

Abstract
A majority of nanoencapsulated drugs that have shown promise in cancer chemotherapy are administered intravenously. Development of effective oral nanoformulations presents a very challenging medical goal. Here, we describe successful applications of innovative polymeric nanogels in the form of conjugates with activated nucleoside analogs for oral administration in cancer chemotherapy. Previously, we reported the synthesis of amphiphilic polyvinyl alcohol and dextrin-based nanogel conjugates with the phosphorylated 5-FU nucleoside Floxuridine and demonstrated their enhanced activity against regular and drug-resistant cancers[1]. In this study, we synthesized and evaluated oral applications of nanogel conjugates of a protected Gemcitabine, the drug never used in oral therapies. These conjugates were able to quickly release an active form of the drug (Gemcitabine 5′-mono-, di- and triphosphates) by specific enzymatic activities, or slowly during hydrolysis. Gemcitabine conjugates demonstrated up to 127 times higher in vitro efficacy than the free drug against various cancer cells, including the lines resistant to nucleoside analogs. Surprisingly, these nanogel-drug conjugates were relatively stable in gastric conditions and able to actively penetrate through the gastrointestinal barrier based on permeability studies in Caco-2 cell model. In tumor xenograft models of several drug-resistant human cancers, we observed an efficient inhibition of tumor growth and extended the life-span of the animals by 4 times that of the control with orally treated Gemcitabine- or Floxuridine-nanogel conjugates. Thus, we have demonstrated a potential of therapeutic nanogel conjugates with the activated and stabilized Gemcitabine as a successful oral drug form against Gemcitabine-resistant and other drug-resistant tumors.
Keywords: Nanogel-drug conjugates, Gemcitabine, phosphorylated nucleoside analogs, oral drug administration, drug-resistant animal tumor xenografts
1. Introduction
Nucleoside analogs (NAs) are first-line therapeutics in the treatment of many cancers. Most cancer patients develop drug resistance to NAs and require escalated dose schedules during therapy, which induce severe side effects such as cardiac [2–4] or hematological toxicities like a secondary leukemia [5]. Drug resistance continues to be a major medical problem and a limiting factor in successful cancer treatment. For that reason, the development of new approaches to overcome drug resistance and reduce side effects of chemotherapy is in urgent demand. To make the existing therapies more effective, NAs can be utilized in combination with other anticancer drugs against chemoresistant tumors. The major advantage of NAs is their non-cross resistance with other classes of drugs [6, 7]. NAs interfere with RNA or DNA synthesis and cellular targets involved in the metabolism of physiological nucleosides [8]. After entering cancer cells as inactive compounds via membrane nucleoside transporters, they are phosphorylated by cellular nucleoside kinases and converted in active nucleoside 5′-mono-, di-, and triphosphates. The key mechanisms of drug resistance to NAs are associated with nucleoside transporter deficiency [9, 10], reduced nucleoside kinase activity [11], over-expression of multidrug resistance proteins (P-gp, MDR, BCRP)[12], or modifications in apoptotic pathways [13]. Recently, we introduced a new strategy of overcoming drug resistance to NAs in cancer cells by application of active 5′-triphosphates of NAs encapsulated into cationic nanogels [14]. This approach was further advanced by the development of biodegradable conjugates of phosphorylated NAs with amphiphilic nanogels and demonstrated an excellent activity against regular and drug-resistant cancer cells [1]. These nanogel-drug conjugates were synthesized using polyvinyl alcohol (PVA), a biocompatible mucoadhesive polymer that is potentially suitable for the fabrication of oral drugs [15]. Oral delivery is the most convenient and patient-friendly route of drug administration. However, problems associated with short gastric residence time, poor drug absorption and instability in the gastrointestinal (GI) tract sometimes limit oral potential of many cancer therapeutics. Application of drug delivery systems (liposomes, nanoparticles, etc) can potentially open novel venues to overcoming these problems [16]. Unfortunately, bile acids in the GI tract are excellent solvents for phospholipids and ruptures the liposomes, one of the most advanced drug carriers [17]. Nanoparticles are more stable in the GI tract, but have poor GI permeability and, therefore, these carriers have never been applied in oral therapies. Only a limited number of oral prodrugs are presently used in cancer chemotherapy [18]. A major problem with Gemcitabine, Cytarabine and other cytosine drugs is oral instability due to the rapid deamination by cytidine deaminase in vivo. In the present paper, we examine the application of encapsulated Gemcitabine in activated and protected form in mucoadhesive nanogels in order to increase the drug stability, GI adsorption, and activity against drug-resistant tumors for successful oral administration.
2. Materials and Methods
2.1. Materials
Chemical reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO) and Alfa Aesar (Wardhill, MA) with the highest available purity and used without purification unless otherwise stated. Nucleoside analogs: Gemcitabine (Beta Pharma Inc., Brandford, CT), Floxuridine (SynQuest Laboratories, Alachua, FL) and Cytarabine (3b Mediacl Systems, Inc, Libertyville, IL); 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), snake venom phosphodiesterase 1, type VI, from Carotalus adamanteus and alkaline phosphatase, poly(vinyl alcohol), Mw 31,000, and dextrin, Mw 9,000 (Sigma, St. Louis, MO) were used in the study. CPVA-p4FLOX was synthesized as earlier described [1]. Proton NMR spectra were recorded using a 500 MHz Varian NMR-spectrometer and tetramethylsilane as a standard. Hydrodynamic diameter, polydispersity, and aggregation stability was measured by dynamic light scattering (DLS) using Zetasizer Nano-ZS90 (Malvern Instruments, Southborough, MA). UV-absorbance was measured by Biophotometer (Eppendorf, Hamburg, Germany) and NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA).
2.2. Cells
Human pancreatic adenocarcinoma MIA PaCa-2 and Capan-1 cells (a kind gift from Dr. Surinder Batra, UNMC) were maintained in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, 1% L-glutamine and 1% penicillin-streptomycin at 37°C in humidified 5%CO2 atmosphere. Floxuridine-resistant MDA-MB-231/FLOX cells were selected by a stepwise increase of the drug concentration in wild-type cells over three months’ period. These cells were grown in DMEM: Nutrient mixture F-12 (DMEM/F12) with 10% FBS, 1% L-glutamine and 1% penicillin-streptomycin in the presence of 10 μM Floxuridine. Gemcitabine-resistant human follicular lymphoma RL7/G cell line, which is characterized by a reduced level of deoxycytidine kinase, was a kind gift from Dr. F. Bontemps (De Duve Institute, Brussels, Belgium). These cells were maintained in the presence of 2 μM Gemcitabine. Cytarabine-resistant human T-cell leukemia CEM/araC/8 cell line, which is deficient in the nucleoside transport, was a generous gift from Dr. C. Galmarini (University Lyon-Sud, Oullins, France). The cells were grown in the presence of 0.5 μM cytarabine. All drug-resistant cell lines were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, and 2% penicillin-streptomycin at 37°C in humidified 5%CO2 atmosphere. CacoReady kit for an in vitro evaluation of drug permeability in Caco-2 monolayers was purchased from ADMEcell (Emeryville, CA) and used according to the manufacturer’s protocol.
2.3. Synthesis of cholesterol conjugates
PVA was grafted with cholesterol according to the procedures described below. Briefly, 5.0 g of PVA (Mw 31,000 Da) was dried over phosphorus oxide in vacuo and dissolved in 50 mL of anhydrous DMSO at 70°C. Triethylamine (1.9 mmol) was added to the cooled solution (25°C) followed by 0.71g (1.61 mmol) of cholesteryl chloroformate, and the reaction was continued overnight at 25°C. The reaction mixture was concentrated in vacuo and dialyzed (MWCO 3.5 kDa) against 20% aqueous ethanol three times for 24 hours. The product (CPVA) was concentrated in vacuo and freeze-dried with a yield of 80%. 1H-NMR spectrum (d6-DMSO): 0.65 (s, 18H), 0.83–0.85(dd, J=5.0, 1.6 Hz, 36H), 0.89(d, J=4.8 Hz, 18H), 0.94(s, 18H), 1.07–1.98(m, 1582H), 3.84(m, 704H), 4.35(12H and OH), 5.28(s, 6H). The dextrin-cholesterol conjugate (CDEX, Mw 9 kDa) was synthesized with a yield of 76% as previously described [1]. 1H-NMR spectrum (d6-DMSO); δ: 0.65 (s, 18H), 0.83–0.85(dd, J=5.0, 1.6 Hz, 36H), 0.89–1.51(m, 211H), 3.24–3.64(m, 333H), 4.28–5.10 (m, 122H), 5.23(s, 6H), 5.37–5.62(m, 55H).
2.4. Synthesis of N-protected Gemcitabine
2′, 2′-difluorodeoxycytidine (0.79 g, 3.0 mmol) was co-evaporated with anhydrous pyridine (15mL × 3) and re-suspended in 3 mL of the solvent. A solution of trimethylchlorosilane (1.95 g, 18.0 mmol) in 10 mL anhydrous pyridine was then added to the nucleoside at 4°C. The reaction mixture was vigorously stirred for 2 hours at 25°C, and then butyryl chloride (0.48 g, 4.5 mmol) was added dropwise. The reaction was continued for additional 2 hours, and then the precipitate was filtered. The combined filtrates were evaporated to dryness and treated with a dioxane-water mixture (5:3 v/v) for 6 h at 25°C. The reaction progress was monitored by TLC on silicagel in methanol-dichloromethane (1:9 v/v). The concentrated in vacuo product was purified by flash chromatography on silica gel column using a concentration gradient of methanol (0 to 10% v/v) in dichloromethane as an eluent. Fractions with N-acylated Gemcitabine were collected and concentrated in vacuo with a yield of 74%. 1H-NMR spectrum (d6-DMSO): 1.0 (d, J=1 Hz, 6H), 2.71 (sept, J=6.5 Hz, 1H), 3.62 (m, 1H), 3.78(m, 1H), 3.86 (m, 1H), 4.21 (m, 1H), 5.27(d, J=5.5, 1H), 6.14(t, J= 7.5 Hz, 1H), 6.29(s, 1H), 7.28(d, J=7.5 Hz, 1H), 8.23(d, J=7.5 Hz, 1H), 10.97(s, 1H).
2.5. Preparation of polymeric drug conjugates
The phosphorylating reagent, 2-cyanoethyl bis-imidazolyl phosphate, CNEtOP(O)Im2 was synthesized as previously described [1]. CPVA (3.3 g) was dried by co-evaporation with anhydrous toluene (15 mL × 3) and dissolved in 33 mL anhydrous dimethylformamide (DMF). The solution was treated with a 2M solution of CNEtOP(O)Im2 in anhydrous DMF (2 mL) for 30 min at 25°C. Then, a 1M solution of n-butylammonium salt of pyrophosphate in anhydrous DMF (4 mL) was added, and the reaction mixture was incubated for 1 h at 25°C. In a separate flask, N-acyl Gemcitabine (696 mg, 2 mmol) was treated with a 2M solution of CNEtOP(O)Im2 in anhydrous DMF (1 mL) at 4°C, and the reaction mixture was allowed to stand for 20 min at 25°C. Then, both reaction mixtures of phosphorylated CPVA and nucleoside were mixed together and stirred for 40 min at 25°C. After the treatment with 1 mL of methanol and overnight incubation at 4°C, the insoluble material was removed by filtration, and the product was purified by dialysis (MWCO 3,5kDa) against 20% aqueous ethanol three times over 24 h to remove un-reacted starting materials and by-products. The CPVA-p4GEMC conjugate was isolated by precipitating as a sodium salt in 2% sodium perchlorate in acetone, washed by acetone and dried in vacuo. The CDEX-p4GEMC conjugate was obtained by the same method. Nucleoside content in both products was measured by UV absorbance of N-acylated Gemcitabine (ε268=9476): CPVA-p4GEMC, 0.385 μmol/mg, and CDEX-p4GEMC, 0.39 μmol/mg. 1H-NMR spectrum (d6-DMSO): δ: 0.64–0.93 (m, 90H), 1.06 (d, J=1 Hz, 72H), 1.32–1.51(m, 1582H), 2.71 (m, 12H), 3.66–4.66(m, 1462H), 3.65 (m, 12H), 5.30 (m, 12H), 6.17(m, 12H), 6.31(m, 12H), 7.30(d, J=7.5 Hz, 12H), 8.23(d, J=7.5 Hz, 12H).
2.6. Particle size and zeta-potential measurements
The hydrodynamic diameter, size distribution, and zeta-potential of nanogel particles were measured by DLS following the ultrasonication of nanogel-drug conjugates (0.1 mg/mL) for 1 h at 4°C and centrifugation for 4 min at 12,000 g. The size distribution was characterized by polydispersity index. Zeta-potential was calculated based on electrophoretic mobility measurements performed with an electrical field strength of 15–18 V cm−1 at 25°C using the instrument software. The data reported in Table 1 represents an average of three measurements.
Table 1.
Physicochemical characteristics of nanogels and drug conjugates
| Polymeric conjugate* | dh, nm (volume averaged) | PDI | ζ, mV |
|---|---|---|---|
| CPVA | 35 ± 1.3 | 0.36 | 0 ± 3.7 |
| CPVA-p4FLOX | 42 ± 6.4 | 0.41 | −8.5 ± 3.6 |
| CPVA-p4GEMC | 30 ± 2.4 | 0.25 | −17.9 ± 7.5 |
| CDEX | 44 ± 8.3 | 0.46 | −8 ± 4.0 |
| CDEX-p4GEMC | 34 ± 0.4 | 0.32 | −11.2 ± 7.0 |
Particle size (dh), polydispersity index (PDI) and zeta-potential (ζ) were measured in 1% solutions in water after 2h sonication. The results are average values ± SD of three measurements.
2.7. Enzymatic hydrolysis
We tested enzymatic degradation of nanogel-drug conjugates in three enzymatic systems (phosphodiesterase, alkaline phosphatase and liver homogenate). The enzymatic stability of conjugates, the kinetics and products of drug release were assayed as follows.
(1) Phosphodiesterase
50 μL of the reaction mixture containing 100 mM Tris-HCl (pH 8.75), 2 mM MgCl2, 0.05 mg of snake venom phosphodiesterase (VPDE), and 0.1 mg nanogel sample was incubated at 37°C, and 5 μL aliquots were taken after specified time periods and quenched with 1.5 μL of 1M HCl before HPLC analysis.
(2) Alkaline phosphatase
50 μL of the reaction mixture containing 0.1 mg nanogel sample were incubated in 0.8 M TEAB (pH 8) containing 10, 7, 5 and 2 units of alkaline phosphatase for 40 min at 37°C. Samples were quenched as before and analyzed by HPLC.
(3) Liver homogenate
One mouse liver was rinsed with tris-EDTA buffer, pH 8, and homogenized in 2.5 ml of the buffer using Tissuemiser (Thermo Fisher Scientific, Waltham, MA.). Nanogel samples (0.25 mg) were incubated with 0.5 mL of the homogenate at 37°C for different time periods and centrifuged (5 min at 12,000 g). Nucleoside/nucleotide fraction was extracted with phenol-chloroform-isoamyl alcohol (25:24:1) and chloroform. The aqueous phase was recovered and lyophilized, and samples were analyzed by HPLC.
(4) HPLC analysis
Ascentis C18 column (10 μm, 15cm × 4.6mm) was used for ion-pair HPLC with UV detection at a flow rate of 1 mL/min. The elution was performed with in 40 mM KH2PO4, 0.2% tetrabutylammonium hydroxide, pH 7.0, in a linear gradient of concentration of acetonitrile (0 to 30% acetonitrile in 20 min). Corresponding nucleoside 5′-mono- and triphosphates have been used as standards.
2.8. In vitro drug release
In vitro drug release was studied using simulated gastric fluids (SGF) or simulated intestinal fluid (SIF). Briefly, SGF was prepared by dissolving 3.2 g/L of pepsin from porcine stomach mucosa (800–2500 units per mg of protein) in 34 mM NaCl and 84 mM HCl containing water with adjustment of final pH to 1.2 with 0.2 M HCl. SIF was prepared by dissolving 10 g/L of pancreatin in 50 mM of monobasic potassium phosphate and 15 mM NaOH containing water with adjustment of final pH to 6.8 with 0.2 M HCl. Nanogel samples (1 mg) were placed in small dialysis tubes (MWCO 3500) contained 1 mL of SGF or SIF and incubated in small 4 mL vials containing same solution at 37 °C and slow magnetic stirring. Drug release was monitored by ion-pair HPLC (see section 2.7 above), 0.2 ml aliquots withdrawn periodically from these vials.
2.9. Cell viability assay
Cytotoxicity of nanogel-drug conjugates was evaluated in cancer cell lines by a standard MTT assay. Briefly, the attached cancer cells (Capan-1, MIA Paca-2, or Floxuridine-resistant MDA-MB-231/F) were seeded at a density of 3,000 cells per 200 μL growth medium per well in flat-bottom 96-well plates and allowed to attach overnight; the same amount of suspension cancer cells (Gemcitabine-resistant RL7/G, or Cytarabine-resistant CEM/araC/8) was placed in round-bottom 96-well plates. Samples of drugs, nanogel-drug conjugates or empty carriers were added to cells and incubated in full medium for 72 h at 37°C. The metabolic activity of cancer cells in each sample was determined by adding 20 μL of a MTT stock solution (5 mg/mL) in sterile PBS buffer to each well. The samples were then incubated for 2 h at 37°C, the medium and the MTT dye were washed out by PBS, and 100 μL of extraction buffer (20% w/v SDS in DMF/water, 1:1, pH 4.7) was added to each well. Samples were incubated for 24 h at 37°C. Optical absorbance was measured at 560 nm using a Model 680 microplate reader (BioRad, Hercules, CA). Cytotoxicity was expressed as percentile cell viability at different drug/nanogel concentrations compared to untreated cells. All samples were analyzed by an average of 8 measurements (means ± SEM). The IC50 (concentration of the 50% cell survival) values were calculated from the plotted curves.
2.10. Caco-2 cell permeability assay
Transcellular transport studies were performed using CacoReady kit (ADMEcell, Emeryville, CA). It consisted of differentiated Caco-2 cell cultures (day 14 of differentiation) on polycarbonate micro-porous filters in HTS transwell-24 plates (6.5 mm diameter, 0.33 cm2 area and 0.4 μm pore diameter). The shipping media of Caco-2 ready plate was replaced with a standard Caco-2 cell culture media (DMEM, Lonza Walkersville, MD) according to manufactures protocol. The transport buffer contains 5.4 mM KCl, 0.44 mM KH2PO4, 0.137 mM NaCl, 0.33 mM Na2HPO, 1.1 mM MgCl2x6H2O, 0.13 mM CaCl2x2H2O, 5 mM D-glucose (pH 6.5). The TEER was measured prior to each experiment to assess the integrity of Caco-2 monolayers using an EVOM epithelial voltmeter with Endohm electrode (World precision Instruments, Sarasota, FL). The TEER values 1660 Ωcm2 showed that cell monolayers have formed tight junctions. Also, Lucifer Yellow dye (Sigma-Aldrich, St. Louis, MO) was used as an internal marker of paracellular permeability (normal retention >99%).
The basal side of the chamber was filled with 750 μL and the apical side with 180 μl of nanogel/drug sample containing Lucifer Yellow (100 μM) in the transport buffer. The cell monolayers were then incubated for 4 h; aliquots were taken every hour from the basolateral side, and the volume was replaced with fresh buffer. All experiments were carried out at 37°C. The apical-to-basolateral permeability coefficient (Papp in cm/sec) was calculated according to following equation:
where dQ/dt – the amount of nanogel/drug in basolateral compartment as a function of time (mg/min), A - the monolayer area (cm2), C0 - the initial concentration of nanogel/drug in apical compartment (mg/mL).
2.11. Tumor growth inhibition
Subcutaneous tumor xenografts were established in female nu/nu mice aged 6–8 weeks (Charles River Laboratories, Wilmington, MA). Animal studies were carried out according to the principles of animal care outlined by the National Institutes of Health, and protocols were approved by the Institutional Animal Care and Use Committee at the University of Nebraska Medical Center. The animals were randomly divided into groups of five per cage and maintained under sterile conditions in controlled environment. All manipulations with animals were performed in a sterile laminar hood using sterile solutions.
Briefly, approximately 5 × 106 cells re-suspended in 400 μL of serum-free medium containing 20% Matrigel (Becton-Dickinson, San Diego, CA) were injected subcutaneously into the right flanks of nu-nu nude mice. When tumors could be palpitated, mice were randomly divided into treatment groups and solutions of CPVA-p4GEMC and Gemcitabine were administered via oral gavage (50 μL) twice a week at doses 23 mg/kg (by GEMC content) and 30 mg/kg[19], respectively. Alternatively, a CPVA-p4FLOX (0.16 mg/mL) solution in drinking water was administered ad libitum. Tumor volume was measured by digital calipers and calculated based on the equation: , where L and W are length and width of tumor (mm).
3. Results and discussion
3.1. Synthesis of nanogel conjugates
We synthesized Gemcitabine conjugates with nanogels based on a mucoadhesive polymer polyvinyl alcohol (PVA, Mw 31 kDa) and a biodegradable polysaccharide dextrin (DEX, Mw 9 kDa). Drug delivery in nanocarriers covered by bioadhesive polymers like PVA can enhance drug absorption in the GI tract, as well as their ability to escape drug efflux transporters [20, 21]. Both polymers have a uniquely neutral polyhydroxylated structure enhancing their circulation time due to the insignificant interactions with macrophages and blood cells [22]. When modified with hydrophobic cholesterol moieties, these hydrophilic polymers become amphiphilic and assemble into compact nanogels by self-aggregation of cholesterol groups after ultrasonication in aqueous media [30]. But the most important new property of cholesteryl-polymers and nanogels is a significant increase of their affinity to cellular membrane and closely associated with this affinity, is the facilitation of the transport mechanism of hydrophilic drugs across the cellular membrane.
Cholesterol moieties attached to PVA (CPVA) and dextrin (CDEX) was quantified by NMR analysis based on proton signals of the cholesterol moiety at δ=0.6–2.4 ppm as described earlier [1]. According to these data, the conjugates contained 6 (CPVA) and 4 (CDEX) cholesterol moieties per polymer molecule. Then, they were used to covalently attach N-acylated Gemcitabine (GEMC) through a biodegradable tetraphosphate linker using a convenient phosphorylation reagent 2-cyanoethyl bis-imidazolyl phosphate (Scheme 1). This approach allowed us to deliver the activated phosphorylated nucleoside analogs into cancer cells. During the synthesis, an exocyclic amino group of Gemcitabine was protected by acylation with isobutyryl anhydride in order to increase the yield and purity of nanogel-drug conjugates. The second goal was to prevent the fast enzymatic deamination of Gemcitabine into an inactive deoxyuridine derivative in vivo [23]. The isobutyryl group was the less stable amide protecting group for cytosine nucleosides, so it would be easily released to activate the drug. Also, this modification can potentially enhance the drug’s stability in the GI tract. To obtain N-acylated Gemcitabine, we used a transient O-protection of Gemcitabine 1 with trimethylsilyl groups and in situ reaction with butyryl chloride in anhydrous pyridine (Scheme 1) [24]. Trimethylsilyl groups were hydrolyzed in aqueous conditions to give N-isobutyryl Gemcitabine 2 with a 74% yield. CPVA-p4GEMC conjugate was synthesized by the same method that was used in the synthesis of CPVA-p4FLOX [1]. We used the CNEtOP(O)Im2 phosphorylating reagent in order to obtain separately an imidazole-activated nucleoside 5′-phosphate 3, and triphosphorylated polymers from CPVA or CDEX 4 (Scheme 1). The polymers were initially treated with CNEtOP(O)Im2 and then, with pyrophosphate in the form of tetrabutylammonium salt. Activated Gemcitabine 5′-phosphate was then added in situ to form the covalent polymer-drug conjugate 5 (Scheme 1). Purified conjugates were characterized by 1H-NMR and UV spectroscopy as shown in supplementary materials (Figure SM1). Gemcitabine content was determined by characteristic benzyl group protons in aromatic region (7.30 and 8.25 ppm) for Gemcitabine and methylene protons (1.32–1.52 ppm) for PVA as a reference peak. According to 1H-NMR data, CPVA-p4GEMC conjugate contained 6 cholesterol and 14 Gemcitabine molecules. This amount accounts for 14% drug loading in the CPVA-p4GEMC conjugate. The drug amount in the conjugate measured by UV absorbance was equal to 0.3–0.4 μmol/mg giving very close to 14% drug content (Figure SM1-B). This conjugate was stable for at least 48 h at pH 1 as determined by dialysis. UV spectra before dialysis (SM2-b) and after dialysis (SM2-c,d) show the same absorbance maximum as in N-isobutyryl Gemcitabine (SM2-a); thus, the nucleoside protection is intact in gastrointestinal conditions.
Scheme 1.

Synthetic steps in preparation of CPVA-p4GEMC conjugates.
3.2. Particle Size
The size and surface charge of nanogel-drug conjugates was measured by dynamic light scattering. Particle morphology and homogeneity was observed by transmission electron microscopy (TEM). The CPVA- and CDEX-drug conjugates formed small nanoparticles after sonication in aqueous solution with a hydrodynamic diameter of 30–44 nm and low negative zeta-potential (Table 1, Figure 1). The negative charge confirms the presence of phosphate groups on the surface of CPVA- and CDEX nanogels, although a significant part remains hidden inside the polymer network. Cholesterol-decorated polymers could form small self-assembled intramolecular micelles with hydrophobic cholesterol core only with a significant input of energy. The hydrodynamic diameter, a measure of particle size in aqueous solution, depended on the nanogel sonication intensity and time. The smallest, supposedly monomolecular particles could form only after 2 h-sonication (Table 1). The size of nanoparticles was found important for their efficient uptake by gut-associated tissues following oral administration. Earlier studies have demonstrated that nanocarriers with a well-defined size of 30–50 nm had the most rapid and reproducible mucosal permeability [25]. Once macromolecular drugs or nanocarriers are absorbed in the GI tract, they can transfer by blood circulation and accumulate in tumors through neovasculature due to the enhanced permeability and retention effect [26]. Their tumoral and cellular accumulation is also size-dependent. The optimal size range to achieve greater cellular internalization of nanoencapsulated drugs inducing apoptosis in cancer cells was found to be between 40 and 50 nm [27, 28].
Figure 1.
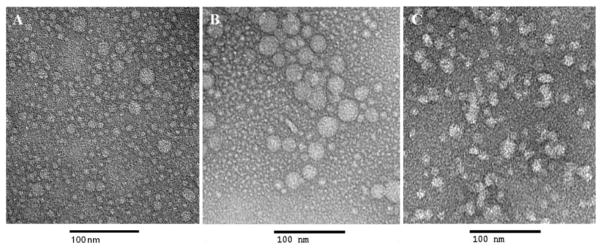
Transmission electron microscopy (TEM) image of empty CPVA nanogels (A), and nanogel-drug conjugates: CPVA-p4FLOX (B) and CPVA-p4GEMC (C). Polymer conjugates were dissolved in water and sonicate for 30 minutes. Samples were stained with vanadate. The bar is 100 nm.
3.3. Gastrointestinal drug release
An important feature of conjugated therapeutics is the drug release rate which is dependent on many factors, such as linker stability to hydrolysis and enzymatic activities in tissues. We monitored the drug release from CPVA-p4GEMC in media simulating the GI conditions. The stability of nanogel-drug conjugates in the GI tract depends on the presence of gastric enzymes and their interaction with nanogels. Here, we evaluated the release of Gemcitabine in simulated gastric fluid (SGF, pH 1.2) and in simulated intestinal fluid (SIF, pH 6.8) [29]. Drug release was monitored for 5 h in SGF and 12 h in SIF; these time periods are generally accepted as the maximum transit times in stomach and intestine. Following incubation at 37°C, the released Gemcitabine was isolated by ultrafiltration and analyzed by HPLC. The CPVA-p4GEMC nanogel showed a negligible Gemcitabine release in those conditions (<1%); so no drug loss is expected during oral administration of these nanocarriers. However, once nanogel-drug conjugates are absorbed by blood vessels, they become susceptible to other enzymatic activities including cellular enzymes [23]. The most common nucleolytic enzymes in circulation are phosphodiesterases, while in mammalian cells there are also nucleotide phosphatases, which can increase, by many-fold, the rate of drug release from the nanogel-drug conjugates and induce the therapeutic activity of NAs in cancer cells [30].
3.4. Enzymatic hydrolysis
Earlier, we demonstrated a slow pH-dependent mechanism of drug release from nanogel-drug conjugates [1]. Here, we investigated how the activity of ubiquitous enzymes, alkaline phosphatase and phosphodiesterase affect the Gemcitabine release in vitro due to the hydrolysis of tetraphosphate linker. Snake venom phosphodiesterase (VPDE) catalyzes hydrolysis of esterified 5′-phosphates and release of free nucleoside 5′-phosphates, while alkaline phosphatase (AP) hydrolyzes polyphosphates by cleaving phosphate bonds and release corresponding 5′-mono-, di- or triphosphates of nucleosides. The degradation process was analyzed by ion-pair HPLC [14]. AP treatment showed partial hydrolysis of CPVA-p4GEMC (elution time: 28–30 min) (Figure 2A) leading to mono- di and triphosphates as final products (Figure 2B) at low enzyme concentration, and to a complete drug release at high enzyme concentration (Figure 2C). With VPDE, we observed disappearance CPVA-p4GEMC and the formation of nucleoside 5′-monophosphate (elution time: 12–14 min) (Figure 2E). The release of Gemcitabine 5′-phosphate by both VPDE and AP was faster in the beginning and slower thereafter, obviously because of the longer time required for the enzyme to reach nucleotides inside the nanogels (Figure 3). Evidently, the hydrolysis with AP was faster compared to VPDE (82% drug release within 4 min) (Figure 3B). The hydrolysis of nanogel-drug conjugate by mouse liver homogenate resulted in the fast formation of deoxyuridine (based on UV spectrum) as a final product (Figure 2F). The deoxyuridine is not active species and significantly less toxic compound than gemcitabine, therefore, it should not cause side effects in liver.
Figure 2.
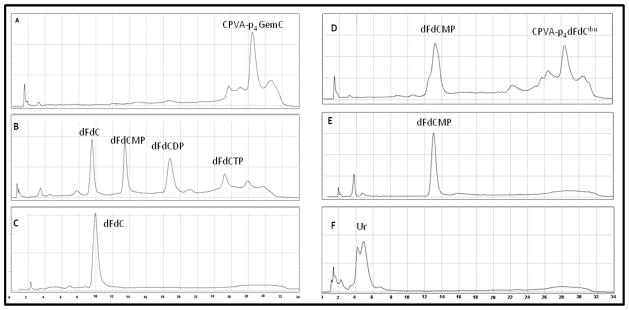
Drug release from nanogel-drug conjugates in result of enzymatic hydrolysis. (A) Gemcitabine (Gem, dFdC)-conjugated nanogel CPVA-p4GemC, (B) the conjugate treated for 40 min with 5 units of alkaline phosphatase (AP), and (C) the same as (B), but treated with 2x AP; (D) the conjugate treated with 2 units of snake venom phosphodiesterase (VPDE) for 4 h, and (E) the same as (D), but treated for 24 h; (F) the conjugate treated for 15 min with freshly extracted mouse liver homogenate. Ion-pair reverse phase HPLC analysis profiles of nucleoside 5′-phosphates are shown (dFdCMP, 5′-mono phosphate; dFdCDP, 5′-di phosphate; dFdCTP, 5′-tri phosphate); UR, deoxyuridine.
Figure 3.
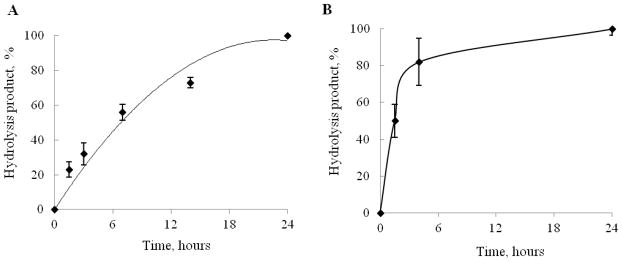
Enzymatic hydrolysis of CPVA-p4GEMC nanogel (at the concentration equivalent to 5 mM GEMC) by snake venom phosphodiesterase, VPDE (A), or alkaline phosphatase, AP (B). In both studies, the nanogel-drug conjugate was incubated with 5 units of enzyme, and samples were analyzed by ion-pair reverse phase HPLC at different time points. The shown data are means ± SEM (n = 3).
3.5. Cytotoxicity assay
Cytotoxicity of activated Gemcitabine and Floxuridine in nanogel conjugates was evaluated in normal and drug-resistant cell lines in comparison with the corresponding NAs. Drug-resistant lines of human follicular lymphoma RL7/G (resistant to Gemcitabine), T-cell leukemia CEM/araC/8 (resistant to Cytarabine), and breast carcinoma MDA-MB-231/FLOX (resistant to Floxuridine) were previously developed by in vitro selection at escalating doses of NAs. These cells also served in the establishment of subcutaneous tumor xenograft animal models. Aggressive human pancreatic carcinoma cell lines, a metastatic Capan-1 cells, and MIAPaCa-2 cells with medium resistance to Gemcitabine were also tested in the cytotoxicity studies.
According to dose-response curves (Figure 4) and calculated IC50 values (Table 2), both CPVA-p4GEMC and CDEX-p4GEMC nanogel-drug conjugates showed higher cytotoxicity compared to free gemcitabine (IC50 600 and 150 μM vs. 19 mM, respectively) in Gemcitabine-resistant RL7/G cells (Figure 4A,B). Thus, nanogel-drug conjugates demonstrated 32–127-fold higher efficacy compared to free Gemcitabine in this cell line. Similarly, CPVA-p4GEMC and CDEX-p4GEMC nanogel-drug conjugates showed 10–17 times higher therapeutic efficacy compared to Gemcitabine (IC50 30 and 50 μM vs. 512 μM, respectively) in Cytarabine-resistant CEM/araC/8 cells (Figure 4C,D, Table 2). These cell lines have different mechanisms of drug resistance, but nanogel-drug conjugates in both cases demonstrated excellent anticancer activity. Previously, we also reported the cross-efficacy of nanogels loaded with activated Gemcitabine 5′-triphosphate against Cytarabine-resistant cancer cell lines [37]. Nanodelivery of the protected drug allowed us to bypass nucleoside transporters and increase the accumulation of intact Gemcitabine in active phosphorylated form, circumventing many mechanisms of drug resistance. In many circumstances, Gemcitabine demonstrate mostly cytostatic effects, while treatment with nanogel-Gemcitabine conjugate usually resulted in complete cell death. In medium Gemcitabine-resistant MIA PaCa-2 cells, CPVA-p4GEMC and CDEX-p4GEMC nanogel-drug conjugates showed 3–25 times higher therapeutic efficacy than free Gemcitabine (IC50 0.16 μM and 0.02 μM vs. 0.52 μM, respectively) as shown in Figure 4E. These data were consistent with our previous results obtained for Floxuridine-nanogel conjugates [1]. For comparison, the CPVA-p4FLOX nanogel-drug conjugate demonstrated 11 times higher cytotoxicity than Floxuridine in the treatment of Floxuridine-resistant MDA-MB-231/FLOX cells (IC50 25 μM vs. 280 μM, respectively) as shown in Figure 4F. Importantly, new Gemcitabine-nanogel conjugates also demonstrated a 7–13 times higher efficacy than Floxuridine (IC50 22 and 40 μM vs. 280 μM, respectively) in Floxuridine-resistant MDA-MB-231/FLOX cells. Empty nanogels without drugs did not exhibit cytotoxicity at concentrations up to 600 times higher than concentrations corresponding to the observed IC50 values. In summary, a significant increase in cytotoxicity was observed by using activated drug-nanogel conjugates in the treatment of cancer cells with resistance to NAs.
Figure 4.

In vitro growth inhibition effect of CPVA-p4GEMC, CDEX-p4GEMC and free nucleoside analogs in different drug-resistant cancer cell lines. (A, B) Cytotoxicity of nanogel-GEMC conjugates vs. Gemcitabine in Gemcitabine-resistant human follicular lymphoma RL7/G cells; (C, D) Cytotoxicity of nanogel-GEMC conjugates in Cytarabine-resistant T-cell leukemia CEM/araC/8 cells compared to free Gemcitabine; (E) Cytotoxicity of CPVA-p4GEMC vs. Gemcitabine in medium Gemcitabine-resistant pancreatic adenocarcinoma MIA PaCa-2 cells; and (F) Cytotoxicity of CPVA-p4FLOX vs. Floxuridine in Floxuridine-resistant human breast carcinoma, MDA-MB-231/FLOX cells. In the MTT assay, cells were incubated for 72 h (n=8). Data shown are means ± SEM. In all samples P was <0.05 compared to free drug.
Table 2.
Cytotoxicity of nanogel-drug conjugates in cancer cells
| Drug Formulation | IC50(μM)*
|
||||
|---|---|---|---|---|---|
| RL7/G | CEM/araC/8 | MIA PaCa-2 | Capan-1 | MDA-MB-231/F | |
| Floxuridine (FLOX) | 8130 | 49 | 2.5 | 1.5 | 280 |
| Cytarabine (ARAC) | - | 1240 | - | - | - |
| Gemcitabine (GEM) | 19,000 | 512 | 0.515 | 2 | - |
| CPVA-p4GEMC | 600 | 30 | 0.16 | 1.5 | - |
| EF | 32 | 17 | 3 | 1 | - |
| CDEX-p4GEMC | 150 | 50 | 0.022 | 0.9 | - |
| EF | 127 | 10 | 23 | 2 | - |
| CPVA-p4FLOX | 3802 | 4.5 | 0.08 | - | 25 |
| EF | 2.1 | 11 | 31 | - | 11 |
Cytotoxicity was measured by MTT assay (72-h treatment). IC50 of CPVA and CDEX polymers in all cell lines were > 5mg/mL. The values in bold letters indicate the cytotoxicity enhancement factor (EF) as compared to a nucleoside analog: EF = IC50 (drug)/IC50(drug conjugate).
3.6. Caco-2 permeability assay
We evaluated GI permeability of nanogel-drug conjugates and calculated their permeability coefficients in an in vitro Caco-2 cellular model of the GI barrier [31, 32]. The human colon adenocarcinoma Caco-2 cells form confluent monolayers with tight junctions and exhibit properties similar to those of the GI epithelium[33]. CPVA-p4GEMC, CDEX-p4GEMC and CPVA-p4FLOX nanogels and empty carriers were labeled with Rhodamine isocyanate (RITC) and used in monitoring the transcellular transport across Caco-2 monolayers. The nanogels integrity was determined by monolayer resistant TEER values and Lucifer Yellow dye permeability before and during the experiment. TEER values were greater than 1660 Ωcm2 and the Lucifer Yellow transport was less than 0.7% over 1 h period, which corresponds to the values recommended in published protocols. Transport of free nucleosides (FLOX and GEM) was measured by UV spectrophotometric methods. We observed effective transport of nanogel-drug conjugates and empty carriers across the Caco-2 monolayers (Figure 5A). Based on permeability coefficients, the transfer of nanogels exceeded free drugs by 4.7 times (FLOX conjugates vs. FLOX) and by 5.7 times (GEMC conjugates vs. GEM) [34]. CPVA-p4GEMC transport was more efficient compared to CPVA-p4FLOX or CDEX-p4GEMC. The more efficient transfer was potentially due to an additional hydrophobicity of CPVA-p4GEMC conjugate as a result of higher cholesterol modifications of CPVA and iso-butyryl protective group in the conjugated Gemcitabine. Effect of hydrophobicity on gastrointestinal transport of drugs was mentioned previously [35]. According to this effect, the nanogel-Gemcitabine conjugate and the non-phosphorylated nanocarrier without drug transported most efficiently in the Caco-2 model (Fig. 5B). Compounds with a Papp coefficient between 10−6 and 10−5 have 20–70% of predicted in vivo absorption (medium permeability). The observed Papp values for all nanogel-drug conjugates demonstrated a significant improvement in drug transfer over NAs. Rapid phosphorolytic cleavage and intestinal degradation results in low bioavailability of Floxuridine and other NAs after oral administration[34]. In contrast, our data show that nanogel-drug conjugates have increased solubility and good stability during intestinal absorption. These results demonstrate a great potential of nanogel-drug conjugates for oral delivery applications.
Figure 5.
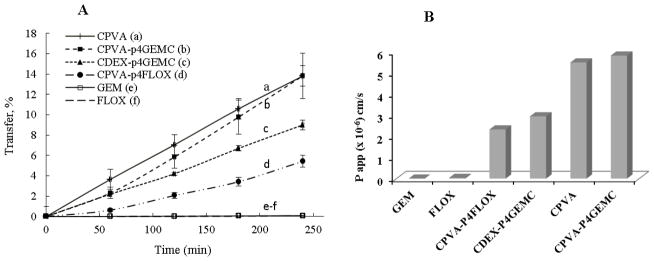
(A) Gastrointestinal (GI) transfer efficacy of nanogel-drug conjugates, empty nanogels and free drugs in Caco-2 cell monolayers. (A) Percentage of drug transfer across the Caco-2 cell monolayer: (a) empty nanogel without drug, (b–d) nanogel-drug conjugates, and (e–f) free nucleoside analogs, Floxuridine and Gemcitabine (60–240 min incubation at 37°C). Data represents means ± SEM (n=3). (B) Permeability coefficients of nanogel-drug conjugates and free drugs calculated from the transfer data. Papp were compared for each formulation to determine their GI transport efficacy.
3.7. In vivo tumor growth inhibition
To evaluate therapeutic efficacy of nanogel-drug conjugates, we studied tumor growth inhibition in subcutaneous tumor xenograft animal models of the aggressive Gemcitabine-resistant human follicular lymphoma (RL7/G) and Cytarabine-resistant T-cell leukemia (CEM/araC/8). These cell lines demonstrated high sensitivity to CPVA-p4GEMC and CPVA-p4FLOX nanogels (Table 2). Palpable tumors formed 7–10 days after inoculation and their size was regularly measured by digital calipers. Animals randomly separated into three groups (n = 5–6) received CPVA-p4GEMC by oral gavage at doses equivalent to 23 mg/kg two times a week for 4–5 consecutive weeks. We observed a significant two-fold decrease of very aggressive RL/G tumor growth in animals treated by the nanogel-drug conjugate compared to control animals (Figure 6A). The differences in tumor growth between control and CPVA-p4GEMC-treated groups were found statistically significant (P<0.05) from Day 24 for RL7/G xenograft model (P = 0.082 on Day 20). The Kaplan-Meier survival curves for animals with the CEM/araC/8 tumors demonstrated a nearly 4-fold extension of their lifespan due to the treatment with CPVA-p4GEMC nanogel as compared to control group (Figure 6B). Oral gavage of Gemcitabine at the corresponding doses resulted in relatively low effect (<50% increase in lifespan). Evidently, it was due to the Gemcitabine instability in gastrointestinal medium.
Figure 6.

Tumor growth inhibition in s.c. tumor xenograft mouse models after oral administration of CPVA-p4GEMC and CPVA-p4FLOX nanogel-drug conjugates. Gemcitabine-resistant human follicular lymphoma RL7/G xenograft model (A, C) and Cytarabine-resistant human T-cell leukemia CEM/araC/8 xenograft model (B, D) were used in the study. Animals were treated by oral gavage twice a week with CPVA-p4GEMC at a drug dose of 23 mg/kg (A, B), or with CPVA-p4FLOX solutions (equivalent to 0.16 mg FLOX/mL) ad libitum. In all experiments female nude mice beared s.c. tumors on the right lateral flank (groups with n=5–10). Data show statistical significance (P<0.05) between control (a) and nanogel-treated (b) groups starting from Day 20 (A), 20 (C) and 23(D).
For comparison, we report here also the tumor growth inhibition data obtained after oral administration of the CPVA-p4FLOX nanogel-drug conjugate. In these experiments, both nanogel-drug conjugate and drug were dissolved in drinking water and administered ad libitum (0.16 mg FLOX/ml). This route of administration is analogous to oral gavage. After the treatment, we observed also a statistically significant 3-fold inhibition of tumor growth in the Gemcitabine-resistant RL/G tumor xenograft model (Figure 6C). In the Cytarabine-resistant CEM/araC/8 tumor xenograft model, the effect of the CPVA-p4FLOX nanogel was also strong compared to control group (Figure 6D). The differences in tumor growth between control and CPVA-p4FLOX-treated groups were found statistically significant (P<0.05) from Day 20 for RL7/G xenograft model (P = 0.135 on Day 16) and from Day 23 for CEM/araC/8 xenograft model (P = 0.069 on Day 20).
We obtained similar results in our earlier experiments, when CPVA-p4FLOX was administered via peritumoral injections [1]. In summary, these experiments demonstrate equal efficacy of nanogel-drug conjugates using different routes of administration. Evidence suggests the activated polymeric NAs are more efficient against drug-resistant tumors and their administration results in significant extension of the animal’s lifespan. We are positive that the proposed polymer prodrugs, such as nanogel conjugates of activated NAs, represent are promising candidates for cancer treatment, which could significantly enhance therapeutic efficacy and reduce non-specific toxicities of cytotoxic nucleoside analogs in therapies against drug-resistant tumors.
Conclusions
We demonstrated that nanogel conjugates of active phosphorylated nucleoside analogs represent effective oral drug formulations, including Gemcitabine that was not administered orally in cancer chemotherapy. Gemcitabine used in the nanogel-conjugated and protected form was significantly more effective against aggressive cancer cells (e.g. pancreatic tumor and lymphoma) and many drug-resistant cancers. The conjugated drug showed suitable absorption in the GI model (Caco-2 cells) and ca. 5 times higher permeability compared to free Gemcitabine. We also demonstrated that nanogel-drug conjugates were stable in GI tract and were able to release active drugs as a result of enzymatic activities in cancer cells. Altogether, the efficient transport and drug release in active form resulted in substantial tumor growth inhibition in drug-resistant tumor xenograft animal models after oral administration. Thus, this novel nanogel-drug system proved to be a promising oral formulation of anticancer nucleoside analogs.
Supplementary Material
Acknowledgments
The financial support from National Cancer Institute (R01 CA136921 for S.V.V.) is gratefully acknowledged. The authors thank Tom Bargar for assistance with TEM microscopy of (Electron Microscopy Core Facility, UNMC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Senanayake TH, Warren G, Vinogradov SV. Novel anticancer polymeric conjugates of activated nucleoside analogues. Bioconjug Chem. 2011;22:1983–1993. doi: 10.1021/bc200173e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hale JP, Lewis IJ. Anthracyclines: cardiotoxicity and its prevention. Arch Dis Child. 1994;71:457–462. doi: 10.1136/adc.71.5.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones LW, Haykowsky MJ, Swartz JJ, Douglas PS, Mackey JR. Early breast cancer therapy and cardiovascular injury. J Am Coll Cardiol. 2007;50:1435–1441. doi: 10.1016/j.jacc.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 4.Von Hoff DD, Layard MW, Basa P, Davis HL, Jr, Von Hoff AL, Rozencweig M, Muggia FM. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91:710–717. doi: 10.7326/0003-4819-91-5-710. [DOI] [PubMed] [Google Scholar]
- 5.Lipshultz SE, Colan SD, Gelber RD, Perez-Atayde AR, Sallan SE, Sanders SP. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;324:808–815. doi: 10.1056/NEJM199103213241205. [DOI] [PubMed] [Google Scholar]
- 6.Moreno-Aspitia A, Perez EA. Anthracycline- and/or taxane-resistant breast cancer: results of a literature review to determine the clinical challenges and current treatment trends. Clin Ther. 2009;31:1619–1640. doi: 10.1016/j.clinthera.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Schmid P, Krocker J, Jehn C, Michniewicz K, Lehenbauer-Dehm S, Eggemann H, Heilmann V, Kummel S, Schulz CO, Dieing A, Wischnewsky MB, Hauptmann S, Elling D, Possinger K, Flath B. Primary chemotherapy with gemcitabine as prolonged infusion, non-pegylated liposomal doxorubicin and docetaxel in patients with early breast cancer: final results of a phase II trial. Ann Oncol. 2005;16:1624–1631. doi: 10.1093/annonc/mdi321. [DOI] [PubMed] [Google Scholar]
- 8.Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002;3:415–424. doi: 10.1016/s1470-2045(02)00788-x. [DOI] [PubMed] [Google Scholar]
- 9.Gati WP, Paterson AR, Belch AR, Chlumecky V, Larratt LM, Mant MJ, Turner AR. Es nucleoside transporter content of acute leukemia cells: role in cell sensitivity to cytarabine (araC) Leuk Lymphoma. 1998;32:45–54. doi: 10.3109/10428199809059245. [DOI] [PubMed] [Google Scholar]
- 10.Spratlin J, Sangha R, Glubrecht D, Dabbagh L, Young JD, Dumontet C, Cass C, Lai R, Mackey JR. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin Cancer Res. 2004;10:6956–6961. doi: 10.1158/1078-0432.CCR-04-0224. [DOI] [PubMed] [Google Scholar]
- 11.Kakihara T, Fukuda T, Tanaka A, Emura I, Kishi K, Asami K, Uchiyama M. Expression of deoxycytidine kinase (dCK) gene in leukemic cells in childhood: decreased expression of dCK gene in relapsed leukemia. Leuk Lymphoma. 1998;31:405–409. doi: 10.3109/10428199809059234. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Visser F, King KM, Baldwin SA, Young JD, Cass CE. The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev. 2007;26:85–110. doi: 10.1007/s10555-007-9044-4. [DOI] [PubMed] [Google Scholar]
- 13.Galmarini CM, Voorzanger N, Falette N, Jordheim L, Cros E, Puisieux A, Dumontet C. Influence of p53 and p21(WAF1) expression on sensitivity of cancer cells to cladribine. Biochem Pharmacol. 2003;65:121–129. doi: 10.1016/s0006-2952(02)01448-x. [DOI] [PubMed] [Google Scholar]
- 14.Vinogradov SV, Zeman AD, Batrakova EV, Kabanov AV. Polyplex Nanogel formulations for drug delivery of cytotoxic nucleoside analogs. J Control Release. 2005;107:143–157. doi: 10.1016/j.jconrel.2005.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JW, Park JH, Robinson JR. Bioadhesive-based dosage forms: the next generation. J Pharm Sci. 2000;89:850–866. doi: 10.1002/1520-6017(200007)89:7<850::AID-JPS2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 16.Kreuter J. Evaluation of nanoparticles as drug-delivery systems. II: Comparison of the body distribution of nanoparticles with the body distribution of microspheres (diameter greater than 1 micron), liposomes, and emulsions. Pharm Acta Helv. 1983;58:217–226. [PubMed] [Google Scholar]
- 17.Kreuter J. Liposomes and nanoparticles as vehicles for antibiotics. Infection. 1991;19(Suppl 4):S224–228. doi: 10.1007/BF01644038. [DOI] [PubMed] [Google Scholar]
- 18.Koukourakis GV, Zacharias G, Tsalafoutas J, Theodoridis D, Kouloulias V. Capecitabine for locally advanced and metastatic colorectal cancer: A review. World J Gastrointest Oncol. 2010;2:311–321. doi: 10.4251/wjgo.v2.i8.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veltkamp SA, Jansen RS, Callies S, Pluim D, Visseren-Grul CM, Rosing H, Kloeker-Rhoades S, Andre VA, Beijnen JH, Slapak CA, Schellens JH. Oral administration of gemcitabine in patients with refractory tumors: a clinical and pharmacologic study. Clin Cancer Res. 2008;14:3477–3486. doi: 10.1158/1078-0432.CCR-07-4521. [DOI] [PubMed] [Google Scholar]
- 20.Mu L, Feng SS. Vitamin E TPGS used as emulsifier in the solvent evaporation/extraction technique for fabrication of polymeric nanospheres for controlled release of paclitaxel (Taxol) J Control Release. 2002;80:129–144. doi: 10.1016/s0168-3659(02)00025-1. [DOI] [PubMed] [Google Scholar]
- 21.Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26:2713–2722. doi: 10.1016/j.biomaterials.2004.07.050. [DOI] [PubMed] [Google Scholar]
- 22.Yamaoka T, Tabata Y, Ikada Y. Comparison of body distribution of poly(vinyl alcohol) with other water-soluble polymers after intravenous administration. J Pharm Pharmacol. 1995;47:479–486. doi: 10.1111/j.2042-7158.1995.tb05835.x. [DOI] [PubMed] [Google Scholar]
- 23.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 24.Uznanski B, Grajkowski A, Wilk A. The isopropoxyacetic group for convenient base protection during solid-support synthesis of oligodeoxyribonucleotides and their triester analogs. Nucleic Acids Res. 1989;17:4863–4871. doi: 10.1093/nar/17.12.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosn B, van de Ven AL, Tam J, Gillenwater A, Sokolov KV, Richards-Kortum R, Roy K. Efficient mucosal delivery of optical contrast agents using imidazole-modified chitosan. J Biomed Opt. 2010;15:015003. doi: 10.1117/1.3309739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug Chem. 2010;21:797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 27.Jiang W, Kim BY, Rutka JT, Chan WC. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 28.Chithrani BD, Ghazani AA, Chan WC. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 29.Das D, Lin S. Double-coated poly (butylcynanoacrylate) nanoparticulate delivery systems for brain targeting of dalargin via oral administration. J Pharm Sci. 2005;94:1343–1353. doi: 10.1002/jps.20357. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Diaz M, Avalos M, Cameselle JC. Methanol esterification reactions catalyzed by snake venom and bovine intestinal 5′-nucleotide phosphodiesterases. Formation of nucleoside 5′-monophosphate methyl esters from guanosine 5′-triphosphate and other nucleoside 5′-polyphosphates. Eur J Biochem. 1991;196:451–457. doi: 10.1111/j.1432-1033.1991.tb15836.x. [DOI] [PubMed] [Google Scholar]
- 31.Artursson P. Epithelial transport of drugs in cell culture. I: A model for studying the passive diffusion of drugs over intestinal absorptive (Caco-2) cells. J Pharm Sci. 1990;79:476–482. doi: 10.1002/jps.2600790604. [DOI] [PubMed] [Google Scholar]
- 32.Chong S, Dando SA, Morrison RA. Evaluation of Biocoat intestinal epithelium differentiation environment (3-day cultured Caco-2 cells) as an absorption screening model with improved productivity. Pharm Res. 1997;14:1835–1837. doi: 10.1023/a:1012112820371. [DOI] [PubMed] [Google Scholar]
- 33.Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96:736–749. [PubMed] [Google Scholar]
- 34.Landowski CP, Song X, Lorenzi PL, Hilfinger JM, Amidon GL. Floxuridine amino acid ester prodrugs: enhancing Caco-2 permeability and resistance to glycosidic bond metabolism. Pharm Res. 2005;22:1510–1518. doi: 10.1007/s11095-005-6156-9. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi M, Tomita M. Mechanistical analysis for drug permeability through intestinal membrane. Drug Metab Pharmacokinet. 2007;22:67–77. doi: 10.2133/dmpk.22.67. [DOI] [PubMed] [Google Scholar]
- 36.Makhey VD, Guo A, Norris DA, Hu P, Yan J, Sinko PJ. Characterization of the regional intestinal kinetics of drug efflux in rat and human intestine and in Caco-2 cells. Pharm Res. 1998;15:1160–1167. doi: 10.1023/a:1011971303880. [DOI] [PubMed] [Google Scholar]
- 37.Galmarini CM, Warren G, Senanayake MT, Vinogradov SV. Efficient overcoming of drug resistance to anticancer nucleoside analogs by nanodelivery of active phosphorylated drugs. Int J Pharm. 2010;395:281–289. doi: 10.1016/j.ijpharm.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.