

Abstract
We present a DNA library preparation method that has allowed us to reconstruct a high coverage (30X) genome sequence of a Denisovan, an extinct relative of Neandertals. The quality of this genome allows a direct estimation of Denisovan heterozygosity indicating that genetic diversity in these archaic hominins was extremely low. It also allows tentative dating of the specimen on the basis of “missing evolution” in its genome, detailed measurements of Denisovan and Neandertal admixture into present-day human populations, and the generation of a near-complete catalog of genetic changes that swept to high frequency in modern humans since their divergence from Denisovans.
Draft genome sequences have been recovered from two archaic human groups, Neandertals (1) and Denisovans (2). While Neandertals are defined by distinct morphological features and occur in the fossil record of Europe, Western and Central Asia from at least 230,000 until about 30,000 years ago (3), Denisovans are known only from a distal manual phalanx and two molars, all excavated at Denisova Cave in the Altai Mountains in southern Siberia (2, 4, 5). The draft nuclear genome sequence retrieved from the Denisovan phalanx revealed that Denisovans are a sister group to Neandertals (2), with the Denisovan nuclear genome sequence falling outside Neandertal genetic diversity, suggesting an independent population history that differs from that of Neanderthals. Also, whereas a genetic contribution from Neandertal to the present-day human gene pool is present in all populations outside Africa, a contribution from Denisovans is found exclusively in island Southeast Asia and Oceania (6).
Both published archaic genome sequences are of low coverage; 1.9-fold genomic coverage from the Denisovan phalanx and a total of 1.3-fold derived from three Croatian Neandertals. As a consequence, many positions in the genomes are affected by sequencing errors or nucleotide misincorporations caused by DNA damage. Previous attempts to generate a genome sequence of high coverage from an archaic human have been hampered by the high levels of environmental contamination. The fraction of hominin endogenous DNA is commonly smaller than 1% and rarely approaches 5% (1, 7), making shotgun sequencing of the entire genome economically and logistically impractical. The only known exception is the Denisovan phalanx, which contains ~70% endogenous DNA. However, an extremely small fragment of this specimen is available to us, and the absolute number of endogenous molecules that could be recovered from the sample was too low to generate high genomic coverage.
A single-stranded library preparation method
DNA libraries for sequencing are normally prepared from double-stranded DNA (Fig. 1). However, for ancient DNA the use of single-stranded DNA may be advantageous as it will double its representation in the library. Furthermore, in a single-stranded DNA library, double-stranded molecules that carry modifications on one strand that prevent their incorporation into double-stranded DNA libraries could still be represented by the unmodified strand. We therefore devised a single-stranded library preparation method wherein the ancient DNA is dephosphorylated, heat denatured, and ligated to a biotinylated adaptor oligonucleotide, which allows its immobilization on streptavidin-coated beads (Fig. 1). A primer hybridized to the adaptor is then used to copy the original strand with a DNA polymerase. Finally, a second adaptor is joined to the copied strand by blunt-end ligation and the library molecules are released from the beads. The entire protocol is devoid of DNA purification steps, which inevitably cause loss of material.
Figure 1.

For single-stranded library preparation, ancient DNA molecules are dephosphorylated and heat-denatured. A biotinylated adaptor oligonucleotide is ligated to 3′-ends of the molecules, which are immobilized on streptavidin-coated beads and copied by extension of a primer hybridized to the adaptor. One strand of a double-stranded adaptor is then ligated to the newly synthesized strand. Finally, the beads are destroyed by heat to release the library molecules (not shown).
We applied this method to aliquots of the two DNA extracts (as well as side fractions) that were previously generated from the 40 mg of bone that comprised the entire inner part of the phalanx (2, 8). Comparisons of these newly generated libraries to the two libraries generated in the previous study (2) show at least a 6-fold and 22-fold increase in the recovery of library molecules (8), which is particularly pronounced for longer molecules (Fig. S4).
In addition to improved sequence yield, the single-strand library protocol reveals new aspects of DNA fragmentation and modification patterns (8). Since the ends of both DNA strands are left intact, it reveals that strand breakage occurs preferentially before and after guanine residues (Fig. S6), suggesting that guanine nucleotides are frequently lost from ancient DNA, possibly as the result of depurination. It also reveals that deamination of cytosine residues occurs with almost equal frequencies at both ends of the ancient DNA molecules. Since deamination is hypothesized to be frequent in single-stranded DNA overhangs (9, 10), this suggests that 5′- and 3′-overhangs occur at similar lengths and frequencies in ancient DNA.
Genome sequencing
We sequenced these libraries from both ends using Illumina’s Genome Analyzer IIx and included reads for two indexes (11), which were added in the clean room to exclude the possibility of downstream contamination with modern DNA libraries (1). Sequences longer than 35 bp were aligned to the human reference genome (GRCh37/1000 Genome project release) and the chimpanzee genome (CGSC 2.1/UCSC pantro2 release) with BWA (12). After removal of PCR duplicates, insertions/deletions and genotypes were called with GATK (8, 13). The three Denisovan libraries yielded 82.2 gigabases of non-duplicate sequence aligned to the human genome (8). Together with previous data (2) this provides about 31-fold coverage of the ~1.86 gigabases of the human autosomal genome to which short sequences can be confidently mapped (8). We also sequenced the genomes of eleven present-day individuals: a San, Mbuti, Mandenka, Yoruba and Dinka from Africa; a French and Sardinian from Europe; a Han, Dai and Papuan from Asia; and a Karitiana from South America. DNA from these individuals was barcoded, pooled and sequenced to ~24 to 33-fold genomic coverage (8). Because the samples were pooled, sequencing errors are the same across samples and are not expected to bias inferences about population relationships.
Genome quality
We used three independent measures to estimate human contamination in the Denisovan genome sequence (8). First, on the basis of a ~4,100-fold coverage of the Denisovan mitochondrial (mt) genome we estimate that 0.35% (95% confidence interval (C.I.) 0.33% – 0.36%) of fragments that overlap positions where the Denisovan mtDNA differs from most present-day humans show the modern human variant. Second, using the fact that the Denisovan phalanx comes from a female (2), we infer male human DNA contamination to be 0.07% (C.I. 0.05% – 0.09%) from alignments to the Y-chromosome. Third, a maximum-likelihood quantification of autosomal contamination gives an estimate of 0.22% (C.I. 0.22 – 0.23%). We conclude that less than 0.5% of the hominin sequences determined are extraneous to the bone (i.e. contamination from present-day humans).
Coverage of the genome is fairly uniform with 99.93% of the ‘mappable’ positions covered by at least one, 99.43% by at least ten, and 92.93% by at least 20 independent DNA sequences (8). High-quality genotypes (genotype quality >= 40) could be determined for 97.64% of the positions. While coverage in libraries prepared from ancient samples with previous methods are biased towards GC-rich sequences (14), the coverage of the libraries prepared with the single-stranded method from the Denisovan individual is similar to the eleven present-day human genomes (prepared from double-stranded DNA) in that coverage is positively correlated with AT-content (Fig. S12).
To estimate average per-base error rates in the Denisovan genome we counted differences between the sequenced DNA fragments and regions of the human genome that are highly conserved within primates (approximately 5.6 million bases, (8)). The error rate is 0.13% for the Denisovan genome, 0.17% to 0.19% for the genome sequences from the eleven present-day humans, and 1.2 – 1.7% for the two trios sequenced by the 1000 Genomes Pilot project (Table S11). The lower Denisovan error rate is likely due to consensus-calling from duplicate reads representing the same DNA fragments, and from overlap-merging of paired-end reads.
Molecular estimates of divergence and fossil age
We estimated the average DNA sequence divergence of all pair-wise combinations of the Denisovan genome and the 11 present-day humans as a fraction of the branch leading from the human-chimpanzee ancestor to present-day humans (Fig. 2, (8)). Assuming a human-chimpanzee average DNA sequence divergence of 6.5 million years ago (15), the Denisova-present-day human divergence is approximately 800,000 years, close to our previous estimate (2).
Figure 2.
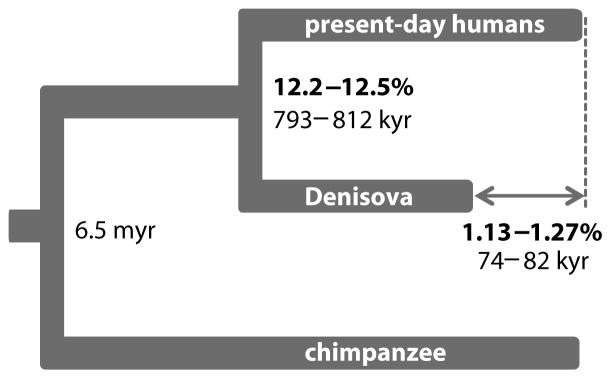
Average sequence divergence and branch length differences between the Denisovan genome and 11 present-day humans represented as a tree. Divergence is reported as fraction of the branch leading from human to the common ancestor with chimpanzee, and in years, assuming a human-chimpanzee divergence of 6.5 million years ago.
We next estimated the divergence of the archaic and modern human populations, which must be more recent than the DNA sequence divergence. To do this, we identified sites that are variable in a present-day West African individual, who is not affected by Denisovan or Neandertal gene flow, and counted how often the Denisovan and Neandertal genomes carry derived alleles not present in chimpanzee (1). From this, we estimate the population divergence between Denisovans and present-day humans to be 170,000–700,000 years (8). This is wider than our previous estimate (1), largely because it takes into account recent studies that broaden the range of plausible estimates for human mutation rates and thus the human-chimpanzee divergence date.
When comparing the number of substitutions inferred to have occurred between the human-chimpanzee ancestor and the Denisovan and present-day human genomes, the number for the Denisovan genome is 1.16% lower (1.13 – 1.27%; Fig. 2, (8)). This presumably reflects the age of the Denisovan bone, which had less time to accumulate changes than present-day humans. Assuming 6.5 million years of sequence divergence between humans and chimpanzees, the shortening of the Denisovan branch allows the bone to be tentatively dated to between 74,000 and 82,000 years before present, in general agreement with the archaeological dates (2). However, we caution that multiple sources of error may affect this estimate (8). For example, the numbers of substitutions inferred to have occurred to the present-day human sequences vary by up to one-fifth of the reduction estimated for the Denisovan bone. Nevertheless, the results suggest that in the future it will be possible to determine dates of fossils based on genome sequences.
Denisovan and Neandertal gene flow
To visualize the relationship between Denisova and the eleven present-day humans, we used TreeMix, which simultaneously infers a tree of relationships and “migration events” (16) (Fig. 3). This method estimates that 6.0% of the genomes of present-day Papuans derive from Denisovans (8). While this procedure does not provide a perfect fit to the data (for example, it does not model Neandertal admixture), it agrees with our previous finding that Denisovans have contributed to the genomes of present-day Melanesians, Australian Aborigines, and other SouthEast Asian islanders (2, 6).
Figure 3.

Maximum likelihood tree relating the Denisovan genome and the genomes of eleven present-day humans, allowing one migration event (shown in yellow).
We tested whether Denisovans share more derived alleles with any of the 11 present-day humans (8). To increase the power to detect gene flow, we used a new approach, ‘enhanced’ D-statistics, which restricts the analysis to alleles that are not present in 35 African genomes and are thus more likely to come from archaic humans. This confirms that Denisovans share more alleles with Papuans than with mainland Eurasians (Fig. 4A, Table S24). However, in contrast to a recent study proposing more allele sharing between Denisova and populations from southern China, such as the Dai, than with populations from northern China, such as the Han (17), we find less Denisovan allele sharing with the Dai than with the Han (although non-significantly so, Z = −0.9) (Fig. 4B; Table S25). Further analysis shows that if Denisovans contributed any DNA to the Dai, it represents less than 0.1% of their genomes today (Table S26).
Figure 4.
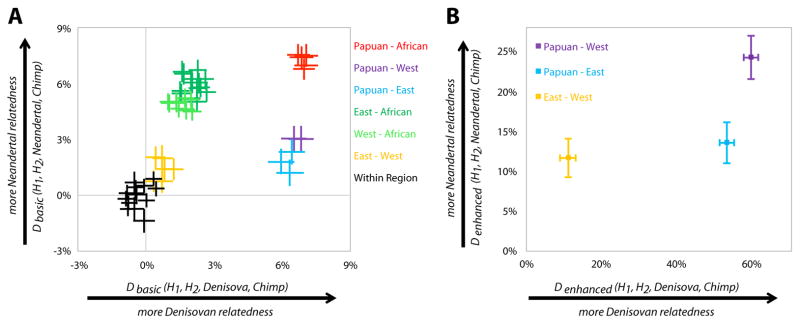
(A) Sharing of derived alleles among present-day humans, Denisovans and Neandertals. We compare all possible pairs of 11 present-day humans {H1, H2} in their “D-statistics”, which measure the rate at which they share derived alleles with Denisovans (x-axis) and Neandertals (y-axis). Each point reports ±1 standard error bars from a Block Jackknife. D-statistics are color-coded by geographic region. The D-statistic is not the same as the mixture proportion; it is also affected by quantities like the amount of shared genetic drift between the samples being compared. (B) Sharing of derived alleles that are absent in Africans among present-day humans, Denisovans and Neandertals. We enhance the power of the D-statistics by restricting to sites where 35 sub-Saharan African samples have the ancestral allele, and pooling modern humans by region to increase resolution (bars again give one standard error). Eastern non-African populations have significantly more archaic ancestry than European populations (Z=5.3 and Z=4.8 for the tests based on the Denisovan and Neandertal D-statistics, respectively).
Interestingly, we find that Denisovans share more alleles with the three populations from eastern Asia and South America (Dai, Han, and Karitiana) than with the two European populations (French and Sardinian) (Z=5.3). However, this does not appear to be due to Denisovan gene flow into the ancestors of present-day Asians, since the excess archaic material is more closely related to Neandertals than to Denisovans (Table S27). We estimate that the proportion of Neandertal ancestry in Europe is 24% lower than in eastern Asia and South America (95% C.I. 12–36%). One possible explanation is that there were at least two independent Neandertal gene flow events into modern humans (18). An alternative explanation is a single Neandertal gene flow event followed by dilution of the Neandertal proportion in the ancestors of Europeans due to later migration out of Africa. However, this would require about 24% of the present-day European gene pool to be derived from African migrations subsequent to the Neandertal admixture.
Strikingly, Papuans share more alleles with the Denisovan genome on the autosomes than on the X chromosome (P=0.01 by a two-sided test) (Table S28). One possible explanation for this finding is that the gene flow into Papuan ancestors involved primarily Denisovan males. Another explanation is population substructure combined with predominantly female migration among the ancestors of modern humans as they encountered Denisovans (thus diluting the Denisovan component on chromosome X) (19). A third possibility is natural selection against hybrid incompatibility alleles, which are known to be concentrated on chromosome X (20). We note that some autosomes (e.g. chromosome 11) also have less Denisovan ancestry (Table S30), suggesting that factors such as hybrid incompatibility may be at play.
Denisovan genetic diversity
The high quality of the Denisovan genome allowed us to measure its heterozygosity, i.e. the fraction of nucleotide sites that are different between a person’s maternal and paternal genomes (Fig. 5A). Several methods indicate that the Denisovan heterozygosity is about 0.022% (8). This is ~20% of the heterozygosity seen in the Africans, ~26–33% of that in the Eurasians, and 36% of that in the Karitiana, a South American population with extremely low heterozygosity (21). Since we find no evidence for unusually long stretches of homozygosity in the Denisovan genome (8), this is not due to inbreeding among the immediate ancestors of the Denisovan individual. We thus conclude that genetic diversity of the population to which the Denisovan individual belonged was very low compared to present-day humans.
Figure 5.

(A) Heterozygosity shown by the distribution of the number of bases matching the human reference genome at sites sampled to 20-fold coverage. The Y-axis is scaled to show the peak representing heterozygous sites in the center. (B) Inference of population size change over time using variation in the time since the most recent common ancestors across the genome shows that Denisovans have had a small population size over the last few hundred thousand years compared with modern humans, but a similar demographic history earlier. The y-axis specifies a number proportional to the population size Ne. The x-axis specifies time in units of divergence per base pair (along the top in years, assuming rates of 0.5×10−9 to 1.0×10−9 per year). Thin red lines around the Denisovan curve represent 100 bootstraps, thus showing the uncertainty of the inference. (C) The small population size in Denisovans is reflected in a greater accumulation of non-synonymous sites (normalized by the number of synonymous sites), whether measured in terms of heterozygous sites in Denisovans vs. modern humans (ratio 2.0 –2.5), or the accumulation of divergent sites on the Denisovan lineage divided by modern human lineages (ratio 1.5–2.0). The analysis is restricted to non-synonymous sites predicted to have a possibly or probably damaging effect on protein structure or function.
To estimate how Denisovan and modern human population sizes have changed over time we applied a Markovian coalescent model (22) to all genomes analyzed. This shows that present-day human genomes share similar population size changes, in particular a more than two-fold increase in size before 125,000–250,000 years ago (depending on the mutation rates assumed (23), Fig. 5B). Denisovans, in contrast, show a drastic decline in size at the time when the modern human population began to expand.
A prediction from a small ancestral Denisovan population size is that natural selection would be less effective in weeding out slightly deleterious mutations. We therefore estimated the ratio of non-synonymous substitutions that are predicted to have an effect on protein function to synonymous substitutions (those that do not change amino acids) in the genomes analyzed and found it to be on average 1.5–2.5 times higher in Denisovans than in the present-day humans, depending on the class of sites and populations to which Denisovans are compared (Fig. 5C, (8)). This is consistent with Denisovans having a smaller population size than modern humans, resulting in less efficient removal of deleterious mutation.
Denisovan genomic features
Since almost no phenotypic information exists about Denisovans, it is of some interest that in agreement with a previous study (24) the Denisovan individual carried alleles that in present-day humans are associated with dark skin, brown hair and brown eyes (Table S58, (8)). We also identified nucleotide changes specific to this Denisovan individual and not shared with any present-day human (8). However, since we have access to only a single Denisovan individual, we expect that only a subset of these would have been shared among all Denisovans.
Of more relevance may be examination of aspects of the Denisovan karyotype. The great apes have 24 pairs of chromosomes while humans have 23. This difference is caused by a fusion of two acrocentric chromosomes that formed the metacentric human chromosome 2 (25), and resulted in the unique head-to-head joining of the telomeric hexameric repeat GGGGTT. A difference in karyotype would likely have reduced the fertility of any offspring of Denisovans and modern humans. We searched all DNA fragments sequenced from the Denisovan individual and identified twelve fragments containing joined repeats. By contrast, reads from several chimpanzees and bonobos failed to yield any such fragments (8). We conclude that Denisovans and modern humans (and presumably Neandertals) shared a karyotype consisting of 46 chromosomes.
Features unique to modern humans
Genome sequences of archaic human genomes allow the identification of derived genomic features that became fixed or nearly fixed in modern humans after the divergence from their archaic relatives. The previous Denisovan and Neandertal genomes (1, 2) allowed less than half of all such features to be assessed with confidence. The current Denisovan genome enables us to generate an essentially complete catalog of recent changes in the human genome accessible with short read technology (26). In total, we identified 111,812 single nucleotide changes (SNCs) and 9,499 insertions and deletions where modern humans are fixed for the derived state while the Denisovan individual carried the ancestral, i.e. ape-like, variant (8). This is a relatively small number. We identified 260 human-specific SNCs that cause fixed amino acid substitutions in well-defined human genes, 72 fixed SNCs that affect splice sites, and 35 SNCs that affect well-defined motifs inside regulatory regions.
One way to identify changes that may have functional consequences is to focus on sites that are highly conserved among primates and that have changed on the modern human lineage after separation from Denisovan ancestors. We note that among the 23 most conserved positions affected by amino acid changes (primate conservation score ≥ 0.95), eight affect genes that are associated with brain function or nervous system development (NOVA1, SLITRK1, KATNA1, LUZP1, ARHGAP32, ADSL, HTR2B, CBTNAP2). Four of these are involved in axonal and dendritic growth (SLITRK1, KATNA1) and synaptic transmission (ARHGAP32, HTR2B) and two have been implicated in autism (ADSL, CBTNAP2). CNTNAP2 is also associated with susceptibility to language disorders (27) and is particularly noteworthy as it is one of the few genes known to be regulated by FOXP2, a transcription factor involved in language and speech development as well as synaptic plasticity (28). It is thus tempting to speculate that crucial aspects of synaptic transmission may have changed in modern humans.
Our limited understanding of how genes relate to phenotypes makes it impossible to predict the functional consequences of these changes. However, diseases caused by mutations in genes offer clues as to which organ systems particular genes may affect. Of the 34 genes with clear associations with human diseases that carry fixed substitutions changing the encoded amino acids in present-day humans, four (HPS5, GGCX, ERCC5, ZMPSTE24) affect the skin and six (RP1L1, GGCX, FRMD7, ABCA4, VCAN, CRYBB3) affect the eye. Thus, particular aspects of the physiology of the skin and the eye may have changed recently in human history. Another fixed difference occurs in EVC2, which when mutated causes Ellis-Van Creveld syndrome. Among other symptoms, this syndrome includes taurodontism, an enlargement of the dental pulp cavity and fusion of the roots, a trait that is common in teeth of Neandertals and other archaic humans. A Denisovan molar found in the cave has an enlarged pulp cavity but lacks fused roots (2). This suggests that the mutation in EVC2, perhaps in conjunction with mutations in other genes, has caused a change in dental morphology in modern humans.
We also examined duplicated regions larger than 9 kilobase pairs (kbp) in the Denisovan and the present-day human genomes, and found the majority of them to be shared (8). However, we find ten regions that are expanded in all present-day humans but not in the Denisovan genome. Notably, one of these overlaps a segmental duplication associated with a pericentric inversion of chromosome 18. In contrast to humans, the Denisovan genome harbors only a partial duplication of this region, suggesting that a deletion occurred in the Denisovan lineage. However, we are unable to resolve if the pericentric inversion is indeed present in Denisovans.
Implications for archaic and modern human history
It is striking that genetic diversity among Denisovans was low although they were present in Siberia as well as presumably in Southeast Asia where they interacted with the ancestors of present-day Melanesians (6). Only future research can show how wide their geographic range was at any one time in their history. However, it is likely that they have expanded from a small population size with not enough time elapsing for genetic diversity to correspondingly increase. When technical improvements such as the one presented here will make it possible to sequence a Neandertal genome to a quality comparable to the Denisovan and modern genomes, it will be important to clarify whether the temporal trajectory of Neandertal effective population size matches that of the Denisovans. If that is the case, it is likely that the low Denisovan diversity reflects the expansion out of Africa of a population ancestral to both Denisovans and Neandertals, a possibility that seems compatible with the dates for population divergences and population size changes presented.
By providing a comprehensive catalog of features that became fixed in modern humans after their separation from their closest archaic relatives, this work will eventually lead to a better understanding of the biological differences that existed between the groups. This should ultimately aid in determining how it was that modern humans came to expand dramatically in population size as well as cultural complexity while archaic humans eventually dwindled in numbers and became physically extinct.
Supplementary Material
Acknowledgments
Raw Denisovan sequence data is available from the European Nucleotide Archive (ENA) under study accession ERP001519. Raw Denisovan sequence and alignment data are available at http://cdna.eva.mpg.de/denisova/ and as a public data set via Amazon Web Services (AWS) at http://aws.amazon.com/datasets/2357. The present-day human sequences are available from the Short Read Archive under accession SRA047577 .
We thank D. Falush, P. Johnson, J. Krause, M. Lachmann, S. Sawyer, L. Vigilant and B. Viola for comments, help and suggestions; Ayinuer Aximu, Barbara Höber, Barbara Höffner, Antje Weihmann, T. Kratzer, R. Roesch for expert technical assistance; R. Schultz for help with data management and M. Schreiber for improvement of graphics.
The Presidential Innovation Fund of the Max Planck Society made this project possible. The U.S. National Science Foundation provided a HOMINID grant #1032255 to NP and DR. PHS is supported by an HHMI International Student Fellowship. FR is supported by a DAAD study scholarship. E.E.E. is on the scientific advisory boards for Pacific Biosciences, Inc., SynapDx Corp, and DNAnexus, Inc.
Footnotes
Materials and Methods
References and Notes
- 1.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reich D, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hublin JJ. Out of Africa: modern human origins special feature: the origin of Neandertals. Proc Natl Acad Sci USA. 2009;106:16022. doi: 10.1073/pnas.0904119106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krause J, et al. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature. 2010;464:894. doi: 10.1038/nature08976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibbons A. Paleoanthropology. Who were the Denisovans? Science. 2011;333:1084. doi: 10.1126/science.333.6046.1084. [DOI] [PubMed] [Google Scholar]
- 6.Reich D, et al. Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. Am J Hum Genet. 2011;89:516. doi: 10.1016/j.ajhg.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burbano HA, et al. Targeted investigation of the Neandertal genome by array-based sequence capture. Science. 2010;328:723. doi: 10.1126/science.1188046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Materials and methods are available as supplementary material on Science Online.
- 9.Briggs AW, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci USA. 2007;104:14616. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orlando L, et al. True single-molecule DNA sequencing of a pleistocene horse bone. Genome Res. 2011;21:1705. doi: 10.1101/gr.122747.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012;40:e3. doi: 10.1093/nar/gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green RE, et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134:416. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodman M. The genomic record of Humankind’s evolutionary roots. Am J Hum Genet. 1999;64:31. doi: 10.1086/302218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickrell J, Pritchard J. Inference of population splits and mixtures from genome-wide allele frequency data. 2012 doi: 10.1371/journal.pgen.1002967. Available from Nature Precedings < http://hdl.handle.net/10101/npre.2012.6956.1>. [DOI] [PMC free article] [PubMed]
- 17.Skoglund P, Jakobsson M. Archaic human ancestry in East Asia. Proc Natl Acad Sci USA. 2011;108:18301. doi: 10.1073/pnas.1108181108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Currat M, Excoffier L. Strong reproductive isolation between humans and Neanderthals inferred from observed patterns of introgression. Proc Natl Acad Sci USA. 2011;108:15129. doi: 10.1073/pnas.1107450108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petit RJ, Excoffier L. Gene flow and species delimitation. Trends Ecol Evol. 2009;24:386. doi: 10.1016/j.tree.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Coyne JA, Orr HA. Two rules of speciation. In: Otte D, Endler JA, editors. Speciation and its Consequences. 1989. pp. 180–207. [Google Scholar]
- 21.Kidd JR, Black FL, Weiss KM, Balazs I, Kidd KK. Studies of three Amerindian populations using nuclear DNA polymorphisms. Hum Biol. 1991;63:775. [PubMed] [Google Scholar]
- 22.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conrad DF, et al. Variation in genome-wide mutation rates within and between human families. Nature Genet. 2011;43:712. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerqueira CC, et al. Predicting homo pigmentation phenotype through genomic data: From neanderthal to James Watson. American journal of human biology : the official journal of the Human Biology Council. 2012 doi: 10.1002/ajhb.22263. [DOI] [PubMed] [Google Scholar]
- 25.IJdo JW, Baldini A, Ward DC, Reeders ST, Wells RA. Origin of human chromosome 2: an ancestral telomere-telomere fusion. Proc Natl Acad Sci USA. 1991;88:9051. doi: 10.1073/pnas.88.20.9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.1000 Genomes Project Consortium, A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vernes SC, et al. A Functional Genetic Link between Distinct Developmental Language Disorders. New England Journal of Medicine. 2008;359:2337. doi: 10.1056/NEJMoa0802828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enard W, et al. A humanized version of Foxp2 affects cortico-basal ganglia circuits in mice. Cell. 2009;137:961. doi: 10.1016/j.cell.2009.03.041. [DOI] [PubMed] [Google Scholar]
- 29.Briggs AW, et al. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 2010;38:e87. doi: 10.1093/nar/gkp1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohland N, Hofreiter M. Ancient DNA extraction from bones and teeth. Nature Protoc. 2007:1756. doi: 10.1038/nprot.2007.247. [DOI] [PubMed] [Google Scholar]
- 31.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blondal T, et al. Isolation and characterization of a thermostable RNA ligase 1 from a Thermus scotoductus bacteriophage TS2126 with good single-stranded DNA ligation properties. Nucleic Acids Res. 2005;33:135. doi: 10.1093/nar/gki149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li TW, Weeks KM. Structure-independent and quantitative ligation of single-stranded DNA. Anal Biochem. 2006;349:242. doi: 10.1016/j.ab.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010;2010 doi: 10.1101/pdb.prot5448. pdb prot5448. [DOI] [PubMed] [Google Scholar]
- 36.Meyer M, et al. From micrograms to picograms: quantitative PCR reduces the material demands of high-throughput sequencing. Nucleic Acids Res. 2008;36:e5. doi: 10.1093/nar/gkm1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dabney J, Meyer M. Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques. 2012;52:87. doi: 10.2144/000113809. [DOI] [PubMed] [Google Scholar]
- 38.Varshney U, van de Sande JH. Specificities and kinetics of uracil excision from uracil-containing DNA oligomers by Escherichia coli uracil DNA glycosylase. Biochemistry. 1991;30:4055. doi: 10.1021/bi00230a033. [DOI] [PubMed] [Google Scholar]
- 39.Krause J, et al. A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr Biol. 2010;20:231. doi: 10.1016/j.cub.2009.11.068. [DOI] [PubMed] [Google Scholar]
- 40.Sawyer S, Krause J, Guschanski K, Savolainen V, Pääbo S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS One. 2012;7:e34131. doi: 10.1371/journal.pone.0034131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang D, Hatahet Z, Melamede RJ, Kow YW, Wallace SS. Characterization of Escherichia coli endonuclease VIII. J Biol Chem. 1997;272:32230. doi: 10.1074/jbc.272.51.32230. [DOI] [PubMed] [Google Scholar]
- 42.Briggs AW, et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325:318. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 43.Kircher M, Stenzel U, Kelso J. Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome biology. 2009;10:R83. doi: 10.1186/gb-2009-10-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kircher M. Analysis of high-throughput ancient DNA sequencing data. Methods in molecular biology. 2012;840:197. doi: 10.1007/978-1-61779-516-9_23. [DOI] [PubMed] [Google Scholar]
- 45.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cann HM, et al. A human genome diversity cell line panel. Science. 2002;296:261. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- 47.Rohland N, Reich D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 2012;22:939. doi: 10.1101/gr.128124.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paten B, Herrero J, Beal K, Fitzgerald S, Birney E. Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 2008;18:1814. doi: 10.1101/gr.076554.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paten B, et al. Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome Res. 2008;18:1829. doi: 10.1101/gr.076521.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McVicker G, Gordon D, Davis C, Green P. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genetics. 2009;5:e1000471. doi: 10.1371/journal.pgen.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McVicker G, Green P. Genomic signatures of germline gene expression. Genome Res. 2010;20:1503. doi: 10.1101/gr.106666.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujita PA, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosenbloom KR, et al. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res. 2012;40:D912. doi: 10.1093/nar/gkr1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2010. [Google Scholar]
- 56.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 58.Hodgkinson A, Eyre-Walker A. Variation in the mutation rate across mammalian genomes. Nature Rev Genet. 2011;12:756. doi: 10.1038/nrg3098. [DOI] [PubMed] [Google Scholar]
- 59.Presgraves DC, Yi SV. Doubts about complex speciation between humans and chimpanzees. Trends Ecol Evol. 2009;24:533. doi: 10.1016/j.tree.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scally A, et al. Insights into hominid evolution from the gorilla genome sequence. Nature. 2012;483:169. doi: 10.1038/nature10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Makova KD, Li WH. Strong male-driven evolution of DNA sequences in humans and apes. Nature. 2002;416:624. doi: 10.1038/416624a. [DOI] [PubMed] [Google Scholar]
- 62.Pool JE, Nielsen R. Population size changes reshape genomic patterns of diversity. Evolution. 2007;61:3001. doi: 10.1111/j.1558-5646.2007.00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duret L, Arndt PF. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genetics. 2008;4:e1000071. doi: 10.1371/journal.pgen.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keinan A, Mullikin JC, Patterson N, Reich D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nature Genet. 2007;39:1251. doi: 10.1038/ng2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wall JD, Lohmueller KE, Plagnol V. Detecting ancient admixture and estimating demographic parameters in multiple human populations. Mol Biol Evol. 2009;26:1823. doi: 10.1093/molbev/msp096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schaffner SF, et al. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bischoff JL, et al. High-resolution U-series dates from the Sima de los Huesos hominids yields 600(−66)(+infinity) kyrs: implications for the 66 evolution of the early Neanderthal lineage. J Archaeol Sci. 2007;34:763. [Google Scholar]
- 68.Stringer C. The status of Homo heidelbergensis (Schoetensack 1908) Evolutionary Anthropology. 2012;21:101. doi: 10.1002/evan.21311. [DOI] [PubMed] [Google Scholar]
- 69.Busing FMTA, Meijer E, Van Der Leeden R. Delete-m jackknife for unequal m. Stat Comput. 1999;9:3. [Google Scholar]
- 70.Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28:2239. doi: 10.1093/molbev/msr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moorjani P, et al. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genetics. 2011;7:e1001373. doi: 10.1371/journal.pgen.1001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seielstad MT, Minch E, Cavalli-Sforza LL. Genetic evidence for a higher female migration rate in humans. Nature Genet. 1998;20:278. doi: 10.1038/3088. [DOI] [PubMed] [Google Scholar]
- 73.Kayser M, et al. Independent histories of human Y chromosomes from Melanesia and Australia. Am J Hum Genet. 2001;68:173. doi: 10.1086/316949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilkins JF, Marlowe FW. Sex-biased migration in humans: what should we expect from genetic data? BioEssays. 2006;28:290. doi: 10.1002/bies.20378. [DOI] [PubMed] [Google Scholar]
- 75.Tucker PK, Sage RD, Warner J, Wilson AC, Eicher EM. Abrupt Cline for Sex-Chromosomes in a Hybrid Zone between 2 Species of Mice. Evolution. 1992;46:1146. doi: 10.1111/j.1558-5646.1992.tb00625.x. [DOI] [PubMed] [Google Scholar]
- 76.Patterson N, Richter DJ, Gnerre S, Lander ES, Reich D. Genetic evidence for complex speciation of humans and chimpanzees. Nature. 2006;441:1103. doi: 10.1038/nature04789. [DOI] [PubMed] [Google Scholar]
- 77.Hammer MF, et al. The ratio of human X chromosome to autosome diversity is positively correlated with genetic distance from genes. Nature Genet. 2010;42:830. doi: 10.1038/ng.651. [DOI] [PubMed] [Google Scholar]
- 78.Gottipati S, Arbiza L, Siepel A, Clark AG, Keinan A. Analyses of X-linked and autosomal genetic variation in population-scale whole genome sequencing. Nature Genet. 2011;43:741. doi: 10.1038/ng.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bailey JA, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 80.Alkan C, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nature Genet. 2009;41:1061. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sudmant PH, et al. Diversity of human copy number variation and multicopy genes. Science. 2010;330:641. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Conrad DF, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 84.Prüfer K, et al. The bonobo genome compared with the chimpanzee and human genomes. Nature. 2012;486:527. doi: 10.1038/nature11128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ventura M, et al. Gorilla genome structural variation reveals evolutionary parallelisms with chimpanzee. Genome Res. 2011;21:1640. doi: 10.1101/gr.124461.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Locke DP, et al. Comparative and demographic analysis of orang-utan genomes. Nature. 2011;469:529. doi: 10.1038/nature09687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sturm RA, et al. A single SNP in an evolutionary conserved region within intron 86 of the HERC2 gene determines human blue-brown eye color. Am J Hum Genet. 2008;82:424. doi: 10.1016/j.ajhg.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eiberg H, et al. Blue eye color in humans may be caused by a perfectly associated founder mutation in a regulatory element located within the HERC2 gene inhibiting OCA2 expression. Hum Genet. 2008;123:177. doi: 10.1007/s00439-007-0460-x. [DOI] [PubMed] [Google Scholar]
- 89.Dennehey BK, Gutches DG, McConkey EH, Krauter KS. Inversion, duplication, and changes in gene context are associated with human chromosome 18 evolution. Genomics. 2004;83:493. doi: 10.1016/j.ygeno.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 90.Goidts V, Szamalek JM, Hameister H, Kehrer-Sawatzki H. Segmental duplication associated with the human-specific inversion of chromosome 18: a further example of the impact of segmental duplications on karyotype and genome evolution in primates. Hum Genet. 2004;115:116. doi: 10.1007/s00439-004-1120-z. [DOI] [PubMed] [Google Scholar]
- 91.Yunis JJ, Prakash O. The origin of man: a chromosomal pictorial legacy. Science. 1982;215:1525. doi: 10.1126/science.7063861. [DOI] [PubMed] [Google Scholar]
- 92.Haubold B, Pfaffelhuber P, Lynch M. mlRho - a program for estimating the population mutation and recombination rates from shotgun-sequenced diploid genomes. Mol Ecol. 2010;19(Suppl 1):277. doi: 10.1111/j.1365-294X.2009.04482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karolchik D, et al. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36:D773. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Howrigan D, Simonson M, Keller M. Detecting autozygosity through runs of homozygosity: A comparison of three autozygosity detection algorithms. BMC Genomics. 2011;12:460. doi: 10.1186/1471-2164-12-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kirin M, et al. Genomic Runs of Homozygosity Record Population History and Consanguinity. PloS One. 2010;5:e13996. doi: 10.1371/journal.pone.0013996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hernandez RD, et al. Classic selective sweeps were rare in recent human evolution. Science. 2011;331:920. doi: 10.1126/science.1198878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lohmueller KE, et al. Natural selection affects multiple aspects of genetic variation at putatively neutral sites across the human genome. PLoS genetics. 2011;7:e1002326. doi: 10.1371/journal.pgen.1002326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7:248. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rhesus Macaque Genome Sequencing and Analysis Consortium. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- 101.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genet. 2011;43:491. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McLaren W, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Desseyn JL, Aubert JP, Porchet N, Laine A. Evolution of the large secreted gel-forming mucins. Mol Biol Evol. 2000;17:1175. doi: 10.1093/oxfordjournals.molbev.a026400. [DOI] [PubMed] [Google Scholar]
- 106.Niimura Y, Nei M. Evolutionary changes of the number of olfactory receptor genes in the human and mouse lineages. Gene. 2005;346:23. doi: 10.1016/j.gene.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 107.Zhang Q, et al. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nature Genet. 2003;33:145. doi: 10.1038/ng1087. [DOI] [PubMed] [Google Scholar]
- 108.Faber PW, et al. Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet. 1998;7:1463. doi: 10.1093/hmg/7.9.1463. [DOI] [PubMed] [Google Scholar]
- 109.Macdonald ME, et al. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntingtons-Disease Chromosomes. Cell. 1993;72:971. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 110.Durfee LA, Lyon N, Seo K, Huibregtse JM. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Molecular Cell. 2010;38:722. doi: 10.1016/j.molcel.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tang Y, et al. Herc5 attenuates influenza A virus by catalyzing ISGylation of viral NS1 protein. J Immunol. 2010;184:5777. doi: 10.4049/jimmunol.0903588. [DOI] [PubMed] [Google Scholar]
- 112.Wang JB, et al. Human mu opiate receptor. cDNA and genomic clones, pharmacologic characterization and chromosomal assignment. FEBS Letters. 1994;338:217. doi: 10.1016/0014-5793(94)80368-4. [DOI] [PubMed] [Google Scholar]
- 113.Quillen EE, et al. OPRM1 and EGFR contribute to skin pigmentation differences between Indigenous Americans and Europeans. Hum Genet. 2012;131:1073. doi: 10.1007/s00439-011-1135-1. [DOI] [PubMed] [Google Scholar]
- 114.Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L. Natural selection has driven population differentiation in modern humans. Nature Genet. 2008;40:340. doi: 10.1038/ng.78. [DOI] [PubMed] [Google Scholar]
- 115.Dolphin CT, Shephard EA, Povey S, Smith RL, Phillips IR. Cloning, primary sequence and chromosomal localization of human FMO2, a new member of the flavin-containing mono-oxygenase family. Biochem J. 1992;287(Pt 1):261. doi: 10.1042/bj2870261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dolphin CT. The Flavin-containing Monooxygenase 2 Gene (FMO2) of Humans, but Not of Other Primates, Encodes a Truncated, Nonfunctional Protein. J Biol Chem. 1998;273:30599. doi: 10.1074/jbc.273.46.30599. [DOI] [PubMed] [Google Scholar]
- 117.Saleh M, et al. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004;429:75. doi: 10.1038/nature02451. [DOI] [PubMed] [Google Scholar]
- 118.Xue Y, et al. Spread of an inactive form of caspase-12 in humans is due to recent positive selection. Am J Hum Genet. 2006;78:659. doi: 10.1086/503116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Prüfer K, et al. FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics. 2007;8:41. doi: 10.1186/1471-2105-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4:1073. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 121.Galdzicka M, et al. A new gene, EVC2, is mutated in Ellis-van Creveld syndrome. Molecular Genetics and Metabolism. 2002;77:291. doi: 10.1016/s1096-7192(02)00178-6. [DOI] [PubMed] [Google Scholar]
- 122.Ruiz-Perez VL, et al. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am J Hum Genet. 2003;72:728. doi: 10.1086/368063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shen W, Han D, Zhang J, Zhao H, Feng H. Two novel heterozygous mutations of EVC2 cause a mild phenotype of Ellis-van Creveld syndrome in a Chinese family. American journal of medical genetics Part A. 2011;155A:2131. doi: 10.1002/ajmg.a.34125. [DOI] [PubMed] [Google Scholar]
- 124.Brkic H, Filipovic I. The meaning of taurodontism in oral surgery--case report. Acta stomatologica Croatica. 1991;25:123. [PubMed] [Google Scholar]
- 125.Neville BW, Damm DD, Allen CM, Bouquot JE. Oral & Maxillofacial Pathology. 5. W.B. Saunders; Philadelphia: 2002. [Google Scholar]
- 126.Jafarzadeh H, Azarpazhooh A, Mayhall JT. Taurodontism: a review of the condition and endodontic treatment challenges. International Endodontic Journal. 2008;41:375. doi: 10.1111/j.1365-2591.2008.01388.x. [DOI] [PubMed] [Google Scholar]
- 127.Barker BC. Taurodontism: the incidence and possible significance of the trait. Australian Dental Journal. 1976;21:272. doi: 10.1111/j.1834-7819.1976.tb05763.x. [DOI] [PubMed] [Google Scholar]
- 128.Glancy M, et al. Transmitted duplication of 8p23.1–8 p23.2 associated with speech delay, autism and learning difficulties. Eur J Hum Genet. 2009;17:37. doi: 10.1038/ejhg.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gibbons RJ, Suthers GK, Wilkie AO, Buckle VJ, Higgs DR. X-linked alpha-thalassemia/mental retardation (ATR-X) syndrome: localization to Xq12-q21.31 by X inactivation and linkage analysis. Am J Hum Genet. 1992;51:1136. [PMC free article] [PubMed] [Google Scholar]
- 130.Alarcón M, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Arking DE, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008;82:160. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519. doi: 10.1038/35097076. [DOI] [PubMed] [Google Scholar]
- 133.Strauss KA, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. The New England Journal of Medicine. 2006;354:1370. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 134.Jaeken J, Van den Berghe G. An infantile autistic syndrome characterised by the presence of succinylpurines in body fluids. Lancet. 1984;2:1058. [PubMed] [Google Scholar]
- 135.Stone RL, et al. A mutation in adenylosuccinate lyase associated with mental retardation and autistic features. Nature Genet. 1992;1:59. doi: 10.1038/ng0492-59. [DOI] [PubMed] [Google Scholar]
- 136.Stenson PD, et al. The Human Gene Mutation Database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Human Genomics. 2009;4:69. doi: 10.1186/1479-7364-4-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Matsumura K, Ervasti JM, Ohlendieck K, Kahl S, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 138.Roberds SL, et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell. 1994;78:625. doi: 10.1016/0092-8674(94)90527-4. [DOI] [PubMed] [Google Scholar]
- 139.Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 140.Stenirri S, et al. Denaturing HPLC profiling of the ABCA4 gene for reliable detection of allelic variations. Clin Chem. 2004;50:1336. doi: 10.1373/clinchem.2004.033241. [DOI] [PubMed] [Google Scholar]
- 141.Liu Y, et al. The human inward rectifier K(+) channel subunit kir5.1 (KCNJ16) maps to chromosome 17q25 and is expressed in kidney and pancreas. Cytogenetics and Cell Genetics. 2000;90:60. doi: 10.1159/000015662. [DOI] [PubMed] [Google Scholar]
- 142.Ellerman DA, et al. Izumo is part of a multiprotein family whose members form large complexes on mammalian sperm. Molecular Reproduction and Development. 2009;76:1188. doi: 10.1002/mrd.21092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Long KR, Trofatter JA, Ramesh V, McCormick MK, Buckler AJ. Cloning and characterization of a novel human clathrin heavy chain gene (CLTCL) Genomics. 1996;35:466. doi: 10.1006/geno.1996.0386. [DOI] [PubMed] [Google Scholar]
- 144.Desmaze C, et al. Physical mapping by FISH of the DiGeorge critical region (DGCR): involvement of the region in familial cases. Am J H Genet. 1993;53:1239. [PMC free article] [PubMed] [Google Scholar]
- 145.Wray GA. The evolutionary significance of cis-regulatory mutations. Nature Rev Genet. 2007;8:206. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- 146.Gruber JD, Vogel K, Kalay G, Wittkopp PJ. Contrasting Properties of Gene-Specific Regulatory, Coding, and Copy Number Mutations in Saccharomyces cerevisiae: Frequency, Effects, and Dominance. PLoS Genetics. 2012;8:e1002497. doi: 10.1371/journal.pgen.1002497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lim LP, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 148.Selbach M, et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 149.Pasquinelli AE, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- 150.Somel M, et al. MicroRNA-driven developmental remodeling in the brain distinguishes humans from other primates. PLoS Biology. 2011;9:e1001214. doi: 10.1371/journal.pbio.1001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Morin RD, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bryen JC, et al. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2008;36:D102. doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bao L, Zhou M, Cui Y. CTCFBSDB: a CTCF-binding site database for characterization of vertebrate genomic insulators. Nucleic Acids Res. 2008;36:D83. doi: 10.1093/nar/gkm875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Raab JR, Kamakaka RT. Insulators and promoters: closer than we think. Nature Rev Genet. 2010;11:439. doi: 10.1038/nrg2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hardy J, Singleton A. Genomewide association studies and human disease. The New England Journal of Medicine. 2009;360:1759. doi: 10.1056/NEJMra0808700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Manolio TA. Genomewide association studies and assessment of the risk of disease. The New England Journal of Medicine. 2010;363:166. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 157.Becker KG, Barnes KC, Bright TJ, Wang SA. The genetic association database. Nature Genet. 2004;36:431. doi: 10.1038/ng0504-431. [DOI] [PubMed] [Google Scholar]
- 158.Cariaso M, Lennon G. SNPedia: a wiki supporting personal genome annotation, interpretation and analysis. Nucleic Acids Res. 2012;40:D1308. doi: 10.1093/nar/gkr798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Buckanovich RJ, Yang YY, Darnell RB. The onconeural antigen Nova-1 is a neuron-specific RNA-binding protein, the activity of which is inhibited by paraneoplastic antibodies. J Neurosci. 1996;16:1114. doi: 10.1523/JNEUROSCI.16-03-01114.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Park JA, Kim KC. Expression patterns of PRDM10 during mouse embryonic development. BMB Rep. 2010;43:29. doi: 10.5483/bmbrep.2010.43.1.029. [DOI] [PubMed] [Google Scholar]
- 161.Lohi H, et al. Functional characterization of three novel tissue-specific anion exchangers SLC26A7,-A8, and-A9. J Biol Chem. 2002;277:14246. doi: 10.1074/jbc.M111802200. [DOI] [PubMed] [Google Scholar]
- 162.Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133:653. doi: 10.1016/j.cell.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mayr MI, et al. The human kinesin Kif18A is a motile microtubule depolymerase essential for chromosome congression. Curr Biol. 2007;17:488. doi: 10.1016/j.cub.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 164.Abelson JF, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science. 2005;310:317. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- 165.Hsu CY, et al. LUZP deficiency affects neural tube closure during brain development. Biochemical and Biophysical Research Communications. 2008;376:466. doi: 10.1016/j.bbrc.2008.08.170. [DOI] [PubMed] [Google Scholar]
- 166.Pisareva VP, Pisarev AV, Komar AA, Hellen CU, Pestova TV. Translation initiation on mammalian mRNAs with structured 5′UTRs requires DExH-box protein DHX29. Cell. 2008;135:1237. doi: 10.1016/j.cell.2008.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Okabe T, et al. RICS, a novel GTPase-activating protein for Cdc42 and Rac1, is involved in the beta-catenin-N-cadherin and N-methyl-D-aspartate receptor signaling. J Biol Chem. 2003;278:9920. doi: 10.1074/jbc.M208872200. [DOI] [PubMed] [Google Scholar]
- 168.Bevilacqua L, et al. A population-specific HTR2B stop codon predisposes to severe impulsivity. 2010;468:1061. doi: 10.1038/nature09629. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nature. 2011;470 [Google Scholar]
- 169.Doly S, et al. Serotonin 5-HT2B receptors are required for 3,4-methylenedioxymethamphetamine-induced hyperlocomotion and 5-HT release in vivo and in vitro. J Neurosci. 2008;28:2933. doi: 10.1523/JNEUROSCI.5723-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Speicher DW, Weglarz L, DeSilva TM. Properties of human red cell spectrin heterodimer (side-to-side) assembly and identification of an essential nucleation site. J Biol Chem. 1992;267:14775. [PubMed] [Google Scholar]
- 171.Jones SS, D’Andrea AD, Haines LL, Wong GG. Human erythropoietin receptor: cloning, expression, and biologic characterization. Blood. 1990;76:31. [PubMed] [Google Scholar]
- 172.Longmore GD, Lodish HF. An activating mutation in the murine erythropoietin receptor induces erythroleukemia in mice: a cytokine receptor superfamily oncogene. Cell. 1991;67:1089. doi: 10.1016/0092-8674(91)90286-8. [DOI] [PubMed] [Google Scholar]
- 173.Yu X, Lin CS, Costantini F, Noguchi CT. The human erythropoietin receptor gene rescues erythropoiesis and developmental defects in the erythropoietin receptor null mouse. Blood. 2001;98:475. doi: 10.1182/blood.v98.2.475. [DOI] [PubMed] [Google Scholar]
- 174.Rubinsztein DC, Easton DF. Apolipoprotein E genetic variation and Alzheimer’s disease. a meta-analysis. Dementia and Geriatric Cognitive Disorders. 1999;10:199. doi: 10.1159/000017120. [DOI] [PubMed] [Google Scholar]
- 175.Pohjalainen T, et al. The A1 allele of the human D2 dopamine receptor gene predicts low D2 receptor availability in healthy volunteers. Molecular psychiatry. 1998;3:256. doi: 10.1038/sj.mp.4000350. [DOI] [PubMed] [Google Scholar]
- 176.Lucht M, et al. Influence of DRD2 and ANKK1 genotypes on apomorphine-induced growth hormone (GH) response in alcohol-dependent patients. Progress in neuro-psychopharmacology & biological psychiatry. 2010;34:45. doi: 10.1016/j.pnpbp.2009.08.024. [DOI] [PubMed] [Google Scholar]
- 177.David SP, et al. Bupropion efficacy for smoking cessation is influenced by the DRD2 Taq1A polymorphism: analysis of pooled data from two clinical trials. Nicotine & tobacco research. 2007;9:1251. doi: 10.1080/14622200701705027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Burton P, Clayton D, Cardon L, Craddock N, Deloukas P. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Frayling TM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lyssenko V, et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. The Journal of clinical investigation. 2007;117:2155. doi: 10.1172/JCI30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Heinz A, Smolka MN. The effects of catechol O-methyltransferase genotype on brain activation elicited by affective stimuli and cognitive tasks. Reviews in the Neurosciences. 2006;17:359. doi: 10.1515/revneuro.2006.17.3.359. [DOI] [PubMed] [Google Scholar]
- 182.Frank MJ, Doll BB, Oas-Terpstra J, Moreno F. Prefrontal and striatal dopaminergic genes predict individual differences in exploration and exploitation. Nature Neuroscience. 2009;12:1062. doi: 10.1038/nn.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pálmason H, et al. Attention-deficit/hyperactivity disorder phenotype is influenced by a functional catechol-O-methyltransferase variant. Journal of Neural Transmission. 2010;117:259. doi: 10.1007/s00702-009-0338-2. [DOI] [PubMed] [Google Scholar]
- 184.Gupta M, et al. Genetic susceptibility to schizophrenia: role of dopaminergic pathway gene polymorphisms. Pharmacogenomics. 2009;10:277. doi: 10.2217/14622416.10.2.277. [DOI] [PubMed] [Google Scholar]
- 185.Duffy DL, et al. A three-single-nucleotide polymorphism haplotype in intron 1 of OCA2 explains most human eye-color variation. Am J Hum Genet. 2007;80:241. doi: 10.1086/510885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Frändberg PA, Doufexis M, Kapas S, Chhájlani V. Human pigmentation phenotype: a point mutation generates nonfunctional MSH receptor. Biochemical and Biophysical Research Communications. 1998;245:490. doi: 10.1006/bbrc.1998.8459. [DOI] [PubMed] [Google Scholar]
- 187.Yoshiura K-i, et al. A SNP in the ABCC11 gene is the determinant of human earwax type. Nature Genet. 2006;38:324. doi: 10.1038/ng1733. [DOI] [PubMed] [Google Scholar]
- 188.Enattah NS, et al. Identification of a variant associated with adult-type hypolactasia. Nature Genet. 2002;30:233. doi: 10.1038/ng826. [DOI] [PubMed] [Google Scholar]
- 189.Bersaglieri T, et al. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004;74:1111. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mulcare CA, et al. The T allele of a single-nucleotide polymorphism 13.9 kb upstream of the lactase gene (LCT) (C-13.9kbT) does not predict or cause the lactase-persistence phenotype in Africans. Am J Hum Genet. 2004;74:1102. doi: 10.1086/421050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tishkoff SA, et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nature Genet. 2007;39:31. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mou C, et al. Enhanced ectodysplasin-A receptor (EDAR) signaling alters multiple fiber characteristics to produce the East Asian hair form. Human Mutation. 2008;29:1405. doi: 10.1002/humu.20795. [DOI] [PubMed] [Google Scholar]
- 193.Kimura R, et al. A common variation in EDAR is a genetic determinant of shovel-shaped incisors. Am J Hum Genet. 2009;85:528. doi: 10.1016/j.ajhg.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rodrigues SM, Saslow LR, Garcia N, John OP, Keltner D. Oxytocin receptor genetic variation relates to empathy and stress reactivity in humans. Proc Natl Acad Sci USA. 2009;106:21437. doi: 10.1073/pnas.0909579106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Roth SM, et al. The ACTN3 R577X nonsense allele is under-represented in elite-level strength athletes. Eur J Hum Genet. 2008;16:391. doi: 10.1038/sj.ejhg.5201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ellis JA, Stebbing M, Harrap SB. Polymorphism of the androgen receptor gene is associated with male pattern baldness. The Journal of Investigative Dermatology. 2001;116:452. doi: 10.1046/j.1523-1747.2001.01261.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.