

Abstract
The past 15 years have seen an explosion in our understanding of how cells replicate damaged DNA and how this can lead to mutagenesis. The Y-family DNA polymerases lie at the heart of this process, which is commonly known as translesion synthesis. This family of polymerases has unique features that enable them to synthesize DNA past damaged bases. However, as they exhibit low fidelity when copying undamaged DNA, it is essential that they are only called into play when they are absolutely required. Several layers of regulation ensure that this is achieved.
A finely tuned and complex molecular machine replicates DNA with very high efficiency and astonishing fidelity. However, the price paid for this is that the machinery is easily disturbed by damage on the DNA template. Despite the plethora of mechanisms for repairing DNA, it is likely that the replication machinery will encounter lesions in the DNA template during each cell cycle. The catalytic site of the replicative DNA polymerases is compact and intolerant of most DNA lesions. As a consequence, DNA synthesis arrests at most forms of DNA damage. This poses a considerable problem for the cell — it must replicate the damaged DNA before mitosis so that a complete copy of the genome is passed to both daughter cells. The solution adopted by cells in every branch of life is known as DNA damage tolerance: DNA is synthesized past the damaged bases, which can be subsequently excised after the replication fork has passed and they are safely located within duplex DNA. Direct replication past DNA damage in these circumstances — a process known as translesion synthesis (TLS) — is carried out by specialized DNA polymerases, the most abundant class of which are those belonging to the Y-family1. As detailed in TABLE 1, there are two member s of this family in Escherichia coli and Saccharomyces cerevisiae and four members in mammalian cells.
Table 1.
The Y-family polymerases from the three domains of life and their key properties
| Gene name | Protein name | Properties |
|---|---|---|
| Escherichia coli | ||
|
| ||
| dinB | Pol IV | • Creates –1 frameshifts when overexpressed in vivo |
| • Bypasses N2-dG adducts efficiently and accurately | ||
| • Involved in TLS of alkylation damage in vivo | ||
|
| ||
| umuD and umuC | Pol V | • Major TLS polymerase in E. coli; is able to traverse a vast array of lesions |
| • Unique among Y-family polymerases in that it is comprised of a heterotrimer of UmuD′ (~24 kDa) and UmuC (~48 kDa) to form an ~72 kDa UmuD′2C complex | ||
| • Interacts with RecA and ATP to form the highly active Pol V Mut polymerase | ||
|
| ||
| Sulfolobus solfataricus | ||
|
| ||
| dpo4 | Dpo4 | • Archaeal orthologue of E.coli Pol IV and human Pol κ |
| • Multiple crystal structures of Dpo4 in the process of bypassing a variety of DNA lesions have been solved | ||
|
| ||
| Saccharomyces cerevisiae | ||
|
| ||
| REV1 | Rev1 | • Specifically incorporates dCMP opposite abasic sites, template G and a limited number of other, minor groove adducts |
| • Interacts with the B-family TLS polymerase Pol ζ (which is comprised of Rev3 and Rev7) to stimulate Pol ζ-dependent TLS in vivo | ||
|
| ||
| RAD30 | Pol η | • First Y-family polymerase to be shown to be a bona fide DNA polymerase |
| • Can bypass a T-T CPD accurately and efficiently | ||
|
| ||
| Homo sapiens | ||
|
| ||
| REV1 | REV1 | • Like the yeast Rev1 protein, it specifically incorporates dCMP opposite dG and abasic sites |
| • Acts as a scaffold protein that interacts with the Y-family polymerases Pol η, Pol ι and Pol κ, as well as the B-family TLS polymerase Pol ζ (which is comprised of Rev3 and Rev7) | ||
| • Generates mutations at G-C base pairs during immunoglobulin gene somatic hypermutation | ||
|
| ||
| POLH (also known as XPV and RAD30A) | Pol η | • Bypasses a T-T CPD efficiently and with the same accuracy as undamaged DNA |
| • Defects lead to XPV | ||
| • Accumulates in replication foci | ||
| • Is subject to ubiquitylation and phosphorylation | ||
| • Generates mutations at A-T base pairs during immunoglobulin gene somatic hypermutation | ||
|
| ||
| POLI (also known as RAD30B) | Pol ι | • In human cells protein is 715 aa in length, but an additional in-frame 25 aa, conserved from frogs to humans, produces a 740 aa protein as a minor species (B. Coull and A.R.L., unpublished observations) |
| • Has unique replication fidelity, replicating template dA reasonably accurately, but replicating template dT in a highly error-prone manner | ||
| • Accumulates in replication foci, but resides in these foci for a shorter time than Pol η | ||
|
| ||
| POLK (also known as DINB1) | Pol κ | • Enzyme is prone to making –1 frameshift mutations, but can accurately and efficiently bypass a number of N2-dG lesions |
| • Has additional roles in the repair synthesis step of nucleotide excision repair | ||
aa, amino acid; CPD, cyclobutane pyrimidine dimer; Pol, DNA polymerase; TLS, translesion synthesis; XPV, variant form of xeroderma pigmentosum.
Drawing on examples across all domains of life, this Review examines the principles that govern the ability of the Y-family polymerases to synthesize past damaged DNA and yet allow the cell to restrict the potentially damaging mutagenic activity of these enzymes. After a brief historical introduction, we focus on the underlying biochemical and structural features of the family and then examine the mechanisms that control their activity. Finally, we summarize the roles of the Y-family polymerases in a number of other processes to illustrate how their properties have been co-opted to meet specialized needs within a cell.
A brief history
Special mechanisms for replicating past DNA damage were first identified in the late 1960s by Rupp and Howard-Flanders2, who found that after they irradiated E. coli with ultraviolet (UV) light, gaps were formed in newly synthesized DNA and subsequently sealed. Although most of these gaps were filled-in by a recombination-mediated `damage avoidance' mechanism, the idea that some of them were filled by an error-prone DNA polymerase was mooted in the early 1970s3. The reversionless (REV) loci in budding yeast4 and UV nonmutable (umu) loci in E. coli5,6 were postulated to be involved in this error-prone bypass pathway, and they were thought (erroneously as it turned out) to somehow lower the fidelity of the replicative polymerases (Supplementary information S1 (box)) to allow them to replicate past damaged bases. At about the same time, the variant form of xeroderma pigmentosum (XPV) was shown to be caused by a reduced ability to make intact daughter DNA strands following UV light irradiation7.
It was more than two decades later that the products of these and related genes were identified. In yeast, the products of the REV1 and REV3 genes were found to be a dCMP transferase8 and DNA polymerase ζ (Pol ζ), respectively. Pol ζ is a B-family polymerase that is capable of bypassing the most common UV light-induced lesion, the cyclobutane pyrimidine dimer (CPD), with about 10% efficiency9. Subsequently, Rad30 was also shown to be a bona fide DNA polymerase (termed Pol η) that could bypass cis-syn cyclobutane T-T dimers efficiently and relatively accurately10. By the end of 1999, a human homologue of Rad30 had been identified and was shown to be the product of the gene that is defective in XPV cells11,12. At roughly the same time, the E. coli DinB protein and UmuD′2C complex were also shown to be bona fide TLS polymerases called E. coli Pol IV13 and Pol V14,15, respectively. Shortly thereafter, a second human orthologue of yeast Rad30 and a human orthologue of E. coli DinB were identified and shown to encode the TLS polymerases Pol ι16–19 and Pol κ20–23, respectively. Thus, within a period of about 18 months, the field underwent a seismic change from having no clear understanding of the mechanisms of TLS to defining an enzymatic process that is facilitated by specialized DNA polymerases conserved from bacteria to humans. The basic steps of TLS, as shown in FIG. 1 and described in more detail below, involve several `polymerase switches'. First, the stalled replicative polymerase must be displaced and replaced with a TLS polymerase, which inserts either correct or incorrect bases opposite the lesion. Extension from the (mis)incorporated base may be performed by the same polymerase or a second TLS polymerase. Finally, when base-pairing has been restored beyond the lesion, the replicative polymerase regains control18,24,25.
Figure 1. The basic mechanism of translesion synthesis.
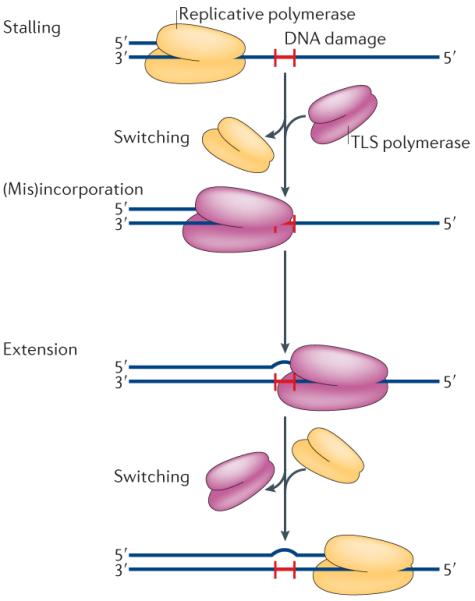
Translesion synthesis (TLS) is a multi-step process. The replicative DNA polymerase stalls at a site of DNA damage (red `H'), then a TLS polymerase (or polymerases) is recruited to the primer terminus and correct, or incorrect, bases are inserted opposite the lesion. The inserted base is subsequently extended to complete TLS131. All TLS polymerases are distributive and dissociate from the primer terminus after synthesizing a short tract of DNA. They are then quickly replaced by the cell's replicative polymerase, which completes genome duplication.
The presence of these low-fidelity polymerases in all domains of life, and indeed their expansion in higher organisms, suggests an essential evolutionarily conserved role26. All cells are exposed to DNA insults from both endogenous and exogenous sources, and the Y-family DNA polymerases allow them to tolerate potentially lethal damage. However, sometimes, but not always, tolerance is accompanied by unwanted mutagenesis, which can have deleterious consequences. It is crucial therefore to strictly regulate the activity of the Y-family polymerase s so that they are not deployed inappropriately.
Structural and biochemical features
The key differences between Y-family polymerases (TABLE 1) and most other polymerases are their ability to replicate past damaged bases and their reduced fidelity on undamaged templates. The features of the polymerases that confer these properties are described in this section.
DNA polymerase fidelity
The structural features of the replicative polymerases are geared towards maximizing replication efficiency and fidelity (Supplementary information S1 (box)). Surprisingly, polymerase fidelity, which is defined as the ratio of incorporation of the correct over incorrect nucleotide, is determined largely by the efficiency of incorporation of the correct nucleotide rather than efficiency of incorrect nucleotide insertion, which varies relatively little between polymerases27. The general structure of the replicative DNA polymerases has been likened to a right hand with palm, finger and thumb domains28 (FIG. 2a): the catalytic carboxylate–metal ion complex sits in the palm domain while the mobile finger and thumb domains grasp the template and primer to create a tight active site that is intolerant of misalignment between the template base and incoming nucleotide29,30 (FIG. 2a). The finger domain undergoes a significant conformational change during the catalytic cycle, as explained in Supplementary information S1 (box).
Figure 2. Structures of the Y-family DNA polymerases.
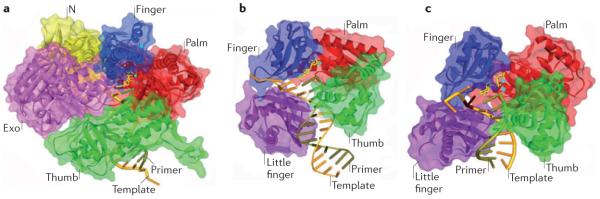
a | Structure of the B-family DNA polymerase from the bacteriophage RB69, with DNA and the incoming dCTP opposite dG (Protein Data Bank (PDB) code 3NCI)132 shown for comparison to the Y-family structures. The main domains are colour coded (palm = red, thumb = green, finger = blue, exonuclease (Exo) = purple, amino-terminal domain (N) = yellow). b | Crystal structure of the Sulfolobus solfataricus Y-family polymerase Dpo4 in a ternary complex with DNA and incoming ddADP (PDB code 1JX4)33. The additional little-finger domain is shown in purple. Note the absence of an exonuclease domain. c | Crystal structure of the human Y-family DNA polymerase Pol η in a ternary complex with a cyclobutane pyrimidine dimer (CPD). In this view, the 3′T of the CPD is in the active site and is correctly paired with incoming dATP (PDB code 3MR3)42. The template strand is shown in rust colour, and the primer is olive green. The incoming dNTP is shown in yellow. The burgundy stick represents the position of the CPD. The small blue spheres represent the metal ions. The protein backbone is represented by ribbon surrounded by semi-transparent solvent accessible surface. This figure was kindly created by A. Vaisman (US National Institute of Child Health and Human Development/US National Institutes of Health) using the Discovery Studio Visualizer program.
General features of the catalytic domains
Despite poor sequence conservation with the replicative polymerases, the structures of Y-family polymerase catalytic domains reveal a similar overall `right hand' topology. However, their active site is more capacious than that of the replicative polymerases, allowing the accommodation of a bulky adduct on the template base. The finger and thumb domains are stubbier, making fewer DNA contacts with both the DNA and incoming nucleotide, which contributes to the enzymes' decreased processivity and poorer fidelity. The Y-family polymerases have an additional domain, termed the `little finger', that also mediates DNA contacts close to the lesion site. Indeed, this domain has been implicated in polymerase selectivity for certain lesions31. These features allow nucleotide incorporation opposite damaged bases, but they also militate against accurate and processive replication, and the mutagenicity of the Y-family polymerases is further compounded by a lack of the 3′–5′ proofreading exonuclease activity that is characteristic of replicative polymerase s (compare FIG. 2b,c with FIG. 2a).
Y-family acrobatics
Within these general principles, the Y-family polymerases adopt an impressive array of novel mechanisms to replicate over a diverse range of DNA lesions, an ability that extends even to bypassing short stretches of non-DNA carbon chain32. Next, we illustrate some of these approaches and features by examining how the structural solutions adopted by Y-family members explain their biochemical properties and ability to replicate particular lesions (TABLE 1). As mentioned previously and shown in FIG. 1, TLS is a multi-step process involving (mis)incorporation opposite the DNA lesion and subsequent extension past the lesion (FIG. 1). The polymerases that are thought to be involved in these different steps in E. coli, S. cerevisiae and human cells are detailed in FIG. 3. The eukaryotic B-family enzyme Pol ζ is especially suited to carry out the extension past the DNA lesion.
Figure 3. Evolutionarily conserved roles of DNA polymerases in translesion synthesis.

Replicative polymerases, such as Escherichia coli DNA polymerase III (Pol III; which is made up of a core, β-clamp and γ-complex) or eukaryotic Pol ε, stall at the site of a DNA lesion (represented in the figure as TT). Translesion synthesis (TLS) polymerases are recruited to the site via interactions with the replicative sliding processivity clamp (the β-clamp in E. coli and PCNA in eukaryotes). E. coli has a choice of two Y-family polymerases, Pol IV (also known as DinB) and Pol V Mut (which is comprised of UmuD′2 C–RecA–ATP133). Saccharomyces cerevisiae also has a choice of two Y-family polymerases, Rev1 and Pol η (which is the product of the RAD30 gene), as well as the B-family TLS polymerase Pol ζ (which is a heterodimer of the Rev3 and Rev7 proteins). In humans, at least five TLS polymerases can be recruited to sites of arrested replication, including Pol η, Pol ι, Pol κ, REV1 and Pol ζ. The likelihood that this insertion step is error-prone, as shown for E. coli and S. cerevisiae, or error-free, as shown for humans, will depend on the nature of the DNA lesion and the polymerase utilized. The extension step may be facilitated by the same enzyme that performed the (mis)insertion or by a completely different polymerase. Once the nascent DNA chain has been extended beyond the lesion, the TLS polymerase is replaced by the cell's replicative DNA polymerase.
Dpo4: a model for Pol IV and Pol κ-like polymerases
Sulfolobus solfataricus Dpo4 (FIG. 4) was the first Y-family DNA polymerase to be crystallized in a ternary complex with DNA and incoming nucleoside triphosphate33. In the intervening 10 years, Dpo4 has been crystallized in the process of facilitating TLS past a broad range of DNA lesions. These structures indicate that Dpo4 is very flexible and can accommodate a plethora of lesions in its active site through various contortions of the primer, template or incoming nucleoside triphosphate. Interestingly, however, the only lesion that Dpo4 bypasses very efficiently is an abasic site. Efficient bypass is achieved by displacement of the abasic site into an extra-helical position, with the 5′ undamaged base serving as the template for synthesis34. This results in a −1 frameshift mutation if the primer template does not realign after TLS and provides the structural basis for the high frequency of −1 bp mutations generated by Dpo4 and its orthologues, which include E. coli Pol IV35 and human Pol κ36.
Figure 4. Domains of the Y-family polymerases.
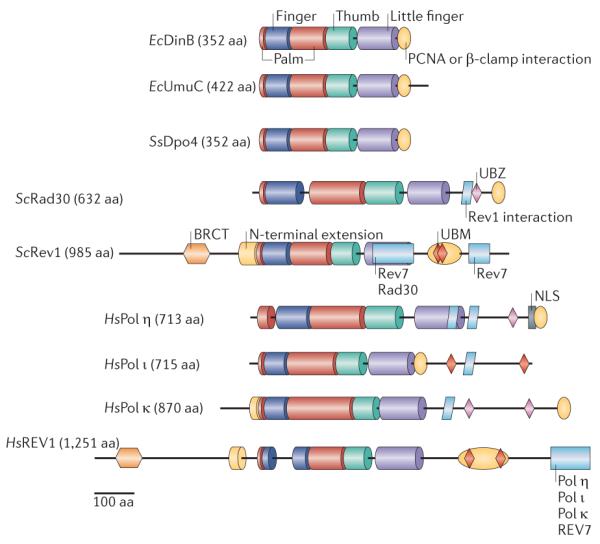
The active sites of the polymerases (Pols) are contained within the palm, thumb, finger and little-finger domains, colour coded as in FIG. 2. The additional domains involved in localization and regulation are BRCT (breast cancer-associated protein-1 carboxy-terminal domain), NLS (nuclear localization signal) and UBM and UBZ (ubiquitin-binding motifs). Species abbreviations: Ec, Escherichia coli; Hs, Homo sapiens; Sc, Saccharomyces cerevisiae; Ss, Sulfolobus solfataricus. aa, amino acid.
Dpo4 also uses a similar mechanism of looping out the much larger benzo[a]pyrene diol-epoxide (BPDE) adduct on dexoyguanosine37. The bulky lesion is flipped into a structural gap between the little-finger domain and the core domains so that the correct geometry for TLS occurs. Archaeal Dpo4 and human Pol κ have strikingly similar structures despite rather poor sequence conservation, with all domains superimposable30. However, subtle differences exist. In particular, the gap between the little-finger domain and the catalytic core is enlarged, which helps to explain why these enzymes are able to bypass a BPDE lesion in vitro23 and in vivo38.
Molecular splinting by Pol η
Pol η is particularly efficient at replicating CPDs. It can do so alone and with high efficiency, incorporating A-A opposite a cis-syn T-T CPD with similar accuracy to unmodified T-T10. This provides a ready explanation for the features of Pol η-defective XPV cells and patients. In the absence of Pol η, other polymerases presumably substitute but with lower accuracy, resulting in higher UV light-induced mutagenesis and carcinogenesis. Evidence suggests that the likely candidates are Pol ι or Pol κ in combination with Pol ζ39,40. A recent set of structures showing yeast and human Pol η in the act of bypassing a CPD reveal a number of features that enable this polymerase to be so effective at replicating this common lesion41,42. The active site of Pol η is particularly large and is able to accommodate both of the linked thymine bases of the template (FIG. 2c). The CPD is further stabilized such that the linked Ts can pair with the correct incoming dA, thereby allowing the polymerase to extract the correct coding information from the lesion. Because the CPD remains in the duplex after replication, it will continue to introduce distortion, which could contribute to slippage and frameshifts as the newly replicated duplex emerges from the catalytic site. Pol η counters this by providing a continuous positively charged molecular surface, created by a specialized β-strand in the little-finger domain, which acts to splint the newly synthesized duplex into a stable B-form. Thus, the enzyme is not only able to accurately synthesize across T-T CPDs, it also ensures the reading frame is maintained. Finally, when the CPD emerges from the active site and the cell is in danger of replicating undamaged DNA with low fidelity, steric clashes ensure that the DNA is displaced from the enzyme when three bases have been inserted beyond the lesion42.
Pol ι's structure confers its unique mutagenic signature
Human Pol ι is unique among Y-family polymerases in exhibiting a 105-fold difference in fidelity depending upon the template base. When copying dA, the enzyme efficiently incorporates the correct base dT, with a respectable misincorporation fidelity of 1–2 × 10−4. However, when replicating dT, the enzyme misinserts dG 3–10 times more frequently than the correct dA. How can an enzyme be reasonably accurate when copying dA but highly error-prone when copying dT? The answer lies in the unique active site of Pol ι and, in particular, key residues in the finger domain that restrict positioning of the templating base. Structural studies reveal that template A is driven into a syn conformation (rather than the normal anti conformation) by Gln59 and Lys60 from the finger domain. In such a conformation, there are few hydrogen-bonding opportunities with any incoming nucleotide other than dT in an anti conformation (so-called `Hoogsteen' pairing)43. The same finger domain residues that are important for accurate replication of template dA are also responsible for the high misincorporation seen at template dT44. Side chains protrude into the active site of Pol ι and restrict its size. As a consequence, the template dT is always held in the anti conformation irrespective of the incoming nucleoside triphosphate. While incoming dA is in the syn conformation and exhibits reduced base stacking, misincorporated dG is in an anti conformation, and the mispair is further stabilized by hydrogen bonds on Gln59 (REF. 44). This restrictive feature of the active site facilitates the correct replication of the important oxidative lesion 8-oxoguanine45. 8-oxoguanine can adopt two alternate conformations (anti or syn) and, as a consequence, with most polymerases it can pair equally well with either dC or dA. However, Pol ι restricts it to the syn conformation, preventing the dual coding properties of the lesion by inhibiting the syn–anti conformational equilibrium and promoting formation of the most stable and correct base pair with dC46. These properties may explain the participation of Pol ι in a specialized TLS pathway within the mechanism of base excision repair47.
Base flipping during deoxycytidyl transfer by REV1
The catalytic activity of REV1 is, unusually, restricted to the insertion of dC opposite template dG8,48 or a limited range of lesions, including abasic sites and bulky N2-dG adducts49,50. Although loss of the catalytic activity of REV1 has no discernable defect on the survival of DT40 cells following DNA damage51 or on murine development52, it has recently been shown to be required for the ability of budding yeast to survive exposure to 4-nitroquinolin e-1-oxide (4-NQO)53. Furthermore, the mutation spectrum at abasic sites is altered in mutants lacking the catalytic activity of REV1 generated during immunoglobulin gene somatic hypermutation in vertebrate cells52,54,55 or during abasic site bypass in yeast cells56. This confirms an in vivo role for the dCMP transferase activity of REV1. The crystal structure57 revealed that although the enzyme is able to detect the presence of a template dG and select for an incoming dC, it does not do this by detecting correct base pairing. Instead, the template dG is swung out of the helix and temporarily coordinated by a specialized loop within the little-finger domain. The space previously occupied by the template G, or provided by an abasic site, is instead filled by Arg324 of REV1, which forms hydrogen bonds with the incoming dCTP. This mechanism therefore allows the bypass of bulky dG adducts while retaining the specificity for incorporation of the correct dC base.
Regulation of the Y-family polymerases
The biochemical activities of the Y-family polymerases promote cell survival by allowing the release of replication blocks, but they are also potentially deleterious to the cell. Thus, their access to sites of DNA synthesis must be carefully regulated to avoid unscheduled or extensive mutagenic polymerization. Managing the balance of appropriate recruitment and undesirable activity is a problem facing all organisms, and despite the apparently different approaches taken to regulate translesion polymerases in prokaryotes and eukaryotes, common themes emerge. In all organisms there appears to exist a range of regulatory mechanisms defined at one end by a simple `mass action' approach, in which the number of active polymerase molecules is regulated, through to exquisitely specific control of protein–protein and protein–DNA interactions. In this section we will explore the strategies for regulating the Y-family polymerases from prokaryotes to mammals.
Regulation of cellular concentrations
The conceptually simplest method for regulating the action of the Y-family polymerases is to adjust their cellular concentrations (FIG. 5A). This approach is central to the SOS response in E. coli, during which ~40 genes are upregulated by DNA damage58. Among these genes are those encoding the two Y-family polymerases DinB (Pol IV) and UmuDC (Pol V)58 (FIG. 4). However, transcriptional regulation alone is just one mechanism that E. coli uses to keep the polymerases under control. Error-prone Pol V, which is made up of hetero-trimeric UmuD′2C, is subject to multiple further levels of regulation. First, both UmuD and UmuC proteins are rapidly degraded by the Lon protease59. Any molecules of UmuD that escape proteolysis need to be post-translationally processed by cleavage of the 24 amino-terminal amino acids to a mutagenically active form, UmuD′60. Both UmuD and UmuD′ form homodimers but prefer to heterodimerize, and in the heterodimer context, UmuD′ is specifically degraded by the ClpXP protease59. Thus, mutagenically active homodimeric UmuD′ only accumulates in a cell after severe DNA damage. This delays the appearance of error-prone Pol V (UmuD′2C) until ~45 minutes after the initial DNA damage, thereby giving the cell time to repair the damage via error-free repair mechanisms before utilizing the error-prone TLS polymerase to traverse the damage.
Figure 5. Different levels of regulation of the Y-family polymerases.

A | Different ways of regulating the cellular concentrations of DNA polymerase η (Pol η). This can be achieved by regulation of transcription (Aa) and subsequent mono-ubiquitylation by PIRH2 (REF. 86), which can lead to poly-ubiquitylation and degradation by the proteasome (Ab) or to inactivation following a conformational change83,86 (Ac). The ubiquitin (Ub) can be removed by a de-ubiquitinase enzyme (Dub). B | Accumulation in replication factories. A nucleus in which Pol η is localized in replication factories is shown (top) with a schematic of polymerases and replication forks that are present in the factories (bottom). C | Recruitment of Pol η by ubiquitylated PCNA at a gap behind the fork (Ca) or by REV1 close to the fork (Cb).
By comparison, transcriptional and translational regulation of Y-family polymerases in eukaryotes is less coordinated and does not appear to have the central role it does in E. coli. One exception is the profound cell cycle regulation of Rev1 in S. cerevisiae, in which Rev1 levels increase ~50-fold during G2 phase61. This is achieved mostly at the level of protein stability, as levels of REV1 transcript vary by no more than threefold between G1 phase and G2 phase61,62. However, it is again likely that controlling the number of polymerase molecules provides only a crude degree of regulation and that low level s do not preclude activity.
Subcellular concentrations at replication foci
A further refinement to the regulation of polymerase levels is to concentrate the enzymes locally in the vicinity of distressed replication forks. This approach seems to be particularly important for vertebrate cells, in which the polymerases accumulate in subnuclear foci that can be observed using indirect immunofluorescence or fluorescent protein tagging after DNA damage (FIG. 5B). These foci, which have been termed `replication factories', are thought to contain multiple replication forks, including forks stalled by DNA damage63,64. Over the past few years, subnuclear focus formation has been increasingly used as a surrogate for enzyme function. However, the link between the two is far from clear. For instance, mutations in the carboxy-terminal motifs of Pol η (FIG. 4 and below) drastically reduce its accumulation in foci but have minimal effects on the ability of the protein to complement the UV light sensitivity of an XPV (Pol η-deficient) cell line65. This suggests that there may be two stages to the recruitment of the Y-family polymerases. The first stage promotes an increase in the local concentration of the enzyme in the vicinity of distressed replication forks, whereas the second stage, as discussed in the following section, results in the selection and loading of the polymerase onto the end of the growing DNA strand to initiate TLS. These two processes are interdependent but distinct, and we believe that this distinction is conceptually helpful when examining more specific mechanisms that are likely to control polymerase access to a particular primer terminus, be it at a replication fork or post-replicative gap.
Regulation at stalled forks
As already noted, the relatively simple `mass action' approaches to regulating the Y-family polymerases are unlikely to provide sufficiently tight regulation to be safe. Even formation of high local concentrations within replication factories is likely to be unsatisfactory on its own, as each `factory' is a large structure and contains multiple forks (FIG. 5B). Consequently, it is also necessary to have more specific signals indicating which replication forks require assistance (FIG. 5C). A common feature of impeded replication is the exposure of single-stranded DNA (ssDNA) near the stalled fork, and in both prokaryotes and eukaryotes this is likely to be the common denominator for signalling replication distress. Although the mechanisms linking ssDNA to polymerase activation are quite distinct in E. coli and in eukaryotes, they achieve the same end, namely the focusing of translesion polymerase activity to where it is needed.
Access to the clamp and PCNA ubiquitylation
Fully effective activity by all DNA polymerases requires them to be tethered to the DNA through interaction with the sliding clamp (β-clamp in prokaryotes, PCNA in eukaryotes) (FIGS 1,3). Eukaryotes appear to have exploited this interaction as a mechanism for regulating which type of polymerase — replicative or translesion — has access to the primer terminus. Many proteins potentially interact with PCNA via a motif designated as a PIP box66, and Pol η, Pol ι and Pol κ all possess PIP box motifs in their non-catalytic C-terminal extensions (FIG. 4). The control of which enzymes are bound to PCNA at the primer terminus is partly determined by their widely differing association and dissociation constants, but further specificity is provided by post-translational modification of PCNA itself, principally by ubiquitin and SUMO67. Thus, regulation of these modifications is likely to contribute to controlling the interaction of PCNA with potential client proteins, including the Y-family polymerases.
Replication arrest leads to the exposure of ssDNA, and PCNA mono-ubiquitylation provides a key link between this event and the recruitment of the TLS polymerases. PCNA mono-ubiquitylation is principally mediated by the E3 ubiquitin ligase RAD18 acting in concert with the E2 ubiquitin-conjugating enzyme RAD6 (REFS 68–71), although a number of other proteins have also now been implicated in creation of this modification72,73. Crucially, the activity of RAD18 in ubiquitylating PCNA is stimulated by its association with ssDNA coated with the ssDNA binding protein RPA74.
As well as the PIP boxes, all eukaryotic Y-family polymerases have UBM or UBZ ubiquitin-binding motifs (FIG. 4), which increase their affinity for ubiquitylated PCNA at the site of blocked replication75–77 and thereby increase the probability of them being used to replicate the lesion. The use of the term `probability' in this context is important. The recruitment of polymerases into foci, and presumably also to the primer terminus, is highly dynamic. For Pol η and Pol ι, the residence time in subnuclear foci is less than 1 second, and ubiquitin binding appears to modulate the residence time of individual molecules in the vicinity of the fork78.
Although PCNA ubiquitylation is central to the regulation of TLS, evidence from chicken DT40 cells and mice carrying a K164R mutation in PCNA has revealed that a component of TLS is independent of PCNA ubiquitylation in vertebrates79–82. As discussed below, at least in DT40 cells, this PCNA ubiquitylation-independent TLS seems to be largely dependent on REV1 (REFS 81,83).
The role of REV1
REV1 has an additional, key, non-catalytic role in the control of TLS that in vertebrates is partly independent of PCNA ubiquitylation81,83. The last 100 or so amino acids of vertebrate REV1 bind Pol κ, Pol η and Pol ι84–86, as well as Pol ζ through its REV7 subunit87, and disruption of this domain leads to an effectively null phenotype51. REV1 also interacts with PCNA, suggesting that its role could be as an adaptor between the clamp and the other polymerases, although there is some dispute about which sequences are involved in its interaction with PCNA51,88–90. The role of the two UBMs of REV1 is enigmatic. The REV1 UBMs bind ubiquitin, and one of them is needed for cell survival and mutagenesis following DNA damage91, but PCNA ubiquitylation is not required for REV1 function at the replication fork83. This structural role of Rev1 in coordinating TLS polymerases with PCNA seems to be conserved in S. cerevisiae, although the way in which it interacts with Pol η and Rev7 is different90,92,93 (FIG. 4).
Post-translational modifications
An additional level of complexity is introduced by the observation that the Y-family polymerases can themselves be post-translationally modified by ubiquitylation, phosphorylation and possibly SUMOylation75,91,94–97. The most well-characterize d of these modifications is the ubiquitylation of Pol η (FIG. 5A). Pol η is ubiquitylated in its C terminus95 by the E3 ubiquitin ligase PIRH2 (also known as RCHY1)98 and, interestingly, this ubiquitylation inhibits its interaction with PCNA95,98. Pol η ubiquitylation is reduced following DNA damage, suggesting an additional level of control of Pol η recruitment. It remains to be determined whether this mode of regulation applies to other Y-family polymerases but, in addition, there are reports of damage-induced phosphorylation of Rev1 in S. cerevisiae94,96 and of Pol η in human cells97. Although the biological importance of Rev1 phosphorylation remains to be established, Pol η phosphorylation has a clear role in facilitating TLS97. Taken together, these findings suggest that the regulatory mechanisms of these enzymes are rather more complex than initially thought. Indeed, there are reports of a number of other proteins that have been implicated in the recruitment of Y-family polymerases to the replication fork99–103 and in regulating the ubiquitylation of PCNA104–107.
Polymerase selection
Despite this complexity, it seems that a basic principle for the recruitment of the Y-family polymerases to sites of arrested replication is to control the affinity of the enzymes for PCNA, or for other structures near the fork, through modulation of the number of points of contact. None of these recruitment mechanisms, however, provides any clear specificity in terms of which polymerase is recruited to a specific lesion. This remains an important question. One obvious selector is the lesion itself. It is self-evident that that once a set of polymerases is recruited to the vicinity of an arrested fork, a polymerase can only be utilized if it can accommodate the damaged template-primer in its active site and carry out a catalytic step before dissociating. Selection will thus be determined by normal enzyme kinetic parameters. The various levels of regulation are summarized schematically in FIG. 5.
Timing of lesion bypass
It has been debated for many years whether TLS occurs at the replication fork or whether gaps are left to be sealed post-replicatively behind the fork; early experiments in both E. coli2 and mammalian cells108 suggested the latter and indeed gave rise to the term post-replication repair to describe the process. With the discovery of the Y-family polymerases, there was an assumption that they acted at the replication fork, although this has not been shown experimentally. More recent data have provided direct visual evidence for the existence of post-replicative gaps behind the forks in UV light-irradiated S. cerevisiae109, and elegant genetic studies, in which Rad18, Rev3 or Pol η was induced only in G2 phase, have indicated that TLS can actually occur in G2 phase after the bulk of the DNA has been replicated110,111. It is evident, therefore, that TLS can occur post-replicatively. Although there are likely to be topological differences between TLS at or behind the fork, this does not necessarily imply major mechanistic differences. At the fork, if TLS is to occur, it must take place before the replicative helicase has run sufficiently far ahead to allow downstream re-initiation beyond the lesion, whereas the converse is true for TLS behind the fork. The differences may merely be kinetic — that is, they may depend on which of these processes occurs first — and this will in turn be dependent on the factors that we have described in the previous sections. However, genetic studies in UV light-irradiated DT40 cells have suggested that REV1 is involved in promoting TLS at the fork, whereas PCNA ubiquitylation is required behind the fork (FIG. 5C), suggesting that there are likely to be subtle mechanistic as well as kinetic differences83. This suggestion has been supported by a recent study showing altered mutation spectra originating from TLS taking place in S phase as opposed to G2 phase in mammalian cells112.
`Non-canonical' roles
In addition to their roles in the replication of damaged DNA, Y-family polymerases have been co-opted into a number of other related processes (FIG. 6). During development of the immune response, the antibody genes of vertebrates exhibit a particularly high rate of focused mutagenesis, known as somatic hypermutation, which is driven by activation-induced deaminase (AID)113. Although AID can only deaminate dC to dU, its action gives rise to mutations at all four bases in a series of reactions that crucially depend on the Y-family polymerases114. The dU formed by the action of AID is removed by uracil DNA glycosylase (UNG), resulting in an abasic site. Direct replication of this abasic site involves REV1 and generates mutations at dG-dC base pairs52,54,55. Recognition of dU can also result in the formation of a single-strand gap, and the filling of these gaps by Pol η results in mutations at dA–dT base pairs115,116 (FIG. 6a).
Figure 6. Non-canonical roles of Y-family polymerases.
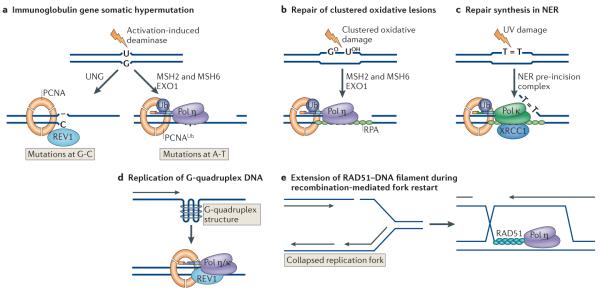
a | The first step in somatic hypermutation at immunoglobulin loci is the deamination of cytosine to uracil by activation-induced deaminase. Removal of uracil by the uracil DNA glycosylase (UNG) generates an abasic site. Replication past the abasic site can result in REV1 inserting C, generating a C-G to G-C transversion. An alternative process involving the mismatch repair proteins MSH2 and MSH6 and exonuclease 1 (EXO1) leads to the formation of a single-strand gap, ubiquitylation of PCNA and recruitment of DNA polymerase η (Pol η), generating mutations at A-T base pairs. b | In a possibly related process, Pol η, recruited by ubiquitylated PCNA, is required for synthesis of gaps generated on removal of clustered oxidative damage (GO, 8-oxoguanine; UOH, 5-hydroxyuracil), in a process that also involves MSH2, MSH6 and EXO1. c | Repair synthesis during nucleotide excision repair (NER) is partially dependent on Pol κ being recruited to the repair site by ubiquitylated PCNA and the scaffold protein XRCC1 (XPG is a nuclease that cleaves 3′ to the damage). d | Y-family polymerases are needed to replicate unusual structures such as G quadruplexes in DNA. e | Homologous recombination involves strand invasion mediated by RAD51 and the formation of a D-loop. Extension of the invading strand in an in vitro system requires Pol η.
Other examples of non-canonical roles occur during excision repair processes. Clusters of damaged bases pose a particular problem to cells, as excision repair can be affected by adjacent base lesions. Pol η has recently been shown to play a part in the repair synthesis of clustered oxidative lesions117 (FIG. 6b). Nucleotide excision repair is the central mechanism by which helixdistorting base lesions, including CPDs, are removed. It involves the recognition and excision of a single-strand tract of DNA containing the damaged base, followed by resynthesis using the undamaged strand as a template. Previously, it had been assumed that this synthesis was carried out by the replicative polymerases, but recently Pol κ was also shown to be involved, particularly during conditions of low deoxyribonucleotide pools118 (FIG. 6c).
REV1 has another non-canonical role (alongside several specialized DNA helicases) in the replication of DNA that is capable of forming a particular secondary structure, the G quadruplex, which can block replicative DNA synthesis119,120. Additionally, Pol η and Pol κ have roles in replicating past unusual structures121, especially at fragile sites122 (FIG. 6d) and, finally, there is evidence for a role for Pol η in some forms of homologous recombination123,124 (FIG. 6e).
Conclusion and perspectives
The past 15 years have seen substantial advances in our understanding of how the Y-family polymerases are able to replicate such a wide range of damaged DNA structures, and we have also learned much about the genetics of how these enzymes are controlled. However, there remain a number of future challenges that need to be addressed if we are to have a full understanding of the mechanism and consequences of the replication of damage d DNA, three of which we highlight here.
Despite the impressive array of structures of the catalytic centres of the Y-family polymerases, there remain a number of important structures that have not yet been solved: for example, that of multi-subunit E. coli Pol V. Most notably, we have only incomplete information on the complexes formed by the Y-family polymerases in vivo and practically no structural information on their non-catalytic extensions (FIG. 4), with the exception of isolated ubiquitin-binding domains77,125,126.
The advent of techniques for visualizing structures below the resolution imposed by the diffraction of visible light127, along with the development of techniques for monitoring the behaviour of single or small ensembles of molecules in living cells, may well provide a parallel approach to understanding the molecular choreography of the Y-family polymerases and their accessory factors.
The Y-family polymerases are responsible for the majority of mutagenic events and hence play a central part in carcinogenesis. However, the only member with a proven link to cancer is Pol η, although polymorphisms in other Y-family polymerases have been linked to the development of certain tumours128. Inhibition of the Y-family polymerases results in a combination of hyper-sensitivity to killing by DNA damaging agents and, in some cases, reduced damage-induced mutation, suggesting that inhibiting their function may not only sensitize tumours to DNA damaging chemotherapeutics but also reduce the incidence of chemotherapy-induced secondary tumours, thus making them attractive cancer drug targets. However, it is only very recent work that has really begun to test these ideas in whole animal models. The work of the Walker and Henman laboratories has shown that inhibition of TLS can not only sensitize a model tumour in vivo to chemotherapy129 but also prevent the acquisition of resistance to chemotherapy130. Further studies of this sort will be essential to determine the promise and limitations of manipulation of TLS as a potential approach in treating tumours, limiting tumour resistance and preventing the development of secondary malignancies following chemotherapy.
Supplementary Material
Damage tolerance
A general term for mechanisms that allow the replication machinery to complete replication despite the presence of DNA damage and without removing the damage.
Replication fork
The point on a replicating DNA molecule where replication is actually taking place and one parental strand generates two daughter strands in a Y-like structure.
Variant form of xeroderma pigmentosum
(XPV). An autosomal recessive disorder that is characterized by increased sensitivity to sunlight and risk of skin cancer, which is due to mutations in DNA polymerase-ε.
dCMP transferase
An enzyme activity that adds dCMP onto the end of a growing DNA chain without the need for a template.
B-family polymerase
On the basis of phylogenetic relationships, DNA polymerases (Pols) can be classified into different families. B-family polymerases include Escherichia coli Pol II and eukaryotic Pol α, Pol δ, Pol ε and Pol ζ.
Cyclobutane pyrimidine dimer
(CPD). The most common ultraviolet light photoproduct, in which two adjacent pyrimidines are joined together by a cyclobutane ring between their 5 and 6 positions. T-T dimers are the most abundant CPDs, although C-T, T-C and C-C dimers are also possible. Several isomers of a CPD can exist. The most common isomer is a cis-syn CPD.
Processivity
The ability of a polymerase to extend a growing DNA chain before it dissociates.
Non-DNA carbon chain
This simply refers to the replacement of a stretch of DNA with a saturated aliphatic chain. It is, of course, not something that is found in vivo, but the ability of translesion synthesis polymerases to bypass such a structure illustrates their broad adaptability when dealing with problematic regions of the genome.
Abasic site
A site in DNA where the base has been lost by hydrolysis of the base–sugar (glycosidic) bond.
B-form
The most common form of the DNA double helix in aqueous environments. In this form, the bases are perpendicular to the axis of the helix.
Syn conformation
Nucleotides can exist in two conformations, anti or syn, depending upon rotation of the base around the glycosidic bond. In order for normal base pairing to occur in DNA, the nucleotide is usually in the anti conformation.
DT40 cells
A B cell line derived from an avian leukosis virus-induced bursal lymphoma in a white leghorn chicken.
SOS response
Following DNA damage to Escherichia coli, and a few related species of bacteria, 40–50 genes are induced, including several DNA repair proteins. This is known as the SOS response.
Replication factories
The clustering of replication and repair proteins into discrete subnuclear foci, which are visible under the light microscope, suggests that multiple replication forks can group together in what has been termed a replication factory.
PCNA
(Proliferating cell nuclear antigen). The trimeric sliding clamp that tethers DNA polymerases and other factors to DNA in a sequence-independent manner, such that they can readily translocate up and down the duplex DNA. The prokaryotic version, which is almost structurally identical, is a dimeric complex called the β-clamp.
Ubiquitin
A 76-amino-acid peptide that can be conjugated to proteins either singly (monoubiquitylation) or in a variety of chains (poly-ubiquitylation). It tags proteins for degradation but is now known to also control cell physiology principally through modulating protein–protein interactions.
SUMO
(Small ubiquitin-like modifier). A post-translation modification related to ubiquitin. Like ubiquitylation, SUMOylation can alter interactions with other proteins.
E3 ubiquitin ligase
An enzyme that catalyses the transfer of ubiquitin from the E2 ubiquitin-conjugating enzyme to the target protein.
E2 ubiquitin-conjugating enzyme
A class of enzymes responsible, in conjunction with an E3 ubiquitin ligase, for attaching ubiquitin to target proteins. Ubiquitin, after activation, is transferred to an E2 ubiquitin-conjugating enzyme, which then interacts with an E3 ubiquitin ligase to effect ubiquitin transfer to the target protein.
RPA
(Replication protein A). Eukaryotic protein that binds specifically to single-stranded DNA.
Activation-induced deaminase
(AID). An enzyme that hydrolytically deaminates cytosines in nucleic acids, resulting in the substitution of cytosines with uracils.
Uracil DNA glycosylase
(UNG). An enzyme that disrupts the glycosidic bond between uracil in DNA and the deoxyribose sugar, generating free uracil and an abasic site in the DNA.
G quadruplex
A class of DNA secondary structure formed in regions of G-rich DNA of the general sequence G3–5-L1–7-G3–5-L1–7-G3–5-L1–7-G3–5 (where L can be any base). The structures are characterized by stacks of planar arrays of four Hoogsteen-bonded dG bases coordinated by a monovalent metal ion.
Acknowledgements
We thank A. Vaisman (US National Institute of Child Health and Human Development (NICHD)/US National Institutes of Health (NIH)) for help in generating figure 2. J.E.S. is funded by the Medical Research Council, Association for International Cancer Research and The Fanconi Anaemia Research Fund; A.R.L. is funded by a Medical Research Council programme grant; and R.W. is funded by the NICHD/NIH Intramural Research Program.
Footnotes
Competing interests statement The authors declare no competing financial interests.
References
- 1.Ohmori H, et al. The Y-family of DNA polymerases. Mol. Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 2.Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J. Mol. Biol. 1968;31:291–304. doi: 10.1016/0022-2836(68)90445-2. [DOI] [PubMed] [Google Scholar]; First demonstration of gaps in newly synthesized DNA in E. coli.
- 3.Radman M. In: Molecular and Environmental Aspects of Mutagenesis. Prakash L, Sherman F, Miller MW, Lawrence CW, Tabor HW, editors. Charles, C. Thomas; Springfield, Illinois: 1974. pp. 128–142. [Google Scholar]
- 4.Lemontt JF. Mutants of yeast defective in mutation induced by ultraviolet light. Genetics. 1971;68:21–33. doi: 10.1093/genetics/68.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kato T, Shinoura Y. Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol. Gen. Genet. 1977;156:121–131. doi: 10.1007/BF00283484. [DOI] [PubMed] [Google Scholar]
- 6.Steinborn G. Uvm mutants of Escherichia coli K12 deficient in UV mutagenesis. I. Isolation of uvm mutants and their phenotypical characterization in DNA repair and mutagenesis. Mol. Gen. Genet. 1978;165:87–93. doi: 10.1007/BF00270380. [DOI] [PubMed] [Google Scholar]
- 7.Lehmann AR, et al. Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV-irradiation. Proc. Natl Acad. Sci. USA. 1975;72:219–223. doi: 10.1073/pnas.72.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson JR, Lawrence CW, Hinkle DC. Deoxycytidyl transferase activity of yeast REV1 protein. Nature. 1996;382:729–731. doi: 10.1038/382729a0. [DOI] [PubMed] [Google Scholar]; Discovery that Rev1 is a dCMP transferase.
- 9.Nelson JR, Lawrence CW, Hinkle DC. Thymine-thymine dimer bypass by yeast DNA polymerase ζ. Science. 1996;272:1646–1649. doi: 10.1126/science.272.5268.1646. [DOI] [PubMed] [Google Scholar]
- 10.Johnson RE, Prakash S, Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science. 1999;283:1001–1004. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- 11.Masutani C, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 12.Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]; References 11 and 12 detail the discovery that the gene encoding Pol η is defective in XPV cells.
- 13.Wagner J, et al. The dinB gene encodes a novel, E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol. Cell. 1999;4:281–286. doi: 10.1016/s1097-2765(00)80376-7. [DOI] [PubMed] [Google Scholar]
- 14.Reuven NB, Arad G, Maor-Shoshani A, Livneh Z. The mutagenesis protein UmuC is a DNA polymerase activated by UmuD′, RecA, and SSB and is specialized for translesion replication. J. Biol. Chem. 1999;274:31763–31766. doi: 10.1074/jbc.274.45.31763. [DOI] [PubMed] [Google Scholar]
- 15.Tang M, et al. UmuD′2C is an error-prone DNA polymerase, Escherichia coli pol V. Proc. Natl Acad. Sci. USA. 1999;96:8919–8924. doi: 10.1073/pnas.96.16.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonald JP, Levine AS, Woodgate R. The Saccharomyces cerevisiae RAD30 gene, a homologue of Escherichia coli dinB and umuC, is DNA damage inducible and functions in a novel error-free postreplication repair mechanism. Genetics. 1997;147:1557–1568. doi: 10.1093/genetics/147.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tissier A, McDonald JP, Frank EG, Woodgate R. polι, a remarkably error-prone human DNA polymerase. Genes Dev. 2000;14:1642–1650. [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature. 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Yuan F, Wu X, Wang Z. Preferential incorporation of G opposite template T by the low-fidelity human DNA polymerase ι. Mol. Cell. Biol. 2000;20:7099–7108. doi: 10.1128/mcb.20.19.7099-7108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogi T, Kato T, Jr, Kato T, Ohmori H. Mutation enhancement by DINB1, a mammalian homologue of the Escherichia coli mutagenesis protein dinB. Genes Cells. 1999;4:607–618. doi: 10.1046/j.1365-2443.1999.00289.x. [DOI] [PubMed] [Google Scholar]
- 21.Gerlach VL, et al. Human and mouse homologs of Escherichia coli DinB (DNA polymerase IV), members of the UmuC/DinB superfamily. Proc. Natl Acad. Sci. USA. 1999;96:11922–11927. doi: 10.1073/pnas.96.21.11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohashi E, et al. Error-prone bypass of certain DNA lesions by the human DNA polymerase κ. Genes Dev. 2000;14:1589–1594. [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, et al. Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woodgate R. Evolution of the two-step model for UV-mutagenesis. Mutat. Res. 2001;485:83–92. doi: 10.1016/s0921-8777(00)00076-8. [DOI] [PubMed] [Google Scholar]
- 25.Shachar S, et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009;28:383–393. doi: 10.1038/emboj.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woodgate R. A plethora of lesion-replicating DNA polymerases. Genes Dev. 1999;13:2191–2195. doi: 10.1101/gad.13.17.2191. [DOI] [PubMed] [Google Scholar]
- 27.Beard WA, Shock DD, Vande Berg BJ, Wilson SH. Efficiency of correct nucleotide insertion governs DNA polymerase fidelity. J. Biol. Chem. 2002;277:47393–47398. doi: 10.1074/jbc.M210036200. [DOI] [PubMed] [Google Scholar]
- 28.Ollis DL, Brick P, Hamlin R, Xuong NG, Steitz TA. Structure of large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature. 1985;313:762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 29.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 30.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc. Natl Acad. Sci. USA. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boudsocq F, et al. Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J. Biol. Chem. 2004;279:32932–32940. doi: 10.1074/jbc.M405249200. [DOI] [PubMed] [Google Scholar]
- 32.Adar S, Livneh Z. Translesion DNA synthesis across non-DNA segments in cultured human cells. DNA Repair. 2006;5:479–490. doi: 10.1016/j.dnarep.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 34.Ling H, Boudsocq F, Woodgate R, Yang W. Snapshots of replication through an abasic lesion; structural basis for base substitutions and frameshifts. Mol. Cell. 2004;13:751–762. doi: 10.1016/s1097-2765(04)00101-7. [DOI] [PubMed] [Google Scholar]
- 35.Kim SR, et al. Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an overexpression of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA. Proc. Natl Acad. Sci. USA. 1997;94:13792–13797. doi: 10.1073/pnas.94.25.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohashi E, et al. Fidelity and processivity of DNA synthesis by DNA polymerase κ, the product of the human DINB1 gene. J. Biol. Chem. 2000;275:39786–39684. doi: 10.1074/jbc.M005309200. [DOI] [PubMed] [Google Scholar]
- 37.Bauer J, et al. A structural gap in Dpo4 supports mutagenic bypass of a major benzo[a]pyrene dG adduct in DNA through template misalignment. Proc. Natl Acad. Sci. USA. 2007;104:14905–14910. doi: 10.1073/pnas.0700717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogi T, Shinkai Y, Tanaka K, Ohmori H. Polκ protects mammalian cells against the lethal and mutagenic effects of polycyclic hydrocarbons. Proc. Natl Acad. Sci. USA. 2002;99:15548–15553. doi: 10.1073/pnas.222377899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, et al. Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase ε, DNA polymerase ι causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007;67:3018–3026. doi: 10.1158/0008-5472.CAN-06-3073. [DOI] [PubMed] [Google Scholar]
- 40.Ziv O, Geacintov N, Nakajima S, Yasui A, Livneh Z. DNA polymerase ζ cooperates with polymerases κ and ι in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients. Proc. Natl Acad. Sci. USA. 2009 Jun 29; doi: 10.1073/pnas.0812548106. doi:10.1073/pnas.0812548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silverstein TD, et al. Structural basis for the suppression of skin cancers by DNA polymerase η. Nature. 2010;465:1039–1043. doi: 10.1038/nature09104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biertümpfel C, et al. Structure and mechanism of human DNA polymerase η. Nature. 2010;465:1044–1048. doi: 10.1038/nature09196. [DOI] [PMC free article] [PubMed] [Google Scholar]; The `molecular splint' structure of Pol η provides the mechanism for correct base-pairing of T-T CPDs.
- 43.Nair DT, Johnson RE, Prakash S, Prakash L, Aggarwal AK. Replication by human DNA polymerase ι occurs by Hoogsteen base-pairing. Nature. 2004;430:377–380. doi: 10.1038/nature02692. [DOI] [PubMed] [Google Scholar]
- 44.Kirouac KN, Ling H. Structural basis of error-prone replication and stalling at a thymine base by human DNA polymerase ι. EMBO J. 2009;28:1644–1654. doi: 10.1038/emboj.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vaisman A, Woodgate R. Unique misinsertion specificity of polι may decrease the mutagenic potential of deaminated cytosines. EMBO J. 2001;20:6520–6529. doi: 10.1093/emboj/20.22.6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirouac KN, Ling H. Unique active site promotes error-free replication opposite an 8-oxo-guanine lesion by human DNA polymerase ι. Proc. Natl Acad. Sci. USA. 2011;108:3210–3215. doi: 10.1073/pnas.1013909108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petta TB, et al. Human DNA polymerase ι protects cells against oxidative stress. EMBO J. 2008;27:2883–2895. doi: 10.1038/emboj.2008.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haracska L, Prakash S, Prakash L. Yeast Rev1 protein is a G template-specific DNA polymerase. J. Biol. Chem. 2002;277:15546–15551. doi: 10.1074/jbc.M112146200. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, et al. Response of human REV1 to different DNA damage: preferential dCMP insertion opposite the lesion. Nucleic Acids Res. 2002;30:1630–1638. doi: 10.1093/nar/30.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Washington MT, et al. Efficient and error-free replication past a minor-groove N2-guanine adduct by the sequential action of yeast Rev1 and DNA polymerase ζ. Mol. Cell. Biol. 2004;24:6900–6906. doi: 10.1128/MCB.24.16.6900-6906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ross AL, Simpson LJ, Sale JE. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 2005;33:1280–1289. doi: 10.1093/nar/gki279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masuda K, et al. A critical role for REV1 in regulating the induction of C:G. transitions and A:T mutations during Ig gene hypermutation. J. Immunol. 2009;183:1846–1850. doi: 10.4049/jimmunol.0901240. [DOI] [PubMed] [Google Scholar]
- 53.Wiltrout ME, Walker GC. The DNA polymerase activity of Saccharomyces cerevisiae Rev1 is biologically significant. Genetics. 2011;187:21–35. doi: 10.1534/genetics.110.124172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ross AL, Sale JE. The catalytic activity of REV1 is employed during immunoglobulin gene diversification in DT40. Mol. Immunol. 2006;43:1587–1594. doi: 10.1016/j.molimm.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 55.Jansen JG, et al. Strand-biased defect in C/G. transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J. Exp. Med. 2006;203:319–323. doi: 10.1084/jem.20052227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim N, Mudrak SV, Jinks-Robertson S. The dCMP transferase activity of yeast Rev1 is biologically relevant during the bypass of endogenously generated AP sites. DNA Repair. 2011;10:1262–1271. doi: 10.1016/j.dnarep.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005;309:2219–2222. doi: 10.1126/science.1116336. [DOI] [PubMed] [Google Scholar]; The structure of Rev1 reveals the mechanism for pairing incoming dCTP with an arginine moiety.
- 58.Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. Ch. 14-15. Am. Soc. for Microbiology; Washington, DC: 1995. [Google Scholar]
- 59.Frank EG, Ennis DG, Gonzalez M, Levine AS, Woodgate R. Regulation of SOS mutagenesis by proteolysis. Proc. Natl Acad. Sci. USA. 1996;93:10291–10296. doi: 10.1073/pnas.93.19.10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nohmi T, Battista JR, Dodson LA, Walker GC. RecA-mediated cleavage activates UmuD for mutagenesis: mechanistic relationship between transcriptional derepression and posttranslational activation. Proc. Natl Acad. Sci. USA. 1988;85:1816–1820. doi: 10.1073/pnas.85.6.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G2/M phase rather than S phase. Proc. Natl Acad. Sci. USA. 2006;103:8971–8976. doi: 10.1073/pnas.0510167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wiltrout ME, Walker GC. Proteasomal regulation of the mutagenic translesion DNA polymerase, Saccharomyces cerevisiae Rev1. DNA Repair. 2011;10:169–175. doi: 10.1016/j.dnarep.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bravo R, Macdonald-Bravo H. Existence of two populations of cyclin/proliferating cell nuclear antigen during the cell cycle: association with DNA replication sites. J. Cell Biol. 1987;105:1549–1554. doi: 10.1083/jcb.105.4.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kannouche P, et al. Domain structure, localization and function of DNA polymerase η, defective in xeroderma pigmentosum variant cells. Genes Dev. 2001;15:158–172. doi: 10.1101/gad.187501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gueranger Q, et al. Role of DNA polymerases η, ι and ζ in UV resistance and UV-induced mutagenesis in a human cell line. DNA Repair. 2008;7:1551–1562. doi: 10.1016/j.dnarep.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 66.Naryzhny SN. Proliferating cell nuclear antigen: a proteomics view. Cell. Mol. Life Sci. 2008;65:3789–3808. doi: 10.1007/s00018-008-8305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 68.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]; Discovery that PCNA is ubiquitylated when replication forks are blocked by damage in S. cerevisiae.
- 69.Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–191. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- 70.Haracska L, Torres-Ramos C, Johnson R, Prakash S, Prakash L. Opposing effects of ubiquitin conjugation and SUMO modification of PCNA on replicational bypass of DNA lesions in Saccharomyces cerevisiae. Mol. Cell. Biol. 2004;24:4267–4274. doi: 10.1128/MCB.24.10.4267-4274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase η with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 72.Terai K, Abbas T, Jazaeri AA, Dutta A. CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol. Cell. 2010;37:143–149. doi: 10.1016/j.molcel.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang S, et al. PCNA is ubiquitinated by RNF8. Cell Cycle. 2008;7:3399–3404. doi: 10.4161/cc.7.21.6949. [DOI] [PubMed] [Google Scholar]
- 74.Davies AA, Huttner D, Daigaku Y, Chen S, Ulrich HD. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein A. Mol. Cell. 2008;29:625–636. doi: 10.1016/j.molcel.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bienko M, et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]; Discovery that all eukaryotic Y-family polymerases have ubiquitin-binding motifs.
- 76.Plosky B, et al. Controlling the subcellular localization of DNA polymerases ι and η via interactions with ubiquitin. EMBO J. 2006;25:2847–2855. doi: 10.1038/sj.emboj.7601178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bomar MG, et al. Unconventional ubiquitin recognition by the ubiquitin-binding motif within the Y family DNA polymerases iota and Rev1. Mol. Cell. 2010;37:408–417. doi: 10.1016/j.molcel.2009.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sabbioneda S, et al. Effect of proliferating cell nuclear antigen ubiquitination and chromatin structure on the dynamic properties of the Y-family DNA polymerases. Mol. Biol. Cell. 2008;19:5193–5202. doi: 10.1091/mbc.E08-07-0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Arakawa H, et al. A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 2006;4:e366. doi: 10.1371/journal.pbio.0040366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Langerak P, Nygren AO, Krijger PH, van den Berk PC, Jacobs H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2007;204:1989–1998. doi: 10.1084/jem.20070902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Szüts D, Marcus AP, Himoto M, Iwai S, Sale JE. REV1 restrains DNA polymerase ζ to ensure frame fidelity during translesion synthesis of UV photoproducts in vivo. Nucleic Acids Res. 2008;36:6767–6780. doi: 10.1093/nar/gkn651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hendel A, et al. PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 2011;7:e1002262. doi: 10.1371/journal.pgen.1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell. 2008;30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 84.Guo C, et al. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003;22:6621–6630. doi: 10.1093/emboj/cdg626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohashi E, et al. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells. 2004;9:523–531. doi: 10.1111/j.1356-9597.2004.00747.x. [DOI] [PubMed] [Google Scholar]
- 86.Tissier A, et al. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase polη and REVl protein. DNA Repair. 2004;3:1503–1514. doi: 10.1016/j.dnarep.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 87.Murakumo Y, et al. Interactions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7. J. Biol. Chem. 2001;276:35644–35651. doi: 10.1074/jbc.M102051200. [DOI] [PubMed] [Google Scholar]
- 88.Guo C, et al. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell. 2006;23:265–271. doi: 10.1016/j.molcel.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 89.de Groote FH, et al. The Rev1 translesion synthesis polymerase has multiple distinct DNA binding modes. DNA Repair. 2011;10:915–925. doi: 10.1016/j.dnarep.2011.04.033. [DOI] [PubMed] [Google Scholar]
- 90.Wood A, Garg P, Burgers PM. A Ubiquitin-binding motif in the translesion DNA polymerase rev1 mediates its essential functional interaction with ubiquitinated PCNA in response to DNA damage. J. Biol. Chem. 2007;282:20256–20263. doi: 10.1074/jbc.M702366200. [DOI] [PubMed] [Google Scholar]
- 91.Guo C, et al. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol. Cell. Biol. 2006;26:8892–8900. doi: 10.1128/MCB.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Acharya N, Haracska L, Prakash S, Prakash L. Complex formation of yeast Rev1 with DNA polymerase η. Mol. Cell. Biol. 2007;27:8401–8408. doi: 10.1128/MCB.01478-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.D'Souza S, Waters L, Walker G. Novel conserved motifs in Rev1 C-terminus are required for mutagenic DNA damage tolerance. DNA Repair. 2008;7:1455–1470. doi: 10.1016/j.dnarep.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sabbioneda S, Bortolomai I, Giannattasio M, Plevani P, Muzi-Falconi M. Yeast Rev1 is cell cycle regulated, phosphorylated in response to DNA damage and its binding to chromosomes is dependent upon MEC1. DNA Repair. 2007;6:121–127. doi: 10.1016/j.dnarep.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 95.Bienko M, et al. Regulation of translesion synthesis DNA polymerase ε by monoubiquitination. Mol. Cell. 2010;37:396–407. doi: 10.1016/j.molcel.2009.12.039. [DOI] [PubMed] [Google Scholar]
- 96.Pages V, Santa Maria SR, Prakash L, Prakash S. Role of DNA damage-induced replication checkpoint in promoting lesion bypass by translesion synthesis in yeast. Genes Dev. 2009;23:1438–1449. doi: 10.1101/gad.1793409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Goehler T, Sabbioneda S, Green CM, Lehmann AR. ATR-mediated phosphorylation of DNA polymerase η is needed for efficient recovery from UV damage. J. Cell Biol. 2011;192:219–227. doi: 10.1083/jcb.201008076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jung YS, Hakem A, Hakem R, Chen X. Pirh2 E3 ubiquitin ligase monoubiquitinates DNA polymerase η to suppress translesion DNA synthesis. Mol. Cell. Biol. 2011;31:3997–4006. doi: 10.1128/MCB.05808-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang M, de Calignon A, Nicolas A, Galibert F. POL32, a subunit of the Saccharomyces cerevisiae DNA polymerase δ, defines a link between DNA replication and the mutagenic bypass repair pathway. Curr. Genet. 2000;38:178–187. doi: 10.1007/s002940000149. [DOI] [PubMed] [Google Scholar]
- 100.Kai M, Wang TS. Checkpoint activation regulates mutagenic translesion synthesis. Genes Dev. 2003;17:64–76. doi: 10.1101/gad.1043203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sabbioneda S, et al. The 9-1-1 checkpoint clamp physically interacts with polζ and is partially required for spontaneous polζ-dependent mutagenesis in Saccharomyces cerevisiae. J. Biol. Chem. 2005;280:38657–38665. doi: 10.1074/jbc.M507638200. [DOI] [PubMed] [Google Scholar]
- 102.Acharya N, Johnson R, Pages V, Prakash L, Prakash S. Yeast Rev1 protein promotes complex formation of DNA polymerase ζ with Pol32 subunit of DNA polymerase δ. Proc. Natl Acad. Sci. USA. 2009;106:9631–9636. doi: 10.1073/pnas.0902175106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tissier A, et al. Crosstalk between replicative and translesional DNA polymerases: PDIP38 interacts directly with Polε. DNA Repair. 2010;9:922–928. doi: 10.1016/j.dnarep.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 104.Huang TT, et al. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nature Cell Biol. 2006;8:341–347. doi: 10.1038/ncb1378. [DOI] [PubMed] [Google Scholar]
- 105.Goehler T, Munoz IM, Rouse J, Blow JJ. PTIP/Swift is required for efficient PCNA ubiquitination in response to DNA damage. DNA Repair. 2008;7:775–787. doi: 10.1016/j.dnarep.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 106.Yang XH, Shiotani B, Classon M, Zou L. Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev. 2008;22:1147–1152. doi: 10.1101/gad.1632808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Terai K, Abbas T, Jazaeri AA, Dutta A. CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol. Cell. 2010;37:143–149. doi: 10.1016/j.molcel.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lehmann AR. Postreplication repair of DNA in ultraviolet-irradiated mammalian cells. J. Mol. Biol. 1972;66:319–337. doi: 10.1016/0022-2836(72)90418-4. [DOI] [PubMed] [Google Scholar]
- 109.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]; Visualization by electron microscopy of ssDNA both at and behind the replication fork in UV light-irradiated cells.
- 110.Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010;465:951–955. doi: 10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Karras GI, Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 2010;141:255–267. doi: 10.1016/j.cell.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 112.Diamant N, et al. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res. 2012;40:170–180. doi: 10.1093/nar/gkr596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 114.Sale JE, et al. Timing matters: error-prone gap filling and translesion synthesis in immunoglobulin gene hypermutation. Phil. Trans. R. Soc. B. 2009;364:595–603. doi: 10.1098/rstb.2008.0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zeng X, et al. DNA polymerase η is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nature Immunol. 2001;2:537–541. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 116.Delbos F, Aoufouchi S, Faili A, Weill JC, Reynaud CA. DNA polymerase η is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 2007;204:17–23. doi: 10.1084/jem.20062131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zlatanou A, et al. The hMsh2-hMsh6 complex acts in concert with monoubiquitinated PCNA and pol η in response to oxidative DNA damage in human cells. Mol. Cell. 2011;43:649–662. doi: 10.1016/j.molcel.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 118.Ogi T, et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell. 2010;37:714–727. doi: 10.1016/j.molcel.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 119.Sarkies P, Reams C, Simpson LJ, Sale JE. Epigenetic instability due to defective replication of structured DNA. Mol. Cell. 2010;40:703–713. doi: 10.1016/j.molcel.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sarkies P, et al. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2011 Oct 22; doi: 10.1093/nar/gkr868. doi:10.1093/nar/gkr868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Betous R, et al. Role of TLS DNA polymerases η and κ in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 2009;48:369–378. doi: 10.1002/mc.20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rey L, et al. Human DNA polymerase η is required for common fragile site stability during unperturbed DNA replication. Mol. Cell. Biol. 2009;29:3344–3354. doi: 10.1128/MCB.00115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kawamoto T, et al. Dual roles for DNA polymerase η in homologous DNA recombination and translesion DNA synthesis. Mol. Cell. 2005;20:793–799. doi: 10.1016/j.molcel.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 124.McIlwraith MJ, et al. Human DNA polymerase η promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol. Cell. 2005;20:783–792. doi: 10.1016/j.molcel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 125.Bomar MG, Pai MT, Tzeng SR, Li SS, Zhou P. Structure of the ubiquitin-binding zinc finger domain of human DNA Y-polymerase η. EMBO Rep. 2007;8:247–251. doi: 10.1038/sj.embor.7400901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Burschowsky D, et al. Structural analysis of the conserved ubiquitin-binding motifs (UBMs) of the translesion polymerase ι in complex with ubiquitin. J. Biol. Chem. 2011;286:1364–1373. doi: 10.1074/jbc.M110.135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Huang B, Bates M, Zhuang X. Super-resolution fluorescence microscopy. Ann. Rev. Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Luedeke M, et al. Predisposition for TMPRSS2-ERG fusion in prostate cancer by variants in DNA repair genes. Cancer Epidemiol. Biomarkers Prev. 2009;18:3030–3035. doi: 10.1158/1055-9965.EPI-09-0772. [DOI] [PubMed] [Google Scholar]
- 129.Doles J, et al. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc. Natl Acad. Sci. USA. 2010;107:20786–20791. doi: 10.1073/pnas.1011409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xie K, Doles J, Hemann MT, Walker GC. Error-prone translesion synthesis mediates acquired chemoresistance. Proc. Natl Acad. Sci. USA. 2010;107:20792–20797. doi: 10.1073/pnas.1011412107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bridges BA, Woodgate R. The two-step model of bacterial UV mutagenesis. Mutat. Res. 1985;150:133–139. doi: 10.1016/0027-5107(85)90110-1. [DOI] [PubMed] [Google Scholar]
- 132.Wang M, et al. Insights into base selectivity from the 1.8 Ă resolution structure of an RB69 DNA polymerase ternary complex. Biochemistry. 2011;50:581–590. doi: 10.1021/bi101192f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jiang Q, Karata K, Woodgate R, Cox MM, Goodman MF. The active form of DNA polymerase V is UmuD′2C-RecA-ATP. Nature. 2009;460:359–363. doi: 10.1038/nature08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.