2H), and sulphonic (RSO3H) form. Although the pKa of most thiol group on free cysteine is between 8 and 9, the surrounding environment of certain reactive cysteine residues can be substantially modified to result in reduced pKa as low as 4 to 5. These reactive cysteine residues (RS-) are easily oxidized to RSOH. RSOH is unstable and can undergo further oxidation into RSO2H. Furthermore, under greater oxidative stress than that for generating RSOH and RSO2H, RSO3H is generated. Although the generation of RSOH and RSO2H is readily reversible, formation of RSO3H is irreversible (Figure 4). Using computational methods and proteomic approaches, it is suggested that RS- might exist in more than 500 proteins, allowing mtROS to modulate a wide variety of protein targets in the cells [85,86].
Figure 3.
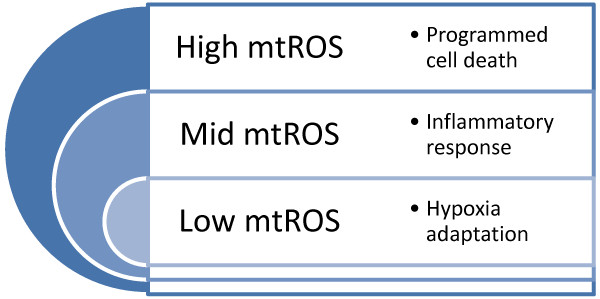
Signaling of mtROS. At low levels, mtROS participate in the process of hypoxia adaptation by regulating the stability of hypoxia-inducible factor 1α (HIF-1α); moderate levels of mtROS are involved in regulating the production of proinflammatory cytokines by directly activating the inflammasome and mitogen-activated protein kinase (MAPK); high levels of mtROS are capable of inducing apoptosis and autophagy by oxidation of the mitochondrial pores and autophagy-specific gene 4 (ATG4) respectively.
Figure 4.
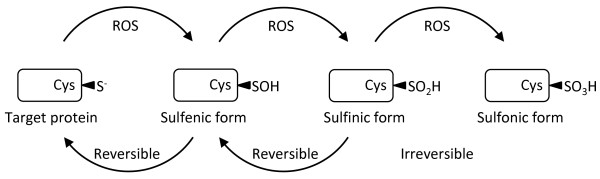
Modification of proteins by ROS. ROS can oxidize specific reactive cysteine (Cys) residues within target proteins generating sulfenic form (RSOH) of proteins. RSOH is unstable and can be further oxidized to sulfinic form (RSO2H). Under greater oxidative stress, sulfonic form (RSO3H) can be generated. Although the formation of RSOH and RSO2H is reversible, generation of RSO3H is irreversible.
Low mtROS
Accumulating evidence suggests that mtROS released under hypoxic conditions regulates HIF-1α. HIF-1α is a heterodimeric protein composed of an α subunit and a β subunit [87], the latter being constitutively expressed. The stability of the α subunit, however, is regulated by oxygen levels such that, it is stabilized under hypoxic conditions while it undergoes proteasomal degradation under normoxic conditions [83]. The picture is becoming clear that, HIF-1α is stabilized in response to mtROS and then feedback and inhibit the production of mtROS [83]. The latter feedback activity is suggested by recent identification of a mitochondrial gene, NADH dehydrogenase [ubiquinone] 1 alpha subcomplex, 4-like 2 (NDUFA4L2) that serves as a direct HIF-1α target [88]. Employing NDUFA4L2-silenced and NDUFA4L2 knockout cells, it has been demonstrated that inhibiting mtROS generation via NDUFA4L2 upregulation induced by HIF-1α is an essential cellular adaption process during hypoxia.
Moderate mtROS
Several recent studies unveil the fact that mtROS act as essential signaling molecules that regulate inflammatory process. On one hand, one member of the cytosolic nucleotide binding and oligomerization domain (NOD)-like receptor (NLR) family, pyrin domain containing 3 (NLRP3)-containing inflammasome (caspase-1 activating protein complex) is shown to be activated by mtROS [5]. The NLRP3 inflammasome is a multiprotein complex consisting of the sensor protein NLRP3, the adaptor protein ASC, and the inflammatory protease precursor pre-caspase-1 [89]. Although several sensor proteins including NLRP3, NLRC4 (NLR family, CARD-containing 4), AIM2 (Absent in melanoma 2), and NLRP6 (NOD-like receptor family pyrin domain containing 6) have been shown to form inflammasomes with caspase-1, the NLRP3 inflammasome has drawn the most attention due to its association with the onset and pathogenesis of numerous inflammatory diseases [90]. Conformational changes in NLRP3 lead to the assembly of the inflammasome and activation of caspase-1, which promotes the maturation and secretion of proinflammatory cytokines IL-1β and IL-18. Besides its role in regulating inflammatory processes, the NLRP3 inflammasome also drives a form of inflammatory cell death, termed pyroptosis [91]. In contrast to non-inflammatory apoptosis, pyroptosis causes local inflammation, as it is linked to the caspase-1 cleavage of pre-IL-1 β and pre-IL-18 and release of mature IL-1β and IL-18 [92]. While apoptotic cells retain their plasma membrane integrity until the final stage of apoptosis, pores rapidly form in the plasma membrane of pyroptotic cells. Such pores provide a direct route for the release of inflammatory molecules including IL-1β and IL-18 [93]. Of note, it has been shown that autophagy blockage results in accumulation of dysfunctional, mtROS generating mitochondria, which also activates the NLRP3 inflammasome [5]. In addition, both NLRP3 and its adaptor ASC co-localize with endoplasmic reticulum (ER) and mitochondria upon inflammasome stimulation. However, it remains unknown whether NLRP3 is the direct target of mtROS. Indeed, this idea is challenged by the observation that ROS inhibitors block priming instead of activation of the NLRP3 inflammasome [94]. Priming of the NLRP3 inflammasome controls the threshold of the inflammasome activation, which involves induction of pro-IL-1β and NLRP3 expression [89]. In this regard, mtROS may be involved in induction of NLRP3 transcripts upstream of post-translational NLRP3 activation. Based on the differences of tissues in the expression levels of NLRP3 and other inflammasome components, we proposed novel “three-tier model and inflammation privilege” for explaining the readiness of tissues in the onset of inflammation in response to stimuli [95].
On the other hand, it has been shown that mtROS cause oxidation and inactivation of MAPK phosphatases, which results in sustained MAPK activation [96]. In addition, inhibition of mtROS production attenuates MAPK activation and production of IL-6 induced by LPS whereas macrophages lacking inflammasome components produce normal levels of IL-6 after LPS stimulation comparable to that of wild-type macrophages [97]. Thus, in a critical mechanistic divergence, mtROS could also regulate inflammasome-independent proinflammatory cytokines such as IL-6 by affecting transcription factor pathways. Indeed, mtROS also appear to activate NF-κB and induce the upregulation of cell surface adhesion molecules in endothelial cells as a part of endothelial cell activation program [67,98].
High mtROS
Similar to the caspase-1 activating inflammasomes, activation of the structurally related protein complex termed apoptosome also requires mtROS [99]. Apoptosome is an oligomeric structure that is assembled when apoptosis activating factor (APAF)-1 interacts with cytochrome c released from mitochondria, and the activation of apoptosome initiates apoptosis by recruiting and activating pre-caspase-9 [100]. MtROS contribute to apoptosome activation by oxidation of the mitochondrial pores, which leads to cytochrome c release [99]. If mtROS are involved in both inflammasome and apoptosome activation, what mechanisms regulate the choice of the signaling pathways? One possibility is that the intensity or duration of mtROS release determines the ultimate biological outcome, with high levels of mtROS capable of inducing apoptotic cell death.
Recent evidence yet suggests the involvement of mtROS in the induction of autophagy, another form of programmed cell death [101]. Starvation of cells stimulates formation of ROS, which localizes with mitochondria. Treatment of cells with antioxidant agents abolishes starvation-induced autophagy. Moreover, a specific reactive cysteine residue on autophagy regulatory protein, autophagy-specific gene 4 (ATG4) is shown to be ROS-sensitive. This new working model in which mtROS oxidize ATG4 and induce autophagy requires further examination.
Detection of mtROS by fluorescent probes
Recent progress on characterization of mtROS has been greatly benefited from new advances in using fluorescence probes in the detection of mtROS. Fluorescent dyes including dichlorodihydrofluorescein (DCF) and dihydroethidium (DHE) have been widely used to detect intracellular ROS in early studies. While DCF is the most widely used probe for detecting intracellular H2O2, DHE is the most frequently used fluorescent indicator for O2 .-. In conjugation with mitochondria-specific markers such as MitoTracker using confocal microscopy, one can determine whether ROS are generated from mitochondria [102-104]. To minimize the effect in subtracting the fluorescence signals coming from non-mitochondrial organelles, some fluorescence indicators have been modified to target mitochondria specifically. The most common way is through the use of lipophilic cations, which are attracted to the negative potential environment caused by the proton gradient across the inner mitochondrial membrane.
MitoSOX is a triphenylphosphonium (TPP+)-linked DHE compound. It exploits the steep electrochemical gradient across the mitochondrial inner membrane to enrich the TPP-tag fluorescence more than a 100-fold within the mitochondria compared with the cytosol [105]. MitoSOX has been effectively used to directly detect mitochondria-derived O2.- in various cell types [106]. However, the reactions of both DHE and MitoSOX with ROS yield two fluorescent products, one of which is O2.- specific, while the other is formed in response to general oxidative stress. Thus, fluorescence microscopy or related flow cytometric techniques are not sufficient to measure the superoxide-specific hydroxylated products using both classical DHE and novel MitoSOX. High-performance liquid chromatography (HPLC) methodologies are required to separate and identify these products [78,107] (Table 2).
Table 2.
Comparison of different detection methods of mtROS
| Methods | Drugs | Detected species | Principle | Limits |
|---|---|---|---|---|
| Fluorescent probes |
MitoSOX |
Superoxide |
Attaching a hydrophobic cation to dihydroethidium. |
Two fluorescent products, one of which is superoxide specific. The two products need to be separated by HPLC. |
| |
MitoPY1 |
Hydrogen peroxide |
Attaching a hydrophobic cation to a phenylboronate ester. |
It also reacts with peroxynitrite. |
| |
MitoAR MitoHR |
Hydroxyl radical |
Reaction with hydroxyl radical cleaves off the quenching motif. |
They also react with peroxynitrite. |
| Mitochondrial inhibitors |
Rotenone Antimycin |
General ROS |
Rotenone is complex I inhibitor and antimycin is complex III inhibitor. |
Rotenone either decrease or increase ROS; Interruption of the ETC. |
| Mitochondrial antioxidant |
MitoQ MitoTEMPO MitoVitE |
General ROS |
Attaching a hydrophobic cation to antioxidants. |
- |
| Electron spin resonance | MitoDEPMPO | Free radicals | Free radicals have unpaired electrons, and thus are paramagnetic and detectable by ESR. | Lower sensitivity than fluorescent probes. |
Appending a TPP + motif to a phenylboronate ester generates MitoPY1 [108], which is a biologically compatible probe for detecting mitochondria-derived H2O2. MitoPY1 selectively responds to rises in H2O2 levels by a significant fluorescence increase detected by both confocal microscopy and flow cytometry methods [108]. It should be noted though: the phenylboronate moiety in MitoPY1 also reacts with ONOO-, so MitoPY1 will respond to this molecule as well.
Highly reactive oxygen species (hROS), including hydroxyl radical (.OH), hypochlorous acid (HOCl), and ONOO-, are generated secondary to mtROS formation (O2 .-, H2O2) [109]. They are highly toxic by directly oxidizing nucleic acids, proteins, and lipids in the cell. .OH originates from H2O2 through Fenton chemistry in the presence of iron or copper centers which are prevalent in the mitochondria. Likewise, NO produced by mitochondrial NOX can combine with O2 - to form ONOO-. In addition, myeloperoxidases (MPOs) catalyze the reaction of H2O2 into HOCl, which possibly could diffuse into and damage mitochondria. As such, several probes have been developed to detect these mitochondrial hROS. For example, MitoAR and MitoHR are developed by attaching rhodamine-like fluorophores (which functions similarly as TPP motif) to either a 4-amino-phenyl aryl ether or a 4-hydroxy aryl ether group, respectively. The ether motif on both MitoAR and MitoHR quench the fluorescence emission. Reaction with mitochondrial hROS cleaves off the quenching motif and results in the highly fluorescent rhodamine type reporters [110]. MitoAR mainly detects .OH and HOCl while MitoHR is most sensitive to .OH, while both of these two probes also react with ONOO- in a slower rate.
Detection of mtROS by other methods
ETC inhibitors and mitochondria-targeted antioxidant
Combination of traditional ROS detection method (DHE fluorescence) with ETC inhibitors can also help to identify mtROS [111-114]. This approach is instrumental, but the results obtained from these inhibitors are not always consistent. For example, when intact cells are used, complex I inhibitor rotenone has been shown to either decrease or increase ROS production [104,115]. Another potential problem with mitochondrial inhibitors is their interruption of the ETC, which may alter cell metabolism such as ATP synthesis. In this regard, mitochondrial-targeted antioxidants represent a promising tool in studying mtROS. A series of antioxidants have been chemically bonded to TPP to scavenge mtROS, including α-tocopherol (MitoVitE) [104], nitroxides (MitoTEMPO) [78], and ubiquinol (MitoQ) [114]. Among these, MitoQ is the best characterized mitochondria-targeted antioxidant in animal studies [34,105,116]. The ubiquinol form of MitoQ is oxidized by mtROS to the ubiquinone form, which is quickly re-reduced by complex II in the ETC, restoring its mtROS scavenging capacity. In addition, the oral administration of MitoQ is safe for at least 24 weeks in mice and rats [117]. For these reasons, orally administered MitoQ has been tested in a number of in vivo animal studies and shown protection against oxidative stress in a variety of CVD, including: hypertension [118], cardiac ischemia reperfusion (I/R) injury [119], kidney damage in type I diabetes [120], sepsis [121,122], and endothelial damage by nitroglycerin [123].
ESR
In some experiments, electron spin resonance (ESR) spectroscopy is used to study mtROS [74,124]. Free radicals such as superoxide have unpaired electrons, and thus are paramagnetic and detectable by ESR. The application of different spin probes, cell permeable versus nonpermeable, allows one to localize and distinguish mtROS versus non-mtROS and intracellular versus extracellular ROS. Another advantage of ESR assay is that cells or tissue samples can be frozen and analyzed later. In addition, ESR has potential in determining the chemical identities of ROS. For example, MitoDEPMPO has been developed as a mitochondria-targeted nitrone traps for ESR detection of mitochondria-derived superoxide [125]. Moreover, it can be used to analyze cells in suspension. However, the sensitivity of ESR assay is lower than that of methods based on fluorescence. Moreover, ESR assay cannot be applied for ROS detection and determination of intracellular distribution in a single cell (Table 2).
MtROS in vascular cells
Endothelial cells (ECs)
Even when plenty of oxygen is presented, ECs rely heavily on glycolysis rather than OXPHOS-mitochondrial respiration to generate ATP [126]. Nevertheless, ECs still contain functional mitochondria, in which oxidative phosphorylation continues [127,128]. Thus, the primary function of mitochondria in ECs may be the regulated generation of ROS for cells signaling purpose, but not generation of ATP for energy production. If this is true, ECs could modulate mtROS production without jeopardizing their energy needs. Indeed, mtROS are involved in regulating a variety of important endothelial functions under basal conditions whereas activating proinflammatory pathways in response to cardiovascular risk factors in ECs [129].
Under normal physiological conditions, mtROS are capable of regulating vascular homeostasis. Firstly, it has been demonstrated that vascular endothelial growth factor (VEGF) promotes endothelial migration through mtROS in cultured human umbilical vein endothelial cells [130]. Endothelial migration is critical in a variety of physiological conditions including wound healing and vascular repair. VEGF increases mitochondrial metabolism and mtROS production. Furthermore, mitochondria-targeted antioxidant prevents VEGF-induced endothelial migration. Secondly, mtROS contribute to endothelium-dependent vasodilation [131]. The endothelium regulates vascular homeostasis in response to shear stress by synthesizing vasodilators such as NO. Using ESR and histochemofluorescence methods, it has been shown that shear flow increases the production of ROS in human coronary resistance arteries, which can be blocked by mitochondrial complex I inhibitor rotenone. Moreover, complex I and complex III inhibitors, but not NADPH oxidase inhibitors, markedly blocked flow-induced dilation. Notably, ROS formation in response to flow after endothelial denudation is significantly decreased, suggesting an important role of endothelium in flow-induced mtROS formation. Thirdly, mtROS are also of importance in hypoxia-induced adaptation response in ECs. At low O2 concentrations, mitochondria of HUVECs (human umbilical vein endothelial cells) have been shown to generate ROS for activation of enzymes such as AMPK (AMP-activated protein kinase) because: 1) AMPK activation coincides with the hypoxia condition at which ROS is produced; 2) antioxidants can rescue hypoxia-induced AMPK activation; and 3) AMPK activation does not occur in ρ0 HUVECs devoid of mitochondria. Under the challenge of cardiovascular risk factors, however, excessive mtROS are produced in endothelial mitochondria. Vascular stressors, as diverse as oxidized lipids, high glucose, and angiotensin II, can all induce mtROS in ECs. (Figure 5, Table 3).
Figure 5.

Role of endothelial mtROS in atherosclerosis. Pathologic stressors as diverse as oxidized low-density lipoproteins (oxLDL), glucose, and Angiotensin II are all capable of inducing mtROS in endothelial cells through their receptors. Excessive mtROS then directly bind to NO and induce endothelial cell dysfunction. Overproduction of mtROS also leads to activation of proinflammatory transcription factors such as nuclear factor kappa B (NFκB). This in turn increases the expression of adhesion molecules and production of inflammatory cytokines in endothelial cells, both of which contribute to the development of atherosclerosis. LOX-1: lectin-type oxidized LDL receptor 1; TLRs: Toll-like receptors; AT1R: Angiotensin II receptor, type 1; GLUT4: glucose transporter type 4.
Table 3.
Identified pathologic stressors that induce mtROS in endothelial cells (ECs)
| Cell type | Treatment | Dose | Time | PMID |
|---|---|---|---|---|
| Bovine aortic ECs |
Oxidized LDL |
200 μg/ml |
30 min |
15805232 |
| Bovine aortic ECs |
Electrophilic lipids |
4 μM |
4 h |
16387790 |
| Human umbilical vein ECs |
LysoPC |
5 μmol/L |
60 min |
16651638 |
| Porcine aortic ECs |
Glycated LDL |
100 μg/ml |
2 h |
20036735 |
| Bovine aortic ECs |
Glucose |
30 mM |
2 h |
10783895 |
| Human aorta ECs |
Glucose |
25 mM |
7 d |
12600878 |
| bEnd3 microvascular ECs |
Glucose |
40 mM |
7 d |
21808008 |
| Human umbilical vein ECs |
Low Glucose |
≤ 80 mg/dL |
5 min |
22207730 |
| Bovine aortic ECs |
Angiotensin II |
200 nmol/L |
4 h |
18096818 |
| Human aortic ECs |
Angiotensin II |
200 nmol/L |
4 h |
20448215 |
| Human aortic ECs |
Homocysteine |
150 μM |
24 h |
21672628 |
| Human umbilical vein ECs |
Hypoxia |
14 mmHg |
30 min |
11950692 |
| Human umbilical vein ECs |
Hypoxia |
0 mmHg |
24 h |
12165534 |
| Porcine aortic ECs |
Hypoxia |
0 mmHg |
1 h |
12690038 |
| Murine pulmonary ECs |
Thrombin |
1 mU/mL |
100 s |
17724077 |
| Human umbilical vein ECs |
PAR1-AP |
50 μmol/L |
1 h |
18983479 |
| Human umbilical vein ECs |
TNF-α |
1 ng/mL |
1 h |
11415943 |
| Murine pulmonary ECs |
TNF-α |
1 ng/mL |
10 min |
21519143 |
| Bovine aortic ECs |
Leptin |
10 ng/mL |
45 min |
11342529 |
| Human coronary artery ECs |
Resistin |
40/80 ng/mL |
24 h |
20435848 |
| Human umbilical vein ECs | VEGF | 50 ng/mL | 5 min | 21653897 |
PMID: PubMed ID.
Oxidized low density lipoprotein (oxLDL). Substantial evidence suggests that the retention and oxidative modification of LDL and subsequently activation of ECs initiates atherosclerotic lesion formation [132]. OxLDL triggers the expression of adhesion molecules and secretion of chemokines by ECs, which drive immune cell infiltration. In this sense, oxLDL has been shown to induce mtROS in ECs in vitro [102,133,134]. Using confocal microscopy, it has been demonstrated that a significant proportion of oxLDL-induced cellular ROS are co-localized to mitochondria. Moreover, ECs that are deficient in functional mitochondria show a substantial decrease in cellular ROS formation stimulated by oxLDL [102]. The precise mechanisms whereby oxLDL induces excessive mtROS generation in ECs remains poorly defined. It has been suggested that oxLDL significantly reduces oxygen consumption and enzyme activity in the mitochondrial ETC [133]. OxLDL also increases mitochondrial membrane potential and reduces SOD2 protein levels [134]. Moreover, c-Jun N-terminal kinases (JNK, one of the MAPKs) small interference RNA (siJNK) reduces oxLDL induced mtROS production substantially. OxLDL contains highly heterogeneous mixtures of biologically active substances [135]. Lysophosphatidylcholine (LysoPC, one of the active component derived from oxLDL) accounts for nearly 50% of the phosphatidylcholine equivalents in oxLDL and is considered as a critical factor that contributes to the proatherogenic activity of oxLDL [136]. As such, one study suggests that ROS production by lysoPC occurs predominantly in the mitochondria and is associated with an increase in mitochondrial Ca2+ uptake [104].
Hyperglycemia-high glucose. Hyperglycemia is a key cardiovascular risk factor for patients with type 2 diabetes [137]. High glucose is the first identified pathogenic stressor that induces mtROS in ECs [111]. High glucose-induced DCF fluorescence in bovine aortic ECs is prevented by several factors including an inhibitor of the ETC complex, an uncoupler of oxidative phosphorylation, uncoupling protein-1 and SOD2 [111]. Furthermore, normalizing levels of mtROS after treatment of cells with each of these agents prevents high glucose-induced activation of protein kinase C, formation of advanced glycation end-products, sorbitol accumulation and NFκB activation [111]. Later on, other studies report similar results [112,138]. One study shows that inhibition of ROS production by uncoupling of the ETC significantly reduces high glucose-mediated induction of IL-8 expression in human aortic ECs [112]. Another study links high glucose-dependent mtROS to increased consumption of H2S [138]. Interestingly, traditional pharmacological drugs including anti-inflammatory Sirt1 activator resveratrol, anti-inflammatory/anti-cancer drug cannabidiol, and hypolipidemic drug simvastatin have been shown to prevent high glucose-induced mtROS [139-141]. The measurement of MitoSOX fluorescence shows that resveratrol attenuates high glucose-induced mtROS production in human coronary arterial ECs. The authors propose that resveratrol, via a pathway that involves the upregulation of antioxidant defense mechanism, attenuates mtROS production [139]. Another paper shows that high glucose markedly increases mtROS, NF-κB activation, upregulation of iNOS, and EC adhesion molecules intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), transendothelial migration of monocytes, and monocyte-endothelial adhesion in human coronary artery ECs. Remarkably, all the above mentioned effects induced by high glucose are attenuated by cannabidiol pretreatment [140]. Similarly, Simvastatin decreases high glucose-induced mtROS in bovine retinal capillary ECs and exerts protective effects against early retinal vascular damage in diabetic rats [141]. A recent study proposes that acute exposure to low glucose also increases mtROS production in human umbilical vein ECs [142]. Interestingly, anti-diabetic drug Metformin can reverse low glucose-induced endothelial dysfunction through inhibiting excessive mtROS production.
Angiotensin II. Ang II is another pathogenic stressor that mediates endothelial dysfunction and promotes vascular inflammation and atherogenesis [143]. Ang II treatment of bovine aortic ECs is shown to significantly increase mtROS production detected by ESR. This effect is associated with decreased endothelial NO availability [74]. Later on, the same group confirms this result using MitoSOX fluorescent probe. Interestingly, supplementation of human aorta ECs with the mitochondria-targeted antioxidant mitoTEMPO abolishes the MitoSOX signal after Ang II stimulation [78]. In addition, mitoTEMPO also prevents the loss of endothelial NO caused by Ang II both in cultured ECs and intact mice. Furthermore, treatment of hypertensive mice with mitoTEMPO after onset of Ang II-induced hypertension significantly reduces blood pressure and substantially improves endothelium-dependent vasodilation.
Macrophages (MΦ) and dendritic cells (DCs)
The mononuclear phagocytic system consists of monocytes, MΦ, and DCs [144]. These cells all originate from the same haematopoietic precursors located in the bone marrow and their main functions are phagocytosis, cytokine secretion and antigen presentation. MΦ and DCs precursors are released into the circulation as monocytes, and within several days they exit the circulation through the endothelium into body tissues and differentiate into mature MΦ and DCs.
The phagocytic response of MΦ involves the production of ROS via NADPH-oxidase-dependent respiratory burst. However, recent studies have suggested that mtROS also have an important role in MΦ innate immune response [145]. Activation of a subset of Toll-like receptors (TLR1, TLR2, and TLR4) results in the recruitment of mitochondria to MΦ phagosomes and increased production of mtROS. This augmentation of mtROS production involves the engagement of a TLR signaling adaptor, tumor necrosis factor receptor-associated factor 6 (TRAF6), and the protein evolutionarily conserved signaling intermediate in Toll pathways) in the mitochondria. ECSIT is implicated in mitochondrial ETC assembly. Interaction between the two molecules then leads to ECSIT ubiquitination and enrichment around mitochondria, resulting in increased mtROS production. Additionally, scavenging mtROS in MΦ by expressing catalase in the mitochondria results in defective bacterial killing, confirming the important role of mtROS in MΦ bactericidal activity.
DCs are potent antigen-presenting cells, capable of inducing T and B responses as well as immune tolerance. Compare to its precursor monocytes, DCs exhibit significantly larger number of mitochondria and higher endogenous respiratory activity [146]. Complex I inhibitor rotenone prevents the increase in mitochondrial number as well as DC differentiation. Moreover, rotenone and catalase treatment both inhibit growth factor-induced mtROS in DCs, indicating that the differentiation of DC can be regulated by mtROS in DCs.
Smooth muscle cells (SMCs)
Vascular SMCs migrate and proliferate when they are stimulated by cytokines and fibrogenic factors [147]. MtROS play an important role in this process as demonstrated by the finding that they mediate the activation of Akt/NFκB signaling pathway in response to 4-hydrocynonenal (4-HNE) stimulation in vascular SMCs [148]. In addition, mtROS in SMCs also play a role in cold-induced constriction of cutaneous arteries [149]. Cold constricts cutaneous arteries by selectively increasing the activity of α2-adrenoceptors (α2-ARs). Complex I inhibitor rotenone can abolish cold-induced increase in α2-ARs activity and dramatically inhibits cold-induced constriction response as well. In the atherosclerotic disease setting, thickened plaques render vascular SMCs prone to be hypoxic because of poor perfusion. Complex I inhibitor rotenone again abolishes hypoxia-induced HIF-1α protein expression and ROS generation, suggesting a critical role of mtROS in this pathological condition [150].
MtROS and cancers
Mitochondria lie at the center of the metabolic theory of cancer [151]. Normally, differentiated cells primarily rely on mitochondrial respiration to produce ATP in the presence of oxygen (generating 36 mol. ATP/mol. glucose). Only under limited oxygen availability will healthy cells rely on anaerobic glycolysis as their energy source (generating 2 mol. ATP/mol. glucose). However, most cancer cells adopt the way of “aerobic glycolysis” for energy production [151]. This phenomenon, also known as the Warburg effect [152], is initially confusing to scientists as it is highly energy inefficient. However, it was later found that cancer cells have other important metabolic requirements that extend beyond the generation of ATP. On one aspect, cancer cells are particularly challenged in dealing with oxidative stress [153]. In highly proliferative cancer cells, the presence of oncogenic mutations promotes aberrant metabolism and protein translation, resulting in increased rates of ROS production. It was found that the Warburg effect is beneficial to transformed cells by upregulating antioxidant systems to counteract the accumulation of ROS. One key glycolytic enzyme – pyruvate kinase, plays an important role in this process: tumor cells express exclusively the embryonic M2 isoform of this enzyme (PKM2) [154] and an increase in intracellular ROS can inhibit PKM2 by oxidation of one reactive cysteine residue in this enzyme [155]. This inhibition of PKM2 then leads to the production of reducing equivalents to detoxify ROS by diverting glucose metabolism into pentose phosphate pathway [155]. By doing this, the regulatory properties of PKM2 provide cancer cells the protection against excessive mtROS production commonly seen in cancer [156].
MtROS and hypertension
Hypertension is associated with increased ROS production in several key target organs, including the vasculature, the kidney, and the central nervous system, which all contribute to the regulation of blood pressure [157]. Ang II, the hormone commonly implicated in hypertension, is shown to increase ROS production in these sites. A key role of NADPH oxidase in this process has been demonstrated both in vitro and in vivo [158]. However, later studies indicates that Ang II activation of NADPH oxidase further leads to mitochondrial dysfunction and increased mtROS production [74]. Importantly, mice transgenic for Trx2, the mitochondrial antioxidant enzyme, are shown to resist the development of Ang II-induced hypertension and endothelial dysfunction [159]. Moreover, Ang II-induced hypertension is also significantly attenuated by either overexpressing SOD2 or treatment with mitoTEMPO [78]. These studies strongly demonstrate the potential of antioxidant strategies specifically targeting mitochondria as therapy in hypertension and possibly other diseases.
MtROS and atherosclerosis
Multiple lines of in vivo experimental data indicate that excessive mtROS within vasculature promote the development of atherosclerosis. ApoE−/− mice that are deficient in SOD2, a mitochondria-specific antioxidant enzyme, exhibit accelerated atherogenesis at arterial branching points [160]. SOD2 is also shown to protect against endothelial dysfunction in carotid artery of ApoE−/− mice [20]. Notably, transgenic overexpression of Trx2, another mitochondrial antioxidant enzyme, improves endothelial function and reduces atherosclerotic lesions in ApoE−/− mice in part by reducing oxidative stress and increasing NO bioavailability [27]. There is still limited knowledge of the involvement of mtROS in human atherogenesis; however, epidemiologic data suggest that genetic nucleotide polymorphisms leading to reduced SOD2 function are associated with increased atherosclerotic risk [161]. In addition, there is significantly increased mtDNA damage in atherosclerotic human arterial specimens compared to that of normal human arterial tissue [160]. Indeed, increased mtDNA damage is also a shared phenotype of multiple diseases including neurological degenerative disease [162] and cancer [163]. As mtDNA contains genes that encode critical structural subunits for three of the four protein complexes of the ETC (complex I, III, and IV) (Table 1) [164], mtDNA damage will lead to increases in mtROS generation and the extent of mtDNA damage is an index of the levels of mtROS.
MtROS and other inflammatory diseases
It has long been recognized that ROS are implicated in many inflammatory diseases other than atherosclerosis, including multiple sclerosis [165], rheumatoid arthritis [166], thyroiditis [167], and type 1 diabetes [168]. Very recently, however, it has been identified that mitochondria-derived ROS rather than NADPH oxidase-derived ROS promote the production of proinflammatory cytokines in TNFR1-associated periodic syndrome (TRAPS) [6]. TRAPS is an autoinflammatory disorder associated with enhanced innate immune responsiveness. It is caused by mutations of the gene encoding type 1 TNF receptor (TNFR1), which leads to aberrant activation of MAPKs [169]. It has been found that mtROS, mitochondrial oxidative capacity, and Δψm are all increased in human patient cells and mouse cells harboring TRAPS-related TNFR1 mutations [6]. Moreover, scavenging mtROS using MitoQ abolishes inflammatory cytokine production after LPS stimulation in these cells, highlighting the potential of targeting mtROS as novel therapy for TRAPS and other inflammatory diseases.
Conclusions
Oxidative stress has long been recognized as a major player in the development of atherosclerosis and other inflammatory diseases. This has led to the enthusiastic use of antioxidants in the prevention and treatment of diseases. However, the results of randomized clinical trials of Vitamin C and Vitamin E have been disappointing [170-173]. If indeed oxidative stress is so crucial in the development and manifestations of atherosclerosis-related diseases including myocardial infarction and stroke, why have so many clinical trials failed to demonstrate its therapeutic efficacy? One possibility may be related to the fact that only a small proportion of known antioxidants in vivo are actually located in the mitochondria. Given the fact that mtROS directly drive proinflammatory cytokine production, we can speculate that specific targeting of mtROS may result in better outcome in combating chronic inflammatory diseases such as atherosclerosis. However, in order to translate this knowledge from benchtop to bedside, future studies that fully characterize the biochemistry of mtROS and the specific role mtROS plays in these inflammatory diseases need to be performed. We hope that our review will encourage investigators to enter this important field of research and to accelerate the pace of translational medicine and therapeutics.
Abbreviations
4HNE: 4-hydrocynonenal (4-HNE); ADP: adenosine diphosphate; AIM2: Absent in melanoma 2; AMPK: AMP-activated protein kinase; Ang II: Angiotensin II; APAF: Apoptosis activating factor; ApoE: Apolipoprotein E; ATG4: Autophagy-specific gene 4; ATP: Adenosine triphosphate; BH4: Tetrahydrobiopterin; CaM: Calmodulin; COX: Cytochrome c oxidase subunit; CPT1: Carnitine palmitoyl transferase I; CREB: cAMP-responsive transcription factor; CVD: Cardiovascular disease; DCF: Dichlorodihydrofluorescein; DCs: Dendritic cells; DHE: Dihydroethidium; ECs: Endothelial cells; ECSIT: Evolutionarily conserved signaling intermediate in Toll pathways; eNOS: Endothelial NOS; ER: Endoplasmic reticulum; ERRα: Estrogen-related receptor alpha; ESR: Electron spin resonance; ETC: Electron transport chain; FADH2: Flavin adenine dinucleotide; FAO: Fatty acid oxidation; FCCP: carbonyl cyanide p-(tri-fluromethoxy)phenyl-hydrazone; GPx: Glutathione peroxidase; GR: gLutathione reductase; GSH: Glutathione; GSSG: Oxidized glutathione; HIF-1α: Hypoxia-inducible factor 1α;HPLC: High-performance liquid chromatography; hROS: Highly reactive oxygen species; HUVECs: Human umbilical vein endothelial cells; ICAM-1: Intercellular adhesion molecule-1; IFN-γ: Interferon-γ; IL: Interleukin; iNOS: Inducible NOS; IRF: Interferon regulatory transcription factor; JNK: c-Jun N-terminal kinases; LPS: Lipopolysaccharide; LysoPC: Lysophosphatidylcholine; MAPK: Mitogen-activated protein kinases; MEF2D: Myocyte enhancer factor-2D; mitoTFs: Mitochondrial TFs; MPOs: Myeloperoxidases; mtDNA: Mitochondrial DNA; mtROS: Mitochondrial ROS; MΦ: Macrophages; NADH: nicotine adenine dinucleotide; NADPH: Nicotinamide adenine dinucleotide phosphate; NDUFA4L2: NADH dehydrogenase [ubiquinone] 1 alpha subcomplex, 4-like 2; NF-κB: Nuclear factor kappa B; NLRC4: NLR family, CARD-containing 4; NLRP3: NOD-like receptor family, pyrin domain containing 3; NLRP6: NOD-like receptor family, pyrin domain containing 6; nNOS: Neuronal NOS; NOS: Nitric oxide synthase; NOX: NADPH oxidase; oxLDL: Oxidized low density lipoprotein; OXPHOS: Oxidative phosphorylation; PGC1α: Peroxisome-proliferator-activated receptor-γ coactivator 1α; PKA: Protein kinase A; PKM2: Pyruvate kinase M2 isozyme; PPAR: Peroxisome proliferator-activated receptor; RLRs: RIG-I-like receptors; ROS: Reactive oxygen species; SMCs: Smooth muscle cells; SNARE: N-ethylmaleimide-sensitive factor attachment protein receptor; SOD: Superoxide dismutase; SRC: Spare respiratory capacity; STAT: Signal transducer and activator of transcription; TFs: Transcription factors; TLR: Toll-like receptors; TNF: Tumor necrosis factor; TNFR1: Type 1 TNF receptor; TPP+: Triphenylphosphonium; TRAF6: Tumor necrosis factor receptor-associated factor 6; Trx: Thioredoxin; UCPs: Uncoupling proteins; VCAM-1: Vascular cell adhesion molecule-1; XO: Xanthine oxidase; α2-ARs: α 2-adrenoceptors; Δψm: Mitochondrial membrane potential
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
XL carried out the primary literature search and drafted the manuscript. PF, JM, and ETC provided material input and helped revising the manuscript. HW and XFY conceived the study and provided field expertise. All authors read and approved the final manuscript.
Contributor Information
Xinyuan Li, Email: xinyuan.li@temple.edu.
Pu Fang, Email: pu.fang@temple.edu.
Jietang Mai, Email: jietang.mai@temple.edu.
Eric T Choi, Email: eric.choi@tuhs.temple.edu.
Hong Wang, Email: hong.wang@temple.edu.
Xiao-feng Yang, Email: xfyang@temple.edu.
Acknowledgments
This work was partially supported by the National Institutes of Health Grants HL094451 and HL108910 (XFY), HL67033, HL82774 and HL77288 (HW), and the American Heart Association Fellowship AHA 11PRE7610011 (JM).
References
- Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- Block K, Gorin Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat Rev Cancer. 2012;12:627–637. doi: 10.1038/nrc3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2010;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, Sack MN, Kastner DL, Siegel RM. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) J Exp Med. 2011;208:519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima Y, Chen YX, Kinukawa N, Sueishi K. Distributions of diffuse intimal thickening in human arteries: preferential expression in atherosclerosis-prone arteries from an early age. Virchows Arch. 2002;441:279–288. doi: 10.1007/s00428-002-0605-1. [DOI] [PubMed] [Google Scholar]
- Strong JP, Malcom GT, McMahan CA, Tracy RE, Newman WP 3rd, Herderick EE, Cornhill JF. Prevalence and extent of atherosclerosis in adolescents and young adults: implications for prevention from the Pathobiological Determinants of Atherosclerosis in Youth Study. Jama. 1999;281:727–735. doi: 10.1001/jama.281.8.727. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–473. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H2023–H2031. doi: 10.1152/ajpheart.01283.2006. [DOI] [PubMed] [Google Scholar]
- Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16:1323–1367. doi: 10.1089/ars.2011.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp J. Mitochondria: Sovereign of inflammation? Eur J Immunol. 2011;41:1196–1202. doi: 10.1002/eji.201141436. [DOI] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, O'Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010;1797:865–877. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Canali R, Rettori D, Kaplowitz N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol Pharmacol. 2003;64:1136–1144. doi: 10.1124/mol.64.5.1136. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Ohashi M, Runge MS, Faraci FM, Heistad DD. MnSOD deficiency increases endothelial dysfunction in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2331–2336. doi: 10.1161/01.ATV.0000238347.77590.c9. [DOI] [PubMed] [Google Scholar]
- Radi R, Turrens JF, Chang LY, Bush KM, Crapo JD, Freeman BA. Detection of catalase in rat heart mitochondria. J Biol Chem. 1991;266:22028–22034. [PubMed] [Google Scholar]
- Yang H, Roberts LJ, Shi MJ, Zhou LC, Ballard BR, Richardson A, Guo ZM. Retardation of atherosclerosis by overexpression of catalase or both Cu/Zn-superoxide dismutase and catalase in mice lacking apolipoprotein E. Circ Res. 2004;95:1075–1081. doi: 10.1161/01.RES.0000149564.49410.0d. [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Torzewski M, Ochsenhirt V, Kleschyov AL, Oelze M, Daiber A, Li H, Rossmann H, Tsimikas S, Reifenberg K, Cheng F. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:850–857. doi: 10.1161/01.ATV.0000258809.47285.07. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, Kubota T, Sunagawa K, Hasegawa Y, Kurihara T. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2006;113:1779–1786. doi: 10.1161/CIRCULATIONAHA.105.582239. [DOI] [PubMed] [Google Scholar]
- Zhang H, Luo Y, Zhang W, He Y, Dai S, Zhang R, Huang Y, Bernatchez P, Giordano FJ, Shadel G. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am J Pathol. 2007;170:1108–1120. doi: 10.2353/ajpath.2007.060960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Cai J, Go YM, Johnson JM, Martin WD, Hansen JM, Jones DP. Identification of thioredoxin-2 as a regulator of the mitochondrial permeability transition. Toxicol Sci. 2008;105:44–50. doi: 10.1093/toxsci/kfn116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol RJ, McKim JM Jr, Goff MC, Ruyle SZ, Devereaux MW, Han D, Packer L, Everson G. Vitamin E reduces oxidant injury to mitochondria and the hepatotoxicity of taurochenodeoxycholic acid in the rat. Gastroenterology. 1998;114:164–174. doi: 10.1016/s0016-5085(98)70644-4. [DOI] [PubMed] [Google Scholar]
- Gilgun-Sherki Y, Melamed E, Offen D. Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology. 2001;40:959–975. doi: 10.1016/s0028-3908(01)00019-3. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- Smith RA, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. Eur J Biochem. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- Davidson SM. Endothelial mitochondria and heart disease. Cardiovasc Res. 2010;88:58–66. doi: 10.1093/cvr/cvq195. [DOI] [PubMed] [Google Scholar]
- Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RA, Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- Ballinger SW. Mitochondrial dysfunction in cardiovascular disease. Free Radic Biol Med. 2005;38:1278–1295. doi: 10.1016/j.freeradbiomed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Toime LJ, Brand MD. Uncoupling protein-3 lowers reactive oxygen species production in isolated mitochondria. Free Radic Biol Med. 2010;49:606–611. doi: 10.1016/j.freeradbiomed.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, Shattock MJ. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res. 2006;72:313–321. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Lee KU, Lee IK, Han J, Song DK, Kim YM, Song HS, Kim HS, Lee WJ, Koh EH, Song KH. Effects of recombinant adenovirus-mediated uncoupling protein 2 overexpression on endothelial function and apoptosis. Circ Res. 2005;96:1200–1207. doi: 10.1161/01.RES.0000170075.73039.5b. [DOI] [PubMed] [Google Scholar]
- Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- Andriantsitohaina R, Duluc L, Garcia-Rodriguez JC. Gil-del Valle L, Guevara-Garcia M, Simard G, Soleti R, Su DF, Velasquez-Perez L, Wilson JX, Laher I: Systems biology of antioxidants. Clin Sci (Lond) 2012;123:173–192. doi: 10.1042/CS20110643. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ. Structure-function aspects in the nitric oxide synthases. Annu Rev Pharmacol Toxicol. 1997;37:339–359. doi: 10.1146/annurev.pharmtox.37.1.339. [DOI] [PubMed] [Google Scholar]
- Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- Shiva S. Mitochondria as metabolizers and targets of nitrite. Nitric Oxide. 2010;22:64–74. doi: 10.1016/j.niox.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. 2009;14:1197–1218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci. 2010;1201:183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- Turrens JF, Freeman BA, Levitt JG, Crapo JD. The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch Biochem Biophys. 1982;217:401–410. doi: 10.1016/0003-9861(82)90518-5. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- McLeod CJ, Aziz A, Hoyt RF Jr, McCoy JP Jr, Sack MN. Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. J Biol Chem. 2005;280:33470–33476. doi: 10.1074/jbc.M505258200. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Kondo-Okamoto N. Mitochondria and autophagy: critical interplay between the two homeostats. Biochim Biophys Acta. 1820;2012:595–600. doi: 10.1016/j.bbagen.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294:H570–H578. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- Pangare M, Makino A. Mitochondrial function in vascular endothelial cell in diabetes. J Smooth Muscle Res. 2012;48:1–26. doi: 10.1540/jsmr.48.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanek K, Lesnefsky EJ, Larner AC. Multi-tasking: nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol. 2012;22:429–437. doi: 10.1016/j.tcb.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta. 1813;2011:1263–1268. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA. Sirtuin chemical mechanisms. Biochim Biophys Acta. 1804;2010:1591–1603. doi: 10.1016/j.bbapap.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz M, Steegborn C. Function and regulation of the mitochondrial sirtuin isoform Sirt5 in Mammalia. Biochim Biophys Acta. 1804;2009:1658–1665. doi: 10.1016/j.bbapap.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, Liu G, Wei YS, Cai H, Liu DP, Liang CC. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res. 2008;80:191–199. doi: 10.1093/cvr/cvn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo M, Minamino T, Miyauchi H, Tateno K, Okada S, Moriya J, Komuro I. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol. 2009;29:889–894. doi: 10.1161/ATVBAHA.109.185694. [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Khanduja KL, Bhardwaj A, Kaushik G. : Resveratrol inhibits N-nitrosodiethylamine-induced ornithine decarboxylase and cyclooxygenase in mice. J Nutr Sci Vitaminol (Tokyo) 2004;50:61–65. [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty JJ, Fu MM, Hah JM, Sweet TJ, Faith SA, Booth T. Effect of resveratrol on herpes simplex virus vaginal infection in the mouse. Antiviral Res. 2005;67:155–162. doi: 10.1016/j.antiviral.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- Chu LM, Lassaletta AD, Robich MP, Sellke FW. Resveratrol in the prevention and treatment of coronary artery disease. Curr Atheroscler Rep. 2011;13:439–446. doi: 10.1007/s11883-011-0202-3. [DOI] [PubMed] [Google Scholar]
- Corda S, Laplace C, Vicaut E, Duranteau J. Rapid reactive oxygen species production by mitochondria in endothelial cells exposed to tumor necrosis factor-alpha is mediated by ceramide. Am J Respir Cell Mol Biol. 2001;24:762–768. doi: 10.1165/ajrcmb.24.6.4228. [DOI] [PubMed] [Google Scholar]
- Hawkins BJ, Solt LA, Chowdhury I, Kazi AS, Abid MR, Aird WC, May MJ, Foskett JK, Madesh M. G protein-coupled receptor Ca2 + −linked mitochondrial reactive oxygen species are essential for endothelial/leukocyte adherence. Mol Cell Biol. 2007;27:7582–7593. doi: 10.1128/MCB.00493-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlands DJ, Islam MN, Das SR, Huertas A, Quadri SK, Horiuchi K, Inamdar N, Emin MT, Lindert J, Ten VS. Activation of TNFR1 ectodomain shedding by mitochondrial Ca2+ determines the severity of inflammation in mouse lung microvessels. J Clin Invest. 2011;121:1986–1999. doi: 10.1172/JCI43839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi SI, Edelstein D, Du XL, Kaneda Y, Guzman M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem. 2001;276:25096–25100. doi: 10.1074/jbc.M007383200. [DOI] [PubMed] [Google Scholar]
- Chen C, Jiang J, Lu JM, Chai H, Wang X, Lin PH, Yao Q. Resistin decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2010;299:H193–H201. doi: 10.1152/ajpheart.00431.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn LS, Anderson BG, Conner JD, Wolden-Hanson T. IL-15 Overexpression Promotes Endurance, Oxidative Energy Metabolism, and Muscle PPARdelta, SIRT1, PGC-1alpha, and PGC-1beta Expression in Male Mice. Endocrinology. 2012. p. . [DOI] [PMC free article] [PubMed]
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- Baudry N, Laemmel E, Vicaut E. In vivo reactive oxygen species production induced by ischemia in muscle arterioles of mice: involvement of xanthine oxidase and mitochondria. Am J Physiol Heart Circ Physiol. 2008;294:H821–H828. doi: 10.1152/ajpheart.00378.2007. [DOI] [PubMed] [Google Scholar]
- Ceylan-Isik AF, Guo KK, Carlson EC, Privratsky JR, Liao SJ, Cai L, Chen AF, Ren J. Metallothionein abrogates GTP cyclohydrolase I inhibition-induced cardiac contractile and morphological defects: role of mitochondrial biogenesis. Hypertension. 2009;53:1023–1031. doi: 10.1161/HYPERTENSIONAHA.108.123422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna IR, Taniyama Y, Szocs K, Rocic P, Griendling KK. NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid Redox Signal. 2002;4:899–914. doi: 10.1089/152308602762197443. [DOI] [PubMed] [Google Scholar]
- Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Gertz M, Fischer F, Wolters D, Steegborn C. Activation of the lifespan regulator p66Shc through reversible disulfide bond formation. Proc Natl Acad Sci U S A. 2008;105:5705–5709. doi: 10.1073/pnas.0800691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Cosentino F, Camici GG, Akhmedov A, Vanhoutte PM, Tanner FC, Luscher TF. Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31:2090–2097. doi: 10.1161/ATVBAHA.111.229260. [DOI] [PubMed] [Google Scholar]
- Napoli C, Martin-Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G, Pelicci P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc Natl Acad Sci U S A. 2003;100:2112–2116. doi: 10.1073/pnas.0336359100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomenko DE, Xing W, Adair BM, Thomas DJ, Gladyshev VN. High-throughput identification of catalytic redox-active cysteine residues. Science. 2007;315:387–389. doi: 10.1126/science.1133114. [DOI] [PubMed] [Google Scholar]
- Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tello D, Balsa E, Acosta-Iborra B, Fuertes-Yebra E, Elorza A, Ordonez A, Corral-Escariz M, Soro I, Lopez-Bernardo E, Perales-Clemente E. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011;14:768–779. doi: 10.1016/j.cmet.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M. Emerging inflammasome effector mechanisms. Nat Rev Immunol. 2011;11:213–220. doi: 10.1038/nri2936. [DOI] [PubMed] [Google Scholar]
- Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol. 2012;13:343–351. doi: 10.1038/ni.2224. [DOI] [PubMed] [Google Scholar]
- Kepp O, Galluzzi L, Zitvogel L, Kroemer G. Pyroptosis - a cell death modality of its kind? Eur J Immunol. 2010;40:627–630. doi: 10.1002/eji.200940160. [DOI] [PubMed] [Google Scholar]
- Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187:613–617. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Yan Y, Jiang X, Mai J, Chen NC, Wang H, Yang XF. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int J Immunopathol Pharmacol. 2009;22:311–322. doi: 10.1177/039463200902200208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208:417–420. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pueyo ME, Gonzalez W, Nicoletti A, Savoie F, Arnal JF, Michel JB. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol. 2000;20:645–651. doi: 10.1161/01.atv.20.3.645. [DOI] [PubMed] [Google Scholar]
- Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmijewski JW, Moellering DR, Le Goffe C, Landar A, Ramachandran A, Darley-Usmar VM. Oxidized LDL induces mitochondrially associated reactive oxygen/nitrogen species formation in endothelial cells. Am J Physiol Heart Circ Physiol. 2005;289:H852–H861. doi: 10.1152/ajpheart.00015.2005. [DOI] [PubMed] [Google Scholar]
- Landar A, Zmijewski JW, Dickinson DA, Le Goffe C, Johnson MS, Milne GL, Zanoni G, Vidari G, Morrow JD, Darley-Usmar VM. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am J Physiol Heart Circ Physiol. 2006;290:H1777–H1787. doi: 10.1152/ajpheart.01087.2005. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Zmijewski JW, Takabe W, Umezu-Goto M, Le Goffe C, Sekine A, Landar A, Watanabe A, Aoki J, Arai H. Activation of mitogen-activated protein kinases by lysophosphatidylcholine-induced mitochondrial reactive oxygen species generation in endothelial cells. Am J Pathol. 2006;168:1737–1748. doi: 10.2353/ajpath.2006.050648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Hartley RC, Murphy MP. Mitochondria-targeted small molecule therapeutics and probes. Antioxid Redox Signal. 2011;15:3021–3038. doi: 10.1089/ars.2011.3969. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Hasko G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc. 2007;2:2295–2301. doi: 10.1038/nprot.2007.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc. 2008;130:9638–9639. doi: 10.1021/ja802355u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Srikun D, Chang CJ. Mitochondrial-targeted fluorescent probes for reactive oxygen species. Curr Opin Chem Biol. 2010;14:50–56. doi: 10.1016/j.cbpa.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide Y, Urano Y, Kenmoku S, Kojima H, Nagano T. Design and synthesis of fluorescent probes for selective detection of highly reactive oxygen species in mitochondria of living cells. J Am Chem Soc. 2007;129:10324–10325. doi: 10.1021/ja073220m. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Yeh M, Danziger EC, Hatley ME, Riggan AE, Leitinger N, Berliner JA, Hedrick CC. Glucose regulates monocyte adhesion through endothelial production of interleukin-8. Circ Res. 2003;92:371–377. doi: 10.1161/01.RES.0000061714.74668.5C. [DOI] [PubMed] [Google Scholar]
- Pearlstein DP, Ali MH, Mungai PT, Hynes KL, Gewertz BL, Schumacker PT. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arterioscler Thromb Vasc Biol. 2002;22:566–573. doi: 10.1161/01.atv.0000012262.76205.6a. [DOI] [PubMed] [Google Scholar]
- Schafer M, Schafer C, Ewald N, Piper HM, Noll T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ Res. 2003;92:1010–1015. doi: 10.1161/01.RES.0000070882.81508.FC. [DOI] [PubMed] [Google Scholar]
- Moreira ES, Brasch NE, Yun J. Vitamin B12 protects against superoxide-induced cell injury in human aortic endothelial cells. Free Radic Biol Med. 2011;51:876–883. doi: 10.1016/j.freeradbiomed.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Cuenca S, Cocheme HM, Logan A, Abakumova I, Prime TA, Rose C, Vidal-Puig A, Smith AC, Rubinsztein DC, Fearnley IM. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic Biol Med. 2010;48:161–172. doi: 10.1016/j.freeradbiomed.2009.10.039. [DOI] [PubMed] [Google Scholar]
- Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cocheme HM, Murphy MP, Dominiczak AF. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- Chacko BK, Reily C, Srivastava A, Johnson MS, Ye Y, Ulasova E, Agarwal A, Zinn KR, Murphy MP, Kalyanaraman B, Darley-Usmar V. Prevention of diabetic nephropathy in Ins2(+/)(−)(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem J. 2010;432:9–19. doi: 10.1042/BJ20100308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med. 2008;45:1559–1565. doi: 10.1016/j.freeradbiomed.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Supinski GS, Murphy MP, Callahan LA. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1095–R1102. doi: 10.1152/ajpregu.90902.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplugues JV, Rocha M, Nunez C, Bosca I, Ibiza S, Herance JR, Ortega A, Serrador JM, D'Ocon P, Victor VM. Complex I dysfunction and tolerance to nitroglycerin: an approach based on mitochondrial-targeted antioxidants. Circ Res. 2006;99:1067–1075. doi: 10.1161/01.RES.0000250430.62775.99. [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Kehrer I, Kozlov AV, Haller M, Redl H, Hermann M, Grimm M, Troppmair J. Mitochondrial ROS production under cellular stress: comparison of different detection methods. Anal Bioanal Chem. 2011;400:2383–2390. doi: 10.1007/s00216-011-4764-2. [DOI] [PubMed] [Google Scholar]
- Hardy M, Chalier F, Ouari O, Finet JP, Rockenbauer A, Kalyanaraman B, Tordo P. Mito-DEPMPO synthesized from a novel NH2-reactive DEPMPO spin trap: a new and improved trap for the detection of superoxide. Chem Commun (Camb) 2007. pp. 1083–1085. [DOI] [PubMed]
- Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A. 2006;103:5379–5384. doi: 10.1073/pnas.0601026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin A, Bolender RP, Weibel ER. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J Cell Biol. 1977;72:441–455. doi: 10.1083/jcb.72.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Hill BG, Darley-Usmar VM. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic Biol Med. 2010;48:905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal. 2011;15:1517–1530. doi: 10.1089/ars.2010.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zang QS, Liu Z, Wu Q, Maass D, Dulan G, Shaul PW, Melito L, Frantz DE, Kilgore JA. Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species. Am J Physiol Cell Physiol. 2011;301:C695–C704. doi: 10.1152/ajpcell.00322.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- Roy Chowdhury SK, Sangle GV, Xie X, Stelmack GL, Halayko AJ, Shen GX. Effects of extensively oxidized low-density lipoprotein on mitochondrial function and reactive oxygen species in porcine aortic endothelial cells. Am J Physiol Endocrinol Metab. 2010;298:E89–E98. doi: 10.1152/ajpendo.00433.2009. [DOI] [PubMed] [Google Scholar]
- Takabe W, Li R, Ai L, Yu F, Berliner JA, Hsiai TK. Oxidized low-density lipoprotein-activated c-Jun NH2-terminal kinase regulates manganese superoxide dismutase ubiquitination: implication for mitochondrial redox status and apoptosis. Arterioscler Thromb Vasc Biol. 2010;30:436–441. doi: 10.1161/ATVBAHA.109.202135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Kobayashi T, Kamata K. Role of lysophosphatidylcholine (LPC) in atherosclerosis. Curr Med Chem. 2007;14:3209–3220. doi: 10.2174/092986707782793899. [DOI] [PubMed] [Google Scholar]
- Haffner SJ, Cassells H. Hyperglycemia as a cardiovascular risk factor. Am J Med. 2003;115(Suppl 8A):6S–11S. doi: 10.1016/j.amjmed.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gero D, Szoleczky P, Chang T, Zhou Z, Wu L. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci U S A. 2011;108:13829–13834. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungvari Z, Labinskyy N, Mukhopadhyay P, Pinto JT, Bagi Z, Ballabh P, Zhang C, Pacher P, Csiszar A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H1876–H1881. doi: 10.1152/ajpheart.00375.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Batkai S, Hasko G, Liaudet L, Drel VR, Obrosova IG, Pacher P. Cannabidiol attenuates high glucose-induced endothelial cell inflammatory response and barrier disruption. Am J Physiol Heart Circ Physiol. 2007;293:H610–H619. doi: 10.1152/ajpheart.00236.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Chen H, Wang H, Ke B, Zheng B, Li Q, Li P, Su L, Gu Q, Xu X. Improvement of retinal vascular injury in diabetic rats by statins is associated with the inhibition of mitochondrial reactive oxygen species pathway mediated by peroxisome proliferator-activated receptor gamma coactivator 1alpha. Diabetes. 2010;59:2315–2325. doi: 10.2337/db10-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Alexanian A, Ying R, Kizhakekuttu TJ, Dharmashankar K, Vasquez-Vivar J, Gutterman DD, Widlansky ME. Acute Exposure to Low Glucose Rapidly Induces Endothelial Dysfunction and Mitochondrial Oxidative Stress: Role for AMP Kinase. Arterioscler Thromb Vasc Biol. 2012;32:712–720. doi: 10.1161/ATVBAHA.111.227389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda RM, Peotta VA, Meyrelles SS, Vasquez EC. Evaluation of vascular function in apolipoprotein E knockout mice with angiotensin-dependent renovascular hypertension. Hypertension. 2005;46:932–936. doi: 10.1161/01.HYP.0000182154.61862.52. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Prete A, Zaccagnino P, Di Paola M, Saltarella M, Oliveros Celis C, Nico B, Santoro G, Lorusso M. Role of mitochondria and reactive oxygen species in dendritic cell differentiation and functions. Free Radic Biol Med. 2008;44:1443–1451. doi: 10.1016/j.freeradbiomed.2007.12.037. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100:607–621. doi: 10.1161/01.RES.0000258492.96097.47. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Seo KW, Yun MR, Bae SS, Lee WS, Hong KW, Kim CD. 4-Hydroxynonenal enhances MMP-2 production in vascular smooth muscle cells via mitochondrial ROS-mediated activation of the Akt/NF-kappaB signaling pathways. Free Radic Biol Med. 2008;45:1487–1492. doi: 10.1016/j.freeradbiomed.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Bailey SR, Mitra S, Flavahan S, Flavahan NA. Reactive oxygen species from smooth muscle mitochondria initiate cold-induced constriction of cutaneous arteries. Am J Physiol Heart Circ Physiol. 2005;289:H243–H250. doi: 10.1152/ajpheart.01305.2004. [DOI] [PubMed] [Google Scholar]
- Sato H, Sato M, Kanai H, Uchiyama T, Iso T, Ohyama Y, Sakamoto H, Tamura J, Nagai R, Kurabayashi M. Mitochondrial reactive oxygen species and c-Src play a critical role in hypoxic response in vascular smooth muscle cells. Cardiovasc Res. 2005;67:714–722. doi: 10.1016/j.cardiores.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell. 2010;40:323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Cell biology. Warburg effect and redox balance. Science. 2011;334:1219–1220. doi: 10.1126/science.1215637. [DOI] [PubMed] [Google Scholar]
- Harrison DG, Gongora MC. Oxidative stress and hypertension. Med Clin North Am. 2009;93:621–635. doi: 10.1016/j.mcna.2009.02.015. [DOI] [PubMed] [Google Scholar]
- Lavoie JL, Sigmund CD. Minireview: overview of the renin-angiotensin system–an endocrine and paracrine system. Endocrinology. 2003;144:2179–2183. doi: 10.1210/en.2003-0150. [DOI] [PubMed] [Google Scholar]
- Widder JD, Fraccarollo D, Galuppo P, Hansen JM, Jones DP, Ertl G, Bauersachs J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension. 2009;54:338–344. doi: 10.1161/HYPERTENSIONAHA.108.127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC. Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106:544–549. doi: 10.1161/01.cir.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- Fujimoto H, Taguchi J, Imai Y, Ayabe S, Hashimoto H, Kobayashi H, Ogasawara K, Aizawa T, Yamakado M, Nagai R, Ohno M. Manganese superoxide dismutase polymorphism affects the oxidized low-density lipoprotein-induced apoptosis of macrophages and coronary artery disease. Eur Heart J. 2008;29:1267–1274. doi: 10.1093/eurheartj/ehm500. [DOI] [PubMed] [Google Scholar]
- Ciccone S, Maiani E, Bellusci G, Diederich M, Gonfloni S. Parkinson’S disease: a complex interplay of mitochondrial DNA alterations and oxidative stress. Int J Mol Sci. 2013;14:2388–2409. doi: 10.3390/ijms14022388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Lev D, Gong Y, Wang S, Pollock RE, Wu X, Gu J. Reduced mitochondrial DNA copy number in peripheral blood leukocytes increases the risk of soft tissue sarcoma. Carcinogenesis. 2013. p. . [DOI] [PubMed]
- Krzywanski DM, Moellering DR, Fetterman JL, Dunham-Snary KJ, Sammy MJ, Ballinger SW. The mitochondrial paradigm for cardiovascular disease susceptibility and cellular function: a complementary concept to Mendelian genetics. Lab Invest. 2011;91:1122–1135. doi: 10.1038/labinvest.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol. 2004;251:261–268. doi: 10.1007/s00415-004-0348-9. [DOI] [PubMed] [Google Scholar]
- Filippin LI, Vercelino R, Marroni NP, Xavier RM. Redox signalling and the inflammatory response in rheumatoid arthritis. Clin Exp Immunol. 2008;152:415–422. doi: 10.1111/j.1365-2249.2008.03634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burek CL, Rose NR. Autoimmune thyroiditis and ROS. Autoimmun Rev. 2008;7:530–537. doi: 10.1016/j.autrev.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Chen J, Gusdon AM, Thayer TC, Mathews CE. Role of increased ROS dissipation in prevention of T1D. Ann N Y Acad Sci. 2008;1150:157–166. doi: 10.1196/annals.1447.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon A, Park H, Maddipati R, Lobito AA, Bulua AC, Jackson AJ, Chae JJ, Ettinger R, de Koning HD, Cruz AC. Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc Natl Acad Sci U S A. 2010;107:9801–9806. doi: 10.1073/pnas.0914118107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet. 2003;361:2017–2023. doi: 10.1016/S0140-6736(03)13637-9. [DOI] [PubMed] [Google Scholar]
- Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. Jama. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- Sesso HD, Buring JE, Christen WG, Kurth T, Belanger C, MacFadyen J, Bubes V, Manson JE, Glynn RJ, Gaziano JM. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. Jama. 2008;300:2123–2133. doi: 10.1001/jama.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook NR, Albert CM, Gaziano JM, Zaharris E, MacFadyen J, Danielson E, Buring JE, Manson JE. A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: results from the Women's Antioxidant Cardiovascular Study. Arch Intern Med. 2007;167:1610–1618. doi: 10.1001/archinte.167.15.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]