

Abstract
HNO (nitroxyl) has been found to have many physiological effects in numerous biological processes. Computational investigations have been employed to help understand the structural properties of HNO complexes and HNO reactivities in some interesting biologically relevant systems. The following computational aspects were reviewed in this work: 1) structural and energetic properties of HNO isomers; 2) interactions between HNO and non-metal molecules; 3) structural and spectroscopic properties of HNO metal complexes; 4) HNO reactions with biologically important non-metal systems; 5) involvement of HNO in reactions of metal complexes and metalloproteins. Results indicate that computational investigations are very helpful to elucidate interesting experimental phenomena and provide new insights into unique structural, spectroscopic, and mechanistic properties of HNO involvement in biology.
Keywords: HNO, Computational investigation, Structural properties, Spectroscopic properties, Mechanistic properties
1. Introduction
HNO (nitroxyl) is a very interesting molecule in chemistry and has many physiological effects in numerous biological processes, such as vascular relaxation, enzyme activity regulation, and neurological function regulation [1–6]. Compared to its sibling molecule, the well-known signaling agent nitric oxide (NO), their chemistries are quite different [7]. For instance, HNO and NO can lead to increases in the biochemical messengers cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), respectively [1]. With a more favorable vasodilative effect than NO and an increased contractility effect, HNO donors offer a promising new class of vasodilators and heart failure treatment [8]. HNO has also been suggested as a potential pharmacological treatment for reduction of neuronal damage, for instance, during stroke [1]. It can enhance oxidative stress and neutrophil infiltration [9], and has also been found to enhance blood–brain barrier disruption [10] and induce the release of the calcitonin gene-related product in peptidergic neurons [11]. One of the other biologically significant roles that HNO play is the inhibition of a number of enzymes, such as aldehyde dehydrogenase [12], zinc finger protein poly(ADP-ribose) polymerase [13], and the yeast copper thiolate transcription factor Ace1 [14]. These observations strongly suggest that HNO has significant roles in biology and medicine. It is interesting to note that quite a number of the HNO biological interactions occur with metalloproteins, such as heme proteins [1–3,15–18], Cu,Zn-SOD (superoxide dismutase) [19], and manganese quercetin dioxygenase [20]. However, the atomic level structural and functional details of HNO interactions with biological systems are largely unknown.
Computational investigations of HNO and its interactions with various chemical and biological systems may offer useful information of the structural, spectroscopic, and mechanistic properties of HNO involvement in biology, which may help evaluate possible structural and functional details in such interactions and thus facilitate the research in this field. Therefore, rather than to be exhaustive of all published computational work on HNO, this review tries to highlight some computational results that could assist our understanding of how HNO may interact with biological systems and models. For this purpose, this computational review first summarizes the structural and energetic properties of HNO isomers, then discusses the geometric features and interactions in HNO complexes formed between the stable HNO isomer and non-metal molecules as well as metal complexes, and finally discusses various reactions between HNO and biologically relevant non-metal systems as well as metalloproteins and metal complexes.
2. Structural and energetic properties of HNO isomers
As illustrated in Fig. 1, the potential energy surfaces of both singlet and triplet states of HNO have been reported [21,22]. In the singlet state, the recent ab initio calculations with more than 17,000 data points in the potential energy surface using the sophisticated multi-reference configuration interaction method at the state-averaged complete active space self-consistent field (CASSCF) level [21], show that there are two stable isomers connected via a transition state (TS). This supports previous quantum chemical studies using density functional theory (DFT) methods and ab initio method at the MP2 level [23,24]. Results indicate that the HNO form is the global minimum state in the potential energy surface of the ground state, with the HON isomer of ~40–42 kcal/mol higher in energy, from using various DFT methods, MP2, and other compound methods (G2 and CBS). The HNO form is even more stable than HON from the recent CASSCF calculations, based on the calculated energy difference of 49.79 kcal/mol [21]. The triplet HON is of lower energy compared to the singlet HON, but is still less stable than the ground state singlet HNO by ca. 26–31 kcal/mol [22,25], as shown in Fig. 1. Detailed comparisons of the spin state effect on HNO and its conjugate base NO− have already been well summarized before [1,3,6]. Overall, the large energy differences between different isomers suggest that the 1HNO form is the most relevant chemical structure under physiological conditions. Even in the excited state, the HNO form is still more stable than the HON isomer by 19.94 kcal/mol [21]. As shown in Fig. 1, the HNO form is bent, which was found to be much more stable than the linear HNO structure by ca. 67 kcal/mol from using various high level quantum chemical methods [26].
Fig. 1.

Stable isomers and the transition states. Energies are from Refs. [21,22].
Although different computational investigations consistently show that the bent HNO form is the global minimum and accurate predictions of geometric parameters can be obtained via a good variety of quantum chemical methods, specific computed spectroscopic properties such as NO vibrational frequency (νNO) do exhibit clear dependence on the used methods, as demonstrated in Table 1. These results suggest that some computed properties of HNO are sensitive to the method of choice [21,24,26–29] and therefore, careful methodological studies need to be performed to find out reasonably good methods for specific property calculations for HNO systems. Our recent methodological investigation of >70 method/basis combinations on the structural and vibrational spectroscopic properties of HNO shows that [29], while the optimized NH and NO bond length errors are within 0.02–0.03 Å, the predicted frequency errors can still be as large as ~170 cm−1, compared to experimental data [27,30]. As illustrated in Table 1, it was found that the pure DFT methods (e.g. BLYP, mPWVWN) perform better than the hybrid HF-DFT methods (e.g. B1LYP, B3LYP, mPW1PW91). The best method, mPWVWN, is capable of reproducing the experimental NO frequency within 1 cm−1, and has the accuracy for both structural and spectroscopic data similar to that from using the high level ab initio method CASSCF, see Table 1.
Table 1.
Selected geometric and vibrational frequency results of HNO.
| Method | RNH (Å) | RNO (Å) | ∠HNO (°) | νNO (cm−1) | Reference |
|---|---|---|---|---|---|
| Expt | 1.063 | 1.212 | 108.6 | 1565 | [27,30] |
| MP2/6–31G(d′) | 1.059 | 1.223 | 108.0 | [26] | |
| MP2/6–311G(2d,2p) | 1.047 | 1.227 | 107.3 | 1592 | [24] |
| B1LYP/6–311G(2d,2p) | 1.059 | 1.199 | 108.4 | 1673 | [24] |
| B3LYP/6–311G(2d,2p) | 1.063 | 1.201 | 108.4 | 1658 | [24] |
| mPW1PW91/6–311G(2d,2p) | 1.058 | 1.194 | 108.5 | 1712 | [24] |
| BLYP/6–311G(2d,2p) | 1.082 | 1.218 | 1559 | [28] | |
| mPWVWN/6–311++G(2d,2p) | 1.069 | 1.217 | 108.6 | 1566 | [29] |
| CASSCF/aug-cc-pVQZ | 1.051 | 1.210 | 108.3 | 1568 | [21] |
3. Interactions between HNO and non-metal molecules
HNO can form various kinds of complexes with non-metal molecules, such as CO [31], HNO [32,33], HOX (X = F, Cl, Br, I) [34], CH3X (X = F, Cl, Br) [35], HFSO2 [36], CH3CHO [37], HCONH2 [38], XCHY (X = H, F, Cl, Br, CH3; Y = O, S) [39], C2H2 [40], HArF [41], and O3 [42]. Based on the computational investigations, it is interesting to note that in these complexes, as long as the geometries allow, the most stable isomers prefer to involve both the terminal H and O atoms of HNO in the interactions, as illustrated in Fig. 2 for some typical complexes. This feature suggests that the dual bonding mode of HNO involving two terminal atoms has a stronger stabilizing force than the other modes, which leads to important implications for HNO in proteins (vide infra). Although the interaction partners are small molecules, most of the atoms involved in the HNO interactions in these complexes also exist in biological systems. Therefore, similar interaction modes could also occur when HNO binds with biological systems such as proteins. As seen from Fig. 2, the most common motif is the interaction pair containing a six-membered ring (see Fig. 2 A–E), followed by the interaction pairs involving a seven-membered ring or a five-membered ring (see Fig. 2 F–H), with the non-planar interaction most rarely seen (Fig. 2 I).
Fig. 2.
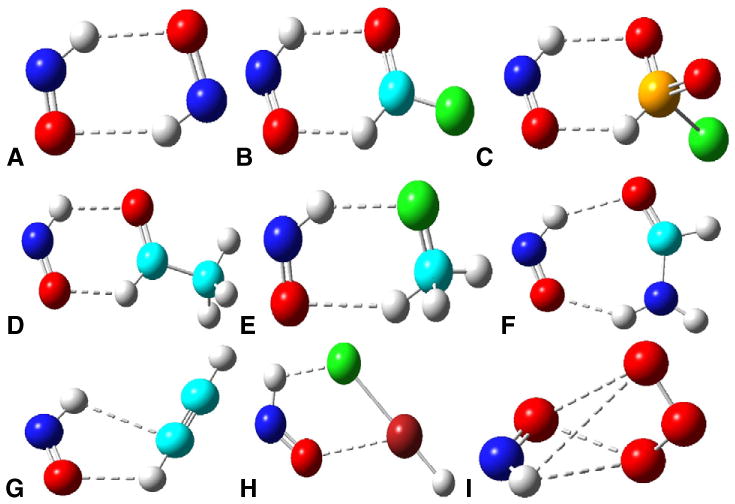
Most stable isomers of non-metal HNO complexes. A) HNO·HNO; B) HNO·FCHO; C) HNO·HFSO2; D) HNO·CH3CHO; E) HNO·CH3F; F) HNO·HCONH2; G) HNO·C2H2; H) HNO·HArF; I) HNO·O3. Atom color scheme: N—blue, O—red, C—cyan, F—green, S—orange, Ar—brown, H—gray. Interactions between HNO and the partner are highlight- ed as dashed lines.
To help understand the nature of the interaction between HNO and these non-metal molecules, the atoms-in-molecules (AIM) theory [43] was used to investigate some of these complexes. According to the AIM theory, every chemical bond has a bond critical point at which the first derivative of the charge density, ρ(r), is zero. The ρ (r) topology is described by a real, symmetric, second-rank Hessian-of-ρ (r) tensor, and the tensor trace is related to the bond interaction energy by a local expression of the virial theorem:
| (1) |
where Δ2ρ(r) is the Laplacian of ρ(r), G(r) and V(r) are the electronic kinetic and electronic potential energy densities, and m is the electron mass, respectively. Negative and positive Δ2ρ(r) values are associated with shared-electron (covalent) interactions and closed-shell (electrostatic) interactions, respectively. In the latter case, one can further evaluate the total energy density, H(r), at the bond critical point:
| (2) |
A negative H(r) is termed partial covalence, while a positive H(r) indicates a purely closed-shell, electrostatic interaction [43,44]. On the basis of the calculated Laplacians of the bond critical point properties (see Table 2), all of the interactions between HNO and its partners are of non-covalent or closed-shell/electrostatic nature. Some strong hydrogen bondings (e.g. in HNO dimer) are of partial covalence due to negative H(r) values, while some other interactions (e.g. in HNO·O3) are of purely electrostatic nature. These results indicate that, although the HNO interactions are basically non-covalent, the strengths vary with specific interaction partners.
Table 2.
Bond critical point properties.
| Complex | Interacting atoms | ρ(r) (au) | Δ2ρ (r) (au) | H(r) (au) | Reference |
|---|---|---|---|---|---|
| HNO·HNO | O…H | 0.0122 | 0.0451 | −0.0016 | [33] |
| HNO·CH3CHO | O…H | 0.0173 | 0.0516 | [37] | |
| O'…H' | 0.0058 | 0.0238 | |||
| HNO·FCHO | O…H | 0.014 | 0.05 | [39] | |
| O'…H' | 0.008 | 0.03 | |||
| HNO·HCONH2 | O…H | 0.021 | 0.069 | [38] | |
| O'…H' | 0.006 | 0.020 | |||
| HNO·C2H2 | O…H | 0.0093 | 0.0304 | [40] | |
| H…C | 0.0092 | 0.0262 | |||
| HNO·HArF | F…H | 0.0302 | 0.1146 | [41] | |
| Ar…O | 0.0079 | 0.0342 | |||
| HNO·O3 | O…H | 0.0081 | 0.0313 | 0.0007 | [42] |
| O…O | 0.0112 | 0.0416 | 0.0008 |
4. Structural and spectroscopic properties of HNO metal complexes
Interestingly, in contrast with the above non-metal HNO complexes which mostly involve the terminal H and O atoms in the interactions, in HNO metal complexes, the interactions basically occur between the central nitrogen atom and the metal center [3,28].
A detailed computational comparison of various possible interaction modes was reported recently on [Fe(Por)(Im)(HNO)] (Por = porphyrinate; Im = imidazole) [28]. As shown in Fig. 3, there are altogether six binding isomers. The three HNO binding modes are all of lower energy compared with the three HON binding modes. The same trend was also seen in the heme system with a thiolate axial ligand [45,46]. However, the energy differences of the HNO and HON heme complexes are smaller than that of HNO and HON. For instance, [Fe(Por)(Im)(HNO)] is more stable than [Fe(Por)(Im)(HON)] by ~22 kcal/mol [28], which is much smaller than the relative stability of HNO vs. HON discussed in Section 2, >40 kcal/mol. This indicates that upon the formation of the heme adduct, there is a stronger stabilization effect for HON than HNO. In fact, a recent DFT investigation showed that the π backbonding is enhanced in the case of HON compared to HNO, as indicated by a 0.078 Å shortened Fe–N distance and correspondingly strengthened Fe–NO vibrational mode [46]. Meanwhile, the enhanced π backbonding moves more charges to the NO moiety's out-of-plane π* orbital, leading to a weak interaction between N and O that makes the NO bond effectively a single bond in the HON heme complex with a much elongated bond length (RNO =1.401 Å) and accordingly much weaker NO vibrational frequency and force constant.
Fig. 3.

[Fe(Por)(Im)(HNO)] isomers. Energy values (in parentheses) are in kJ/mol from B3LYP/6–31G(d) calculations, bond lengths are in Angstroms, and bond angles are in degrees. Atom color scheme: N—blue, O—red, C—green, H—white.
From Ref. [28].
These results together with the early report of lower energy of HNO compared to that of HON, strongly suggest that in biological systems, HNO is the most relevant isomer. It is also interesting to note that, in both HNO and HON binding modes, the N-bonded isomers are always more stable than the corresponding O-bonded isomers, see Fig. 3. This is consistent with the experimental observation that in all of the known HNO metal complexes characterized crystallographically [47–49], the metal binding is through the central N atom in HNO.
Although HNO is known to bind stably with many different metals [47–58], except for the most recent pentacyanoferrate HNO complex [57], all other HNO organometallic compounds are with late transition metals, suggesting that the metal center may play an important role in HNO binding. Given the broad tremendous interest in heme systems, it is interesting to note that while a Ru porphyrin complex with HNO was reported many years ago [56], only very recently the iron porphyrin complex of the conjugate base of HNO (i.e. NO−) was reported with the assistance of a specially designed porphyrin [59], and there is still no report of a successful synthesis of iron porphyrin HNO complex. In contrast, most reported HNO metalloprotein systems are with Fe [1–3,15–18] and the protein environment was found to help stabilize the HNO binding in myoglobin (Mb) [60]. These results suggest that the early transition metal may bind with HNO more weakly than the late transition metal, and thus needs the aid of a special ligand or protein environment. To help evaluate metal center effects to offer general HNO binding trends and reveal the electronic origin of such trends, we recently reported the first systematic computational investigation of metals in different rows and periods in the periodic table on a common porphyrin platform M(Por)(5-MeIm)(HNO) [61], where M and 5-MeIm stand for metal and 5-methylimidazole (a Cβ truncated histidine residue, the heme axial ligand in Mb), respectively. It is interesting to note that all these HNO metal complexes fall into two clusters: one involves stable binding with all d6 metals, and the other contains the non-stable binding with d10 and s0 metals, as shown by the calculated binding Gibbs free energies in Fig. 4. These results provide the first computational evidence that stable HNO metal complexes involve d6 metals, but not d10 or s0 metals, which is consistent with experimental findings [47–58,62,63]. These results also indicate that in the same group, the late transition metal binds with HNO more strongly than the early transition metal, so it is relatively easier to synthesize HNO complexes with late transition metal centers, consistent with more of such complexes reported in the area of organometallic chemistry [47–58]. Moreover, this systematic computational investigation reveals a common principal HNO binding mechanism: metal back-donation to the π-acid HNO. Therefore, in the same period and with the same electronic configuration, the metal with a low oxidation state can provide more back-donation than that with a high oxidation state, and consequently it binds with HNO more strongly, in agreement with the calculated Gibbs free energy trends.
Fig. 4.
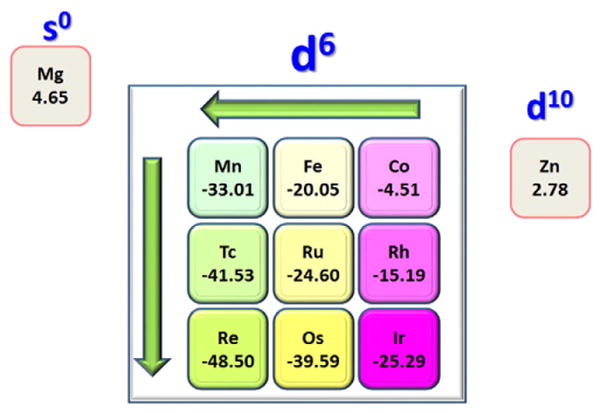
Binding Gibbs free energies (unit: kcal/mol).
From Ref. [61].
Spectroscopic investigations of HNO metalloprotein complexes and synthetic models can provide useful information to help characterize these systems. A recent investigation shows that the proton NMR shift is a sensitive probe of the active sites in several oxygen-binding hemoglobins [62]. This shift also varies with the metal center and ligand set in synthetic HNO metal complexes [47–56], suggesting a useful role in characterizing synthetic HNO metal systems, too. The 15N NMR chemical shifts of the HNO moieties in a number of HNO heme protein complexes also vary with the specific protein environment [62] and they are different from the nitrogen shifts observed in RNO iron porphyrin complexes [64]. In addition, experimental studies suggest that the NO vibrational frequency, νNO, is another useful probe of the metal environment in both the HNO heme protein (MbHNO) [63] and synthetic metal systems [47–56].
Recently quantum chemical calculations were reported to help understand some experimental spectroscopic results. A DFT investigation of MbHNO using the [Fe(Por)(Im)(HNO)] model [28] explained the unusually high Fe–NHNO frequency relative to those of the corresponding {FeNO}6 and {FeNO}7 porphyrinates [63], despite the fact that the Fe–NHNO bond is longer than either of its Fe–NNO counterparts. This was found to result from an extremely small reduced mass of 3.5 amu for νFe–N(HNO) due to the hydrogen atom bonded to NO in MbHNO, compared to those of 15.0–20.8 amu seen in ferric and ferrous NO iron porphyrins. Although it was not mentioned, the computed normal vibrational mode which involves significant H motion and small N movement with an angle to the Fe–N bond, and negligible O displacement, indicate that it is mostly an FeNH bending mode, rather than the typical Fe–NNO stretching mode seen in{FeNO}6 and {FeNO}7 porphyrinates [63]. Interestingly, the bonding of a hydrogen atom to NO was also recognized to be responsible for the peculiar behavior in the νFeN/νNO plot for Fe(II) NO adducts of heme proteins [65]. DFT computational results indicate that, when the nitrogen atom of NO is the hydrogen bond acceptor, an unexpected positive correlation between νFeN and NO was found, in contrast with the normal anticorrelations as observed with CO adducts of heme proteins. The HNO orientation with respect to Im was, however, found to have an insignificant effect on either geometries or vibrations [28]. Compared to NO, HNO was found to have a smaller trans effect on the Im ligand in a recent computational investigation [66].
Calculations on non-heme HNO metal complexes were also reported [58,67,68]. For [Fe(CN)5(HNO)]3−, a reduction product of pentacyanoferrate, DFT calculations predicted two frequencies of 1394 and 1338 cm−1 for νNO modes with some hydrogen participation [67]. These predictions have been recently verified by experimental IR studies, showing 1380 and 1304 cm−1 for the asymmetric and symmetric stretching modes for NO in this complex [57]. Quantum chemical calculations also helped assign its experimental UV/Vis spectrum. More recently, similar to this pentacyanoferrate platform that can bind with NO+, NO, and NO−/HNO, the complexes of pentachloroirodate with these three NO redox partners were also investigated both experimentally and computationally [58]. The computational investigation helped confirm the synthesis of [IrCl5(HNO)]2−based on the IR spectrum. The reduction of the NO moiety from NO+ to NO and then to NO−/HNO results in an elongated NO bond length and reduced NO vibrational frequency, indicating an increased population of electrons in the antibonding NO π* orbital. This trend is similar to that found for NO/HNO heme complexes [29]. In addition, DFT calculations of several experimental spectroscopic results including IR, Mössbauer, and EPR parameters for [Fe(NO)(cyclam-ac)]0 provided a compelling evidence to assign this complex as a {FeNO}8 species, instead of HNO or NOH complexes [68]. Theoretical investigations reveal that HNO acts as σ-donor and π-acceptor in HNO metal systems [51]. Metal coordination was found to strengthen the NH bond while weakening the NO bond, resulting in smaller νNO observed in HNO metal complexes (1335–1493 cm−1) [3] compared to that in HNO alone (1565 cm−1) [30]. Calculations of the hydrogen bond effect on νNO were also reported for a synthetic HNO metal complex Ru(HNO)(‘pybuS4’) (‘pybuS4’ = 2,6-bis(2-mercapto-3,5-di-tert-butylphenylthio)dimethyl-pyridine) [49].
Apart from the above computational investigations that assisted in understanding the experimental spectroscopic properties of HNO metal complexes and related characterization of HNO binding in some heme and non-heme metal systems, accurate computational calculations of some characteristic spectroscopic properties have also helped in the discovery of a unique HNO binding mode in the first stable HNO protein complex—MbHNO [60], as with previous integrated quantum mechanics and spectroscopic studies of protein structures [44,69–74]. For a number of NO/HNO/RNO (R = alkyl and aryl) containing systems, our DFT predictions of the 1H and 15N NMR shifts and NO vibrational frequencies have theory-versus-experiment correlation coefficients R2=0.990, 0.996, and 0.998 respectively, with errors being 1.8–3.1% of the experimental data ranges [29]. In addition, an excellent linear quantitative structure observable relationship was found between NO vibrational frequencies and bond lengths with R2=0.977, which may help deduce the NO bond lengths from experimental NO vibrational data. Interestingly, by comparing experimental 1H and 15N NMR results and NO vibrational frequency in MbHNO with those predicted from various MbHNO models, a dual hydrogen bond model (structure B in Fig. 5) was proposed for the first stable MbHNO isomer with the calculated values of δH (15.10 ppm), δN (664 ppm), and NO (1380 cm−1) in excellent agreement with experimental data of 14.93 ppm, 661 ppm, and 1385 cm−1 [62,63]. Another dual hydrogen bond model (structure A in Fig. 5) was suggested for the second MbHNO isomer with a slightly upfield experimental 1H NMR chemical shift (14.87 ppm) compared to the first MbHNO isomer [75], which again was well reproduced in the calculation: 15.03 ppm. Such kind of dual hydrogen bonding with both terminal H and O atoms in HNO is consistent with the most commonly seen non-covalent interaction mode of HNO with non-metal small molecules as discussed above (Section 3), as well as reported recently in a synthetic HNO metal complex [49]. It should be noted that this kind of dual hydrogen bonding capability of HNO is unique, compared to heme protein complexes with other small molecules such as NO and O2, which only involves one hydrogen bond with the distal histidine residue. As a matter of fact, the dual hydrogen bond mode was found to be critical to account for the experimentally observed stronger binding affinity of Mb to HNO than the native substrate dioxygen [60]. As this opens up the way that the atomic level structure of HNO binding in proteins can be determined with a combined investigation of quantum chemical results and experimental spectroscopic data, it is anticipated that more work will be done in this field to help reveal more HNO bound protein structures and facilitate studies of the related functional roles of HNO in biology.
Fig. 5.
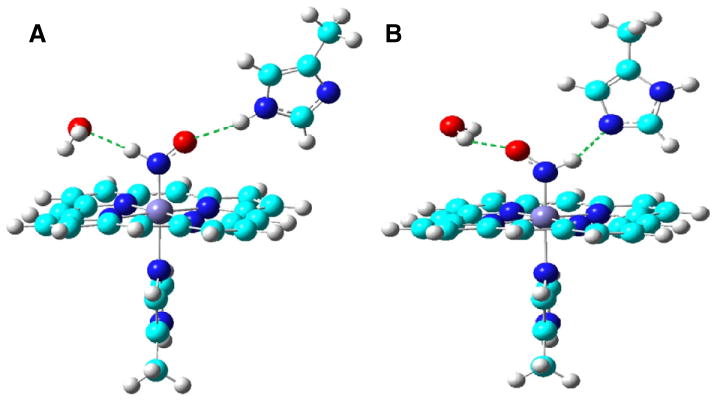
Active site structures of two MbHNO isomers.
From Ref. [60].
5. HNO reactions with biologically important non-metal systems
A more straightforward investigation of HNO's biological roles may be the examination of the HNO reactions with some biologically relevant molecular systems. HNO is known to be involved in numerous chemical reactions. But mechanistic details are usually complicated and quantum chemical calculations may provide useful information to help us understand experimental reactivities.
One area of interest is how HNO is produced from HNO donors, which may help design new HNO donors as HNO has broad biomedical applications [1–6]. Angeli's salt (AS, Na2N2O3) is widely used as an HNO donor to investigate the biological effects of HNO [1]. However, Angeli's salt can produce not only HNO, but also NO, depending on the pH environment of the solution. Experimental studies [76] show that when the pH values are in the range of 8–4, HNO is produced and quickly dimerizes to yield N2O, while in lower pH solutions (< 4), NO is generated. There are actually a number of mechanisms proposed for HNO production, while little is known about the NO production, until recently DFT methods and high accuracy CBS-QB3 method were applied to investigate such HNO and NO generation reactions [25]. As reactions are pH-dependent, all possible monoprotonated and diprotonated species were computationally investigated. As illustrated in Fig. 6, in an aqueous solution of AS titrated with acid, AS is initially protonated to yield an unreactive intermediate 1, which is in equilibrium with the reactive intermediate 7, that decomposes to generate HNO and nitrite at a rate of 5.2 s−1. After the pH is further decreased, 1 is further protonated to give 9, which is in equilibrium with species 20/15, that quickly loses water and forms two NO with a rate constant of 1.7×105s−1. This mechanism was found to well fit the experimental pH dependent rate constant curve with a correlation coefficient R2=0.93 [25]. A similar computational study was reported on a simplified compound of isopropylamine diazeniumdiolate, another widely used HNO donor, which again has a similar pH dependence production of HNO and NO [77]. While these computational investigations help in understanding the production pathways of HNO donors, it should be noted that these studies were done for the isolated HNO donors. The decomposition of AS is also very sensitive to the presence of other molecules in a solution, such as transition metal complexes [78,79]. Hence, analysis of HNO decomposition as isolated might not reflect the pathway in vivo and the kinetics could also change. More work is needed to investigate the details of HNO yielding reactions in the presence of other molecules, such as transition metal complexes [78,79].
Fig. 6.

HNO and NO production from Angeli's salt.
From Ref. [25].
HNO reactivities with some biologically relevant non-metal systems have also been computationally investigated. As shown in Table 3, HNO reaction energies in both gas phase and aqueous solution were reported for a good variety of nucleophiles [80]. These results suggest that HNO is relatively inert towards oxygen-based nucleophiles (reactions 1 and 2). In contrast, the HNO reaction with amine is favorable in both gas phase and solution (reaction 3), with the addition of thiol being particularly facile (reaction 4). As a matter of fact, HNO was found to react with a number of thiols, including thiophenol, glutathione, dithiothreitol, and cysteines in proteins [1].
Table 3.
HNO reaction energies (unit: kcal/mol) [80].
| No. | Reaction | ΔE (gas) | ΔE (H2O) |
|---|---|---|---|
| 1 | HNO+H2O→NH(OH)2 | 7.6 | 8.0 |
| 2 | HNO+CH3OH→NH(OH)(OCH3) | 3.0 | 2.6 |
| 3 | HNO+CH3NH2→NH(OH)(NHCH3) | −9.2 | −10.0 |
| 4 | HNO+CH3SH→NH(OH)(SCH3) | −20.8 | −17.5 |
| 5 | HNO+HNO→NH(OH)(NO) | −37.4 | −40.0 |
| 6 | HNO+3NO− →NH(NO)O− | −50.3 | −39.6 |
Due to the biological significance of HNO reactions with thiols, a recent computational investigation was performed on five different thiols, RSH with R=H, CH3, CF3, Ph, and cysteine to specifically address issues of hydrophobicity of the environment, the availability of a local base, and the identity of the thiol substituent on the HNO reactivities [81]. The first step is the same for different R groups, from RSH to RSNHOH (similar to reaction 4), which then undergoes two different reaction pathways, A and B, to form sulfonamide (RSONH2) and disulfide (RSSR), respectively, as shown in Fig. 7. An interesting finding is that the first putative intermediate N-hydroxysulfenamide (RSNHOH) should be protonated under physiological conditions for R=H, CH3, Ph, and Cys. Both pathways were found to be irreversible in aqueous solution, and thus the competition is controlled kinetically. As shown in Table 4, electron-withdrawing groups were predicted to prefer the production of sulfonamide via pathway A, while electron-donating groups shall help the formation of disul-fide through pathway B. Based on this table, PhSH and Cys are most likely to yield disulfide products, consistent with experimental observations [1,81].
Fig. 7.
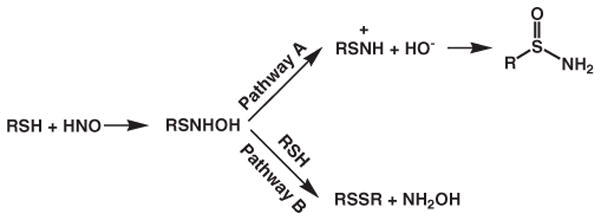
Reaction pathways for HNO reactions with thiols.
Table 4.
Gibbs free energy barriers in HNO reactions with RSH [81].
| R | 3G≠(kcal/mol) | 3G≠(kcal/mol) |
|---|---|---|
| pathway A | pathway B | |
| H | 22.2 | 26.0 |
| CH3 | 19.8 | 13.6 |
| CF3 | 11.8 | 34.7 |
| Ph | 24.9 | 15.1 |
| Cys | 23.0 | ~10 |
Another important reaction relevant to HNO biological effects is its dimerization [1], which may compete with HNO reactions with other biological targets [62]. The recent redetermination [82] of the rate constant of 8×106M−1 s−1 is considerably smaller than the previous report of 2×109M−1 s−1, which indicates that HNO could react with some other molecules prior to the dimerization of itself. The calculated dimerization reaction energy in Table 3 suggests that the dimerization is strongly thermodynamically favored. But kinetic details to the final product of N2O and H2O are quite complicated. Starting from a trans iso- mer of hyponitrous acid (HONNOH) that is more stable than the cis isomer, four reaction pathways were found to yield the same products in the gas phase, depending on the order of two terminal hydrogen transfers from their respective nitrogen atom to the same oxygen atom as well as the conformational alternatives for the transition states [83]. In addition to quantum chemical calculations, ab initio dynamics calculations of the final step of each pathway were also performed to verify the final production formation. However, very recently, the HNO dimerization picture was modified with the information of solution reaction details from DFT investigations [84]. As shown in Fig. 8, N2O formation was suggested to come predominantly from the cis pathway under physiological conditions, with the trans pathway being a minor one. The authors called for new isotope labeled experiments to further test the mechanism. This is a good example to illustrate that HNO reaction details can be different under different conditions.
Fig. 8.

HNO dimerization mechanism. Numbers in parentheses represent pKa values or pK values in case of isomerization reactions. The species marked with frames are unstable with respect to decomposition yielding nitrous oxide.
Modified from Ref. [84].
A recent computational investigation of HNO with O2 in aqueous solution was focused on the characterization of four possible intermediates via both hybrid quantum mechanics and molecular mechanics (QM/MM) calculations and molecular dynamics simulations [85]. The vibrational IR and Raman spectra were predicted for all the computed intermediate species with suggestions for future experiments to discriminate them. However, full reaction details remain to be elucidated.
Overall, it can be seen that mechanistic details in HNO reactions with biologically relevant non-metal systems can be very complicated and computational investigations are helpful to evaluate some possibilities. More computational investigations are needed to address not only more individual HNO reactions, but also some combinations to help understand situations closer to in vivo, as HNO can react with a number of biologically relevant systems [1].
6. HNO reactions with metal complexes and metalloproteins
Theoretical mechanistic investigations have also provided interesting insights to help understand how the HNO metal complexes can be synthesized [4,54,55,67] and what kind of possible roles the HNO metal complexes play in a number of inorganic [4] and bioinorganic [3,45,46,86,87] reactions.
Based on a DFT study [67], the stable two-electron reduction product of [Fe(CN)5(NO)]2− was proposed to be [Fe(CN)5(HNO)]3−, since the protonation sites of the oxygen of NO or axial or equatorial CN all lead to complexes with energies of ca. 21–26 kcal/mol higher. This is in contrast with its one-electron reduction products, among which the protonation of cyanides is more favorable than that of NO. Interestingly, this was confirmed recently in an experimental work [57] and the [Fe(CN)5(HNO)]3− complex has been well characterized by NMR and IR techniques, as well as other spectroscopies.
On another formation mechanism of HNO metal complex by the hydrogen addition to NO, DFT calculations were used to evaluate two different hypothesized pathways from the parent complex RuHCl(CO)(PiPr3)2 with NO [54,55]. In one reaction pathway, two of the parent complexes react with each other to form the 1:1 ratio products: RuCl(HNO)(CO)(PiPr3)2 and RuCl(NO)(CO)(PiPr3)2 (iPr=isopropyl), in which one serves as a H-atom donor while the other is a H-atom acceptor. Although the predicted negative ΔG of −18.8 kcal/mol using PMe3 as the model for PiPr3 may suggest that it could proceed in this way, the transition state of having two such big metal complexes together would entail a large steric penalty. Alternatively, another pathway was proposed, with the parent complex first reacting with NO to form RuHCl(NO)(CO)(PiPr3)2, followed by another NO attack to form the products RuCl(NO)(CO)(PiPr3)2 and HNO (ΔG= −9.9 kcal/mol). After-wards, the HNO can bind favorably with the five-coordinate parent complex to form the six-coordinate product RuCl(HNO)(CO)(PiPr3)2 (ΔG=−21.2 kcal/mol). This avoids the high steric interaction of the first hypothesis, and was therefore favored. Such kind of hydrogen addition to form HNO metal complexes represents a new synthetic approach and has been demonstrated for other systems, such as OsHCl(CO)(PiPr3)2, RuHCl(PPh3)3, RhH(porphyrin), IrHCl2(PR3)2, and PtHClL2 [54,55].
Investigations of the HNO involvement in bioinorganic systems can be traced back to early studies of biological denitrification processes in plants, bacteria, and fungi by nitrite and nitric oxide reductases [2,3]. Computational investigations have been reported on cytochrome P450 nitric oxide reductase (P450nor) [45,46,86,88–90]. The {Fe(HNO)}8 species was found to be a stable intermediate [45] in the catalytic cycle, formed from a direct hydride transfer from NADH to the bound NO moiety [46,86], and it is more stable than the O-protonated {Fe(HON)}8 complex by 26.2 kcal/mol [46] (see Fig. 9). The preferable formation of HNO instead of HON was also confirmed in a more recent QM/MM investigation [86]. This result together with previous calculations of MbHNO models [28] and experimental X-ray structures of HNO metal complexes [47–49] consistently shows that the metal prefers the binding of HNO isomer via the nitrogen atom. As shown in Fig. 9, this species can then undergo another protonation to form an intermediate, which was believed to be the one observed in the experiment [91] due to basicity of the intermediate {Fe(HNO)}8 [46,66]. This idea may be further supported by the good accord between the predicted Fe–N frequency (609 cm−1) in the doubly protonated {Fe(HNOH)}8 species and the experimental observation (596 cm−1). However, the predicted Fe–N frequency of the {Fe(HNO)}8 intermediate, 601 cm−1, is also close to the experimental value. Therefore, future experimental work of NO vibrations is needed to provide a more decisive conclusion as the predicted NO vibrational frequencies are quite different: 1386 cm−1 for {Fe(HNO)}8 and 952 cm−1 for {Fe(HNOH)}8.
Fig. 9.
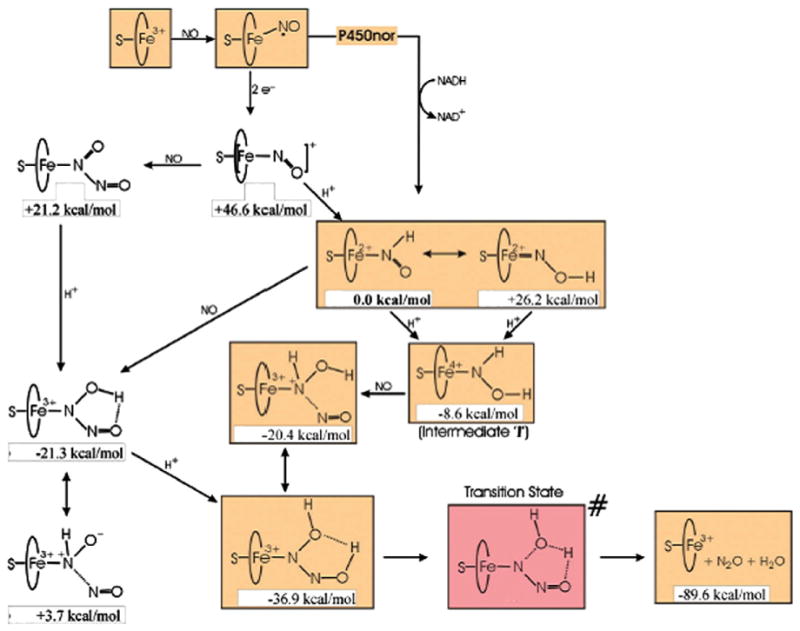
The recently proposed P450nor mechanism.
Modified from Ref. [46].
As regards the possible involvement of HNO in the catalytic cycle of cytochrome c nitrite reductase (ccNiR), a recent computational investigation provides some insights [87]. Because the overall six electron reduction of nitrite to ammonia in this enzyme is complicated, this work is focused on the investigation of geometric and electronic structures of possible intermediates in the catalytic cycle. In contrast with the direct hydride transfer to form the stable {Fe(HNO)}8 intermediate in P450nor, in the case of ccNiR, this HNO bound intermediate was proposed to be produced from two electron reduction steps and a protonation step from the NO bound form [87]. One thing common in these two enzymatic reactions is that the {Fe(NOH)}8 species is again of higher energy than the {Fe(HNO)}8 intermediate (by ~19 kcal/mol), and thus is ruled out. Comparing the proposed mechanisms for P450nor and ccNiR [46,87], it can be seen that not only the formation processes of the {Fe(HNO)}8 intermediate are different, but also the subsequent steps after the {Fe(HNO)}8 intermediate, due to distinctive products in these two enzymes. In ccNiR, the {Fe(HNO)}8 intermediate was proposed to follow two-electron reduction and two protonation steps to yield the bound hydroxylamine species (NH2OH). After another two-electron reduction and two protonation steps, NH2OH is converted to the final products NH3 and H2O.
These mechanistic results suggest that the HNO intermediate could be an important species in NOx metabolism. However, as described above, not all the species or reaction details of the proposed pathways have been investigated, so more computational work is still needed to provide the whole reaction picture to compare with experimental results in order to finally establish the mechanisms.
Apart from the above theoretical bioinorganic studies in which HNO is involved as an intermediate, computational investigations of the coordination of a typical HNO donor and metalloporphyrins were also reported [78,79]. This is of interest in the field of HNO trapping and characterization. Since both HNO and NO are of vital importance in biomedical sciences, chemical agents that can distinguish them and thus help differentiate their biological effect would be very helpful. Recently, several metalloporphyrins have been found to have distinctive reactions with HNO and NO [78,79]. For instance, two manganese(III) porphyrinates can react with HNO in a fast, effi-cient, and selective manner, producing reduced {MnNO}7 complexes, while their reactions with NO are inert or very slow [78]. This kind of reductive nitrosylation was also observed before in heme proteins and ferric porphyrins [1]. More detailed analyses suggest two possible reaction pathways [78,79]: one is that the HNO donor directly delivers HNO to the oxidized metal center and initiate the reductive nitrosylation, and the other route involves catalytic decomposition of the HNO donor via the coordination to metalloporphyrin. The second mechanism is different from the traditional one (the first one) and offers increased reaction rates. The putative intermediate of the coordination of a typical HNO donor, Angeli's salt, to the metal center, was investigated computationally and found to be a stable species in the potential energy surface [78,79]. Interestingly, the energy costs of the bond dissociation leading to the products in the metal-coordinated AS are significantly lower than that in free AS, supporting a catalytic decomposition due to metal coordination [78,79]. However, to fully characterize these mechanisms, more calculations are to be done, which may help future design of more efficient HNO trapping agents.
7. Conclusions
HNO is a very interesting molecule in chemistry and plays significant roles in many biological processes. However, atomic level structural and functional details of HNO interactions with biological systems are largely unknown. Computational investigations have been employed to help understand the structural, spectroscopic, and mechanistic properties of HNO and its complexes in some interesting biologically relevant systems. Prior computational investigations of HNO potential energy surfaces show that the HNO isomer is the global minimum, in both ground and excited states, and the big energy difference between HNO and HON suggests that HNO is the most relevant chemical structure under physiological conditions. Computational studies of HNO complexes with various kinds of non-metal molecules indicate that the interaction is of non-covalent or electrostatic nature and the most preferable interaction mode involves the participation of both terminal H and O atoms of HNO. In contrast, the preferable interaction mode between HNO and a metal center was found to be through the coordination of central N atom in HNO, which is consistent with existing X-ray structures of HNO metal complexes. The HON metal complexes were found to be always of much higher energies than their corresponding HNO metal complexes, which rules out the possible involvement of HON in stable metalloproteins and metal complexes. A recent computational investigation revealed some general HNO binding trends with respect to metal's electronic configuration, position in the periodic table, and oxidation state, and the electronic origin. Computational studies have also assisted in the understanding of experimental spectroscopic data in both heme and non-heme HNO complexes, and the discovery of a unique HNO binding mode in the first stable HNO protein complex—MbHNO. HNO is known to be involved in various kinds of chemical reactions. But mechanistic details are usually complicated and quantum chemical calculations have provided useful information to help us understand experimental reactivities, such as experimental pH dependent production of HNO and NO from some HNO donors, HNO reactivities with various nucleophiles, particularly biologically relevant thiols, and HNO dimerization. Theoretical investigations have also provided interesting insights to help us understand how the HNO metal complexes can be synthesized and what kind of roles the HNO metal complexes play in a number of bioinorganic reactions. These results build an important basis for future work. It is anticipated that more computational investigations will come out to help investigate the HNO binding in biological systems and the mechanistic details of HNO involvement and function in regulating biological targets including both minimum energy states and transition states to address thermodynamic and kinetic issues. More interplay between experimental and computational studies will greatly facilitate our understanding of HNO biological effects and aid future pharmaceutical work that takes advantage of unique HNO biological functions.
Acknowledgments
This work was supported by the NIH grant GM-085774. Y.Z. thanks Liu Yang for her assistance during the preparation of this manuscript.
Abbreviations
- AIM
atoms-in-molecules
- AS
Angeli's salt
- CASSCF
complete active space self-consistent field
- ccNiR
cytochrome c nitrite reductase
- DFT
density functional theory
- QM/MM
quantum mechanics and molecular mechanics
- Im
imidazole
- M
metal
- Mb
myoglobin
- 5-MeIm
5-methylimidazole
- P450nor
cytochrome P450 nitric oxide reductase
- Por
porphyrinate
- TS
transition state
- SOD
superoxide dismutase
References
- 1.Miranda KM. Coord Chem Rev. 2005;249:433–455. [Google Scholar]
- 2.Averill BA. Chem Rev. 1996;96:2951–2964. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- 3.Farmer PJ, Sulc F. J Inorg Biochem. 2005;99:166–184. doi: 10.1016/j.jinorgbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Roncaroli F, Videla M, Slep LD, Olabe JA. Coord Chem Rev. 2007;251:1903–1930. [Google Scholar]
- 5.Flores-Santana W, Switzer C, Ridnour LA, Basudhar D, Mancardi D, Donzelli S, Thomas DD, Miranda KM, Fukuto JM, Wink DA. Arch Pharmacal Res. 2009;32:1139–1153. doi: 10.1007/s12272-009-1805-x. [DOI] [PubMed] [Google Scholar]
- 6.Doctorovich F, Bikiel D, Pellerino J, Suarez SA, Larsen A, Marti MA. Coord Chem Rev. 2011;255:2764–2784. [Google Scholar]
- 7.Wilson EK. Chem Eng News. 2004;82:39–44. [Google Scholar]
- 8.Feelisch M. Proc Natl Acad Sci U S A. 2003;100:4978–4980. doi: 10.1073/pnas.1031571100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma XL, Cao F, Liu GL, Lopez BL, Christopher TA, Fukuto JM, Wink DA, Feelisch M. Proc Natl Acad Sci U S A. 1999;96:14617–14622. doi: 10.1073/pnas.96.25.14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boje KMK, Lakhman SS. J Pharmacol Exp Ther. 2000;293:545–550. [PubMed] [Google Scholar]
- 11.Booth BP, Tabrizi-Fard MA, Fung HL. Biochem Pharmacol. 2000;59:1603–1609. doi: 10.1016/s0006-2952(00)00290-2. [DOI] [PubMed] [Google Scholar]
- 12.Nagasawa HT, Demaster EG, Redfern B, Shirota FN, Goon JW. J Med Chem. 1990;33:3120–3122. doi: 10.1021/jm00174a001. [DOI] [PubMed] [Google Scholar]
- 13.Sidorkina O, Espey MG, Miranda KM, Wink DA, Laval J. Free Radical Biol Med. 2003;35:1431–1438. doi: 10.1016/j.freeradbiomed.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Shinyashiki M, Chiang KT, Switzer CH, Gralla EB, Valentine JS, Thiele DJ, Fukuto JM. Proc Natl Acad Sci U S A. 2000;97:2491–2496. doi: 10.1073/pnas.050586597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rusche KM, Spiering MM, Marletta MA. Biochemistry. 1998;37:15503–15512. doi: 10.1021/bi9813936. [DOI] [PubMed] [Google Scholar]
- 16.Huang JM, Sommers EM, Kim-Shapiro DB, King SB. J Am Chem Soc. 2002;124:3473–3480. doi: 10.1021/ja012271v. [DOI] [PubMed] [Google Scholar]
- 17.Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Feelisch M, Fukuto JM, Wink DA. Proc Natl Acad Sci U S A. 2003;100:9196–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller TW, Cherney MM, Lee AJ, Francoleon NE, Farmer PJ, King SB, Hobbs AJ, Miranda KM, Burstyn JN, Fukuto JM. J Biol Chem. 2009;284:21788–21796. doi: 10.1074/jbc.M109.014282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt H, Hofmann H, Schindler U, Shutenko ZS, Cunningham DD, Feelisch M. Proc Natl Acad Sci U S A. 1996;93:14492–14497. doi: 10.1073/pnas.93.25.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar MR, Zapata A, Ramirez AJ, Bowen SK, Francisco WA, Farmer PJ. Proc Natl Acad Sci U S A. 2011;108:18926–18931. doi: 10.1073/pnas.1111488108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li A, Han H, Xie D. J Chem Phys. 2011;135:104304–104310. doi: 10.1063/1.3632994. [DOI] [PubMed] [Google Scholar]
- 22.Li A, Xie C, Xie D, Guo H. J Chem Phys. 2011;134:194309–194309. doi: 10.1063/1.3592375. [DOI] [PubMed] [Google Scholar]
- 23.Krokidis X, Silvi B, Alikhani ME. Chem Phys Lett. 1998;292:35–45. [Google Scholar]
- 24.Jalbout AF, Darwish AM, Alkahby HY. J Mol Struct (THEOCHEM) 2002;585:199–203. [Google Scholar]
- 25.Dutton AS, Fukuto JM, Houk KN. J Am Chem Soc. 2004;126:3795–3800. doi: 10.1021/ja0391614. [DOI] [PubMed] [Google Scholar]
- 26.Jursic BS. J Mol Struct (THEOCHEM) 1999;492:35–43. [Google Scholar]
- 27.Ellis HB, Ellison GB. J Chem Phys. 1983;78:6541–6558. [Google Scholar]
- 28.Linder DP, Rodgers KR. Inorg Chem. 2005;44:8259–8264. doi: 10.1021/ic0504745. [DOI] [PubMed] [Google Scholar]
- 29.Ling Y, Mills C, Weber R, Yang L, Zhang Y. J Am Chem Soc. 2010;132:1583–1591. doi: 10.1021/ja907342s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clough PN, Thrush BA, Ramsay DA, Stamper JG. Chem Phys Lett. 1973;23:155–156. [Google Scholar]
- 31.Li AY. J Phys Chem A. 2006;110:10805–10816. doi: 10.1021/jp062291p. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Liu W, Li H, Liu J, Yang Y. J Phys Chem A. 2006;110:11760–11764. doi: 10.1021/jp060908x. [DOI] [PubMed] [Google Scholar]
- 33.Solimannejad M, Massahi S, Alkorta I. Chem Phys. 2009;362:1–7. [Google Scholar]
- 34.Solimannejad M, Scheiner S. J Phys Chem A. 2008;112:4120–4124. doi: 10.1021/jp711141x. [DOI] [PubMed] [Google Scholar]
- 35.Solimannejad M, Scheiner S. J Phys Chem A. 2007;111:4431–4435. doi: 10.1021/jp071695u. [DOI] [PubMed] [Google Scholar]
- 36.Liu Y, Liu WQ, Li HY, Yang Y, Cheng S. Int J Quantum Chem. 2007;107:396–402. [Google Scholar]
- 37.Yang Y, Zhang WJ, Gao XM. Int J Quantum Chem. 2006;106:1199–1207. [Google Scholar]
- 38.Liu Y, Liu W, Li H, Yang Y, Cheng S. J Mol Struct (THEOCHEM) 2006;778:49–53. [Google Scholar]
- 39.Trung NT, Hue TT, Nguyen MT. J Phys Chem A. 2009;113:3245–3253. doi: 10.1021/jp810826z. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Liu WQ, Yang Y, Liu JG. Int J Quantum Chem. 2006;106:2122–2128. [Google Scholar]
- 41.Liu Y, Liu W-q, Li H-y, Yang Y, Cheng S. Chin J Chem Phys. 2007;20:37–42. [Google Scholar]
- 42.Solimannejad M, Alkorta I, Elguero J. Chem Phys Lett. 2009;474:253–257. [Google Scholar]
- 43.Bader RFW. J Phys Chem A. 1998;102:7314–7323. [Google Scholar]
- 44.Zhang Y, Oldfield E. J Am Chem Soc. 2008;130:3814–3823. doi: 10.1021/ja075978b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silaghi-Dumitrescu R. Eur J Inorg Chem. 2003:1048–1052. [Google Scholar]
- 46.Lehnert N, Praneeth VKK, Paulat F. J Comput Chem. 2006;27:1338–1351. doi: 10.1002/jcc.20400. [DOI] [PubMed] [Google Scholar]
- 47.Wilson RD, Ibers JA. Inorg Chem. 1979;18:336–343. [Google Scholar]
- 48.Melenkivitz R, Hillhouse GL. Chem Commun. 2002:660–661. doi: 10.1039/b111645b. [DOI] [PubMed] [Google Scholar]
- 49.Sellmann D, Gottschalk-Gaudig T, Haussinger D, Heinemann FW, Hess BA. Chem Eur J. 2001;7:2099–2103. doi: 10.1002/1521-3765(20010518)7:10<2099::aid-chem2099>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 50.Grundy KR, Reed CA, Roper WR. J Chem Soc D Chem Commun. 1970:1501–1502. [Google Scholar]
- 51.Southern JS, Green MT, Hillhouse GL, Guzei IA, Rheingold AL. Inorg Chem. 2001;40:6039–6046. doi: 10.1021/ic010669m. [DOI] [PubMed] [Google Scholar]
- 52.Southern JS, Hillhouse GL, Rheingold AL. J Am Chem Soc. 1997;119:12406–12407. [Google Scholar]
- 53.Melenkivitz R, Southern JS, Hillhouse GL, Concolino TE, Liable-Sands LM, Rheingold AL. J Am Chem Soc. 2002;124:12068–12069. doi: 10.1021/ja0277475. [DOI] [PubMed] [Google Scholar]
- 54.Marchenko AV, Vedernikov AN, Dye DF, Pink M, Zaleski JM, Caulton KG. Inorg Chem. 2002;41:4087–4089. doi: 10.1021/ic025699j. [DOI] [PubMed] [Google Scholar]
- 55.Marchenko AV, Vedernikov AN, Dye DF, Pink M, Zaleski JM, Caulton KG. Inorg Chem. 2004;43:351–360. doi: 10.1021/ic0349407. [DOI] [PubMed] [Google Scholar]
- 56.Lee JY, Richter-Addo GB. J Inorg Biochem. 2004;98:1247–1250. doi: 10.1016/j.jinorgbio.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 57.Montenegro AC, Amorebieta VT, Slep LD, Martin DF, Roncaroli F, Murgida DH, Bari SE, Olabe JA. Angew Chem Int Ed. 2009;48:4213–4216. doi: 10.1002/anie.200806229. [DOI] [PubMed] [Google Scholar]
- 58.Escola N, Bikiel DE, Baggio R, Di Salvo F, Doctorovich F. Inorg Chim Acta. 2011;374:528–539. [Google Scholar]
- 59.Pellegrino J, Bari SE, Bikiel DE, Doctorovich F. J Am Chem Soc. 2010;132:989–995. doi: 10.1021/ja905062w. [DOI] [PubMed] [Google Scholar]
- 60.Yang L, Ling Y, Zhang Y. J Am Chem Soc. 2011;133:13814–13817. doi: 10.1021/ja204072j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang L, Fang WH, Zhang Y. Chem Commun. 2012;48:3842–3844. doi: 10.1039/c2cc31016c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumar MR, Pervitsky D, Chen L, Poulos T, Kundu S, Hargrove MS, Rivera EJ, Diaz A, Colon JL, Farmer PJ. Biochemistry. 2009;48:5018–5025. doi: 10.1021/bi900122r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Immoos CE, Sulc F, Farmer PJ, Czarnecki K, Bocian DF, Levina A, Aitken JB, Armstrong RS, Lay PA. J Am Chem Soc. 2005;127:814–815. doi: 10.1021/ja0433727. [DOI] [PubMed] [Google Scholar]
- 64.Godbout N, Sanders LK, Salzmann R, Havlin RH, Wojdelski M, Oldfield E. J Am Chem Soc. 1999;121:3829–3844. [Google Scholar]
- 65.Xu CL, Spiro TG. J Biol Inorg Chem. 2008;13:613–621. doi: 10.1007/s00775-008-0349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goodrich LE, Paulat F, Praneeth VKK, Lehnert N. Inorg Chem. 2010;49:6293–6316. doi: 10.1021/ic902304a. [DOI] [PubMed] [Google Scholar]
- 67.Lebrero MCG, Scherlis DA, Estiu GL, Olabe JA, Estrin DA. Inorg Chem. 2001;40:4127–4133. doi: 10.1021/ic010140p. [DOI] [PubMed] [Google Scholar]
- 68.Serres RG, Grapperhaus CA, Bothe E, Bill E, Weyhermuller T, Neese F, Wieghardt K. J Am Chem Soc. 2004;126:5138–5153. doi: 10.1021/ja030645+. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Y, Gossman W, Oldfield E. J Am Chem Soc. 2003;125:16387–16396. doi: 10.1021/ja030340v. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, Oldfield E. J Am Chem Soc. 2004;126:4470–4471. doi: 10.1021/ja030664j. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Oldfield E. J Am Chem Soc. 2004;126:9494–9495. doi: 10.1021/ja0401242. [DOI] [PubMed] [Google Scholar]
- 72.Mao JH, Mukherjee S, Zhang Y, Cao R, Sanders JM, Song YC, Zhang YH, Meints GA, Gao YG, Mukkamala D, Hudock MP, Oldfield E. J Am Chem Soc. 2006;128:14485–14497. doi: 10.1021/ja061737c. [DOI] [PubMed] [Google Scholar]
- 73.Ling Y, Davidson VL, Zhang Y. J Phys Chem Lett. 2010;1:2936–2939. doi: 10.1021/jz101159x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fu R, Gupta R, Geng J, Dornevil K, Wang S, Zhang Y, Hendrich MP, Liu A. J Biol Chem. 2011;286:26541–26554. doi: 10.1074/jbc.M111.253237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumar MR, Fukuto JM, Miranda KM, Farmer PJ. Inorg Chem. 2010;49:6283–6292. doi: 10.1021/ic902319d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huges MN, Wimbledon PE. J Chem Soc, Dalton Trans. 1976:703–707. [Google Scholar]
- 77.Dutton AS, Suhrada CP, Miranda KM, Wink DA, Fukuto JM, Houk KN. Inorg Chem. 2006;45:2448–2456. doi: 10.1021/ic051505z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marti MA, Bari SE, Estrin DA, Doctorovich F. J Am Chem Soc. 2005;127:4680–4684. doi: 10.1021/ja044632n. [DOI] [PubMed] [Google Scholar]
- 79.Suarez SA, Marti MA, De Biase PM, Estrin DA, Bari SE, Doctorovich F. Polyhedron. 2007;26:4673–4679. [Google Scholar]
- 80.Bartberger MD, Fukuto JM, Houk KN. Proc Natl Acad Sci U S A. 2001;98:2194–2198. doi: 10.1073/pnas.041481598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sherman MP, Grither WR, McCulla RD. J Org Chem. 2010;75:4014–4024. doi: 10.1021/jo100172t. [DOI] [PubMed] [Google Scholar]
- 82.Shafirovich V, Lymar SV. Proc Natl Acad Sci U S A. 2002;99:7340–7345. doi: 10.1073/pnas.112202099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ruud K, Helgaker T, Uggerud E. J Mol Struct (THEOCHEM) 1997;393:59–71. [Google Scholar]
- 84.Fehling C, Friedrichs G. J Am Chem Soc. 2011;133:17912–17922. doi: 10.1021/ja2075949. [DOI] [PubMed] [Google Scholar]
- 85.Guardia CMA, Gonzalez Lebrero MC, Bari SE, Estrin DA. Chem Phys Lett. 2008;463:112–116. [Google Scholar]
- 86.Kramos B, Menyhard DK, Olah J. J Phys Chem B. 2012;116:872–885. doi: 10.1021/jp2080918. [DOI] [PubMed] [Google Scholar]
- 87.Einsle O, Messerschmidt A, Huber R, Kroneck PMH, Neese F. J Am Chem Soc. 2002;124:11737–11745. doi: 10.1021/ja0206487. [DOI] [PubMed] [Google Scholar]
- 88.Vincent MA, Hillier IH, Ge J. Chem Phys Lett. 2005;407:333–336. [Google Scholar]
- 89.Tsukamoto K, Watanabe T, Nagashima U, Akiyama Y. THEOCHEM J Mol Struct. 2005;732:87–98. [Google Scholar]
- 90.Harris DL. Int J Quantum Chem. 2002;88:183–200. [Google Scholar]
- 91.Obayashi E, Takahashi S, Shiro Y. J Am Chem Soc. 1998;120:12964–12965. [Google Scholar]