

Abstract
Mitochondria are essential organelles that regulate cellular energy homeostasis and cell death. The removal of damaged mitochondria through autophagy, a process called mitophagy, is thus critical for maintaining proper cellular functions. Indeed, mitophagy has been recently proposed to play critical roles in terminal differentiation of red blood cells, paternal mitochondrial degradation, neurodegenerative diseases, and ischemia or drug-induced tissue injury. Removal of damaged mitochondria through autophagy requires two steps: induction of general autophagy and priming of damaged mitochondria for selective autophagic recognition. Recent progress in mitophagy studies reveals that mitochondrial priming is mediated either by the Pink1-Parkin signaling pathway or the mitophagic receptors Nix and Bnip3. In this review, we summarize our current knowledge on the mechanisms of mitophagy. We also discuss the pathophysiological roles of mitophagy and current assays used to monitor mitophagy.
Keywords: autophagy, mitophagy, Nix, Parkin
Introduction
Macroautophagy (hereafter referred to as autophagy) is a genetically programmed, evolutionarily conserved catabolic process that degrades cellular proteins, and damaged or excessive organelles through the formation of a double-membrane structure known as the autophagosome (Mizushima, 2007; Nakatogawa et al., 2009; Yang and Klionsky, 2010). Autophagosomes then fuse with lysosomes to form autolysosomes where the enveloped contents are degraded. Although the exact process of autophagosome formation is still not completely known, >30 autophagy-related genes (Atg) have been identified in yeast, and most of them have mammalian homologues that participate in autophagy or an autophagy-related process (Klionsky et al., 2003; Nakatogawa et al., 2009). Most Atg proteins form multi-molecule complexes to regulate autophagosome formation, which include (1) the ULK1 kinase complex, (2) the Beclin 1-VPS34 class III phosphoinositide 3-kinase (PI3K) complex, (3) the Atg9-Atg2-Atg18 complex, and (4) the Atg5-Atg12-Atg16 and Atg8/LC3 conjugation systems. The important roles of these Atg complexes in regulating autophagy have been extensively reviewed (Ravikumar et al., 2010; Ding et al., 2011; Mizushima and Komatsu, 2011) and will not be discussed here.
Although it was initially thought that autophagy was a non-selective bulk degradation pathway, it is now widely accepted that there are two types of autophagy, non-selective and selective. In response to starvation or nutrient deprivation, non-selective autophagy is activated to provide cells with essential amino acids and nutrients for their survival. In contrast, selective autophagy occurs to specifically remove damaged or excessive organelles or protein aggregates even under nutrient-rich conditions. Thus far, several cargo-specific autophagy processes have been reported in organisms from yeast to mammals, including specific removal of peroxisomes (pexophagy) (Komatsu et al., 2006), endoplasmic reticulum (erphagy) (Reef et al., 2006), ribosomes (ribophagy) (Kraft et al., 2008; MacIntosh and Bassham, 2011), lipid droplets (lipophagy) (Singh et al., 2009; Ding et al., 2010a), invading microbes (xenophagy) (Levine, 2005; Kudchodkar and Levine, 2009), and protein aggregates (Ding et al., 2007; Komatsu et al., 2007; Ding and Yin 2008; Matsumoto et al., 2011). In addition, a well-studied type of selective autophagy is mitophagy, which selectively removes damaged or excessive mitochondria.
De Duve and Wattiaux (1966) first coined the term ‘autophagy’ to describe the process characterized by the cytoplasm-sequestered double-membrane-delimited vesicles, or autophagosomes. When they administrated rats with glucagon, they also observed that there was an increased number of autophagosomes that had sequestrated mitochondria. Xue et al. (1999) also found that mitochondria were often enveloped by double-membrane autophagosomes in nerve growth factor-deprived culture neurons. The same group also found that the entire cellular mitochondria could disappear, likely through autophagy, when the cells were treated with apoptotic stimuli in the presence of general caspase inhibitors. Later on, Lemasters and his colleagues demonstrated that when cultured rat hepatocytes were treated with glucagon in the absence of serum, depolarized mitochondria were often found colocalized with acidic lysosomes and GFP-LC3-positive autophagosomes (Rodriguez-Enriquez et al., 2006; Kim et al., 2007). They used the term ‘mitophagy’ for the selective autophagic process for mitochondria (Rodriguez-Enriquez et al., 2006). There has been rapid progress in the study of mitophagy in the past few years, which has led to a greater understanding of the molecular mechanisms, the pathophysiological role in development and in diseases, and the analytic approaches, which will be reviewed here.
Mechanisms of mitophagy
Mitophagy in yeast
It should be noted that although mitophagy was first observed in rat hepatocytes, the molecular machinery and pathways regulating mitophagy were best documented in yeast. The first evidence that mitophagy is genetically controlled in yeast was reported by Kissova et al. (2004), who identified that Uth1p is required for the autophagic degradation of mitochondria. Uth1p is a member of the so-called SUN family, which was initially found in a genetic screen aiming to identify proteins involved in the regulation of the yeast life span (Austriaco, 1996). Uth1p is mainly localized in the mitochondrial outer membrane and is required for the removal of excessive mitochondria during starvation (Kissova et al., 2004). In addition to Uth1p, it was reported that Aup1p, one of a family of protein phosphatase homologs that localizes to the mitochondrial intermembrane space, is also required for efficient mitophagy in stationary-phase cells (Tal et al., 2007). Interestingly, neither Uth1p nor Aup1p is required for starvation-induced autophagy, suggesting their specificity for mitophagy. Furthermore, deletion of Mdm38, a mitochondrial inner membrane protein that regulates the mitochondrial K+/H+ exchange system, leads to mitochondrial depolarization and fragmentation followed by degradation in the vacuole (Nowikovsky et al., 2007). Blocking fission by the deletion of a profission gene dnm1 prevented mitochondrial fragmentation and also mitophagy. Thus, it appears that mitochondrial fragmentation facilitates mitophagy, a phenomenon that is also observed in mammalian cells (see below). Because there were no externally added drugs or physiological stresses placed on the mdm38Δ cells, this study also suggests that damaged mitochondria themselves could be sufficient to trigger mitophagy. It should also be noted that deletion of Mdm38 leads to mitochondrial depolarization followed by mitophagy. However, whether depolarization is truly required for mitophagy may require further investigation, as treatment of a mitochondria uncoupler, carbonyl cyanide m-chlorophenylhydrazone (CCCP), did not induce mitophagy in wild-type yeast (Wang and Klionsky, 2011).
More recently, two independent groups using genome-wide screening reported that Atg32 is a mitochondrial receptor for mitophagy in the yeast (Kanki et al., 2009; Okamoto et al., 2009). Similar to Uth1p and Aup1p, Atg32 is necessary for mitophagy but not for non-specific autophagy. Atg32 is a 59-kDa protein that has an intramitochondrial domain at its C terminus and a tetrapeptide sequence WQAI in its cytoplasmic domain, which is critical for its interaction with Atg8. The tetrapeptide sequence WXXL is also known to be present in several other Atg8- or LC3-binding partners important for selective autophagy. Atg32 also binds to Atg11, a selective autophagy receptor important for cargo selection (Yorimitsu and Klionsky, 2005) and autophagosome formation by acting as a scaffold protein, which further recruits other autophagy proteins to form the autophagosome. Indeed, these studies found that Atg11 is also required for mitophagy (Kanki et al., 2009; Okamoto et al., 2009). In contrast, Atg19, which is specifically involved in the Cvt pathway, is not required for mitophagy.
It was found that Atg32 is phosphorylated after nitrogen starvation, and the phosphorylation of Atg32 is required for mitophagy (Aoki et al., 2011). Phosphorylation of Atg32 promotes Atg32-Atg11 interaction but does not affect Atg32-Atg8 interaction (Aoki et al., 2011). In yeast, Hog1 is a mitogen-activated protein kinase (MAPK), whose activation is dependent on its phosphorylation by Pbs2, an MAPK kinase. Deletion of either Hog1 or Pbs2 leads to inhibition of Atg32 phosphorylation as well as mitophagy. Unlike Hog1 and Pbs2, which function specifically for mitophagy, Slt2, another MAPK kinase involved in the protein kinase C-cell wall integrity kinase signaling pathway in yeast, is required for the degradation of both mitochondria and peroxisomes (Mao et al., 2011). Slt2, but not Hog1, affects the recruitment of mitochondria to the phagophore assembly site, a critical step in the packaging of cargo for selective degradation (Mao et al., 2011).
To note, the expression of Atg32 is increased in respiratory growth conditions (not fermentable conditions), and N-acetylcysteine (NAC), a precursor of the antioxidant glutathione and a scavenger for free radicals, suppresses Atg32 expression and mitophagy (Okamoto et al., 2009). However, it is not clear whether Atg32-dependent mitophagy is directly mediated by oxidative stress under respiratory conditions because reactive oxygen species (ROS) levels were not determined in this study (Okamoto et al., 2009). Interestingly, it was reported in another study that NAC suppresses mitophagy by fueling the cellular glutathione (GSH) pool rather than scavenging for free radicals (Deffieu et al., 2009). Deprivation of nitrogen in yeast was associated with an early decrease of GSH before mitophagy. How exactly GSH can modulate mitophagy in yeast is not clear, but it is possible a decreased GSH level may alter mitochondrial function and subsequently lead to mitochondrial damage and mitophagy. Moreover, it is still unknown what increased the expression of Atg32 under respiratory conditions. It would be interesting to determine whether there is also a depletion of GSH levels under respiratory conditions or whether other antioxidants in addition to the GSH precursor NAC would also suppress Atg32 expression and mitophagy.
Mitophagy in mammalian cells
Mammalian homologues of Atg32, Uth1p, and Aup1p have not been found. In addition, mammalian mitophagy presents some unique features that may also be specific to mammals. The molecular mechanisms and signaling pathways behind mitophagy regulation only begin to be revealed in mammalian cells. In general, it seems that multiple molecules could be involved and there are both autophagy-dependent and -independent pathways that regulate mitochondrial homeostasis. The possible mitophagy pathways are summarized in Figure 1 and discussed in the following sections.
Figure 1. Proposed models for mitophagy in mammalian cells.
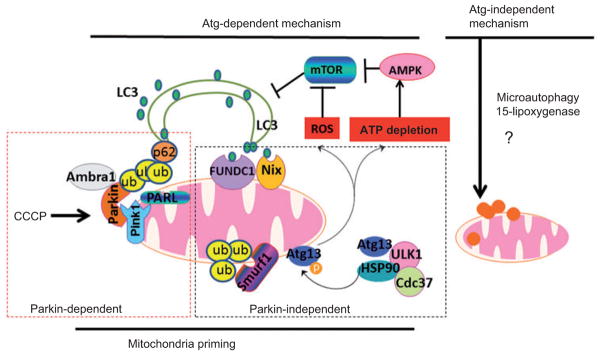
We propose a two-step mitophagy model in mammalian cells: the induction of canonic Atg-dependent macroautophagy and mitochondrial priming. The induction of canonic autophagy requires Atg proteins and furthermore involves mTOR suppression mediated by mitochon-drial damage-generated ROS production and ATP depletion-mediated AMPK activation. Priming of mitochondria is mediated by multiple mechanisms that could be Parkin dependent or Parkin independent. In the presence of Parkin, one common mechanism is that mitochondrial depolarization (e.g., following CCCP treatment) results in impaired PARL-mediated Pink1 cleavage, leading to Pink1 stabilization and Parkin recruitment to the mitochondria. Mitochondria-localized Parkin promotes ubiquitination of outer membrane proteins, which may either be degraded through the proteasome or serve as binding partners for p62. p62 may in turn act as an adaptor molecule through direct interaction with LC3 to recruit autophagosomal membranes to the mitochondria. Parkin can also interact with Ambra1, which in turn activates the PI3K complex around mitochondria to facilitate selective mitophagy. For the Parkin-independent mechanism, damaged mitochondria (particularly under hypoxia conditions) may increase FUNDC1 and Nix expression, which may in turn recruit autophagosomes to mitochondria by direct interaction with LC3 through their LIR domains. Upon mitochondrial depolarization, Smurf1 also targets mitochondria to promote mitophagy, most likely through the ubiquitination of mitochondrial proteins. Hsp90-Cdc37 stabilizes and activates Ulk1, which further phosphorylates Atg13. Phosphorylated Atg13 is recruited to damaged mitochondria to promote mitophagy. Atg-independent mitophagy is less understood, but 15-lipoxygenase has been shown to promote mitochondrial degradation. Direct lysosomal invagination or interaction with damaged mitochon-dria (microautophagy) could also play a role.
Mitochondrial permeability transition
Mitochondrial permeability transition (MPT) has been proposed to be responsible for the mitophagy of depolarized mitochondria in mammalian cells (Lemasters et al., 2002). MPT regulates apoptosis and necrosis in mammalian cells, and is mediated by the permeability transition (PT) pore, which is composed of voltage-dependent anion channel (VDAC) in the outer membrane, the adenine nucleotide translocator (ANT) in the inner membrane, and cyclophilin D (CypD) in the matrix space. In addition, creatine kinase (found in the intermembrane space), hexokinase (outer membrane), and Bax (outer membrane) are thought to be associated with PT pores. Mitochondria become permeable to all solutes up to a molecular mass of about 1500 Da after the onset of MPT, which can lead to mitochondrial depolarization. MPT can trigger the release of proapoptotic mitochondrial intermembrane space proteins into the cytosol, which include cytochrome c, apoptosis-inducing factor, and Smac/Diablo. Cyclosporin A (CsA), an immunosuppressant compound, inhibits MPT through interaction with CypD (Lemasters et al., 2002). When cultured hepatocytes were deprived of nutrition, depolarized mitochondria could be found to colocalize with acidic vesicles, which was suppressed by CsA (Rodriguez-Enriquez et al., 2006). Some of the mitochondria were enveloped by GFP-LC3-positive autophagosomes (Kim et al., 2007). In addition to nutrient deprivation, mitophagy in hepatocytes was also induced by laser-induced photodamage or when hepatocytes underwent differentiation and cytoplasm remodeling under prolonged culture conditions (Kim et al., 2007; Rodriguez-Enriquez et al., 2009). Finally, increased numbers of depolarized mitochondria and mitophagy were observed in fibroblasts derived from patients deficient in coenzyme (CoQ), a small lipophilic molecule critical for the transport of electrons from complexes I and II to complex III in the mitochondrial respiratory chain. Interestingly, CoQ supplementation or CsA treatment attenuated mitophagy in these cells, suggesting that these mitochondrial defects are specifically induced by CoQ deficiency, and that MPT can regulate this type of mitochondrial damage as well as mitophagy (Rodriguez-Hernandez et al., 2009).
Theoretically, removal of damaged mitochondria requires the formation of separate autophagosomes, which may then mobilize to the site where the damaged mitochondria are located. However, the effects on general autophagy by CsA were not determined in these studies. Paradoxically, CsA has been shown to induce general autophagy in cultured primary human renal tubular cells, and in rat kidneys (Pallet et al., 2008) and in the muscle of mice deficient in collagen VI in vivo (Grumati et al., 2010). The mechanisms for CsA-induced autophagy are thought to be mediated by endoplasmic reticulum stress and the regulation of Beclin 1 and Bnip3 expression. Therefore, it is likely that the suppression of mitophagy by CsA observed in these studies may be mainly due to the inhibition of mitochondrial damage and subsequent mitochondrial depolarization by CsA, rather than through general autophagy induction.
Mitochondrial fragmentation
Mitochondria are dynamic organelles constantly undergoing fusion and fission (Westermann, 2010). Mitochondrial fusion is mediated by the profusion genes mitofusin (Mfn) 1 and 2, and optic atrophy 1 (OPA1). Mfn 1 and 2 are large GTPases located on the outer mitochondrial membrane, whereas OPA1 is a dynamin-related GTPase that is essential for inner membrane fusion. Mammalian OPA1 has eight isoforms that are generated by alternative splicing and alternative processing at two cleavage sites located between the N-terminal transmembrane domain and the first heptad repeat. It has been suggested that the rhomboid-related protease, presenilins-associated rhomboid-like (PARL) (Cipolat et al., 2006), or the AAA protease in the matrix, paraplegin, may be responsible for OPA1 processing (Ishihara et al., 2006). However, their roles in regulating OPA1 processing have been challenged because normal OPA1 processing was found in mouse embryonic fibroblasts (MEFs) lacking either PARL or paraplegin (Duvezin-Caubet et al., 2007). OPA1 isoforms are constitutively cleaved by the intermembrane space AAA protease Yme1 to generate the short and long forms of OPA1 (S- and L-OPA1) under normal conditions (Griparic et al., 2007). When cells were treated with CCCP, a mitochondria uncoupler that depolarizes mitochondria, L-OPA1 was further cleaved by an inducible protease OMA1, which is a metalloprotease with a zinc-binding motif. This cleavage resulted in mitochondrial fragmentation by preventing mitochondrial fusion (Ehses et al., 2009; Head et al., 2009).
Similar to mitochondrial fusion, mitochondrial fission requires a dynamin-related GTPase (Drp1) in mammals. Drp1 is a cytosolic protein, but it can be recruited to the surface of mitochondria where it interacts with a partner protein, mitochondrial fission 1 (Fis1), through the adaptor proteins Mdv1 and Caf4 to trigger mitochondrial fission in yeast (Griffin et al., 2005; Westermann, 2010). The mammalian homologue of yeast Fis1 has been identified, which suggests an evolutionarily conserved mechanism of mitochondrial fission (Smirnova et al., 2001; Yoon and Koob, 2003); however, homologues of Mdv1 and Caf4 have not yet been determined. More recent evidence suggests that mitochondria fission factor, a tail-anchored mitochondrial outer membrane protein that physically interacts with Drp1 but not Fis1, is an essential factor for Drp1-mediated mitochondria fission in mammalian cells (Gandre-Babbe and van der Bliek 2008; Otera et al., 2010). Moreover, mitochondria and endoplasmic reticulum are often tightly associated and in physical contact. A recent study reported that mitochondrial fission dynamics, such as Drp1, are localized at the endoplasmic reticulummitochondria contact sites and that the endoplasmic reticulum may play a direct role in the process of mitochondrial fission (Friedman et al., 2011).
It has been suggested that fragmented mitochondria are more readily taken up by autophagosomes due to their smaller size. In nitric oxide-treated cultured cortical neurons, Fis1 was found to be involved in mitophagy (Barsoum et al., 2006). Gomes and Scorrano (2008) found that overexpression of Fis1 itself can induce general autophagy in HeLa cells as demonstrated by increased YFP-LC3 puncta and p62 degradation. Overexpressing a dominant-negative form of Fis1, which had ablated the first α-helix, induced autophagy and large mitochondrial structures. Moreover, a conserved mutation in Fis1 (hFis1K148R) induced mitochondrial fission but was unable to induce mitochondrial dysfunction and did not induce autophagy (Gomes and Scorrano, 2008). These observations suggest that mitochondrial dysfunction, rather than mitochondrial fragmentation, is responsible for the induction of autophagy. Two independent groups recently reported that during starvation, cellular cyclic AMP levels increase and protein kinase A is activated, which in turn phosphorylates Drp1 to prevent its translocation to mitochondria (Gomes et al., 2011; Rambold et al., 2011). As a result, mitochondria are highly elongated in these starved cells. More importantly, elongated mitochondria are spared from autophagic degradation, further supporting the notion that mitochondrial fragmentation can facilitate mitophagy. Using a photo-labeling approach, Twig et al. (2008) found that mitochondria went through constant cycles of fusion and fission, and fission events generated two subsets of daughter mitochondria with either increased or decreased membrane potential. They further found that the daughter mitochondria with higher membrane potential would proceed to fusion, whereas the depolarized daughter mitochondria were unable to proceed to the fusion process and were removed by mitophagy. Overexpression of OPA1 led to increased mitochondria fusion and decreased mitophagy. Therefore, it seems that mitochondrial fission is essential for mitophagy.
Nix and Bnip3
Bnip3 (Bcl-2/E1B-19 kDa interacting protein 3) was first identified in a yeast two-hybrid screen that interacted with adenovirus E1B 19 kDa (Boyd et al., 1994). Bnip3 contains a Bcl-2 homology 3 (BH3) domain and a carboxyl-terminal transmembrane domain, and acts as a pro-apoptotic mitochondrial protein (Chen et al., 1997; Yasuda et al., 1998). Nix/Bnip3L is a homolog of Bnip3, and they share 53–56% amino acid sequence identity (Chen et al., 1999). Both Bnip3 and Nix are inserted into the outer mitochondrial membrane through their C-terminal transmembrane domains, while their N termini are exposed to the cytoplasm. Unlike other BH3-only pro-apoptotic proteins, the transmembrane domain of Bnip3, but not its BH3 domain, is required for mitochondrial targeting and proapoptotic function (Chen et al., 1997; Ray et al., 2000). Moreover, Bnip3 has also been implicated in necrosis and autophagic cell death (Vande Velde et al., 2000; Daido et al., 2004; Azad et al., 2008).
It should be noted that although they are expressed in the liver, skeletal muscle, heart, kidney, and brain even under physiological conditions, Bnip3 and Nix are not ubiquitously expressed under normal conditions (Galvez et al., 2006). Under hypoxia conditions, both Bnip3 and Nix are highly induced. Their expression levels are regulated by hypoxia-inducible factor-1 (HIF-1), and HIF-1 binding to a site on the Bnip3 promoter is enhanced (Bruick, 2000; Guo et al., 2001) in cells during hypoxia. Hypoxia induces mitophagy in cultured MEF cells, and this process requires the HIF-1-dependent expression of BNIP3 (Zhang et al., 2008). As such, under conditions of hypoxia, mitophagy serves as an adaptive metabolic response to prevent increased levels of ROS through removal of damaged mitochondria, and this in turn mitigates cell death (Zhang et al., 2008). In addition to HIF-1, the expression of Bnip3 is also regulated by the Foxo3 transcription factor (Mammucari et al., 2007). During starvation, Foxo3 activity is increased and binds to Bnip3 and Nix promoter regions, which increase their expression in muscle cells (Mammucari et al., 2007). Nix is highly expressed during erythroid differentiation (Schweers et al., 2007), and coincidentally, Foxo3 expression, nuclear localization, and transcriptional activity are also increased (Marinkovic et al., 2007). However, the role of Foxo3 in mitophagy in these models has not been determined.
During red blood cell differentiation and maturation, reticulocytes completely eliminate their mitochondria. In Nix-deficient mice, mitochondrial clearance in reticulocytes is significantly inhibited or retarded, suggesting that Nix is required for the selective elimination of mitochondria (Schweers et al., 2007; Sandoval et al., 2008). Treatment with a mitochondria uncoupler (e.g., CCCP) or a BH3 mimetic (e.g., ABT-737) induces depolarization and restores the sequestration of mitochondria into autophagosomes in Nix-deficient erythroid cells, suggesting that one mechanism for Nix to trigger mitochondrial clearance is likely due to its role in inducing mitochondria depolarization (Sandoval et al., 2008). This notion may also help explain why MPT is involved in hepatocyte mitophagy, because in most cases, the onset of MPT leads to mitochondrial depolarization. We also found that Nix-deficient MEFs are relatively resistant to CCCP-induced mitochondrial depolarization and general autophagy induction (Ding et al., 2010b). Therefore, mitochondrial depolarization seems to be a consistent feature in mammalian mitophagy, although this does not seem to be as crucial in yeast (Tolkovsky, 2009). Nix may have a dual role in what we call the ‘two-step mitophagy’ model. First, Nix can stimulate the induction of autophagy by dissociating the Bcl-2-Beclin 1 complex through competitively binding to Bcl-2 by means of its BH3 domain (Bellot et al., 2009).
In addition, Nix may promote ROS generation. Nix-deficient MEFs have less production of ROS (Ding et al., 2010b). ROS have been implicated in the activation of autophagy (Scherz-Shouval and Elazar, 2011), although the actual mechanisms are not clear. Our studies have indicated that ROS could cause inhibition of the mTOR signaling pathway and thus the induction of autophagy (Ding et al., 2010a,b). Other studies have indicated that ROS could inactivate Atg4 to reduce its de-conjugation activity and consequently increase LC3 association with the autophagosomal membrane (Scherz-Shouval et al., 2007).
The second role of Nix in mitophagy is in the priming of mitochondria for autophagic recognition. Nix is required in CCCP-induced Parkin recruitment to the mitochondria due to its role in mitochondrial depolarization (Ding et al., 2010b). Nix can also directly interact with LC3 and GABARAP (Schwarten et al., 2009; Novak et al., 2010), and may thus be directly involved in the recruitment of the autophagy machinery to the damaged mitochondria.
FUNDC1
In addition to Nix, an outer mitochondrial membrane protein, FUNDC1, has recently been reported to have an essential role in hypoxia-induced mitophagy (Liu et al., 2012). Overexpression of FUNDC1 is sufficient to induce mitophagy in several cell lines, including HeLa, MCF7, and MEFs. FUNDC1-induced mitophagy depends on Atg5, but not Beclin 1, suggesting involvement of a Beclin 1-independent mechanism. FUNDC1 seems to play a specific role in hypoxia-induced mitophagy because knockdown of FUNDC1 has no effect on starvation-induced mitophagy and only has a modest role in FCCP-induced mitophagy. Similar to Nix and yeast Atg32, FUNDC1 interacts with LC3 through the characteristic LIR motif Y(18)XXL(21), which is essential for FUNDC1-mediated mitophagy because mutation of this site inhibits mitophagy. Under normoxia conditions, FUNDC1 is constantly phosphorylated by the Src kinase. Src can be inactivated under hypoxia conditions, leading to the dephosphorylation of FUNDC1, which has a higher affinity binding with LC3 than the phosphorylated form. It seems that the mechanisms of FUNDC1-induced mitophagy are different from Nix- or Bnip3-mediated mitophagy, although all of them are associated with hypoxia conditions. Unlike Nix, which is transcriptionally upregulated through HIF or Foxo3, the mRNA level of FUNDC1 decreases under hypoxia conditions (Guo et al., 2001). Other differences between these mechanisms include weaker binding of Nix with LC3B compared with FUNDC1 (Novak et al., 2010). Moreover, overexpression of Nix or Bnip3 promotes mitochondria-mediated cell death; however, FUNDC1 has no impact on cell viability under hypoxia conditions. Interestingly, it should be noted that the cell lines used in this study have very low expression levels of Parkin, suggesting that FUNDC1-mediated mitophagy could be Parkin independent.
ULK1 and Atg7
Similar mitochondrial clearance defects were observed in Atg7- or ULK1-deficient mice, further confirming the involvement of autophagy in this process (Kundu et al., 2008; Zhang et al., 2009). It should be noted that in all these genetic knockout mouse models, lack of Nix, ULK1, or Atg7 only caused a delayed clearance of mitochondria in reticulocytes; ultimately, the majority of red cells clear their mitochondria and other organelles (Schweers et al., 2007; Kundu et al., 2008; Zhang et al., 2009). However, it seems that Nix plays a greater role than that of ULK1or Atg7, because the extent of mitochondrial clearance was more significantly affected by the loss of Nix than the loss of ULK1 or Atg7 (Kundu et al., 2008; Sandoval et al., 2008; Zhang et al., 2009). In contrast, it was reported that mitochondrial clearance in the reticulocytes of Atg5-knockout mice is normal (Matsui et al., 2006).
Pink1-Parkin signaling pathway
In certain forms of early onset of Parkinson’s disease, there are two genes often found to be mutated, Pink1 and Parkin, which are therefore suspected to be pathogenic. Pink1 encodes the PTEN-induced putative kinase, a serine/threonine kinase with a mitochondrial targeting sequence, whereas Parkin is an E3 ubiquitin ligase. Genetic studies in Drosophila suggest that Pink1 and Parkin may function in the same pathway where Pink1 is upstream of Parkin. This notion is supported by the evidence that genetic deletion of both Pink1 and Parkin leads to the same expressed phenotype as deletion of either one of them. Overexpression of Pink1 failed to rescue the Parkin-knockout phenotypes, whereas overexpression of Parkin partially rescued the phenotype of Pink1 knockout in Drosophila (Clark et al., 2006; Park et al., 2006; Yang et al., 2006).
It is well known that mitochondrial dysfunction is a prominent phenomenon in the pathogenesis of Parkinson’s disease. Therefore, removal of damaged mitochondria through mitophagy would have a great impact in this disease. Narendra et al. (2008) was the first to demonstrate that Parkin can play a critical role in mitophagy in mammalian cells. They found that the intracellular location of Parkin is regulated by mitochondrial function. Parkin normally resides in the cytosol but it translocates to depolarized mitochondria following treatment with CCCP. Mitochondriallocalized Parkin promotes the colocalization of mitochondria with the autophagy marker LC3 (Narendra et al., 2008). Translocation of Parkin to mitochondria is also observed in cells treated with paraquat, which increases oxidative stress, and in cells with mitochondrial DNA (mtDNA) mutations (Narendra et al., 2008; Suen et al., 2010). In each of these cases, there is a loss of mitochondrial membrane potentials.
Pink1 and Parkin physically interact with each other, and the mitochondrial translocation of Parkin is dependent on Pink1. The cellular localization of Pink1 is controversial, with proposed locations including the intermembrane space of mitochondria or the outer mitochondrial membrane (Silvestri et al., 2005; Zhou et al., 2008; Narendra et al., 2010b). The N terminus of Pink1 is required for its import to the mitochondria inner membrane through the Tim23 import pathway (Silvestri et al., 2005).
The level of Pink1 in healthy cells is quite low because it is rapidly cleaved and degraded by PARL at the mitochondrial inner membranes (Jin et al., 2010). The constitutive degradation of PINK1 is inhibited in the absence of PARL. However, Pink1 is stabilized on the outer mitochondrial membrane and forms a large complex with the translocase of the outer membrane where it can recruit Parkin to impaired mitochondria when mitochondrial membrane potential is dissipated (Jin et al., 2010; Lazarou et al., 2012). Therefore, the bioenergetic state of mitochondria can regulate PINK1 levels as well as the subsequent Parkin recruitment to the mitochondria. This unique regulation may allow PINK1 and Parkin to promote the selective and efficient turnover of mitochondria that have become damaged.
Because it has been suggested that the kinase domain of Pink1 faces the cytoplasm (Zhou et al., 2008), it is tempting to hypothesize that the kinase activity of Pink1 may be required for the recruitment of Parkin to mitochondria by direct phosphorylation of Parkin. Indeed, kinase-deficient Pink1 fails to recruit Parkin to mitochondria in Pink1-knockout MEFs. Moreover, it was found that Pink1 directly phosphorylates Parkin on Thr175 and Thr217 at the linker region of Parkin, which promotes Parkin mitochondrial translocation (Kim et al., 2008). However, it is possible that other sites could be phosphorylated by Pink1 or that another kinase may induce Parkin recruitment on mitochondria (Narendra et al., 2010b).
How does Parkin promote mitophagy? Emerging evidence has suggested that ubiquitination plays a critical role in the selective removal of protein aggregates, peroxisomes, ribosomes, and invading bacteria by autophagy (Kirkin et al., 2009). This is mainly achieved through several adapter molecules such as p62 and NBR1, which can directly interact with poly- and mono-ubiquitin and LC3. Because Parkin has E3 ligase activity, it is not surprising to find that Parkin-positive mitochondria are also positive for ubiquitin staining (Ding et al., 2010b). A subset of outer mitochondrial membrane proteins including VDAC and Mfn 1 and 2 are found to be ubiquitinated in a Parkin-dependent manner (Geisler et al., 2010; Gegg et al., 2010). p62 can be recruited to the ubiquitinated mitochondria in CCCP-treated Parkin-positive cells, most likely due to its C-terminal ubiquitin binding domain (Ding et al., 2010b; Geisler et al., 2010; Huang et al., 2011). While p62 can promote Parkin-mediated mitophagy, its role in mitophagy is not essential (Ding et al., 2010b; Geisler et al., 2010; Narendra et al., 2010a; Okatsu et al., 2010; Huang et al., 2011). Parkin-mediated mitochondrial ubiquitination could have different functional roles in mitophagy. Upon mitochondrial membrane depolarization, proteasomes can be recruited to the mitochondria in a Parkin-dependent manner, which leads to the degradation of mitochondrial outer membrane and intermembrane space proteins but not the inner-membrane and matrix proteins (Chan et al., 2011; Yoshii et al., 2011).
Parkin also induces rupture of the outer mitochondrial membrane, an event dependent on proteasomes (Yoshii et al., 2011). In addition, p97, an AAA+ ATPase, also accumulates on mitochondria in a Parkin-dependent manner to promote the degradation of outer mitochondrial membrane proteins and mitophagy (Tanaka et al., 2010). It is possible that this process induces mitochondrial fragmentation through the proteasome-dependent degradation of Mfn 1 and 2 proteins. As noted above, mitochondrial fragmentation facilitates mitophagy. Further supporting this possibility is the finding that Parkin-mediated mitophagy is suppressed in Drp1-knockout MEFs, which have excessively fused mitochondria (Tanaka et al., 2010). However, it has also been found that neither proteasome nor Parkin-induced rupture of outer mitochondrial membrane is obligatory for autophagic degradation of mitochondrial inner membrane and matrix proteins (Yoshii et al., 2011). It is thus still arguable how mitophagy should be defined and whether the proteasome and autophagy work coordinately or independently to accomplish the same task.
In addition to the ubiquitination of mitochondrial proteins, in the vertebrate central nervous system, Parkin also interacts with Ambra1 (activating molecule in Beclin 1-regulated autophagy), a protein that promotes general autophagy by activating the class III phosphatidylinositol 3-kinase complex (Fimia et al., 2007; Van Humbeeck et al., 2011). The Parkin-Ambra1 interaction is increased during prolonged mitochondrial depolarization. Although no evidence for ubiquitination of Ambra1 by Parkin was found, Ambra1 is recruited to perinuclear clusters of depolarized mitochondria in a Parkin-dependent manner, activates autophagy around these mitochondria, and contributes to their selective autophagic clearance (Van Humbeeck et al., 2011). These findings suggest that Parkin can also play a dual role in the two-step mitophagy process, priming the mitochondria through the ubiquitination of mitochondrial proteins on the depolarized mitochondria, and promoting induction of autophagy by interacting with Ambra1 and activating class III PI3K.
SMURF1
In a recent high-content, genome-wide small interfering RNA screen to detect genes required for the colocalization of Sindbis virus capsid protein with autophagolysosomes, Orvedahl et al. (2011) identified 141 candidate genes required for viral autophagy. Among these genes, 96 were also required for Parkin-mediated mitophagy, indicating that autophagic targeting of viral nucleocapsids and autophagic targeting of damaged mitochondria may share common molecular determinants. Furthermore, the group also identified that SMURF1, another ubiquitin E3 ligase, was required for the selective autophagy of damaged mitochondria in CCCP-treated cultured cells. They also found that SMURF1-deficient mice accumulate damaged mitochondria in the heart, brain, and liver. Thus, it seems that in addition to Parkin, other ubiquitin E3 ligases may play important roles in selective autophagy, and this process may depend on the expression levels of different ubiquitin E3 ligases in different tissues.
High mobility group box 1 (HMGB1)
HMGB1 is a chromatin-associated nuclear protein and an extracellular damage-associated molecular pattern molecule. Recent evidence reveals that HMGB1 may also be involved in the mitophagy acting on both the steps of autophagy induction and mitochondrial priming. On one hand, cytosolic HMGB1 directly interacts with the autophagy protein Beclin 1 and increases autophagic flux in response to stimuli that enhance ROS production (Tang et al., 2010). On the other hand, nuclear HMGB1 modulates the expression of heat shock protein β-1 (HSPB1/HSP27). HSPB1 is a cytoskeleton regulator, which is critical for dynamic intracellular trafficking during autophagy and mitophagy. Loss of either HMGB1 or HSPB1 produces phenotypes similar to those characterized by mitochondrial fragmentation with decreased aerobic respiration and ATP production (Tang et al., 2011). However, unlike Nix or Parkin, which directly targets damaged mitochondria and primes the mitochondria for further recognition by autophagy machinery, HMGB1 or HSPB1 regulates the actin cytoskeleton and affects the trafficking of damaged mitochondria to autophagosomes, thereby indirectly affecting mitophagy.
Hsp90-Cdc37 complex
Molecular chaperones, especially members of the heat shock protein 90 (Hsp90) family, are also involved in the regulation of mitochondrial homeostasis. Hsp90 and its related molecule, TRAP-1, interact with CypD (Kang et al. , 2007 ). Treatment with Hsp90 inhibitors induces an expansion of the mitochondrial compartment, accompanied by mitochondrial fragmentation and condensed mitochondrial morphology, ultimately compromising mitochondrial integrity and leading to apoptosis (Kang et al. , 2007 ). A subsequent study revealed that inhibition of Hsp90 induces post-transcriptional accumulation of mitochondrial proteins, which may be related to the impaired proteasome-mediated turnover of mitochondrial proteins (Margineantu et al., 2007). More recently, an Hsp90-Cdc37 chaperone complex was found to interact with Ulk1, stabilizing and activating Ulk1, which in turn is required for the phosphorylation and release of Atg13 from Ulk1 (Joo et al., 2011). The released phosphorylated Atg13 is then recruited to damaged mitochondria and promotes mitophagy. Using a stable isotope labeling by amino acids in cell culture (SILAC)-based mass spectrometric approach, Ser318 of Atg13 was identified as a site of Ulk1 phosphorylation. Interestingly, phosphorylation of Atg13 at Ser318 is only required for mitophagy, but not basal or starvation-induced autophagy, indicating a selective nature for mitophagy. It remains to be studied how the Atg13-targeted mitochondria are degraded by mitophagy, but it seems that Atg13-mediated mitophagy requires Parkin, suggesting that Atg13 could work downstream of Parkin (Joo et al. , 2011 ).
Atg9A and ULK1 complex
It has been generally thought that the link of autophagosomes to damaged mitochondria is through the interaction of LC3 (on the isolation and autophagosome membrane) with a receptor such as p62 or Nix (on the damaged mitochondria). However, a recent study found that after Parkin was recruited on depolarized mitochondria, it induces formation of Atg9A-positive structures as well as ULK1-positive structures closely associated with mitochondria, and these two structures are not dependent on each other (Itakura et al., 2012). Moreover, these Atg9A- and/or ULK1-positive structures were associated with mitochondria in the absence of membrane-bound LC3, suggesting that LC3 and its adaptor molecules may not be essential for the recognition of mitochondria by autophagosomes. Instead, these results suggest that it is possible that selective mitophagy may directly result from the de novo formation of ‘mitophagosomes’ on the damaged mitochondrial membranes. However, it is not known whether these mitophagosomes behave like canonic autophagosomes. For example, whether mitophagosomes also need to fuse with lysosomes and how they further recruit lysosomes are not known and need to be further studied.
15-Lipoxygenase
This enzyme has been shown to be involved in organelle degradation in erythrocytes by increasing the permeability of organelle membranes (van Leyen et al., 1998). Therefore, it seems that both autophagy-dependent and autophagy-independent mechanisms are regulating the mitochondrial homeostasis during erythroid differentiation.
Pathophysiological role of mitophagy
As mitochondria are essential organelles that regulate cellular energy metabolism and cell death, mitochondrial homeostasis has been linked to many pathophysiological conditions and diseases.
Development
As discussed above, mitophagy plays an essential role in erythrocyte differentiation and maturation. Both the autophagy machinery (such as Atg7 and ULK1) and mitochondrial recognition signals for mitophagy (such as Nix) are important in this physiological process. As mentioned, autophagy-independent pathways are likely also involved in the elimination of mitochondria in this process (Schweers et al., 2007; Sandoval et al., 2008).
Perhaps the most intriguing is the role of mitophagy in the destruction of paternal mitochondria in the fertilized oocytes. In almost all eukaryotes, paternal mtDNA and mitochondria themselves are selectively eliminated or degraded from the embryonic cytoplasm; thus, mitochondrial genes are inherited mainly from the maternal parent (Ankel-Simons and Cummins, 1996). In mammals, the ubiquitin proteasome system has been suggested to specifically degrade paternal spermborne mitochondria (which are ubiquitin positive) after they enter the ooplasm during fertilization (Sutovsky et al., 2004). Prohibitin, an inner mitochondrial membrane protein, was further identified as one of the ubiquitinated substrates that make sperm mitochondria recognizable by the ubiquitin-proteasome system in the egg, and likely facilitates sperm mitochondrial degradation after fertilization (Thompson et al., 2003). The expression level of Parkin in the sperm is not known. It would be interesting to see whether the Pink1-Parkin pathway or other E3 ligases such as SMURF1 would be involved in such a process. It would also be interesting to see whether Nix plays a role during the selective removal of paternal mitochondria in mammals.
Several reports now suggest that autophagy or the lysosome is required for the elimination of paternal mitochondria in Caenorhabditis elegans (Al Rawi et al., 2011; Sato and Sato, 2011; Zhou et al., 2011a). In lgg1 (a homologue of yeast Atg8)-deficient zygotes, paternal mitochondria and other membrane structures remain in the first larval stage. In fertilized mouse embryos, many autophagy markers such as LC3, GABRAP, and p62 are found closely associated with paternal ubiquitin-positive mitochondria. Ubiquitin has been suggested to serve as an ‘eat-me tag’ for the selective autophagy of various cellular cargos, including protein aggregates, damaged or excessive organelles, or invading microbes. Strikingly, although sperm mitochondria are often found to be ubiquitin positive, in C. elegans, the paternal mitochondria are not ubiquitinated. Instead, there are many vesicular structures called membranous organelles (MOs) that are ubiquitinated and degraded together with the paternal mitochondria by autophagy (Al Rawi et al., 2011). Therefore, it was suggested that paternal mitochondria are incorporated into autophagosomes owing to their close association with the MOs. It is also possible that the paternal mitochondria in C. elegans may use other tags for selective removal through autophagy in addition to ubiquitin. Collectively, although mitophagy seems to play a role in development, more work is needed in the future to further support such a notion.
Aging
It has been known that autophagy declines during aging, and the expression levels of many autophagy genes or related proteins (such as Atg7, Atg5, Atg4B, and LAMP-2) decreases in the brains and liver of aging humans and mice (Zhang and Cuervo, 2008; Lipinski et al., 2010; Wang et al., 2011). Conditions that activate autophagy, such as caloric restriction, have shown beneficial effects on delaying the aging-related degeneration process. Stimulation of autophagy has been shown to extend the life span in multiple organisms, including yeast, C. elegans, mice, and primates (Kissova et al., 2004; Alberti et al., 2010; Madeo et al., 2010). For example, in yeast, deletion of the mitochondrial membrane protein Uth1p leads to a selective defect in mitophagy and a decreased lifespan (Kissova et al., 2004). In C. elegans, worms that carry the loss-of-function mutation in the insulin-like signaling pathway (daf-2) display extended lifespans and induction of autophagy (Melendez et al., 2003). Although the exact mechanisms by which autophagy may delay aging are not clear, it has been suggested that mitophagy may help reduce the production of mitochondria-derived ROS and remove dysfunctional mitochondria that would generate mtDNA mutations during aging (Lemasters, 2005).
Cancer
Autophagy can act as a tumor suppressor, and loss of autophagy promotes tumorigenesis. An essential autophagy gene, Beclin 1, was frequently found monoallelically deleted in many human cancers such as breast, prostate, and ovarian cancers (Aita et al., 1999). Mice with allelic loss of Beclin 1 are prone to hepatocellular carcinoma, lung adenocarcinoma, mammary hyperplasia, and lymphoma. Many known tumor suppressor genes such as Lkt, Ampk, and Pten are positive regulators of autophagy (Cully et al., 2006; Liang et al., 2007; Degtyarev et al., 2008; Hezel and Bardeesy, 2008), whereas many oncogenes, including the class I PI3K, Akt, and anti-apoptotic Bcl-2 family proteins, suppress autophagy (Maiuri et al., 2009), further supporting the notion that autophagy suppresses tumorigenesis. Mice that have liver-specific loss of Atg7 or Atg5 have accumulated damaged mitochondria and develop liver injury, steatophepatitis, and adenocarcinoma (Inami et al., 2011; Takamura et al., 2011). However, it is likely that autophagy has multiple roles in acting as a tumor suppressor in addition to removal of damaged mitochondria. For example, autophagy may act to buffer metabolic stress caused by limited nutrients or oxygen in the tumor tissues (Rosenfeldt and Ryan, 2011). Future works are needed to further determine the exact role of mitophagy in tumorigenesis.
Neurodegenerative diseases
As discussed above, some familial forms of Parkinson’s disease are provoked by mutations in Pink1 or Parkin, some of which can result in defects in mitophagy. In addition, mitophagy may also play a role in other neurodegenerative diseases including Alzheimer’s disease (AD) and Huntington’s disease (HD). β-Amyloid fragments can target mitochondria and cause mitochondrial dysfunction in AD (Casley et al., 2002). Indeed, there is evidence that mitochondrial cytochrome oxidase is defective in AD (Maurer et al., 2000). Mitochondrial dysfunction has also been associated with HD due to the dysregulation of PGC1-α, an important transcription factor for mitochondrial biogenesis (Cui et al., 2006). However, the mechanisms and the role of mitophagy in AD and HD are unclear and need to be further studied.
Innate immunity
Accumulating evidence now support the idea that autophagy is closely associated with innate immunity. Autophagy plays an important role in defending against invading intra-cellular microbes, including Mycobacterium tuberculosis, Salmonella, Listeria, Shigella, HIV-1, and Sindbis viruses (Deretic, 2012). Autophagy can be regulated by the DAMP molecules such as HMGB1 and IL-1β; Toll-like receptors; Nod-like receptors, including NLRC4, NLRP3, and NLRP4; and RIG-I-like receptors (Deretic, 2012). Other innate immune signaling molecules such as TBK-1 and IKKα/β are also associated with autophagy (Deretic, 2012; Weidberg and Elazar, 2011). Recent evidence suggests that suppression of mitophagy/autophagy leads to the accumulation of damaged, ROS-generating mitochondria, and this in turn activates the NLRP3 inflammasome (Zhou et al., 2011b). In addition to ROS, depletion of LC3B or Beclin 1 promotes the accumulation of dysfunctional mitochondria and cytosolic translocation of mtDNA in macrophages. Cytosolic mtDNA promotes the secretion of IL-1β and IL-18 following stimulation with lipopolysaccharide or ATP (Nakahira et al., 2011). These studies suggest that mitophagy can regulate NALP3-dependent inflammation by preserving mitochondrial integrity.
Tissue injury
It has been well known that the mitochondrion is a central executioner for regulating cell death (Ding and Yin , 2004 ; Green et al., 2011). In response to apoptotic stimuli, apoptotic proteins target mitochondria to cause mitochondrial swelling or membranepermeability changes that result in the release of apoptotic factors such as cytochrome c and Smac. In addition to apoptosis, necrotic stimuli can also damage mitochondria, resulting in the depletion of cellular ATP levels. Moreover, mitochondria are also the major source of ROS that promote cellular damage. In ischemia-reperfusion-induced heart injury, Parkin-mediated mitophagy has a protective role against the cell death of cardiomyocytes (Huang et al., 2011). In contrast, TIGAR (TP53-induced glycolysis and apoptosis regulator), which was exclusively upregulated in ischemic myocardium, suppresses mitophagy and exacerbates cardiac damage after ischemia (Hoshino et al. , 2012). Acetaminophen overdose, one of the most prevalent poisonings worldwide, induces liver damage that features mitochondrial damage-mediated hepatic necrosis (Jaeschke and Bajt , 2006 ). We recently found that mitophagy plays a critical role against acetaminophen-induced liver damage by reducing ROS production (Ni et al., 2012). In addition to regulating cell death, mitochondria also play an important role in regulating lipid homeostasis by burning fat through fatty acid β-oxidation. Decreased autophagic activity is found in the livers of obese mice, and genetic deletion of Atg7 or pharmacological inhibition of autophagy is associated with hepatic steatosis (Singh et al., 2009; Yang et al., 2010; Mei et al., 2011). We also found that induction of autophagy leads to reduced alcohol-induced hepatic steatosis (Ding et al. , 2010a). Although autophagy may selectively remove lipid droplets (lipophagy), it is possible that removal of damaged mitochondria may also contribute to the attenuation of steatosis. Therefore, targeting damaged mitochondria by activating mitophagy may be a novel approach for treating damaged mitochondria-mediated tissue injury in the future.
Analysis of mitophagy
Although mitophagy was first studied in mammalian cells, reliable quantitative methods for specifically monitoring mitophagy in mammalian cells are not yet available. In contrast, several reliable quantitative mitophagy assays have been developed for yeast. Below, we will discuss the current techniques that have been used to monitor mitophagy in both yeast and mammalian cells.
Electron microscopy
Transmission electron microscopy (TEM) is still one of the best approaches to provide direct evidence for mitophagy since autophagy was first detected by TEM in the 1950s. The morphological hallmark of autophagy is the formation of double-membrane autophagosomes that contain cytoplasmic material and/or organelles at various stages of degradation. The autophagosomes eventually fuse with the lysosomes to form matured autolysosomes, which usually have only one limiting membrane. The early stage of mitophagy (autophagosome with engulfed mitochondria) can be readily detected through identification of unique mitochondrial structures such as the cristae (Figure 2A and C). The late stage of mitophagy may be reflected by the single-membrane autolysosomes with residue mitochondria (Figure 2B and D), on the basis of their similar electron density with mitochondria. It would be better to perform immuno-EM for specific mitochondrial markers, such as Tom20 or CypD, to confirm the nature of mitochondria in the late stage of mitophagy.
Figure 2. EM for mitophagy.

Wild-type C57BL/6 mice were treated with acetaminophen (i.p., 500 mg/kg) for 6 h (A, B) or ethanol (gavage, 4.5 g/kg) for 6 h (C, D). Thin liver sections were processed for EM studies. (A and C) An early double-membraned autophagosome envelops mitochondria. (B and D) A late autolysosome contains degrading mitochondria.
Cautionary notes
Pros: EM can directly provide the ‘seeing is believing’ evidence for the presence of mitophagy. Moreover, if one can carefully perform the morphometric analysis, data from EM could be more helpful for mitophagy. Cons: EM can be problematic in quantitative studies because of the limited cell numbers/sections that can be determined. Data generated from EM studies could have big variations, and unbiased selection for samples and scoping are extremely important.
Fluorescence microscopy for mitochondria-autophagosome or mitochondria-lysosome colocalization
Two different approaches can be used to determine mitochondria-autophagosome or mitochondria-lysosome colocalization depending on whether the cell being imaged is live or fixed. For live-cell imaging microscopy, MitoTracker, but not membrane potential sensitive dyes such as tetramethylrhodamine, methyl ester (TMRM) dye, should be chosen. MitoTracker is taken up electrophoretically by mitochondria, dependent on the mitochondrial membrane potential. Unlike TMRM, it covalently binds to mitochondrial proteins and thus remains in mitochondria even if they subsequently depolarize (Elmore et al., 2001). By using a GFP-LC3 and MitoTracker Red cola-beling approach, one could examine the relation between mitochondria and the GFP-LC3-positive autophagosomal structures. For fixed cells, in addition to MitoTracker staining, mitochondria can also be visualized using immunostaining for mitochondrial proteins with antibodies against mitochondrial proteins such as Tom20, VDAC, or the complex IV subunit. Quantification of the colocalization of mitochondria and autophagosomes provide an indication of the degree of sequestration and could function as a marker for mitophagy. Similar to autophagic flux assay, the number of mitochondria and GFP-LC3 colocalized puncta would also be increased in the presence of lysosomal inhibitors such as chloroquine or E64 D plus pepstatin A (Figure 3).
Figure 3. Confocal microscopy for the colocalization of autophagosomes with mitochondria.
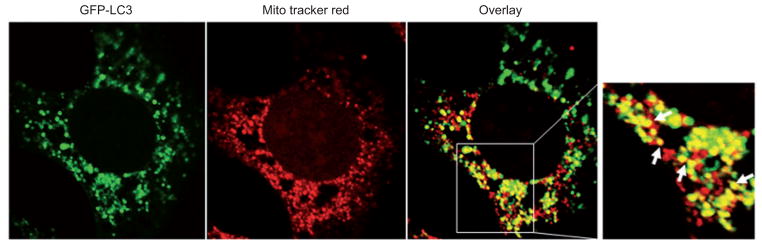
GFP-LC3-expressing MEFs were fi rst loaded with MitoTracker Red (50 nM) for 30 min and then treated with CCCP (30 μM) plus the lyso-somal protease inhibitors E64D (10 μM) and Pepstatin A (10 μM) for 6 h. The cells were fi xed with 4% paraformaldehyde followed by confocal microscopy. Arrows: yellow dots represent colocalized GFP-LC3-positive autophagosomes with red fluorescence-labeled mitochondria.
Because autophagosomes eventually need to fuse with lysosomes, the colocalization of mitochondria with lysosomal markers could alternatively be used to monitor mitophagy. For live-cell imaging microscopy, MitoTracker-stained mitochondria and LysoTracker-stained lysosomes may be used to visualize the process. However, it should be cautioned that LysoTracker will stain all the acidic compartments present and is therefore not specific for autolysosomes. Alternatively, fixed cells can be stained using antibodies specific for mitochondrial proteins and an antibody against the lysosomal-associated membrane protein 1 (LAMP-1) or LAMP-2. The successful fusion of mitochondria-containing autophagosome with the lysosomal structures can be quantified by analyzing the colocalization signals as an indicator for mitophagy.
Cautionary notes
Pros: This assay can assess a large number of cells compared with EM. It can also monitor the dynamic process of mitophagy (mitochondrial depolarization and fission, formation of GFP-LC3-positive autophagosomes, enveloping of mitochondria by GFP-LC3 autophagosomes, and fusion of autophagosomes with lysosomes) in a live cell setting. Cons: Not all GFP-LC3-positive ‘puncta’ are autophagosomes but could instead be GFP-LC3 aggregates. LysoTracker is not specific for lysosomes or autolysosomes. Counting the number of dots that represent colocalized mitochondrial with either GFP-LC3 puncta or lysosomes can be very subjective, and consistent criteria need to be kept in all experiments. Furthermore, mitochondrial colocalization with GFP-LC3 puncta or lysosomal markers does not implicate the mitochondrial degradation process. Addition of lysosomal inhibitors, such as bafilomycin A1 and CQ, in these experiments may provide additional evidence of whether a degradation process is indeed occurring.
Mitochondrial mass
Analyzing the mitochondrial mass has also been used as a quantitative way to monitor the last step of the degradation process of mitophagy. Mitochondrial mass can be measured by a fluorescence-activated cell sorting technique using MitoTracker staining. This technique has been successfully used to monitor mitochondrial elimination during red blood cell maturation (Zhang et al., 2009). Immunostaining for mitochondrial proteins using specific antibodies has also been applied to monitor mitochondrial mass. For example, immunostaining for Tom20 (an outer mitochondrial membrane protein) in CCCP-treated Parkin-positive cells has initially been used as a marker for the loss of mitochondria resulting from mitophagy. However, more recent studies suggest that most outer mitochondrial membrane proteins are degraded by the ubiquitin proteasome system, whereas the matrix proteins are degraded through autophagy, although the exact mechanisms and reasons why different mitochondrial proteins are degraded through different mechanisms are not clear (Yoshii et al., 2011). Therefore, immunostaining for mitochondrial matrix proteins such as CypD and HSP60 would be more appropriate for monitoring mitophagy. In addition, quantitative PCR can be used to quantify mtDNA, such as 16S rRNA, and nuclear DNA, such as hexokinase 2. The mitochondrial-to-nuclear DNA ratio in each sample can consequently be calculated by dividing the value of 16S rRNA by that of hexokinase (Lagouge et al., 2006).
Cautionary notes
Pros: This assay is more objective and quantitative. Cons: MitoTracker staining depends on the mitochondrial membrane potential and may not stain damaged mitochondria. It is still not clear whether autophagy is the only mechanism to remove/degrade damaged mitochondria. It is also not very clear what subsets of mitochondrial proteins are specifically degraded by mitophagy (but not by proteasome), although it is suggested that mitochondrial matrix proteins are likely degraded through mitophagy. Thus, caution needs to be exercised when using these proteins as a marker for mitochondrial mass.
Immunoblot for mitochondrial proteins
Western blot analysis for mitochondrial proteins is another way to quantify mitochondrial mass. However, as discussed above, this should be conducted to cover all mitochondrial compartment proteins, including outer and inner mitochondrial proteins, intermembrane space, and matrix proteins. Only monitoring outer mitochondrial membrane proteins such as Tom20, VDAC, and Mfn 1/2 could be misleading because these proteins could be preferentially degraded by the proteasome. Western blot analysis for mitochondrial matrix proteins would be more appropriate and specific for mitophagy.
Cautionary notes
Pros: This assay is more objective and quantitative. Cons: The cons are similar to those of the assays for mitochondrial mass. As it is not very clear what subsets of mitochondrial proteins are specifically degraded by mitophagy, caution needs to be exercised when interpreting these data.
Om45-GFP processing assay
To quantitatively detect mitophagy in the yeast, Kanki et al. (2009) developed the Om45-GFP processing assay. Om45 is a mitochondrial outer membrane protein, and a GFP-tagged Om45 (GFP at the C terminus of Om45) is correctly localized on this organelle. When mitophagy is induced, mitochondria, along with Om45-GFP, are delivered into the vacuole for degradation. Om45 is proteolytically removed or degraded, whereas the GFP moiety is relatively stable and accumulates in the vacuole. Thus, mitophagy can be monitored and quantified according to the appearance and intensity of free GFP by immunoblot. A similar approach was used in another study in which yeast cells were expressing a mitochondria-targeted dehydrofolate reductase-GFP (Okamoto et al., 2009). However, the free GFP fragments could be difficult to detect in mammalian cells because lysosomal pH is more acidic in mammalian cells than in yeast vacuoles (Ni et al., 2011). Therefore, it remains to be determined whether a similar GFP-tagged mitochondrial protein-processing assay would work in mammalian cells. In contrast to using Western blot analysis for GFP fragments, Yoshii et al. (2011) developed a flow cytometry-based assay using cell lines that are stably expressing GFP proteins fused to either the mitochondrial matrix (subunit 9 of F0-ATPase, Su9-GFP) or an outer mitochondrial protein (GFP-Omp25). They found that the Su9-GFP and GFP-Omp25 signals were largely unaffected in Parkin-untransfected MEFs but significantly decreased in Parkin-transfected wild-type MEFs in a time-dependent manner following CCCP treatment. In either FIP200 KO or Atg5 KO MEFs, CCCP-induced decreases in Su9-GFP but not GFP-omp25 signals were markedly inhibited (Yoshii et al., 2011). These data confirmed the notion discussed above that degradation of matrix proteins but not outer mitochondrial membrane proteins relies on autophagy and also provides a novel reliable quantitative approach to monitor mitophagy in mammalian cells.
Cautionary notes
Pros: The specificity for this assay is high. It is also more objective and quantitative. Cons: The stability of GFP in the lysosomes could vary in different cell types depending on the acidity and activity of their lysosomes.
Citrate synthase activity assay
As an important metabolic power house, mitochondria contain many important enzymes for the citric acid cycle and oxidative phosphorylation. The decrease in mitochondrial content over time due to mitophagy can be measured by analyzing the activities of some mitochondrial enzymes. For example, citrate synthase, a citric acid cycle enzyme, has been used to measure mitochondrial mass in cells and tissue (Watts et al. , 2004 ; Hargreaves et al. , 2007). Disruption of mitochondrial functions such as the inhibition of the electron transport chain or the depletion of mtDNA does not generally affect citrate synthase activity. Therefore, changes of citrate synthase activity are good indicators for mitochondrial content (Watts et al., 2004 ; Hargreaves et al. , 2007 ). In Parkin-overexpressing SH-SY5Y cells, treatment with CCCP induced mitophagy and resulted in decreased mitochondrial mass and citrate synthase activity. The levels of the decreased mitochondrial proteins were strongly correlated with citrate synthase activity (Gegg et al., 2010).
Cautionary notes
Pros: This assay is more objective and quantitative. Cons: It lacks the specificity for mitophagy. It is currently not clear whether other factors also regulate citrate synthase activity.
Future perspectives
Although there have been significant advances recently in our knowledge of selective mitophagy, many questions remain unanswered. The Pink1-Parkin signaling pathways play critical roles in mitophagy in cells/tissues that have high expression levels of Parkin, such as in the mouse brain, thymus, kidney, and muscle (Figure 4 and Table 1). However, cells that have low or undetectable levels of Parkin (Table 1) may utilize Parkin-independent mitophagy pathways. In addition to hypoxia-induced Parkin-independent mitophagy, what are the other Parkin-independent mitophagy pathways? How do the ubiquitin proteasome system and the autophagy machinery complete the degradation of mitochondrial proteins in coordination? Is there a mitochondrial clearance mechanism independent of the canonic autophagic process that requires the Atg genes? Finally, reliable quantitative assays to monitor mitophagy still need to be developed. Answering these questions will not only improve our knowledge regarding general mitochondrial homeostasis, but it may also reveal new avenues for treatment of diseases with dysfunctional mitochondria.
Figure 4. The expression level of Parkin in different mouse tissues.

A 2-month-old Parkin wild-type mouse was sacrifi ced, and different tissues were harvested. Total cell lysates were extracted and subjected to Western blot analysis using an anti-Parkin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Total liver lysate from a Parkin-knockout mouse was used as a negative control.
Table 1.
Parkin expression in different cells.
| Cells/cell lines | Origin | Parkin expression | Reference |
|---|---|---|---|
| HEK293 | Human embryonic kidney | Yes | Narendra et al., 2008; Ding et al., 2010b |
| SH-SY5Y | Human neuroblastoma | Yes | Gegg et al., 2010 |
| Primary neuron | Rat | Yes | Narendra et al., 2010 |
| HeLa | Human cervical cancer | N.D. | Narendra et al., 2008; Ding et al., 2010b |
| MEF | Mouse embryonic fi broblast | N.D. | Narendra et al., 2008; Ding et al., 2010b |
N.D., undetected.
Acknowledgments
We thank Ms. Barbara Fegley (KUMC Electron Microscopy Research Laboratory) for her excellent assistance in the EM studies, Ms. Sheila Tsau and Ms. Jessica Williams for the critical reading of our manuscript, and Dr. Hong-Min Ni for performing the experiments presented in Figure 4. This study was supported in part by the National Institute of Health (NIH) funds R21 AA017421 and R01 AA020518-01, National Center for Research Resources (5P20RR021940-07), and P20 RR016475 from the IDeA Networks of Biomedical Research Excellence (INBRE) program of the National Center for Research Resources (W.X.D). X.M Yin was in part supported by the NIH (R01CA83817). No additional external funding was received for this study.
Biographies

Dr. Wen-Xing Ding received his PhD from the National University of Singapore. He is currently an assistant professor at the Department of Pharmacology, Toxicology and Therapeutics at the University of Kansas Medical Center, USA. Dr. Ding’s Lab focuses on the mechanisms of mitophagy in alcohol and drug-induced liver injury. Their ultimate goal is to investigate the possibilities to attenuate alcohol and drug-induced liver injury by modulating autophagy.

Dr. Xiao-Ming Yin, MD, PhD, is a Louis Y. Mazzini Professor of Pathology at the Department of Pathology and Laboratory Medicine, Indiana University School of Medicine. Dr. Yin’s research is centered on the mechanisms of apoptosis and autophagy in tissue injury and cancer therapy.
References
- Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- Alberti A, Michelet X, Djeddi A, Legouis R. The autophagosomal protein LGG-2 acts synergistically with LGG-1 in dauer formation and longevity in C. elegans. Autophagy. 2010;6:5. doi: 10.4161/auto.6.5.12252. [DOI] [PubMed] [Google Scholar]
- Ankel-Simons F, Cummins JM. Misconceptions about mitochondria and mammalian fertilization: implications for theories on human evolution. Proc Natl Acad Sci USA. 1996;93:13859–13863. doi: 10.1073/pnas.93.24.13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Kanki T, Hirota Y, Kurihara Y, Saigusa T, Uchiumi T, Kang D. Phosphorylation of serine 114 on Atg32 mediates mitophagy. Mol Biol Cell. 2011;22:3206–3217. doi: 10.1091/mbc.E11-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austriaco NR., Jr Review: to bud until death: the genetics of ageing in the yeast, Saccharomyces. Yeast. 1996;12:623–630. doi: 10.1002/(SICI)1097-0061(19960615)12:7%3C623::AID-YEA968%3E3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, Gibson SB. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy. 2008;4:195–204. doi: 10.4161/auto.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, D’Sa-Eipper C, Chinnadurai G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell. 1994;79:341–351. doi: 10.1016/0092-8674(94)90202-x. [DOI] [PubMed] [Google Scholar]
- Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA. 2000;97:9082–9087. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA. β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Ray R, Dubik D, Shi L, Cizeau J, Bleackley RC, Saxena S, Gietz RD, Greenberg AH. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med. 1997;186:1975–1983. doi: 10.1084/jem.186.12.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, Shi L, Dubik D, Greenberg A. Nix and Nip3 form a subfamily of proapoptotic mitochondrial proteins. J Biol Chem. 1999;274:7–10. doi: 10.1074/jbc.274.1.7. [DOI] [PubMed] [Google Scholar]
- Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D’Adamio L, Derks C, et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neuro-degeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- Deffieu M, Bhatia-Kissova I, Salin B, Galinier A, Manon S, Camougrand N. Glutathione participates in the regulation of mitophagy in yeast. J Biol Chem. 2009;284:14828–14837. doi: 10.1074/jbc.M109.005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, Dijk SV, Wu H, Gray DC, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–116. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol. 2012;24:21–31. doi: 10.1016/j.coi.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-α-induced apoptosis in liver injury. J Cell Mol Med. 2004;8:445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:41–150. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010a;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, II, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010b;285:27879–27890. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Manley S, Ni HM. The emerging role of autophagy in alcoholic liver disease. Exp Biol Med (Maywood) 2011;236:546–556. doi: 10.1258/ebm.2011.010360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Koppen M, Wagener J, Zick M, Israel L, Bernacchia A, Jagasia R, Rugarli EI, Imhof A, Neupert W, et al. OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria. Mol Biol Cell. 2007;18:3582–3590. doi: 10.1091/mbc.E07-02-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirschenbaum LA, Dorn GW., II Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J Biol Chem. 2006;281:1442–1448. doi: 10.1074/jbc.M509056200. [DOI] [PubMed] [Google Scholar]
- Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Gomes LC, Scorrano L. High levels of Fis1, a profission mitochondrial protein, trigger autophagy. Biochim Biophys Acta. 2008;1777:860–866. doi: 10.1016/j.bbabio.2008.05.442. [DOI] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin EE, Graumann J, Chan DC. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol. 2005;170:237–248. doi: 10.1083/jcb.200503148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16:1313–1320. doi: 10.1038/nm.2247. [DOI] [PubMed] [Google Scholar]
- Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K, Yu KT, Jaye M, Ivashchenko Y. Hypoxia induces the expression of the proapoptotic gene BNIP3. Cell Death Differ. 2001;8:367–376. doi: 10.1038/sj.cdd.4400810. [DOI] [PubMed] [Google Scholar]
- Hargreaves IP, Duncan AJ, Wu L, Agrawal A, Land JM, Heales SJ. Inhibition of mitochondrial complex IV leads to secondary loss complex II–III activity: implications for the pathogenesis and treatment of mitochondrial encephalomyopathies. Mitochondrion. 2007;7:284–287. doi: 10.1016/j.mito.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hezel AF, Bardeesy N. LKB1; linking cell structure and tumor suppression. Oncogene. 2008;27:6908–6919. doi: 10.1038/onc.2008.342. [DOI] [PubMed] [Google Scholar]
- Hoshino A, Matoba S, Iwai-Kanai E, Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M, Mita Y, et al. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol. 2012;52:175–184. doi: 10.1016/j.yjmcc.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci. 2012 Jan 24; doi: 10.1242/jcs.094110. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, Zhang J, Iyengar R, Jung CH, Suen D, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell. 2011;43:572–585. doi: 10.1016/j.molcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–39074. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Cregg JM, Dunn WA, Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurode-generation in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata JI, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10:602–610. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- Kudchodkar SB, Levine B. Viruses and autophagy. Rev Med Virol. 2009;19:359–378. doi: 10.1002/rmv.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–1502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, Brenner DA. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:769–781. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci USA. 2010;107:14164–14169. doi: 10.1073/pnas.1009485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- MacIntosh GC, Bassham DC. The connection between ribophagy, autophagy and ribosomal RNA decay. Autophagy. 2011;7:662–663. doi: 10.4161/auto.7.6.15447. [DOI] [PubMed] [Google Scholar]
- Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 2010;12:842–846. doi: 10.1038/ncb0910-842. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R, Kroemer G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009;16:87–93. doi: 10.1038/cdd.2008.131. [DOI] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Lisi RD, Sandri C, Zhao J, et al. Foxo3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Mao K, Wang K, Zhao M, Xu T, Klionsky DJ. Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J Cell Biol. 2011;193:755–767. doi: 10.1083/jcb.201102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margineantu DH, Emerson CB, Diaz D, Hockenbery DM. Hsp90 inhibition decreases mitochondrial protein turnover. PLoS One. 2007;2:e1066. doi: 10.1371/journal.pone.0001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinkovic D, Zhang X, Yalcin S, Luciano JP, Brugnara C, Huber T, Ghaffari S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J Clin Invest. 2007;117:2133–2144. doi: 10.1172/JCI31807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M, Yamamoto A, Kuma A, Ohsumi Y, Mizushima N. Organelle degradation during the lens and erythroid differentiation is independent of autophagy. Biochem Biophys Res Commun. 2006;339:485–489. doi: 10.1016/j.bbrc.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Maurer I, Zierz S, Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455–462. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, Copple BL, Ding WX. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339:487–498. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010a;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010b;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011;7:188–204. doi: 10.4161/auto.7.2.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowikovsky K, Reipert S, Devenish RJ, Schweyen RJ. Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ. 2007;14:1647–1656. doi: 10.1038/sj.cdd.4402167. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl A, Sumpter R, Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallet N, Bouvier N, Legendre C, Gilleron J, Codogno P, Beaune P, et al. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy. 2008;4:783–791. doi: 10.4161/auto.6477. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- Ray R, Chen G, Vande Velde C, Cizeau J, Park JH, Reed JC, Gietz RD, Greenberg AH. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem. 2000;275:1439–1448. doi: 10.1074/jbc.275.2.1439. [DOI] [PubMed] [Google Scholar]
- Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, Kimchi A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell. 2006;22:463–475. doi: 10.1016/j.molcel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Kai Y, Maldonado E, Currin RT, Lemasters JJ. Roles of mitophagy and the mitochondrial permeability transition in remodeling of cultured rat hepatocytes. Autophagy. 2009;5:1099–1106. doi: 10.4161/auto.5.8.9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Hernandez A, Cordero MD, Salviati L, Artuch R, Pineda M, Briones P, Izquierdo LG, Cotán D, Navas P, Sánchez-Alcázar JA. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy. 2009;5:19–32. doi: 10.4161/auto.5.1.7174. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32:955–963. doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–1144. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarten M, Mohrluder J, Ma P, Stoldt M, Thielmann Y, Stangler T, Hersch N, Hoffmann B, Merkel R, Willbold D. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy. 2009;5:690–698. doi: 10.4161/auto.5.5.8494. [DOI] [PubMed] [Google Scholar]
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet. 2005;14:3477–3492. doi: 10.1093/hmg/ddi377. [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci USA. 2010;107:11835–11840. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutovsky P, Van Leyen K, McCauley T, Day BN, Sutovsky M. Degradation of paternal mitochondria after fertilization: implications for heteroplasmy, assisted reproductive technologies and mtDNA inheritance. Reprod Biomed Online. 2004;8:24–33. doi: 10.1016/s1472-6483(10)60495-6. [DOI] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J Biol Chem. 2007;282:5617–5624. doi: 10.1074/jbc.M605940200. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ, II, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B, Zeh HJ, III, Lotze MT. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011;13:701–711. doi: 10.1016/j.cmet.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WE, Ramalho-Santos J, Sutovsky P. Ubiquitination of prohibitin in mammalian sperm mitochondria: possible roles in the regulation of mitochondrial inheritance and sperm quality control. Biol Reprod. 2003;69:254–260. doi: 10.1095/biolreprod.102.010975. [DOI] [PubMed] [Google Scholar]
- Tolkovsky AM. Mitophagy. Biochim Biophys Acta. 2009;1793:1508–1515. doi: 10.1016/j.bbamcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B, Vandenberghe W. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci. 2011;31:10249–10261. doi: 10.1523/JNEUROSCI.1917-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leyen K, Duvoisin RM, Engelhardt H, Wiedmann M. A function for lipoxygenase in programmed organelle degradation. Nature. 1998;395:392–395. doi: 10.1038/26500. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011;7:297–300. doi: 10.4161/auto.7.3.14502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Ahn IS, Fischer TD, Byeon JI, Dunn WA, Jr, Behrns KE, Leeuwenburgh C, Kim JS. Autophagy suppresses age-dependent ischemia and reperfusion injury in livers of mice. Gastroenterology. 2011;141:2188–2199. e2186. doi: 10.1053/j.gastro.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JA, Kline JA, Thornton LR, Grattan RM, Brar SS. Metabolic dysfunction and depletion of mitochondria in hearts of septic rats. J Mol Cell Cardiol. 2004;36:141–150. doi: 10.1016/j.yjmcc.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Weidberg H, Elazar Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci Signal. 2011;4:pe39. doi: 10.1126/scisignal.2002355. [DOI] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci. 1999;14:180–198. doi: 10.1006/mcne.1999.0780. [DOI] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Theodorakis P, Subramanian T, Chinnadurai G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J Biol Chem. 1998;273:12415–12421. doi: 10.1074/jbc.273.20.12415. [DOI] [PubMed] [Google Scholar]
- Yoon YG, Koob MD. Efficient cloning and engineering of entire mitochondrial genomes in Escherichia coli and transfer into transcriptionally active mitochondria. Nucleic Acids Res. 2003;31:1407–1415. doi: 10.1093/nar/gkg228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, Klionsky DJ. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol Biol Cell. 2005;16:1593–1605. doi: 10.1091/mbc.E04-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–965. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang J, Randall MS, Loyd MR, Dorsey FC, Kundu M, Cleveland JL, et al. Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood. 2009;114:157–164. doi: 10.1182/blood-2008-04-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci USA. 2008;105:12022–12027. doi: 10.1073/pnas.0802814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li H, Xue D. Elimination of paternal mitochondria through the lysosomal degradation pathway in C. elegans. Cell Res. 2011a;21:1662–1669. doi: 10.1038/cr.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011b;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]