

Abstract
Androgen-insensitive DU145 and PC3 human prostate cancer cells express high levels of specificity protein (Sp) transcription factors Sp1, Sp3 and Sp4, and treatment of cells with methyl 2-cyano-3,11-dioxo-18β-olean-1,12-dien-30-oate (CDODA-Me) inhibited cell growth and downregulated Sp1, Sp3 and Sp4 expression. CDODA-Me (15 mg/kg/d) was a potent inhibitor of tumor growth in a mouse xenograft model (PC3 cells) and also decreased expression of Sp transcription factors in tumors. CDODA-Me-mediated downregulation of Sp1, Sp3 and Sp4 was due to induction of the transcriptional repressor ZBTB4 which competitively binds and displaces Sp transcription factors from GC-rich sites in Sp1, Sp3, Sp4 and Sp-regulated gene promoters. ZBTB4 levels are relatively low in DU145 and PC3 cells due to suppression by microRNA (miR) paralogs that are members of the miR-17-92 (miR-20a/17-5p) and miR-106b-25 (miR-106b/93) clusters. Examination of publically available prostate cancer patient array data showed an inverse relationship between ZBTB4 and miRs-20a/17-5p/106b/93 expression, and increased ZBTB4 in prostate cancer patients was a prognostic factor for increased survival. CDODA-Me induces ZBTB4 in prostate cancer cells through disruption of miR-ZBTB4 interactions and this results in downregulation of pro-oncogenic Sp transcription factors and Sp-regulated genes.
Keywords: ZBTB4, CDODA-Me, Sp, miR-17-92, miR-106b
Introduction
Prostate cancer is the most common tumor in men, and it is estimated that in 2012 241,700 men will be diagnosed with prostate cancer and 28,170 will die from this disease (1). Prostate cancer is highly complex, and androgen deprivation therapy (ADT) by surgical or chemotherapeutic means has been critical for treatment of early and advanced prostate cancers (2-5). ADT therapies are highly variable but are primarily focused on inhibition of androgen synthesis which decreases circulating levels of the endogenous hormone or on blocking the androgen receptor (AR) signaling with various AR agonists (2). Not surprisingly, many patients with prostate cancer develop castrate-resistant prostate cancer (CRPC) which was previously referred to as hormone-refractory or androgen-independent (6). Some CRPC patients may still be responsive to some forms of antiandrogen therapy; however, many new combination therapies that often include a cytotoxic drug such as docetaxel or other taxane derivatives plus various agents that block angiogenesis or growth factor receptor signaling pathways in clinical trials (3-5). Development of new therapies for CRPC is also focused on decreasing the toxic side-effects and increasing efficacy of these treatments, and this includes the use of new mechanism-based drugs that target specific pathways and genes.
The addiction of many tumors to oncogenes such as various kinases including growth factor receptors has led to development of various kinase inhibitors and neutralizing antibodies such as erlotinib (Tarceva®) and trastuzumab (Herceptin®) which are clinically used for cancer chemotherapy (3-5, 7, 8). It has recently been suggested that non-oncogene addiction by cancer cells is also critical for maintaining the tumorigenic state of cells (9-11) and “a large class of non-oncogenes (that) are essential for cancer cell survival and present attractive drug targets” (10).
Specificity protein (Sp) transcription factors Sp1, Sp3 and Sp4 are overexpressed in multiple cancer cell lines and tumors (12-21). Although these factors are important for embryonic and postnatal development (22, 23), there is evidence that Sp1 expression decreases with age (24, 25) and, in animal models, minimal to non-detectable levels of Sp proteins are observed (14, 15). Evidence of a significant role for Sp transcription factors in non-oncogene addiction pathways in cancer cells is supported by RNA interference (RNAi) studies which show that Sp1, Sp3 and Sp4 regulate pro-oncogenic factors that play a role in cell proliferation, survival and angiogenesis. Sp-regulated genes include epidermal growth factor receptor (EGFR), hepatocyte growth factor receptor (c-MET), bcl-2, survivin, vascular endothelial growth factor (VEGF) and its receptors (VEGFR1 and VEGFR2), p65NFκB, and pituitary tumor transforming gene 1 (PTTG1) (13, 18-21, 26, 27). Moreover, knockdown of Sp1, Sp3 and Sp4 in cancer cells also inhibits cell proliferation and G0/G1 to S phase progression and induces apoptosis (15, 18-20, 28).
Methyl 2-cyano-3,11-dioxo-18β-olean-1,12-dien-30-oate (CDODA-Me) is a synthetic triterpenoid methyl ester derived from glycyrrhetinic acid, a major component of licorice extracts (29). In this study, we show that CDODA-Me inhibits androgen-insensitive prostate cancer cell and tumor growth and downregulates Sp1, Sp3 and Sp4 through induction of the Sp repressor ZBTB4 and downregulation of microRNA-20a (miR-20a) and related paralogs that inhibit ZBTB4 expression. MiR-dependent suppression of the Sp repressors ZBTB4 and ZBTB10 are critical for the high expression of Sp transcription factors (30, 31), and disruption of miR-ZBTB4 by CDODA-Me plays an essential role as an inhibitor of androgen-insensitive prostate cancer.
Materials and Methods
Cell lines, antibodies, plasmids and reagents
The PC3 and DU145 prostate cancer cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were initially grown and multiple aliquots were stored at −180°C for future use as required. Cells were purchased more than 6 months ago and were not further tested or authenticated by the authors. Cells were grown in RPMI 1640 medium supplemented with 10% FBS and 1× antibiotic antimycotic solution (Sigma-Aldrich, St.Louis, MO) and maintained at 37°C in the presence of 5% CO2. Sp1 and E2F3 antibodies were purchased from Millipore (Temecula, CA), and Sp3, Sp4, E2F1, E2F2, uPAR, VEGF and β-actin antibodies were obtained from Santa Cruz (Santa Cruz, CA). Survivin and ZBTB4 antibodies were purchased from Cell Signaling (Danvers, MA) and Aviva Systems Biology (San Diego, CA), respectively. Mimics and antisense microRNAs and their counterpart controls were purchased from Dharmacon (Lafayette, CO). siRNAs for Sp1, Sp3 and Sp4 were purchased from Sigma-Aldrich (St. Louis, MO) and scrambled control siRNA from Quiagen (Valencia, CA). Reporter luciferase constructs for pSp1, pSp3, pVEGF and pSurvivin were previously used in our studies (30, 31).
Invasion and scratch (migration) assay
Invasion assay was performed as previously described (31) with addition of indicated compound after 18 hr. For scratch assay, after cells were > 80% confluent in 6-well plates, the scratch was made using a sterile pipette and then treated with control (DMSO) or compound. Cell migration into the scratch was determined after 18 hr (7-8 determinations/treatment).
Cell proliferation, apoptosis and fluorescence-activated cell sorting cell cycle analysis
PC3 and DU145 prostate cancer cells (2 × 104) were plated in 12-well plates, and the next day fresh medium containing either control (DMSO) or the indicated compound was added. Cells were counted at the indicated times using a Coulter Z1 cell counter (Beckman Coulter, Fullerton, CA). For cell cycle analysis, cells were treated with staining solution containing propidium iodine (50 μg/ml) after treatment with the compound for 24 hr. Stained cells were analyzed by a FACS Calibur Flow Cytometer (Becton Dickinsin Systems, Franklin Lakes, NJ). Apoptosis was detected using Annexin V FITC kit (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol.
Quantative real-time PCR analysis for mRNAs and miRNAs
Total RNA was extracted using the mirVanaRNA extraction kit (Applied Biosystems, Carlsbad, CA). The change in expression of miRNAs was quantified using Taqman miRNA kit using either RNU6B or U6 small nuclear RNA as control. For mRNA quantification, SYBR real-time PCR was carried out and normalized to TBP as previously described (31). The following primer sets used for qRT-PCR analysis:
E2F1 (forward): 5′ ATGTTTTCCTGTGCCCTGAG 3′
E2F1 (reverse): 5′ ATCTGTGGTGAGGGATGAGG 3′
E2F2 (forward): 5′ CCTTGGAGGCTACTGACAGC 3′
E2F2 (reverse): 5′ CCACAGGTAGTCGTCCTGGT 3′
E2F3 (forward): 5′ CACCCTGGACCTCAAACTGT 3′
E2F3 (reverse): 5′ AAGTCTTTGGAAGCGGGTTT 3′
TBP (forward):5′ TGCACAGGAGCCAAGAGTGAA-3′
TBP (reverse): 5′-CTTCAGGTGGTCCTCCATGT 3′
Western blotting, immunostaining and luciferase assay
Western blotting was carried out using β-actin as a loading control as previously described (31). Different promoter reporter luciferase constructs, β-galactosidase, ZBTB4 expression plasmid (1 μg/well), or antisense or mimic miRs (150 nMol/well) and their corresponding controls (150 nMol/ml) were cotransfected by Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The luciferase activities were either normalized to β-galactosidase or protein concentration. For Immunostaining, briefly, cells were fixed in 4% paraformaldehyde, permeabilized in PBS with 0.3% Triton X-100 for 10 min, and preincubated for 2 hr with 10% normal goat serum for blocking. Cells were then incubated with Sp1 antibody overnight and incubated with FITC-conjugated secondary antibody. Images were captured with LSM 510 Meta confocal microscope (Carl Zeiss).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using ChIP-IT Express Magnetic Chromatin Immunoprecipitation Kit (Active Motif, Carlsbad, CA) according to manufacturer's protocol. Briefly, PC3 cells (1 × 107 cells) were treated with DMSO or CDODA-Me (5 μM) for 18 hr, and cells were fixed, sonicated and co-immunoprecipitated with Sp1 antibody and IgG control. The immunoprecipitated lysates were reversely DNA crosslinked and then DNA was prepared by proteinase K digestion followed by PCR amplification. The ChIP primer sets used were:
Primer I (forward): 5′ CTTGGAGAGCAAGCGAGTCT 3′
Primer I (reverse): 5′ GGACTCATCCTTACCGCTCA 3′
Primer II (forward): 5′ CCAGCCTTCTTGGTGTGTTT 3′
Primer II (reverse): 5′ CTACTCCCAGGACGGATCAA 3′
Cancer patient gene expression data analysis
Normalized microRNA gene expression data were obtained from Gene Expression Omnibus (accession number GSE23022). Four ZBTB4 prostate cancer patient expression data sets were obtained from the Oncomine data base (32-36). For Kaplan-Meier patient survival analysis by ZBTB4 expression level, Dr. Luo in the University of Pittsburgh kindly provided us clinical data for the analysis (37).
Xenograft studies in athymic mice
Male athymic nude mice (Foxn1nu, ages 6–8 weeks) were purchased from Harlan Laboratories (Indianapolis, IN). PC3 cells (1 × 106) in 1:2 ratio of matrigel (BD Bioscience, San Jose, CA) were injected in the flank of nude mice, and mice were randomly divided into two groups of six animals each. Either corn oil (control) or CDODA-Me (15 mg/kg/day) were orally administered to each group, respectively. The volume and size of xenografted tumors were measured throughout the study: V = LW/2, where L and W were length and width. The selected tissues were further examined by routine H&E staining and RNA analysis.
Immunohistochemistry
Paraffin-embedded tissue sections (5 μmol/L thick) were deparaffinized and endogeneous peroxidase activity was blocked by the use of 2% hydrogen peroxide in PBS for 2 min. Antigen retrieval for Sp1 staining was made after incubation for 30 min in 10 mmol/L sodium citrate buffer (pH=6.0) at 95°C in a steamer followed by cooling to 20°C for 10-15 min. The slides were incubated with a protein blocking solution (VECTASTAIN Elite ABC kit; Vector Laboratories, Burlingame, CA) and stained by manufacturer's protocol with 1:100 dilution of Sp1 antibody (VECTASTAIN Elite ABC kit). The staining was developed by diaminobenzidine reagent (Vector Laboratories) as a brown color and the sections were then counterstained with Gill's hematoxylin.
Results
CDODA-Me (Fig. 1A) inhibits LNCaP prostate cancer cell growth (38), and results in Figures 1A and 1B show that CDODA-Me also inhibited growth of androgen-insensitive PC3 and DU145 prostate cancer cells, increased the percentage of cells in G0/G1 and decreased the percentage in S and G2/M phases of the cell cycle. The antiproliferative activity of CDODA-Me was accompanied by induction of both early and late apoptosis which was determined with an Annexin V-FITC kit (Fig. 1C). We also observed that after treatment of PC3 and DU145 cells with 1.0 or 2.5 μM CDODA-Me for 18 hr, there was a significant decrease in cell invasion using a Boyden chamber assay and in migration using a scratch assay (Fig. 1D).
Figure 1.

Effects of CDODA-Me on prostate cancer cell proliferation, invasion and migration. Cell proliferation (A) and cell cycle progression (B) DU145 or PC3 cells were treated with DMSO or CDODA-Me (1 – 5 μM) for up to 72 hr (A) or 24 hr (B), and the number of cells or their distribution in different phases of the cell cycle were determined as outlined in the Materials and Methods. Apoptosis (C) and migration/invasion (D). Cells were treated with CDODA-Me for 32 hr (apoptosis) or 18 hr (migration/invasion), and the effects on apoptosis, migration and invasion were determined as outlined in the Materials and Methods. Results are expressed as means ± SE for at least three replication determinations, and significantly (p < 0.05) increased (*) or decreased (**) responses are indicated.
Previous studies show that CDODA-Me decreased Sp1, Sp3, Sp4 and Sp-regulated gene expression in colon cancer cells (15), and results in Figure 2A shows that CDODA-Me also decreased Sp1, Sp3 and Sp4 protein levels in PC3 and DU145 prostate cancer cells. Using PC3 cells as a model, cells were treated with DMSO or 2.5 μM CDODA-Me for 1 hr and then stained with DAP-1 and Sp1-FITC alone or in combination. There was an overlap between nuclear DAPI and Sp1 staining in control cells confirming the nuclear location of Sp1 (Fig. 2B). In cells treated with CDODA-Me, there was a significant decrease in nuclear Sp1 staining. CDODA-Me also decreased expression of several Sp-regulated genes including survivin, VEGF andurokinase plasminogen activator receptor (uPAR) and cyclin D1 in both cell lines (Fig. 2C). The role of Sp1 downregulation by CDODA-Me in modulating cell cycle progression was confirmed by RNA interference (RNAi) which showed that knockdown of Sp1 (iSp1) increased the percentage of PC3 and DU145 cells in G0/G1 and decreased their percentage in S and G2/M phases (Fig. 2D), and this was consistent with results obtained for CDODA-Me (Fig. 1B).
Figure 2.
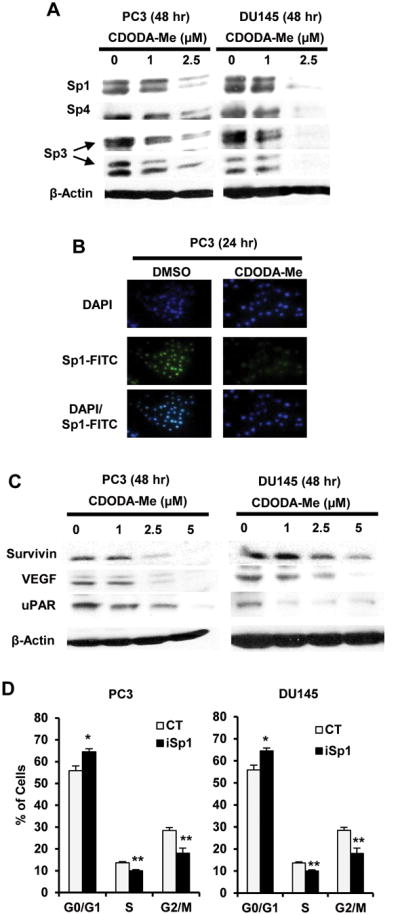
CDODA-Me-dependent regulation of Sp transcription factors. Downregulation of Sp proteins by western blot (A) or immunostaining (B). Cells were treated with CDODA-Me for the indicated times and Sp1, Sp3 and Sp4 were analyzed by western blots or Immunostaining (Sp1 only) as described in the Materials and Methods. (C) Decreased Sp-regulated genes. Cells were treated as described in (A) and Sp-regulated genes were analyzed by western blots. (D) Sp1 knockdown alters cell cycle progression. Cells were transfected with siSp1 oligonucleotide and after 48 hr, cells were treated with 2.5 μM CDODA-Me for 24 hr and analyzed by FACS as described in the Materials and Methods. Results in (D) are expressed as means ± SE for three replicate determinations and significant (p < 0.05) increases (*) or decreases (**) compared to control non-specific oligonucleotide (CT) are indicated.
Recent studies in breast cancer cells showed that high expression of Sp transcription factors is due to miR-dependent suppression of the transcriptional repressor ZBTB4 which is also a prognostic factor for breast cancer patient survival (31). Figure 3A shows that CDODA-Me induced ZBTB4 protein and mRNA expression in PC3 and DU145 cells; moreover, overexpression of ZBTB4 in these cell lines was accompanied by downregulation of Sp1, Sp3, Sp4 and the Sp-regulated genes VEGF, survivin and uPAR (Fig. 3B). ZBTB4 overexpression also decreased luciferase activity in PC3 and DU145 cells transfected with constructs containing GC-rich inserts from the Sp1 (pSp1), Sp3 (pSp3), VEGF (pVEGF), and survivin (pSurvivin) gene promoters (Fig. 3B). Like CDODA-Me (Fig. 1B) and iSp1 (Fig. 2D), overexpression of ZBTB4 also increased the percentage of PC3 and DU145 cells in G0/G1 and decreased the percentage of cells in G2/M and S (minimal) phases of the cell cycle (Fig. 3C). The effects of CDODA-Me on binding of Sp1 and ZBTB4 with the GC-rich proximal region of the Sp1 promoter was investigated in a ChIP assay (Fig. 3D). Sp1 but not ZBTB4 was constitutively bound to the GC-rich region and, after treatment with CDODA-Me, occupation of this site by Sp1 was decreased and this was accompanied by increased occupation by ZBTB4 (Primer I). In contrast, interactions of Sp1 and ZBTB4 with a distal non-GC-rich region of the Sp promoter were not detected (Primer II). These results suggest that the effects of CDODA-Me are consistent with induction of the Sp repressor ZBTB4 which in turn suppresses Sp-regulated genes and responses through competitive displacement of Sp proteins bound to GC-rich sites. Examination of publically available prostate cancer patient array data showed that ZBTB4 is more highly expressed in normal prostate tissue compared to prostate carcinomas, and high expression of ZBTB4 is a prognostic factor for increased relapse-free survival (Suppl. Figs. 1A – 1C) (33-36, 38-41), and similar results were reported for breast cancer patients (31). Future studies will also examine ZBTB4 protein expression in prostate cancer patients.
Figure 3.

ZBTB4 induction and function. (A) Induction of ZBTB4 mRNA and protein. Cells were treated with CDODA-Me for the indicated times, and mRNA and protein levels were determined by real time PCR and western blots, respectively, as outlined in the Materials and Methods. (B) Effects of ZBTB4 overexpression on Sp-regulated proteins and constructs. ZBTB4 (1 μg/well) was overexpressed alone in PC3 and DU145 cells or in combination with several GC-rich Sp-regulated constructs and proteins, and luciferase activity was determined as described in the Materials and Methods. ZBTB4 overexpression modulates the cell cycle (C) and Sp1 binding to the GC-rich Sp1 promoter in a ChIP assay (D). Cells were transfected with ZBTB4 and after 24 hr (C) or 18 hr (D), cells were analyzed by FACS or in a ChIP assay as described in the Materials and Methods. Results are expressed as means ± SE for at least three replication determinations, and significantly (p < 0.05) increased (*) or decreased (**) responses are indicated.
Suppression of ZBTB4 in breast cancer cells has been linked to microRNA-17-5p (miR-17), miR-20a and their paralogs miR-93 and miR-106b, and these miRs were expressed in PC3 and DU145 cells (Fig. 4A). Moreover, the four miR paralogs were all more highly expressed in prostate tumors vs. non-tumor tissues (Fig. 4A) and these results were inversely related to ZBTB4 expression in the same tissues (Suppl. Fig. 1). Treatment of PC3 and DU145 cells with CDODA-Me also decreased expression of the four miR paralogs (Fig. 4B) and this was correlated with the induction of ZBTB4 mRNA and protein observed in these cell lines (Fig. 3A). Moreover, luciferase activity was decreased in PC3 and DU145 cells transfected with constructs containing promoter sequences upstream from miR-93 and miR-106b [pMCM7(-558)] and miR-17-5p and miR-20a [pmiR-17-92(pro1353)] (39, 40) and treated with CDODA-Me (Fig. 4C). Since miR-93 and miR-106b are part of the miR-106b-25 cluster located in the 13th intron of the MCM7 gene, we investigated the effects of CDODA-Me on MCM7 and show that expression of this gene was also decreased in PC3 and DU145 cells. This demonstrates that the miR paralogs are primary targets of CDODA-Me and the mechanism of drug-induced repression of these miRs is currently being investigated.
Figure 4.

MiR paralog expression and regulation in prostate cancer cells. (A) Expression in cells and patients. MiR levels in PC3 and DU145 cells were determined by real time PCR as described in the Materials and Methods, and expression in tumors was obtained from publically available results (GSE23022). Effects of CDODA-Me on miR expression (B) and luciferase activity (C). Cells were treated with 2.5 μM CDODA-Me for 24 hr (B) or transfected with promoter construct from MCM7 gene and miR-17-92 cluster (C) promoters. MiR levels and luciferase activities were determined as outlined in the Materials and Methods. (D) MCM7 expression. Cells were treated with 2.5 μM CDODA-Me for 24 hr and MCM7 mRNA levels were determined by real time PCR as outlined in the Materials and Methods. The significance (p < 0.05) of treatment-related decreases are indicated (**), and the level of significance for the patient data (A) is indicated.
Confirmation of the role of the miR paralogs in repressing ZBTB4 and facilitating high expression of Sp1, Sp3 and Sp4 was further investigated in PC3 cells transfected with miR antagomirs and mimics. Figure 5A shows that miR-93 and miR-106b mimics decreased and miR-93 and miR-106b antagomirs increased luciferase activity in PC3 and DU145 cells transfected with the ZBTB4-3′-UTR(2559 bp) construct which contains the 3′-UTR sequence from the ZBTB4 gene with binding sites for the four miR paralogs (31). Moreover, transfection of the PC3 cells with miR paralog antagomirs also induced ZBTB4 mRNA expression (Fig. 5B). Using miR-106b and miR-93 antagomirs (As-miR-106b and As-miR-93) as models, we also show that transfection of both antagomirs decreased luciferase activity in PC3 and DU145 cells cotransfected with pSp1, pSp3, pVEGF and pSurvivin promoter-reporter constructs (Fig. 5C) and also downregulated expression of Sp1, Sp3, Sp4, VEGF, uPAR and survivin proteins (Fig. 5D).
Figure 5.

MiR mimics and antagomirs and their effects on ZBTB4 and Sp-regulated genes/reporter genes. Interactions with the 3′-UTR of ZBTB4 (A) and induction of ZBTB4 mRNA (B). PC3 cells were transfected with the ZBTB4-UTR-luciferase construct containing the CACUUUA miR binding site (A) and cotransfected with indicated miR mimics (A) or antagomirs (A and B). Luciferase activity or ZBTB4 mRNA expression were determined as outlined in the Materials and Methods. (C) MiRantagomirs decrease luciferase activity (C) and downregulate Sp proteins and Sp-regulated genes (D). Cells were transfected with the antagomirs and, in (C), were cotransfected with GC-rich constructs. Luciferase activity or protein expression was determined as outlined in the Materials and Methods. Results (A – C) are expressed as means ± SE for at least three replicate determinations, and significantly (p < 0.05) induced (*) or decreased (**) responses are indicated.
Both the miR-17-92 and miR-106b-25 clusters are regulated by E2F transcription factors (39-41), and downregulation of the four miR paralogs by CDODA-Me (Fig. 4B) could also be due to either direct or indirect effects on E2Fs. Supplemental Figures 2A and 2B show that CDODA-Me decreased expression of E2F1 and E2F2 but not E2F3 protein and mRNA levels, respectively, in PC3 and DU145 cells. Since CDODA-Me downregulates the four miR paralogs (Fig. 4B) and induces the transcriptional repressor ZBTB4, we overexpressed ZBTB4 in PC3 and DU145 cells and show that ZBTB4 decreased expression of E2F1 and E2F2 but not E2F3 (Suppl. Fig. 2C). This suggests that CDODA-Me indirectly suppresses E2F1 and E2F2 through induction of ZBTB4. Moreover, knockdown of Sp1, Sp3 and Sp4 by RNAi also showed that E2F1 and E2F2 were Sp-regulated genes in PC3 cells (Suppl. Fig. 2D) and similar data were observed in DU145 cells (Suppl. Fig. 2E). CDODA-Me-induced suppression of E2F1 and E2F2 is due to disruption of miR-ZBTB4 interactions and results in downregulation of Sp1, Sp3 and Sp4. Previous reports show that drug-induced downregulation of Sp proteins can be proteasome- or ROS-dependent (12, 15, 28, 42); however, glutathione and proteasome inhibitors did not affect CDODA-Me-induced downregulation of Sp1, Sp3 and Sp4 (Suppl. Fig. 3), and we are currently investigating the mechanisms of CDODA-Me-induced effects on the miR paralogs:ZBTB4-Sp protein axis.
Treatment of male athymic nude mice bearing PC3 cells as xenografts with CDODA-Me (15 mg/kg/d) decreased tumor cell growth and weight (Fig. 6A) and nuclear Sp1, Sp3 and Sp4 staining in tumors (Fig. 6B). Moreover, H&E staining showed a more disorganized and disrupted pattern in treated vs. control tumors. Examination of the tumors showed that CDODA-Me decreased expression of miR-106b, miR-93, miR-17 and miR-20a (Fig. 6C). Figure 6D summarizes the pathways activated by CDODA-Me in which the primary targets are the miR paralogs which serve to suppress ZBTB4 in prostate cancer cells. CDODA-Me induces downregulation of the miRs and induction of ZBTB4 which represses expression Sp transcription factors and the pro-oncogenic Sp-regulated genes and their downstream responses.
Figure 6.

In vivo studies. Inhibition of tumor size and weights (A), H&E and Sp1, Sp3 and Sp4 staining (B), and miR-106b/miR-92 levels (C) in mice treated with CDODA-Me. Athymic nude mice bearing PC3 cells as xenografts were treated with CDODA-Me (15 mg/kg/d) over a period of 21 days, and tumor volumes were estimated. Tumor weights, H&E and Sp1 staining, and miR mRNA levels were determined as outlined in the Materials and Methods. (D) Mode of action for CDODA-Me. Proposed pathways triggered by CDODA-Me in prostate cancer cells/tumors that lead to downregulation of Sp1, Sp3, Sp4 and Sp-regulated genes/responses. Results are expressed as means ± SE for at least three replication determinations, and significantly (p < 0.05) decreased (*) responses are indicated.
Discussion
Sp1, Sp3 and Sp4 transcription factors are prototypical non-oncogenes that are overexpressed in cancer cells and tumors and regulate a battery of genes that play a role in cancer cell growth, survival, angiogenesis and metastasis. Research in this laboratory has identified several anticancer agents that target downregulation of Sp1, Sp3, Sp4 and Sp-regulated genes in cancer cells and these include arsenic trioxide, curcumin, non-steroidal anti-inflammatory drugs (NSAIDs), and several triterpenoids including betulinic acid, CDODA-Me and the structurally-related methyl 2-cyano-3,12-dioxooleana-1,9-dien-28-oate (CDDO-Me) (12-21, 28, 29, 42). The mechanisms associated with drug-induced Sp downregulation are both proteasome-dependent and -independent and also vary with cell context. For example, betulinic acid induces proteasome-dependent downregulation of Sp1, Sp3 and Sp4 in androgen-sensitive LNCaP prostate cancer cells (38), whereas in colon cancer cells, this response is ROS-dependent and proteasome-independent (14). Cell context-dependent differences in pathways leading to Sp downregulation by curcumin and tolfenamic acid have also been observed (16, 17, 19, 21, 43).
CDODA-Me inhibits growth of colon, bladder, prostate and pancreatic cancer cell lines (15, 19, 38) and, although CDODA-Me is a PPARγ agonist, the anticancer activity of this compound in LNCaP cells is PPARγ-independent (38). Like betulinic acid (12), CDODA-Me induces proteasome-dependent degradation of Sp1, Sp3 and Sp4 in LNCaP cells (unpublished results); however, in androgen-insensitive PC3 and DU145 cells, CDODA-Me decreased expression of Sp1, Sp3, Sp4 and Sp-regulated genes and this response was proteasome independent (Suppl. Fig. 3). The effects of CDODA-Me on inhibition of cell growth, invasion and migration, induction of apoptosis, and G0/G1 to S phase arrest (Figs. 1 and 2) were consistent with results obtained by knockdown of Sp transcription factors (Fig. 2D) (15, 18-20, 28). These in vitro results are complemented by in vivo studies showing that CDODA-Me (15 mg/kg) also decreased tumor growth in a PC3 cell xenograft study and downregulated expression of Sp1 (Fig. 6).
A possible explanation for the high expression of Sp1, Sp3 and Sp4 in cancer cell lines is due to miR-dependent inhibition of the transcriptional repressors ZBTB10 and ZBTB4 which can act as Sp-repressors by competitively binding GC-rich promoters resulting in the loss of Sp proteins from these sites (30, 31). We first characterized miR-27a-mediated suppression of ZBTB10 in breast cancer cells showing that both antisense miR-27a or ZBTB10 overexpression repressed expression of Sp1, Sp3 and Sp4 (30). Recent studies also show that miR-20a and related paralogs that are part of the miR-17-92 (miR-20a and miR-17-5p) and miR-106b-25 (miR-106b and miR-93) clusters inhibit ZBTB4 expression (31). MiR-27a inhibits expression of Myt-1, a kinase that arrests cells in G2/M phase, and as-miR-27a or downregulation of miR-27a by CDODA-Me in colon cancer cells arrest cells in G2/M phase of the cell cycle (15). In contrast, both CDODA-Me and Sp1 knockdown inhibited G0/G1 to S phase progression of PC3 and DU145 cells (Figs. 1C and 2D). These results, coupled with relatively low expression of miR-27a, prompted us to examine the effects of CDODA-Me on miR-mediated inhibition of ZBTB4 in prostate cancer cells. ZBTB4 was clearly induced by CDODA-Me in PC3 and DU145 cells (Fig. 3A), and overexpression of ZBTB4 repressed Sp1, Sp3, Sp4 and Sp-regulated genes and reporter genes (Figs. 3B and 3C). CDODA-Me also decreased expression of the four miR paralogs and induced ZBTB4. MiR-106b and miR-93 antagomirs also suppressed Sp and Sp-regulated genes and reporter genes (Figs. 4 and 5) as previously reported for miR-20a and miR-17-5p (31). Previous studies show that E2F transcription factors regulate expression of both miR-17-92 and miR-106b-25 clusters (39-41). CDODA-Me decreased expression of E2F1 and E2F2 in DU145 and PC3 cells; however, the effects of CDODA-Me were not upstream from the miR cluster (Fig. 6D) since knockdown of Sp1, Sp3 and Sp4 by RNAi showed that E2F1 and E2F2 were also Sp-regulated genes (Suppl Figs. 2D and 2E). Drug-induced ROS and ROS-mediated disruption of miR-27a:ZBTB10 has also been linked to Sp downregulation and these effects are reversed after cotreatment with antioxidants (14, 19, 28, 42). In contrast, glutathione did not affect downregulation of Sp1, Sp3 and Sp4 by CDODA-Me (Suppl. Fig. 3) and the mechanisms associated with the effects of CDODA-Me on miR-ZBTB4 regulation and expression of Sp transcription factors is currently being investigated.
In summary, our results show that CDODA-Me-mediated suppression of Sp1, Sp3, Sp4 and Sp-regulated genes is due to disruption of miR-20a/17-5p/106b/93-dependent regulation of ZBTB4 in androgen-insensitive prostate cancer cells, and this pathway contributes to the antitumorigenic activity of this drug. Interestingly, ZBTB4 was more highly expressed in non-tumor vs. prostate tumor tissue (Suppl. Figs. 1A and 1B) (33-36), whereas the inverse expression of the miRs was observed (Fig. 4A), and ZBTB4 overexpression was a prognostic factor for increased prostate cancer patient survival (Suppl. Fig. 1C). These results suggest that “addiction” to non-oncogenic Sp transcription factors by prostate cancer cells is due to miR-dependent suppression of ZBTB4, suggesting that the miRs are important targets for activating downregulation of Sp1, Sp3 and Sp4 (Fig. 6D). Current studies are focused on identifying other agents that target miR-20a and related paralogs. We are also investigating the mechanism of drug-miR interactions that result in the induction of the transcriptional repressor ZBTB4 which has both prognostic and functional significance in prostate cancer and serves to downregulate “non-oncogenic” Sp transcription factors that contribute to the prostate tumor phenotype.
Supplementary Material
Acknowledgments
Funding: This research was supported by the National Institutes of Health (CA136571 to S. Safe) and Texas AgriLife (to S. Safe).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol. 2011;29:3651–8. doi: 10.1200/JCO.2011.35.2005. [DOI] [PubMed] [Google Scholar]
- 3.Dayyani F, Gallick GE, Logothetis CJ, Corn PG. Novel therapies for metastatic castrate-resistant prostate cancer. J Natl Cancer Inst. 2011;103:1665–75. doi: 10.1093/jnci/djr362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asmane I, Ceraline J, Duclos B, Rob L, Litique V, Barthelemy P, et al. New strategies for medical management of castration-resistant prostate cancer. Oncology. 2011;80:1–11. doi: 10.1159/000323495. [DOI] [PubMed] [Google Scholar]
- 5.Attard G, de Bono JS. Translating scientific advancement into clinical benefit for castration-resistant prostate cancer patients. Clin Cancer Res. 2011;17:3867–75. doi: 10.1158/1078-0432.CCR-11-0943. [DOI] [PubMed] [Google Scholar]
- 6.Isaacs JT, Coffey DS. Adaptation versus selection as the mechanism responsible for the relapse of prostatic cancer to androgen ablation therapy as studied in the Dunning R-3327-H adenocarcinoma. Cancer Res. 1981;41:5070–5. [PubMed] [Google Scholar]
- 7.Ellis LM, Hicklin DJ. Resistance to targeted therapies: refining anticancer therapy in the era of molecular oncology. Clin Cancer Res. 2009;15:7471–8. doi: 10.1158/1078-0432.CCR-09-1070. [DOI] [PubMed] [Google Scholar]
- 8.Gupta SC, Kim JH, Prasad S, Aggarwal BB. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 2010;29:405–34. doi: 10.1007/s10555-010-9235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130:986–8. doi: 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma SV, Settleman J. Exploiting the balance between life and death: targeted cancer therapy and “oncogenic shock”. Biochem Pharmacol. 2010;80:666–73. doi: 10.1016/j.bcp.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Chintharlapalli S, Papineni S, Ramaiah SK, Safe S. Betulinic acid inhibits prostate cancer growth through inhibition of specificity protein transcription factors. Cancer Res. 2007;67:2816–23. doi: 10.1158/0008-5472.CAN-06-3735. [DOI] [PubMed] [Google Scholar]
- 13.Chadalapaka G, Jutooru I, Burghardt R, Safe S. Drugs that target specificity proteins downregulate epidermal growth factor receptor in bladder cancer cells. Mol Cancer Res. 2010;8:739–50. doi: 10.1158/1541-7786.MCR-09-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chintharlapalli S, Papineni S, Lei P, Pathi S, Safe S. Betulinic acid inhibits colon cancer cell and tumor growth and induces proteasome-dependent and -independent downregulation of specificity proteins (Sp) transcription factors. BMC Cancer. 2011;11:371. doi: 10.1186/1471-2407-11-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chintharlapalli S, Papineni S, Abdelrahim M, Abudayyeh A, Jutooru I, Chadalapaka G, et al. Oncogenic microRNA-27a is a target for anticancer agent methyl 2-cyano-3,11-dioxo-18beta-olean-1,12-dien-30-oate in colon cancer cells. Int J Cancer. 2009;125:1965–74. doi: 10.1002/ijc.24530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chadalapaka G, Jutooru I, Chintharlapalli S, Papineni S, Smith R, 3rd, Li X, et al. Curcumin decreases specificity protein expression in bladder cancer cells. Cancer Res. 2008;68:5345–54. doi: 10.1158/0008-5472.CAN-07-6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdelrahim M, Baker CH, Abbruzzese JL, Sheikh-Hamad D, Liu S, Cho SD, et al. Regulation of vascular endothelial growth factor receptor-1 expression by specificity proteins 1, 3, and 4 in pancreatic cancer cells. Cancer Res. 2007;67:3286–94. doi: 10.1158/0008-5472.CAN-06-3831. [DOI] [PubMed] [Google Scholar]
- 18.Jutooru I, Chadalapaka G, Sreevalsan S, Lei P, Barhoumi R, Burghardt R, et al. Arsenic trioxide downregulates specificity protein (Sp) transcription factors and inhibits bladder cancer cell and tumor growth. Exp Cell Res. 2010;316:2174–88. doi: 10.1016/j.yexcr.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jutooru I, Chadalapaka G, Lei P, Safe S. Inhibition of NFκB and pancreatic cancer cell and tumor growth by curcumin is dependent on specificity protein down-regulation. J Biol Chem. 2010;285:25332–44. doi: 10.1074/jbc.M109.095240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chintharlapalli S, Papineni S, Lee SO, Lei P, Jin UH, Sherman SI, et al. Inhibition of pituitary tumor-transforming gene-1 in thyroid cancer cells by drugs that decrease specificity proteins. Mol Carcinog. 2011;50:655–67. doi: 10.1002/mc.20738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papineni S, Chintharlapalli S, Abdelrahim M, Lee SO, Burghardt R, Abudayyeh A, et al. Tolfenamic acid inhibits esophageal cancer through repression of specificity proteins and c-Met. Carcinogenesis. 2009;30:1193–201. doi: 10.1093/carcin/bgp092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouwman P, Philipsen S. Regulation of the activity of Sp1-related transcription factors. Mol Cell Endocrinol. 2002;195:27–38. doi: 10.1016/s0303-7207(02)00221-6. [DOI] [PubMed] [Google Scholar]
- 23.Black AR, Black JD, Azizkhan-Clifford J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001;188:143–60. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 24.Ammendola R, Mesuraca M, Russo T, Cimino F. Sp1 DNA binding efficiency is highly reduced in nuclear extracts from aged rat tissues. J Biol Chem. 1992;267:17944–8. [PubMed] [Google Scholar]
- 25.Oh JE, Han JA, Hwang ES. Downregulation of transcription factor, Sp1, during cellular senescence. Biochem Biophys Res Commun. 2007;353:86–91. doi: 10.1016/j.bbrc.2006.11.118. [DOI] [PubMed] [Google Scholar]
- 26.Abdelrahim M, Smith R, 3rd, Burghardt R, Safe S. Role of Sp proteins in regulation of vascular endothelial growth factor expression and proliferation of pancreatic cancer cells. Cancer Res. 2004;64:6740–9. doi: 10.1158/0008-5472.CAN-04-0713. [DOI] [PubMed] [Google Scholar]
- 27.Higgins KJ, Abdelrahim M, Liu S, Yoon K, Safe S. Regulation of vascular endothelial growth factor receptor-2 expression in pancreatic cancer cells by Sp proteins. Biochem Biophys Res Commun. 2006;345:292–301. doi: 10.1016/j.bbrc.2006.04.111. [DOI] [PubMed] [Google Scholar]
- 28.Jutooru I, Chadalapaka G, Abdelrahim M, Basha MR, Samudio I, Konopleva M, et al. Methyl 2-cyano-3,12-dioxooleana-1,9-dien-28-oate decreases specificity protein transcription factors and inhibits pancreatic tumor growth: role of microRNA-27a. Mol Pharmacol. 2010;78:226–36. doi: 10.1124/mol.110.064451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chadalapaka G, Jutooru I, McAlees A, Stefanac T, Safe S. Structure-dependent inhibition of bladder and pancreatic cancer cell growth by 2-substituted glycyrrhetinic and ursolic acid derivatives. Bioorg Med Chem Lett. 2008;18:2633–9. doi: 10.1016/j.bmcl.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mertens-Talcott SU, Chintharlapalli S, Li X, Safe S. The oncogenic microRNA-27a targets genes that regulate specificity protein transcription factors and the G2-M checkpoint in MDA-MB-231 breast cancer cells. Cancer Res. 2007;67:11001–11. doi: 10.1158/0008-5472.CAN-07-2416. [DOI] [PubMed] [Google Scholar]
- 31.Kim K, Chadalapaka G, Lee SO, Yamada D, Sastre-Garau X, Defossez PA, et al. Identification of oncogenic microRNA-17-92/ZBTB4/specificity protein axis in breast cancer. Oncogene. 2011 doi: 10.1038/onc.2011.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A. 2004;101:811–6. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, Dhanasekaran SM, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- 35.Luo JH, Yu YP, Cieply K, Lin F, Deflavia P, Dhir R, et al. Gene expression analysis of prostate cancers. Mol Carcinog. 2002;33:25–35. doi: 10.1002/mc.10018. [DOI] [PubMed] [Google Scholar]
- 36.Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8:393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–9. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 38.Papineni S, Chintharlapalli S, Safe S. Methyl 2-cyano-3,11-dioxo-18 beta-olean-1,12-dien-30-oate is a peroxisome proliferator-activated receptor-gamma agonist that induces receptor-independent apoptosis in LNCaP prostate cancer cells. Mol Pharmacol. 2008;73:553–65. doi: 10.1124/mol.107.041285. [DOI] [PubMed] [Google Scholar]
- 39.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–22. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem. 2007;282:2130–4. doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki S, Adachi A, Hiraiwa A, Ohashi M, Ishibashi M, Kiyono T. Cloning and characterization of human MCM7 promoter. Gene. 1998;216:85–91. doi: 10.1016/s0378-1119(98)00323-0. [DOI] [PubMed] [Google Scholar]
- 42.Pathi SS, Jutooru I, Chadalapaka G, Sreevalsan S, Anand S, Thatcher GR, et al. GT-094, a NO-NSAID, inhibits colon cancer cell growth by activation of a reactive oxygen species-microRNA-27a: ZBTB10-specificity protein pathway. Mol Cancer Res. 2011;9:195–202. doi: 10.1158/1541-7786.MCR-10-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdelrahim M, Baker CH, Abbruzzese JL, Safe S. Tolfenamic acid and pancreatic cancer growth, angiogenesis, and Sp protein degradation. J Natl Cancer Inst. 2006;98:855–68. doi: 10.1093/jnci/djj232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.