

Abstract
Background
Genetic studies have identified numerous genes reproducibly associated with asthma, yet these studies have focused almost entirely on single nucleotide polymorphisms (SNPs), and virtually ignored another highly prevalent form of genetic variation: Copy Number Variants (CNVs).
Objective
To survey the prevalence of CNVs in genes previously associated with asthma, and to assess whether CNVs represent the functional asthma-susceptibility variants at these loci.
Methods
We genotyped 383 asthmatic trios participating in the Childhood Asthma Management Program (CAMP) using a competitive genomic hybridization (CGH) array designed to interrogate 20,092 CNVs. To ensure comprehensive assessment of all potential asthma candidate genes, we purposely used liberal asthma gene inclusion criteria, resulting in consideration of 270 candidate genes previously implicated in asthma. We performed statistical testing using FBAT-CNV.
Results
Copy number variation in asthma candidate genes was prevalent, with 21% of tested genes residing near or within one of 69 CNVs. In 6 instances, the complete candidate gene sequence resides within the CNV boundaries. On average, asthmatic probands carried 6 asthma-candidate CNVs (range 1–29). However, the vast majority of identified CNVs were of rare frequency (< 5%), and were not statistically associated with asthma. Modest evidence for association with asthma was observed for 2 CNVs near NOS1 and SERPINA3. Linkage disequilibrium analysis suggests that CNV effects are unlikely to explain previously detected SNP associations with asthma.
Conclusions
Although a substantial proportion of asthma-susceptibility genes harbor polymorphic CNVs, the majority of these variants do not confer increased asthma risk. The lack of linkage disequilibrium (LD) between CNVs and asthma-associated SNPs suggests that these CNVs are unlikely to represent the functional variant responsible for most known asthma associations.
INTRODUCTION
Genetic mapping of asthma susceptibility single nucleotide polymorphisms (SNPs) has been very fruitful, with identification of over 200 genes associated with asthma, many of these replicated in multiple populations.[1, 2] Most recently, GWAS analyses and meta-analyses, involving more than 10,000 subjects, have identified multiple highly replicated genes, offering insights into previously unexamined asthma pathways.[3, 4] Despite these substantial advances, however, SNPs identified thus far explain only a small fraction of the estimated 36–79% genetic contribution to asthma liability (so-called heritability) estimated from numerous twin studies.[5] This problem of “missing heritability” is not unique to asthma, having been noted for most common, complex genetic traits.[6, 7]
One potential explanation for this “missing heritability” is that until recently, GWAS and most candidate gene studies focused almost entirely on SNPs, and have ignored an entire class of common genetic polymorphism: copy number variants (CNVs). CNVs are large-scale duplications and deletions traditionally defined as spanning at least 1000 bases (1kb), though advances in CNV genotyping lead some to include structural variants as small as 50bp in CNV analyses.[8, 9] Though less frequent in total number than SNPs, CNVs represent a substantial proportion of genetic variation due to their sheer size (often greater than 100kb), and frequently disrupt gene function by overlapping vital coding and/or regulatory gene sequences. Conservative estimates suggest that up to 5% of the human genome is copy-number variable,[10] and integrative genomic analyses demonstrate that CNVs serve as important regulators of gene expression.[11] However, due to technical limitations in high-throughput CNV genotyping, evaluation of CNV in relation to human traits has been largely limited to candidate variant studies.
There are compelling reasons to study CNVs in asthma. The prevalence of CNVs is enriched within genomic regions harboring genes related to the immune response,[12] and most of the established disease-related CNVs identified to date are associated with immune-related diseases, including Crohn’s disease,[13, 14] systemic lupus erythematosis,[15] and rheumatoid arthritis.[16] Moreover, one of the best-characterized disease-CNV associations is that of a common deletion of Glutathione S-Transferase Mu 1 (GSTM1) with asthma and airflow obstruction. The GSTM1-null variant (homozygous null prevalence of ~50% in Caucasian populations) has been repeatedly associated with asthma,[17–20] COPD,[21] and reduced lung function or reduced lung function growth, [20, 22, 23] particularly in the setting of increased oxidative stress. Finally, given that CNVs frequently occur in gene-rich regions[24, 25] and the recognition that diallelic CNVs (i.e. deletions or one-copy gains) are often in strong linkage disequilibrium with common SNPs, [8, 10, 26, 27] it is important to establish whether previously observed SNP associations with asthma could be directly explained by common CNVs.
It is in this context that we set out to evaluate the role of structural genetic variation in asthma using a customized microarray designed specifically for the purpose of genotyping known CNVs. Herein we describe the results of a survey in a family-based cohort of childhood asthmatics of all identified CNVs that map to within 50kb of genes for which prior SNP-association with asthma has been reported. To facilitate comprehensive evaluation, we purposely used a liberal inclusion threshold to investigate the effects of CNV on all potential asthma candidate genes, resulting in consideration of 270 genes.
METHODS
Populations
The Childhood Asthma Management Program (CAMP) is a multi-centered North American clinical trial (NCT00000575) designed to investigate the long-term effects of inhaled anti-inflammatory medications in children with mild to moderate asthma.[28, 29] A diagnosis of asthma was based on methacholine hyperreactivity (PC20 [provocative concentration causing a 20% fall in FEV1] no greater than 12.5 mg/ml) and one or more of the following criteria for at least 6 months in the year before recruitment: (1) asthma symptoms at least two times per week, (2) at least two uses per week of an inhaled bronchodilator, or (3) daily asthma medication. Protocols for collection of baseline phenotypic data have been described in detail elsewhere.[28, 29]
Of the 1,041 children enrolled in the original clinical trial, 968 children and 1,518 of their parents contributed DNA samples. Due to the small number of samples from non-white ethnic groups and concerns regarding genetic heterogeneity, we restricted genotyping to the self-described white subjects and their available parents. In total, 1212 subjects were genotyped, including 420 probands and 383 complete nuclear families.
The Institutional Review Boards of the Brigham and Women’s Hospital and of the other CAMP study centers approved this study (IRB#2004P000996). Informed assent and consent were obtained from the study participants and their parents to collect DNA for genetic studies.
CNV Genotyping
All individuals were genotyped using a custom-designed SurePrint G3 Human CGH 4×180K Microarray (Agilent Technologies, Santa Clara, CA). CNV Regions (CNVRs) were selected for targeting using multiple datasets detailing CNV location and breakpoints, in a tiered approach, favoring high-resolution data. We incorporated CNV regions identified by the Structural Genomic Variation Consortium (based on data from 42 million CGH probes[26]), data from the June 2009 release of the 1000 genomes project,[30] deep sequencing of an individual genome,[31] and a list of segmental duplications[32] and novel insertions.[33] Finally, we incorporated variants identified in the Database of Genomic Variants that were >500bp and <2 Mb in size and did not overlap any other regions.[30, 34] In total, each array interrogates 20,092 highly confident and distinct CNV regions in a single assay, with each CNV region surveyed by 6–9 probes.
DNA samples were labeled using Genomic DNA ULS Labeling Kit (cat. no. 5190-0419, Agilent, Santa Clara, CA) according to the manufacturer’s protocol. These test samples were competitively hybridized to the Agilent array against a well-characterized male HapMap reference sample (NA10851). Purification, hybridization and washing of samples was performed per manufacturer’s protocol (Agilent Technologies, Santa Clara, CA). Arrays were scanned at a 5 micron resolution in an Agilent scanner (G2505C) and processed with Agilent Feature Extraction v10.5.1.1 (Agilent Technologies, Santa Clara, CA). Samples with derivative Log Ratio spread (DLRS) values <0.3 passed quality control. Data quality was very high; only 1 sample failed QC post-hybridization.
We employed the following quality controls and CNV calling algorithms to define high-confidence copy number status. Raw signal intensities of each probe were normalized across the entire array to limit potential bias due to dye normalization and technical errors. Log2 ratios of each probe were calculated using the normalized intensities of the cy5 (sample) and cy3 (reference) channels. We then assessed all probes for variability using the Bioconductor package CNVTools,[35] and eliminated probes without variability. Composite, regional intensities were calculated as the mean log2 ratio for all reliable probes mapping to the previously defined, known CNVRs. From these mean intensities, discrete CNVR copy numbers were calculated using CNVTools, with the largest bin nearest to the log2 = 0 assumed to be the diploid (i.e. 2 copy) representation. For consideration in the current study, we limited our evaluation to those CNVs that we considered high-confidence, where the CNVR had assigned copy-number bins of >.99 posterior-probability in at least 80% of genotyped individuals (see Figure 1). We finally eliminated those CNVs that were in the lowest IQR of variability of log2ratio (range <.88). Thus, 1259 common CNVs (with non-2-copy >5%) and 2535 rare CNVs were analyzed. We tested for r2 with neighboring SNP in PLINK[36], using data from an Illumina 550Kv3 SNP array available in 1056 of the subjects; QC methods for the SNP array have been previously published.[37]
Figure 1.
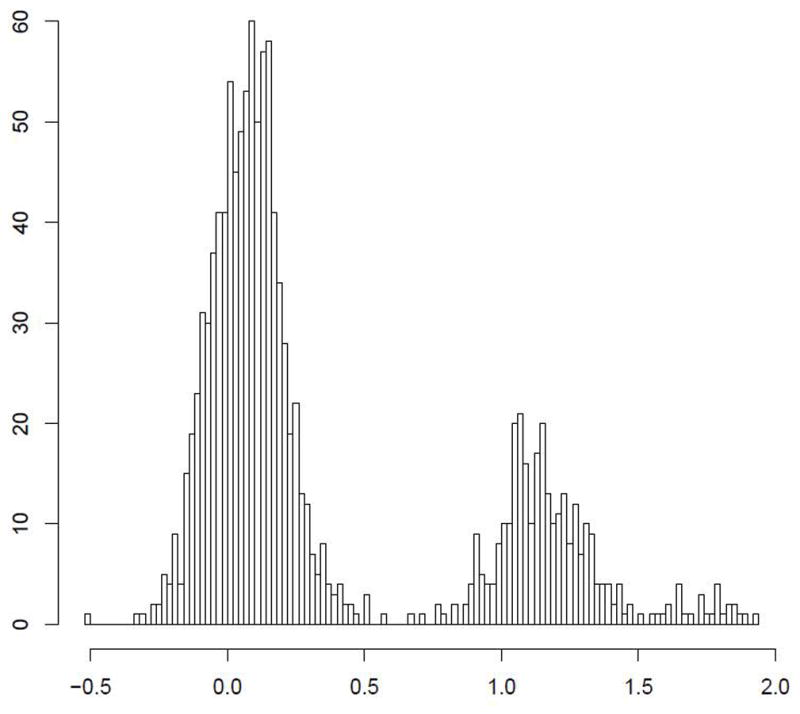
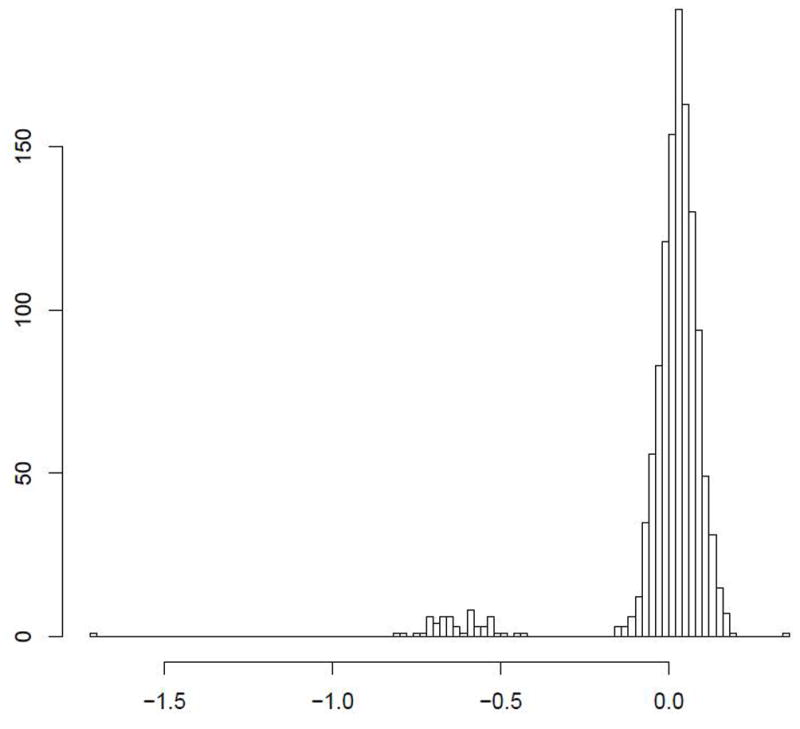
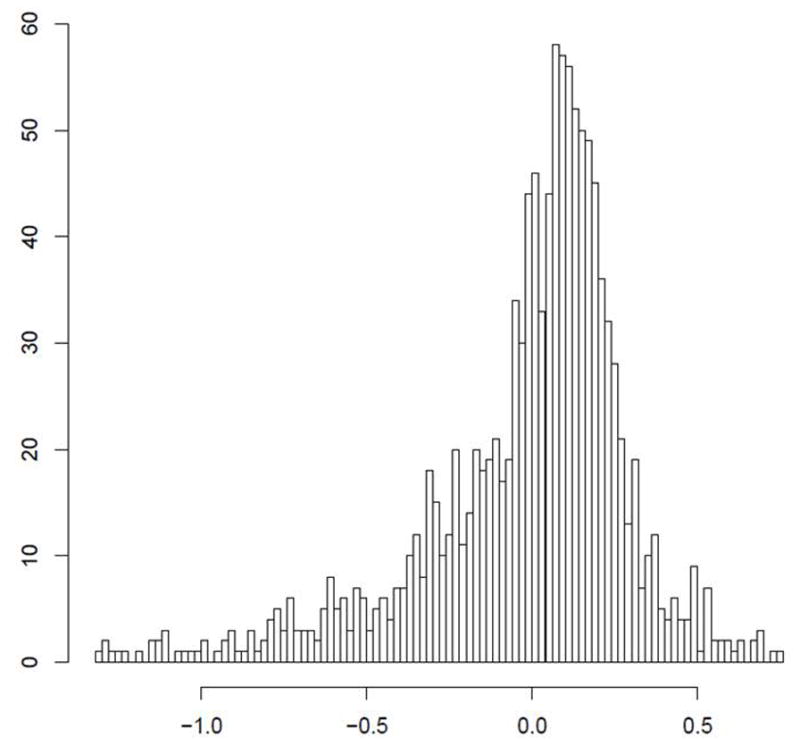
The certainty of CNV copy number call is predicated on discrete separation of signal intensity log2 ratios. Representative examples of high certainty calls: (A) a common 600BP gain in PTPRD; (B) a rare 1.5Kb loss in DPP10 (B) are presented. In comparison, a 1Kb region near IL17RA (C) shows a suggestion of CNV, but bins much less clearly.
Asthma Candidate Selection
We included all previously identified asthma candidate genes, including those compiled in Ober et al.[2], Rogers et al.[37], and the recent review by Wu et al,[38] and novel asthma loci identified in the recent GWAS meta-analyses of the Gabriel[3] and EVE consortia.[4] We excluded candidate genes on the X chromosome, and several immunoglobulin regions with known high rearrangement rates, including T-cell receptors. We also looked at the effects of CNV on a subset of genes with the most robust associations (i.e. those replicated genes identified in GWAS or associated with asthma in >10 populations (N=12 genes, see Supplemental Table 1).
Statistical Methods
We performed family-based quality control assessment, including screening for parent-offspring genotype incompatibilities (Mendelian errors), and a family-based normalization of log2 ratios. Markers with large numbers of Mendelian errors, or those whose subsequent association test was unstable following family-adjustment were excluded from analysis. The physical relationships between genes and CNVs were mapped using the Galaxy workbench.[39, 40] All autosomal CNVs were tested for association with asthma affection status in the 383 complete trios, using discrete copy number calls from CNVTools as a surrogate for copy number. Analyses were performed using FBAT-CNV, which generates a test statistic by comparing expected subject genotype based on the mean of the known parental genotypes (binned CNVRs) to the actual subject genotype.[41] Regions with significant association with asthma (p<.05) were manually reviewed. Power calculations were performed using Quanto.[42]
Technical validation
Genotyping error can be a source of both false positive or false negative results. Using customized quantitative PCR assays (TaqMan assays, Applied Biosystems Inc, Foster City, CA), we were able to validate 46 of 63 (73%) high confidence CNVs, demonstrating high concordance rates between qPCR and Agilent microarray CNV calls.
RESULTS
All but one of 1212 individuals were successfully genotyped using the 180K CGH array. Among 20,092 CNV regions represented on the array, 3,794 were autosomal, binned with high confidence, and polymorphic in the CAMP cohort, including 1,259 with a minor copy-number frequency of at least 5%. A total of 69 polymorphic CNVs mapped to within 50kb of 58 asthma genes (21% of the 270 candidate genes surveyed, Supplemental Table 1). Of these, 6 CNVs overlap the complete coding sequence of one or more genes, 3 overlap at least one exon, 17 are imbedded within an intragenic (intronic) sequence, and 43 are intergenic (Figure 2). On average, these CNVs are rare (median frequency 3%, range .002–.52), with only 27 present in at least 5% of individuals. The majority of CNVs consisted of copy-number losses (40); 23 consisted of a copy-number gain only, while 6 CNVs were more highly polymorphic, with both gains and losses observed. The 420 asthmatic probands in our study carried a median of 6 such CNVs (range 1–29).
Figure 2.
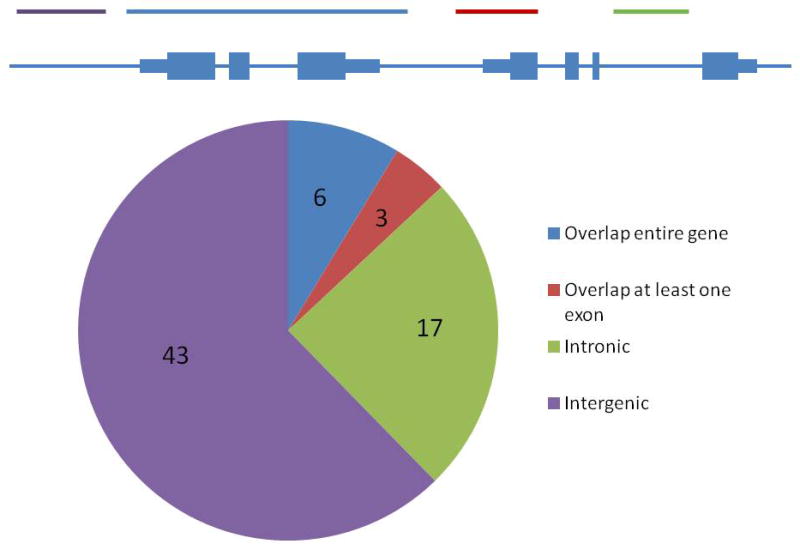
Distribution of 69 CNVs in or near asthma-associated genes
We evaluated the impact of CNV on asthma candidate genes using several approaches. We first assessed whether CNVs were more prevalent among asthma candidate genes in our cohort as compared to the background genome-wide rate. Overall, the prevalence rate among candidate asthma genes of 21% (58/270) was similar to the background genome-wide rate of 23% for RefSeq genes (p=.>.1 by Fisher’s Exact test). Slightly higher (33% prevalence), but not significant (p >.1) enrichment for CNVs was observed upon refining our analysis to the best-validated, high-confidence asthma candidate genes (N=12).
We next performed individual family-based tests of association for each of the 69 identified CNVs in 383 self-reported white asthmatic trios. None of these region achieved statistical significance following adjustment for multiple comparisons. Consistent with our prior studies,13 we saw no association with asthma at the GSTM1 locus. Two candidate CNVRs demonstrated nominal association with asthma at p<0.05, one in NOS1, the other near SERPINA3 (Table 1). These asthma-associated CNVR were both quite rare, with frequency of less than one percent. The most significant association was with a 1.5Kb intronic duplication in the NOS1 gene (Figure 3). This gain was observed 7 times in our cohort, and only in parents, with no transmission to offspring, suggesting that the deletion variant confers decreased susceptibility to asthma. Similarly, a 1.4kB gain downstream of SERPINA3 was seen 5 times in the cohort, again only in parents, suggesting a protective effect.
Table 1.
Top Asthma-associated CNVs
| GeneName | RefSeq | Gene position (Hg18) | CNV location (Hg18) | Relationship | Freq | TDT | Pval |
|---|---|---|---|---|---|---|---|
| NOS1 | NM_001204214 | chr12:116130329-116231598 | chr12:116178484-116179795 | CNV within Gene (intronic) | 0.006 | 0 of 7 | 0.008 |
| SERPINA3 | NM_001085 | chr14:94148466-94160143 | chr14:94170293-94171680 | Downstream of gene | 0.004 | 0 of 5 | 0.025 |
Figure 3.
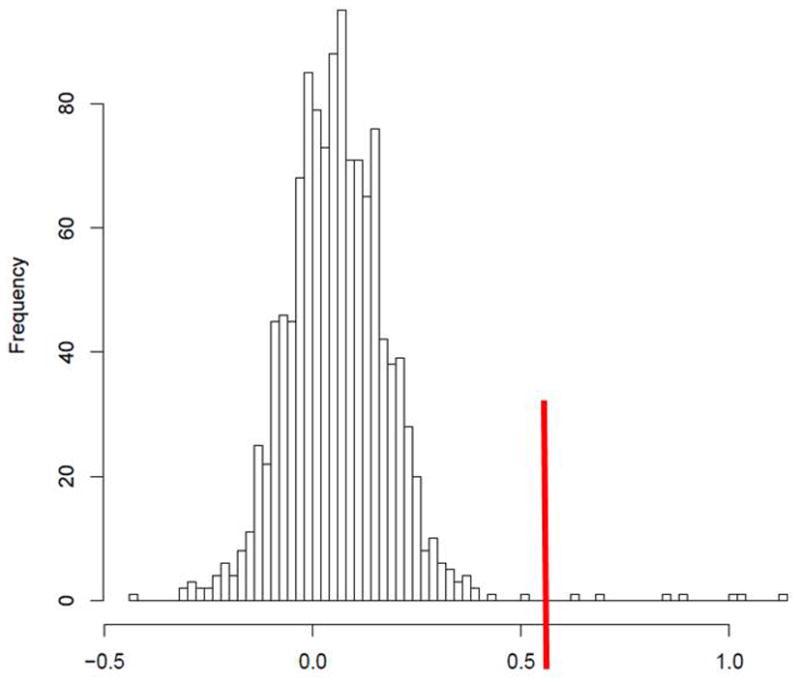

NOS1 CNV location and histogram.
Figure 3A. The 2kb CNV is intronic within NOS1, as shown in red.
Figure 3B. CNVTools calls a gain for all CNVs with log2ratio greater than 0.6, demarcated with a red line in this image. All 7 gains (to the right of the red line) are in parents in the CAMP cohort.
Given the preponderance of rare CNVs showing some association with asthma in our cohort, we wanted to assess the possibility that rare CNVs contribute to asthma liability. Because statistical power in our cohort is low for individual association testing of rare variants, we performed joint association testing of all 42 CNVs with frequency less than 5%, considering the transmission distortion rate for all variants simultaneously. The observed rates of rare CNV transmission did not differ from the null expectation (p = .32), suggesting that rare CNVs in known asthma candidate genes do not contribute substantially to asthma liability.
The genetic and physical relationships between CNV and asthma-associated SNP
The majority of disease-associated SNPs identified by GWAS studies are themselves not functional, disease-causing variants, but rather are in LD with untyped causal variants. In is conceivable that some of these associations are due to indirect tagging of disease-causing CNV. We tested this in our dataset and found no evidence of this: Among the 1211 subjects genotyped using this 180K Agilent platform, 1056 also had SNP genotyping from an Illumina 550Kv3 array available, enabling evaluation of pairwise LD, yet only 3 of the 69 CNVs (near ESR1, TGFBR3, and SOD2) were strongly tagged by a neighboring SNP. Furthermore, in all three instances, both the CNV and the tagging SNP were physically far removed from previously identified asthma SNPs (>20kB in all cases), and the CNV-tagging variants were themselves not in LD with previously associated asthma SNP. Notably, the two rare CNVs that were nominally associated with asthma above were not reliably tagged by SNP on GWAS platforms. Thus, in no instance did we find evidence that CNVs were likely to explain a previously identified asthma association.
DISCUSSION
Substantial advances have been made in recent years in elucidating the genetic underpinnings of asthma, particularly with the very exciting results of recently published GWAS analyses. Yet only a fraction of the over-all heritability of asthma has been explained to date; furthermore, among bonafide asthma genes that have been identified thus far, functional loci are largely still unknown. As-yet unexamined CNVs have the potential to address both of these issues. Using a custom-designed CNV genotyping array, we were able to assess the effects of CNV in known asthma genes. Though we found substantial evidence of structural variation near and within these asthma genes, with more than 20% of genes harboring CNV and asthmatic probands harboring an average of 6 CNV per genome, our analyses suggest that these variants are themselves not association with asthma, and do not explain prior asthma-SNP associations. Within the 58 asthma candidate genes that either contained or were adjacent to a CNV, we identified only 3 cases in which a SNP on a standard GWAS genotyping array was in high LD with our tested CNV, of which none were linked to the known associated asthma SNP. In sum, these results suggest that although CNV is prevalent among asthmatics, the common variants observed in our cohort do not represent the causal variants underlying previously observed SNP associations.
This study represents, to our knowledge, the first systematic evaluation of CNV in asthma genes. Unlike SNP-based genome-wide studies, we used CGH technology targeting more than 20,000 previously identified CNV regions. The fine-scale nature of this technology allows greater confidence in our CNV genotyping than the CNV calls extrapolated from SNP-based genotyping methods, providing greater confidence that our lack of associations did not result from technical inadequacies. Indeed, independent technical validation studies by qPCR confirmed the CNV genotyping accuracy of 74%, consistent with prior studies using CGH. Furthermore, although our sample size of 383 trios is modest, because our analysis was limited to only 69 variants, this study was adequately powered to appreciate modest odds ratios of 1.35–1.8 for common CNVs (80% power to appreciate these ORs in CNV with non-two-copy frequency .05–.50 in 383 trios, with nominal significance level of α .05). While these odds ratios are larger than those attributed to most SNP-based variation (with typical O.R.’s closer to 1.2), the effect size of pathogenic CNVs is frequently higher than that of SNPs, presumably due to their large size physical size and greater likelihood of disrupting gene function.[43] We are therefore confident that the common tested CNVs are not important contributors in asthma pathogenesis. We are underpowered to identify minor increases in risk in the rare CNV variants with MAF <5%, requiring OR >2 to meet significance.
Although our results suggest no role for CNVs in the mechanisms underlying prior asthma associations, it is important to emphasize that we have not excluded a broader role in asthma pathogenesis for structural genetic variation. First, the goal of this study was to specifically explore the relationship of CNV to previously implicated asthma genes, but was not designed as a comprehensive genome-wide study of all genomic regions. Second, the CAMP cohort we studied included children with mild to moderate asthma; given the increasingly recognized heterogeneity in asthma phenotypes,[44] our negative results may not extrapolate to other types of asthmatics (including, adult-onset or more severe asthma, for example). Third, the customized array was designed to evaluate known variants identified in small to modest sized cohorts, sampled without regard to asthma status. As a result, the array would underrepresent highly penetrant asthma variants with low prevalence in the general population. Fourth, though this study was well powered for common variants, studies of rare variants require substantially larger cohorts. Though our pooled analysis of 42 rare variants showed no significant association, formal testing of individual variants in larger cohorts is warranted. We note that the study of rare structural variants poses additional challenges beyond simple statistical power. Population-based CNV calling algorithms are more accurate if the variant is more frequent, as clearer binning of observed log2 intensity ratios into discrete copy-number counts is possible. For rare CNVs such as the ~2,500 with frequency <5% in our dataset, binning is less confident, with only the most extreme outliers classified as variant. Finally, the lower size limit of CNVs targeted by our array was >500 bp; the role of indels, inversions, and very small CNVs on the candidate genes was not explored in this work. Improvements in CNV identification and quantification, using next-generation whole-genome sequencing technology,[8] will eventually address these limitations, enabling complete characterization of the spectrum of structural genetic variation in asthma.
Our analyses identified two copy-number variable regions that exhibited (albeit marginal) association with asthma. Particularly intriguing is the association of a NOS1 intronic CNV. NOS1 encodes the nitric oxide synthase 1, which synthesizes NO from L-Arginine. While present in less than 1% of the cohort, this CNV was noted only in parents, but was never transmitted to asthmatic probands, suggesting a protective genetic effect. Several SNP-based candidate gene studies of NOS1 have reported associations with asthma. However, there is little evidence of reproducibility across studies, and the exact causal variants are not known.[45, 46] The role of CNVs in NOS1 has never been previously assessed. As with any genetic study, the importance of these CNVs in asthma pathogenesis must be confirmed with replication in additional populations.
In summary, we have performed a comprehensive assessment of the role of copy number variation in human asthma genes. Though 21% of asthma candidates are copy-number variable and several rare CNVs were found to be weakly associated with asthma, the vast majority of previously reported asthma-SNP associations do not appear to be due to the effects of neighboring CNVs. Future studies of focusing on genome-wide structural variants beyond those previously associated with asthma are required to search for additional asthma-susceptibility structural genetic variants.
Supplementary Material
Acknowledgments
We thank all subjects for their ongoing participation in this study. We acknowledge the CAMP investigators and research team, supported by the National Heart, Lung, and Blood Institute (NHLBI), for collection of CAMP Genetic Ancillary Study data.
This work is supported by NHLBI grant R01 HL093076, “Structural Genetic Variation in Asthma”. The CAMP Genetics Ancillary Study is supported by the NHLBI, N01 HR16049. Additional support for this research came from grants R01 HL086601, U01 HL065899, P01 HL083069, and K12HL089990 from the National Institutes of Health and the NHLBI. All work on data from the CAMP Genetic Ancillary Study was conducted at the Channing Laboratory and the Brigham and Women’s Hospital under appropriate CAMP policies and human subjects protections. Dr. Rogers is supported the Parker B. Francis Foundation.
Footnotes
CONFLICT OF INTERESTS
The authors declare no conflict of interests.
References
- 1.Meyers DA. Genetics of asthma and allergy: what have we learned? J Allergy Clin Immunol. 126:439–446. doi: 10.1016/j.jaci.2010.07.012. quiz 447–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ober C, Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun. 2006;7:95–100. doi: 10.1038/sj.gene.6364284. [DOI] [PubMed] [Google Scholar]
- 3.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, von Mutius E, Farrall M, Lathrop M, Cookson WO. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, Himes BE, Levin AM, Mathias RA, Hancock DB, Baurley JW, Eng C, Stern DA, Celedon JC, Rafaels N, Capurso D, Conti DV, Roth LA, Soto-Quiros M, Togias A, Li X, Myers RA, Romieu I, Van Den Berg DJ, Hu D, Hansel NN, Hernandez RD, Israel E, Salam MT, Galanter J, Avila PC, Avila L, Rodriquez-Santana JR, Chapela R, Rodriguez-Cintron W, Diette GB, Adkinson NF, Abel RA, Ross KD, Shi M, Faruque MU, Dunston GM, Watson HR, Mantese VJ, Ezurum SC, Liang L, Ruczinski I, Ford JG, Huntsman S, Chung KF, Vora H, Calhoun WJ, Castro M, Sienra-Monge JJ, del Rio-Navarro B, Deichmann KA, Heinzmann A, Wenzel SE, Busse WW, Gern JE, Lemanske RF, Jr, Beaty TH, Bleecker ER, Raby BA, Meyers DA, London SJ, Gilliland FD, Burchard EG, Martinez FD, Weiss ST, Williams LK, Barnes KC, Ober C, Nicolae DL. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. 43:887–892. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandford A, Weir T, Pare P. The genetics of asthma. Am J Respir Crit Care Med. 1996;153:1749–1765. doi: 10.1164/ajrccm.153.6.8665031. [DOI] [PubMed] [Google Scholar]
- 6.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 7.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla A, Conrad DF, Fu Y, Grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Khurana E, Kural D, Lam HY, Leng J, Li R, Li Y, Lin CY, Luo R, Mu XJ, Nemesh J, Peckham HE, Rausch T, Scally A, Shi X, Stromberg MP, Stutz AM, Urban AE, Walker JA, Wu J, Zhang Y, Zhang ZD, Batzer MA, Ding L, Marth GT, McVean G, Sebat J, Snyder M, Wang J, Eichler EE, Gerstein MB, Hurles ME, Lee C, McCarroll SA, Korbel JO. Mapping copy number variation by population-scale genome sequencing. Nature. 470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlattl A, Anders S, Waszak SM, Huber W, Korbel JO. Relating CNVs to transcriptome data at fine resolution: assessment of the effect of variant size, type, and overlap with functional regions. Genome Res. 21:2004–2013. doi: 10.1101/gr.122614.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, Elliott AL, Parkin M, Hubbell E, Webster T, Mei R, Veitch J, Collins PJ, Handsaker R, Lincoln S, Nizzari M, Blume J, Jones KW, Rava R, Daly MJ, Gabriel SB, Altshuler D. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 11.Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, Tyler-Smith C, Carter N, Scherer SW, Tavare S, Deloukas P, Hurles ME, Dermitzakis ET. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper GM, Nickerson DA, Eichler EE. Mutational and selective effects on copy-number variants in the human genome. Nat Genet. 2007;39:S22–29. doi: 10.1038/ng2054. [DOI] [PubMed] [Google Scholar]
- 13.McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, Duerr RH, Silverberg MS, Taylor KD, Rioux JD, Altshuler D, Daly MJ, Xavier RJ. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008 doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prescott NJ, Dominy KM, Kubo M, Lewis CM, Fisher SA, Redon R, Huang N, Stranger BE, Blaszczyk K, Hudspith B, Parkes G, Hosono N, Yamazaki K, Onnie CM, Forbes A, Dermitzakis ET, Nakamura Y, Mansfield JC, Sanderson J, Hurles ME, Roberts RG, Mathew CG. Independent and population-specific association of risk variants at the IRGM locus with Crohn’s disease. Hum Mol Genet. doi: 10.1093/hmg/ddq041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, Hebert M, Jones KN, Shu Y, Kitzmiller K, Blanchong CA, McBride KL, Higgins GC, Rennebohm RM, Rice RR, Hackshaw KV, Roubey RA, Grossman JM, Tsao BP, Birmingham DJ, Rovin BH, Hebert LA, Yu CY. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007;80:1037–1054. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKinney C, Merriman ME, Chapman PT, Gow PJ, Harrison AA, Highton J, Jones PB, McLean L, O’Donnell JL, Pokorny V, Spellerberg M, Stamp L, Willis J, Steer S, Merriman T. Evidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheumatoid arthritis. Ann Rheum Dis. 2007 doi: 10.1136/ard.2007.075028. [DOI] [PubMed] [Google Scholar]
- 17.Lee YL, Hsiue TR, Lee YC, Lin YC, Guo YL. The association between glutathione S-transferase P1, M1 polymorphisms and asthma in Taiwanese schoolchildren. Chest. 2005;128:1156–1162. doi: 10.1378/chest.128.3.1156. [DOI] [PubMed] [Google Scholar]
- 18.Brasch-Andersen C, Christiansen L, Tan Q, Haagerup A, Vestbo J, Kruse TA. Possible gene dosage effect of glutathione-S-transferases on atopic asthma: using real-time PCR for quantification of GSTM1 and GSTT1 gene copy numbers. Hum Mutat. 2004;24:208–214. doi: 10.1002/humu.20074. [DOI] [PubMed] [Google Scholar]
- 19.Ivaschenko TE, Sideleva OG, Baranov VS. Glutathione-S-transferase micro and theta gene polymorphisms as new risk factors of atopic bronchial asthma. J Mol Med. 2002;80:39–43. doi: 10.1007/s001090100274. [DOI] [PubMed] [Google Scholar]
- 20.Rogers AJ, Brasch-Andersen C, Ionita-Laza I, Murphy A, Sharma S, Klanderman BJ, Raby BA. The interaction of glutathione S-transferase M1-null variants with tobacco smoke exposure and the development of childhood asthma. Clin Exp Allergy. 2009;39:1721–1729. doi: 10.1111/j.1365-2222.2009.03372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng SL, Yu CJ, Chen CJ, Yang PC. Genetic polymorphism of epoxide hydrolase and glutathione S-transferase in COPD. Eur Respir J. 2004;23:818–824. doi: 10.1183/09031936.04.00104904. [DOI] [PubMed] [Google Scholar]
- 22.Gilliland FD, Gauderman WJ, Vora H, Rappaport E, Dubeau L. Effects of glutathione-S-transferase M1, T1, and P1 on childhood lung function growth. Am J Respir Crit Care Med. 2002;166:710–716. doi: 10.1164/rccm.2112065. [DOI] [PubMed] [Google Scholar]
- 23.Palmer CN, Doney AS, Lee SP, Murrie I, Ismail T, Macgregor DF, Mukhopadhyay S. Glutathione S-transferase M1 and P1 genotype, passive smoking, and peak expiratory flow in asthma. Pediatrics. 2006;118:710–716. doi: 10.1542/peds.2005-3030. [DOI] [PubMed] [Google Scholar]
- 24.Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 25.Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, Oseroff VV, Albertson DG, Pinkel D, Eichler EE. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AW, Robson S, Stirrups K, Valsesia A, Walter K, Wei J, Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME. Origins and functional impact of copy number variation in the human genome. Nature. 2009 doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet. 2006;38:82–85. doi: 10.1038/ng1695. [DOI] [PubMed] [Google Scholar]
- 28.The Childhood Asthma Management Program (CAMP): design, rationale, and methods. Childhood Asthma Management Program Research Group. Control Clin Trials. 1999;20:91–120. [PubMed] [Google Scholar]
- 29.Long-term effects of budesonide or nedocromil in children with asthma. The Childhood Asthma Management Program Research Group. N Engl J Med. 2000;343:1054–1063. doi: 10.1056/NEJM200010123431501. [DOI] [PubMed] [Google Scholar]
- 30. [Google Scholar]
- 31.Kim JI, Ju YS, Park H, Kim S, Lee S, Yi JH, Mudge J, Miller NA, Hong D, Bell CJ, Kim HS, Chung IS, Lee WC, Lee JS, Seo SH, Yun JY, Woo HN, Lee H, Suh D, Kim HJ, Yavartanoo M, Kwak M, Zheng Y, Lee MK, Kim JY, Gokcumen O, Mills RE, Zaranek AW, Thakuria J, Wu X, Kim RW, Huntley JJ, Luo S, Schroth GP, Wu TD, Kim H, Yang KS, Park WY, Church GM, Lee C, Kingsmore SF, Seo JS. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 33.Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, Pang AW, Shago M, Stockwell TB, Tsiamouri A, Bafna V, Bansal V, Kravitz SA, Busam DA, Beeson KY, McIntosh TC, Remington KA, Abril JF, Gill J, Borman J, Rogers YH, Frazier ME, Scherer SW, Strausberg RL, Venter JC. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 35.Barnes C, Plagnol V, Fitzgerald T, Redon R, Marchini J, Clayton D, Hurles ME. A robust statistical method for case-control association testing with copy number variation. Nat Genet. 2008;40:1245–1252. doi: 10.1038/ng.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rogers AJ, Raby BA, Lasky-Su JA, Murphy A, Lazarus R, Klanderman BJ, Sylvia JS, Ziniti JP, Lange C, Celedon JC, Silverman EK, Weiss ST. Assessing the reproducibility of asthma candidate gene associations, using genome-wide data. Am J Respir Crit Care Med. 2009;179:1084–1090. doi: 10.1164/rccm.200812-1860OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H, Romieu I, Shi M, Hancock DB, Li H, Sienra-Monge JJ, Chiu GY, Xu H, del Rio-Navarro BE, London SJ. Evaluation of candidate genes in a genome-wide association study of childhood asthma in Mexicans. J Allergy Clin Immunol. 125:321–327. e313. doi: 10.1016/j.jaci.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. Chapter 19(Unit 19):10, 11–21. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ionita-Laza I, Perry GH, Raby BA, Klanderman B, Lee C, Laird NM, Weiss ST, Lange C. On the analysis of copy-number variations in genome-wide association studies: a translation of the family-based association test. Genet Epidemiol. 2008;32:273–284. doi: 10.1002/gepi.20302. [DOI] [PubMed] [Google Scholar]
- 42.Gauderman WJ, Morrison JM. Quanto 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies. 2006. [Google Scholar]
- 43.Ionita-Laza I, Rogers AJ, Lange C, Raby BA, Lee C. Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis. Genomics. 2009;93:22–26. doi: 10.1016/j.ygeno.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, D’Agostino R, Jr, Castro M, Curran-Everett D, Fitzpatrick AM, Gaston B, Jarjour NN, Sorkness R, Calhoun WJ, Chung KF, Comhair SA, Dweik RA, Israel E, Peters SP, Busse WW, Erzurum SC, Bleecker ER. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 181:315–323. doi: 10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grasemann H, Yandava CN, Drazen JM. Neuronal NO synthase (NOS1) is a major candidate gene for asthma. Clin Exp Allergy. 1999;29 (Suppl 4):39–41. [PubMed] [Google Scholar]
- 46.Wechsler ME, Grasemann H, Deykin A, Silverman EK, Yandava CN, Israel E, Wand M, Drazen JM. Exhaled nitric oxide in patients with asthma: association with NOS1 genotype. Am J Respir Crit Care Med. 2000;162:2043–2047. doi: 10.1164/ajrccm.162.6.2003089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.