

Abstract
Dynamin 1-like protein (DNM1L) mediates fission of mitochondria and peroxisomes, and dysfunction of DNM1L has been implicated in several neurological disorders. To study the molecular basis of mitochondrial remodelling, we determined the crystal structure of DNM1L that is comprised of a G domain, a bundle signalling element and a stalk. DNM1L assembled via a central stalk interface, and mutations in this interface disrupted dimerization and interfered with membrane binding and mitochondrial targeting. Two sequence stretches at the tip of the stalk were shown to be required for ordered assembly of DNM1L on membranes and its function in mitochondrial fission. In the crystals, DNM1L dimers further assembled via a second, previously undescribed, stalk interface to form a linear filament. Mutations in this interface interfered with liposome tubulation and mitochondrial remodelling. Based on these results and electron microscopy reconstructions, we propose an oligomerization mode for DNM1L which differs from that of dynamin and might be adapted to the remodelling of mitochondria.
Keywords: dynamin 1-like protein, dynamin superfamily GTPases, membrane remodelling, mitochondrial fission, protein structure
Introduction
Mitochondria are dynamic organelles that continuously divide and fuse, thereby forming a tubulovesicular network throughout the cell (Hoppins et al, 2007; Bereiter-Hahn et al, 2008; Benard and Karbowski, 2009; Otera and Mihara, 2011). Mitochondrial dynamics is crucial for the maintenance of mitochondrial DNA, respiratory activity and calcium signalling, and plays a role in the control of embryonic development, apoptosis and neuronal plasticity (Cho et al, 2010; Nunnari and Suomalainen, 2012). As mitochondria cannot be created de novo, a scission mechanism is required for distribution of mitochondria throughout dividing cells.
Human dynamin 1-like protein (DNM1L), an 80-kDa mechano-chemical GTPase of the dynamin superfamily, constitutes a key player in the process of mitochondrial (Smirnova et al, 1998; Labrousse et al, 1999; Smirnova et al, 2001) and peroxisomal (Koch et al, 2003) division. The protein is recruited from the cytosol to the mitochondrial outer membrane (MOM) where it oligomerizes at discrete foci. Some of these foci develop into mitochondrial scission sites (Labrousse et al, 1999; Smirnova et al, 2001). In mammals, recruitment to the MOM involves the endoplasmic reticulum (Friedman et al, 2011) and is thought to be mediated via adaptor proteins, such as mitochondrial fission factor (Mff) (Otera et al, 2010), mitochondrial elongation factor 1/mitochondrial dynamics proteins of 49 and 51 kDa (MIEF1/MiD49/51) (Palmer et al, 2011; Zhao et al, 2011), and mitochondrial fission protein Fis1 (Yoon et al, 2003; Koch et al, 2005). For the yeast homologue of DNM1L, Dnm1, recruitment via similar adaptors (Mdv1, Caf4) was shown to nucleate and promote self-assembly of Dnm1 into helical structures, thereby driving membrane scission (Tieu and Nunnari, 2000; Griffin et al, 2005; Lackner et al, 2009).
Dysfunction of DNM1L has been implicated in mitochondria-based neurological disorders (reviewed in Chen and Chan, 2009). Impaired mitochondrial function and increased fragmentation of mitochondria was reported in Parkinson’s disease patients (Orth and Schapira, 2001; Schon and DiMauro, 2003; Lin and Beal, 2006; Knott et al, 2008; Schapira, 2008). Perturbed mitochondrial morphology in some of these patients might be caused by diminished proteasome-dependent degradation and subsequent accumulation of DNM1L (Wang et al, 2011). Fibroblasts from Alzheimer’s disease patients show significantly elongated mitochondria and a marked decrease in DNM1L expression (Wang et al, 2008). Furthermore, S-nitrosylation of DNM1L caused by the expression of the β-amyloid protein was reported to trigger mitochondrial fission and induce synaptic loss and neuronal damage. This shows the importance of DNM1L also in Alzheimer’s disease (Cho et al, 2009), although these results are controversial (Bossy et al, 2010). Abnormal mitochondrial dynamics and altered DNM1L expression levels were also found in Huntington’s disease patients (Costa et al, 2010; Shirendeb et al, 2012). Finally, a newborn girl with the A395D mutation in DNM1L shows severe developmental defects, such as microcephaly, optic neuropathy, hypoplasia, persistent lactic acidemia and elevated plasma levels of long-chain fatty acids (Waterham et al, 2007).
Recent X-ray structures of dynamin (Chappie et al, 2010; Faelber et al, 2011; Ford et al, 2011) and the dynamin-related antiviral myxovirus resistance protein A (MxA) (Gao et al, 2010, 2011) revealed common architectural principles of eukaryotic dynamins. These proteins are comprised of three distinct domains, the G domain, the bundle signalling element (BSE) and the stalk. The stalk was shown to mediate stable dimerization and dynamic oligomerization of dynamin and MxA into filaments by assembling in a criss-cross fashion via three distinct interfaces (interfaces 1–3). The stalk also mediates regulatory interactions with the BSE of the neighbouring dimer (in both dynamin and MxA), and with the PH domain (in the case of dynamin) (Faelber et al, 2011; Gao et al, 2011). GTP-dependent dimerization of the G domains via a highly conserved surface, the G interface, across the nucleotide-binding site was proposed to mediate contacts between neighbouring dynamin filaments (Mears et al, 2007; Chappie et al, 2010, 2011; Gao et al, 2010; Faelber et al, 2011; Ford et al, 2011). In turn, GTP hydrolysis was shown to trigger a power stroke of the BSE relative to the G domain leading to dynamic rearrangements of the dynamin oligomer (Hinshaw and Schmid, 1995; Marks et al, 2001; Roux et al, 2006; Chappie et al, 2011; Faelber et al, 2011; Ford et al, 2011).
Yeast Dnm1 forms highly ordered oligomers around lipid tubules with an average outer diameter of ∼120 nm (Ingerman et al, 2005; Mears et al, 2011). Electron microscopy (EM) reconstructions of this oligomer in the presence of a non-hydrolysable GTP analogue showed that Dnm1 assembles as a two-start helix with a helical pitch of 28.8 nm around these tubules, that is, two distinct helices with a spacing of about 14.4 nm were evident. Upon GTP hydrolysis, lipid tubules covered by Dnm1 constrict to ∼70 nm (Mears et al, 2011). In contrast, nucleotide-free dynamin oligomerizes around tubulated liposomes as a one-start helix with an outer diameter of 50 nm (Zhang and Hinshaw, 2001; Chen et al, 2004). In the GTP-bound form, this oligomer further constricts to generate an even narrower tubule (Sweitzer and Hinshaw, 1998; Mears et al, 2007). Thus, despite their related sequences, DNM1L and dynamin show differences in their assembly modes at membrane surfaces. To understand these variations, we determined the crystal structure of human DNM1L in the nucleotide-free form and characterized its assembly mode in a structure-function study.
Results
The structure of DNM1L
The full-length isoform 2 of human DNM1L was expressed in bacteria and purified to homogeneity for structural studies (Figure 1A; Supplementary Figure 1A). The purified protein bound to a non-hydrolysable GTP analogue, guanosine 5′-O-[gamma-thio] triphosphate (GTP-γ-S), with a dissociation constant (KD) of 10 μM, and to GDP with a KD of 23 μM (Supplementary Figure 1B). Despite extensive screening efforts in the absence and presence of various nucleotides, no crystals were obtained for this construct.
Figure 1.

Structure of DNM1L. (A) Structure-based domain architecture of human DNM1L. The first and last residue of each domain is labelled as well as the modified residues for crystallization. The classical predicted domain assignment is shown below. (B) Analytical ultracentrifugation sedimentation velocity experiments for DNM1L, DNM1L ΔB-insert, DNM1L 4A and the combined mutant (4A+ΔB). The relative protein concentration c(s) as function of the normalized sedimentation coefficient s20,w is plotted. Dimer peaks for the DNM1L 4A and the combined mutant are indicated. Peaks in the wild-type sedimentation profile could not be assigned to oligomeric species as the protein is in a fast equilibrium between different oligomeric states. (C) Ribbon-type presentation of human DNM1L. Regions not resolved in the crystal structure, such as the B-insert, are indicated by dotted lines. Important structural elements are labelled. (D) The stalks of DNM1L molecules A and C were superimposed. A 17.5° rotation of the G domains around hinge 1 is evident. (E) Side and top view on the DNM1L dimer that assembles via the central stalk interface-2. (F) Close-up view of the dimer interface-2. Hydrogen bonds are indicated as dotted lines.
Mutations in a highly conserved loop at the tip of the stalk were shown to interfere with oligomerization in dynamin and MxA and were required for their crystallization (Gao et al, 2010, 2011; Faelber et al, 2011; Ford et al, 2011). This loop is thought to be part of an assembly interface (interface-3). Also in yeast Dnm1, a mutation in this motif prevents mitochondrial targeting (Ford et al, 2011).
We followed a similar strategy as for dynamin and MxA and introduced the GPRP401-404AAAA mutation in the corresponding loop L2S of DNM1L (from here on referred to as 4A mutation). Analytical ultracentrifugation sedimentation velocity experiments showed that DNM1L in solution was in a dynamic oligomerization equilibrium (Figure 1B). The 4A mutation led to a dimeric DNM1L species that was unable to further oligomerize. Also in analytical gelfiltration right angle light scattering (RALS) experiments, DNM1L displayed a dimer-tetramer equilibrium whereas the 4A mutant formed only dimers (Supplementary Figure 2).
A second loop of 100 amino acids at the tip of the stalk, the B-insert, was predicted to be unstructured. We hypothesized that this loop might interfere with crystallization. Deletion of this loop led to a DNM1L variant that still formed higher order oligomers, although oligomerization appeared somewhat reduced (Figure 1B; Supplementary Figure 2).
Crystals of a construct combining the 4A mutation and the deletion of the B-insert were grown in the absence of nucleotides and diffracted to 3.5 Å resolution. The phase problem was solved by molecular replacement using the isolated nucleotide-free G domain and the stalk of human dynamin 1 (Faelber et al, 2011) as search models (Table I). The asymmetric unit of the crystal contained four DNM1L molecules (chains A–D), and the use of non-crystallographic symmetry during refinement resulted in improved electron density maps. Accordingly, the BSE and several loop regions not present in the initial search model could be built into the electron density (Supplementary Figure 3A). The structure was refined to an Rwork/Rfree of 25.1%/27.6% (Table I).
Table 1. Data collection and refinement statistics.

aData in highest resolution shell are indicated in paranthesis.
When not otherwise indicated, the best defined monomer in the asymmetric unit (chain A) is described in the following. DNM1L has a typical dynamin superfamily architecture composed of a G domain, a BSE and a stalk (annotated as superscript G, B and S) (Figure 1C; Supplementary Figure 4). These domain boundaries deviate from the sequence-predicted domain boundaries (Figure 1A). Thus, both the BSE and stalk contain sequences of the predicted middle domain and the GTPase effector domain (GED), as outlined in detail below.
The amino (N-) terminal G domain of DNM1L is composed of a central β-sheet of eight β-strands surrounded by eight α-helices. Structural elements of the G interface mediating GTP-dependent dimerization in dynamin, including switches I and II and the trans stabilizing loop, are only weakly defined in the electron density. Accordingly, the G domains in the crystals do not form contacts via this G interface, as expected for the nucleotide-free state of a dynamin superfamily member.
Adjacent to the G domain, the BSE is composed of a three helix bundle (Figure 1A and C; Chappie et al, 2009): α1B is derived from the very N-terminus of DNM1L, α2B (previously assigned to the middle domain) follows the G domain at the C-terminus, and α3B (previously assigned to the GED) comprises the very C-terminus of the same DNM1L molecule. These three helices assemble via a hydrophobic core (Supplementary Figure 3A). Furthermore, α1 and α3 of the BSE interact via hydrophilic and hydrophobic contacts with the G domain (Supplementary Figure 3B). The BSE’s central localization within the DNM1L molecule and its architecture involving elements from widely dispersed regions in the DNM1L sequence suggest a function as transmitter of conformational changes from the G domain to the stalk.
The stalk constitutes an elongated, antiparallel four-helix bundle (Figure 1A and C). The first three helices are derived from the predicted middle domain. Helix α1S is subdivided into α1NS, α1MS and α1CS by the two disordered loops, L1NS and L1CS. Compared to MxA, α3S is extended by the conserved loop L4S. α4S at the C-terminus of the stalk (formerly assigned to the GED, Figure 1A) leads the polypeptide chain back to the BSE and G domain.
The B-insert is interspersed between L4S and α4S, at the equivalent sequence position as the PH domain of dynamin (Faelber et al, 2011; Ford et al, 2011) and the membrane binding loop L4 of MxA (von der Malsburg et al, 2011). It is predicted to be unstructured and was proposed to constitute a putative membrane interaction site (Mears et al, 2011), although this hypothesis was later challenged (Zhang et al, 2011; Strack and Cribbs, 2012).
The DNM1L monomers in the asymmetric unit showed two different orientations of the G domain and BSE relative to the stalk, where chains A and B, and chains C and D were similar to each other. However, compared to chains A/B, the G domain—BSE units in chain C/D were 17.5° rotated around the two loops L1BS and L2BS connecting the BSE and stalk (Figure 1D). We previously suggested that these two loops constitute a hinge in dynamin and MxA (hinge 1), which allows for a flexible domain interplay during the mechano-chemical coupling (Faelber et al, 2011; Gao et al, 2011). The two conformations observed in these crystals indicate that such large-scale conformational movements around hinge 1 are indeed possible. The overall structure of DNM1L is similar to the full-length structures of dynamin and MxA (Supplementary Figure 3C). The main differences originate from the different conformations of hinge 1.
DNM1L dimerizes via the stalk
The four DNM1L molecules in the asymmetric unit assembled into two dimers via a symmetric interface of 1000 Å2 in the centre of the stalks (interface-2, Figure 1E and F). This dimerization interface includes hydrophobic contacts in the centre and salt bridges at the periphery, resulting in an X-shaped stalk dimer. Residues in this interface are highly conserved in dynamin and partially conserved in MxA (Supplementary Figure 3D). Accordingly, a similar X-shaped stalk dimer assembled via interface-2 was found in dynamin and MxA (Gao et al, 2010; Faelber et al, 2011; Ford et al, 2011).
To probe the function of this interface for assembly, point mutations were introduced in interface-2. Mutations M482D, N635A, or D638A led to aggregated or partially aggregated protein (Supplementary Figure 2). However, introduction of a charge reversal in the centre of interface-2 by the K642E and E490R mutations interfered with dimerization, resulting in a mostly monomeric species. In contrast, the E490A mutation had no effect on the native assembly (Figure 2A; Supplementary Figure 2).
Figure 2.
Dimerization via the stalk mediates oligomerization and mitochondrial remodelling. (A) Analytical ultracentrifugation sedimentation velocity experiments for DNM1L and the indicated interface-2 mutants as described in Figure 1B. Monomer and dimer peaks for E490R and K642E are indicated. As DNM1L, the E490A mutant undergoes rapid exchange reactions between different oligomeric species. (B) Left panel: Sedimentation experiments and liposome binding assays for DNM1L and DNM1L K642E. Sedimentation experiments were performed in the absence and presence of 2 mM GTP-γ-S in the absence of liposomes. Liposome co-sedimentation assays were carried out in the presence of 2 mM GDP, and in the presence or absence of PS liposomes. Lanes from SDS–PAGE are representative for three independent experiments. Boxed lanes are from the same gel. SN, supernatant. P, pellet fraction. Right panel: Quantification of sedimentation and liposome binding assays (error bars represent the s.e.m.). Bars show the percentage of protein found in the pellet with respect of total protein applied on gel. The statistical significance was calculated with respect to the corresponding DNM1L experiments. ***P<0.001; **P<0.01; *P<0.05 (also for all subsequent statistical analyses). (C) Basal and PS liposome-stimulated GTPase activities of DNM1L and the K642E mutant were determined at 37°C (n=2 for each experiment, error bars represent the s.e.m.). The statistical significance was calculated with respect to the corresponding DNM1L experiments. (D) Negative-stain electron microscopic analysis of DNM1L in the presence of PS liposomes and different nucleotides. The K642E mutant did not show any tubulation in the absence and presence of nucleotides. (E) Cellular localization and mitochondrial morphology studies in mito-dsRed expressing COS-7 cells. Cells depleted of DNM1L by siRNA were co-transfected with GFP, siRNA-resistant GFP-DNM1L or GFP-DNM1L K642E, respectively. Scrambled siRNA and co-transfected GFP were used as a control. Magnified boxed regions and a line scan plot with the relative fluorescence of the indicated GFP constructs and mito-dsRed are shown to the lower right of each subpanel. Scale bars: 50 μm. (F) Western blot showing efficient siRNA-mediated knock down of endogenous DNM1L. Scrambled siRNA was used as a control. Actin was stained as a loading control. Antibody efficiency was monitored using a COS cell lysate and recombinant DNM1L in a separate western blot. (G) FRAP assay for mitochondrial network connectivity. Mito-dsRed in an ROI (d=6 μm) containing multiple mitochondria was photobleached and its fluorescence recovery monitored for 90 s. Curves show mean values from 20 independent experiments under the indicated conditions. Prebleach intensities were normalized. For clarity, only three representative error bars are shown for each experiment.
Source data for this figure is available on the online supplementary information page

To analyse oligomerization of DNM1L, sedimentation assays were performed, as previously established for MxA (Gao et al, 2010). In the nucleotide-free state, DNM1L was found predominantly in the supernatant after ultracentrifugation. Addition of GDP had no effect on the sedimentation whereas addition of GTP-γ-S led to the formation of higher order oligomers which could be sedimented (Figure 2B). The monomeric K642E mutant sedimented only weakly, even after addition of GTP-γ-S, suggesting that higher order oligomerization requires an intact interface-2.
In liposome co-sedimentation assays, DNM1L bound to negatively-charged liposomes composed of phosphatidylserine (PS) (Figure 2B) or of a typical mitochondrial membrane mixture (Supplementary Figure 5A; van Meer et al, 2008). When analysing the effects of different nucleotides, the greatest sedimentation differences in the absence and presence of liposomes were observed with GDP (Supplementary Figure 5A), which was used in all further experiments (Figure 2B). In contrast to DNM1L, the K642E mutant co-sedimented only weakly with liposomes, underpinning the importance of dimerization via interface-2 for further oligomerization and/or liposome binding. We also tested liposome floatation assays to demonstrate membrane binding of DNM1L (Supplementary Figure 5B). However, addition of DNM1L to PS liposomes prevented their floatation. This effect might be caused by extensive remodelling of these liposomes by DNM1L (see below).
DNM1L showed a low basal GTP hydrolysis rate of 1.5/min, which was stimulated ∼10-fold in the presence of PS liposomes (Figure 2C). This stimulated GTPase rate is still 20-fold slower than the maximal reported GTPase rate of dynamin under similar conditions (Stowell et al, 1999; Faelber et al, 2011). Interestingly, this extent of GTPase stimulation was only observed at low (0.5 mM), but not at high (>2 mM) MgCl2 concentrations (Supplementary Figure 5C). This effect is likely related to the observed fusion and aggregation of PS liposomes at higher MgCl2 concentrations (Supplementary Figure 5D). The K642E mutant displayed a similar basal GTPase rate, but only 2.5-fold stimulated GTPase activity at 0.5 mM MgCl2 in the presence of liposomes, confirming the importance of assembly via interface-2 for membrane binding and resulting GTPase activation (Figure 2C).
In the absence of nucleotides, DNM1L deformed PS liposomes into long tubular structures and formed a tight protein coat with pronounced striations around the liposomes, as observed in negative-stain EM (Figure 2D). These oligomers had a diameter of 130–150 nm, similar to that of yeast Dnm1 oligomers (Mears et al, 2011). In the presence of GTP-γ-S, the oligomer constricted to ∼85 nm, and further constricted in the presence of GTP and GDP to ∼75 and 60 nm, respectively, again reminiscent of the situation in yeast Dnm1 (Mears et al, 2011). The K642E mutant was unable to deform liposomes either in the absence or in the presence of nucleotides (Figure 2D; Supplementary Figure 5E), indicating the importance of interface-2 for membrane remodelling.
To analyse the function of interface-2 in vivo, mitochondrial dynamics and morphology were analysed in the monkey kidney-derived fibroblast cell line COS-7 (Figure 2E–G; Friedman et al, 2011). Mitochondria were labelled by expression of a red fluorescent protein targeted to the mitochondrial lumen (mito-dsRed). In control cells, mitochondria showed both punctate and elongated shapes (Figure 2E). siRNA depletion of endogenous DNM1L (Figure 2F) induced the formation of long interconnected mitochondrial networks and perinuclear clusters (Figure 2E), in agreement with previous results (Gandre-Babbe and van der Bliek, 2008). Upon expression of an siRNA resistant eGFP-tagged DNM1L construct, the protein was found in punctate structures that mostly co-localized with mitochondria (Figure 2E; Supplementary Movie 1). Transfection of this construct led to a reduced length of mitochondrial tubes that appeared even more fragmented than in control cells. In contrast, eGFP-tagged DNM1L K642E did not localize to mitochondria but was diffusely distributed throughout the cytosol. Accordingly, the K642E mutant also did not rescue the aberrant mitochondrial phenotype induced by DNM1L depletion (Figure 2E). These results indicate a role of stalk dimerization for the recruitment of DNM1L to the mitochondrial membrane.
To quantify these experiments, fluorescence recovery after photobleaching (FRAP) experiments were conducted (Figure 2G; Mitra and Lippincott-Schwartz, 2010). Mito-dsRed was photobleached in a circular region of interest (ROI) and fluorescence recovery followed over time. In control cells, only a small percentage of fluorescence recovered within 90 s, indicating only marginal connectivity of mitochondria in and outside the ROI (Supplementary Movie 2A). However, upon downregulation of DNM1L by siRNA, fluorescence rapidly recovered to 67% of the pre-bleach value (Figure 2G; Supplementary Movie 2B). In these cells, the elongated mitochondrial network extended beyond the ROI, allowing fast diffusion of dsRed within the mitochondrial lumen. Expression of siRNA-resistant DNM1L in DNM1L-depleted cells decreased the extent of fluorescence recovery to a value slightly lower than that of control cells (Figure 2G; Supplementary Movie 2C). Expression of the K642E mutant, however, did not revert the high FRAP recovery rate in DNM1L-depleted cells (Figure 2G). We conclude that dimerization of the stalk via interface-2 is crucial for the function of DNM1L in catalysing mitochondrial division.
Characterization of the B-insert and the GPRP motif
The GPRP motif in loop L2S and the two termini of the B-insert are located at the tip of the stalk. The B-insert contains most of the reported post-translational modifications (Figure 3A; Supplementary Figure 4). Furthermore, it was previously suggested that the B-insert allosterically modulates DNM1L assembly (Zhang et al, 2011; Strack and Cribbs, 2012).
Figure 3.

Functional characterization of the GPRP motif and the B-insert. (A) Ribbon-type representation of the DNM1L monomer illustrating the positions of the B-insert and the GPRP motif, the known post-translational modifications (green, black and magenta) and disease and functionally inactivating mutations (orange). The first and last visible residues of the B-insert and L2S are indicated. (B) Upper panel: Oligomerization experiments and liposome binding assays as in Figure 2B for DNM1L, DNM1L ΔB-insert, DNM1L 4A and the combined DNM1L variant (4A+ΔB). Examples are representative for three independent experiments. SN, supernatant; P, pellet fraction. Lower panel: Quantification of sedimentation and liposome binding assays. (C) Basal and PS liposome-stimulated GTPase activities of DNM1L and DNM1L mutants were determined at 37°C, as in Figure 2C. (D) Cellular localization and mitochondrial morphology studies, as in Figure 2E. COS-7 cells depleted of DNM1L by siRNA were co-transfected with mito-dsRed and siRNA-resistant DNM1L ΔB-insert and the 4A mutant. Magnified boxed regions and a line scan plot with the relative fluorescence of the indicated eGFP fusion proteins and mito-dsRed are shown at the lower right of each subpanel. Scale bars: 50 μm. (E) Mitochondrial network connectivity quantified by an FRAP assay, as in Figure 2G.
To test the importance of these two structural elements, the 4A and ΔB variants were functionally analysed. In oligomerization assays, the 4A mutant assembled with reduced efficiency compared to DNM1L (Figure 3B). Similarly to the corresponding mutation in MxA (Gao et al, 2010), some residual assembly was still observed in the presence of GTP-γ-S, suggesting that the 4A mutation on its own is not fully disruptive. Liposome co-sedimentation was also diminished. In line with the AUC data (Figure 1B), these observations support a model in which interface-3 mediates oligomerization as a prerequisite for membrane binding.
In oligomerization assays, the ΔB variant sedimented already in the absence of nucleotide (Figure 3B). This might be explained by the lower solubility of this mutant, which was also apparent during its purification. Addition of GTP-γ-S further enhanced sedimentation (Figure 3B). In contrast, GDP binding partially stabilized this protein, as previously reported for other GTPases (Feuerstein et al, 1987). This variant showed no enhanced sedimentation in the presence of liposomes.
Interestingly, the crystallized double mutant (4A+ΔB) did not sediment at all in the absence of nucleotides and presence of GDP. As the 4A mutant, it still showed some residual sedimentation in the presence of GTP-γ-S and liposomes. Whereas the basal GTPase activity of the 4A, ΔB and the (4A+ΔB) variants lay in a similar range to that of DNM1L, none of these proteins showed significant GTPase activation in the presence of PS liposomes (Figure 3C). Furthermore, neither the 4A nor the ΔB variants were able to tubulate liposomes (Supplementary Figure 6). These data indicate an essential role of the GPRP motif and the B-insert for the ordered assembly of DNM1L on membrane surfaces, concomitant with GTPase stimulation.
When expressed in DNM1L-depleted COS-7 cells, both the 4A and ΔB mutants failed to localize to mitochondria (Figure 3D). Instead, the 4A mutant was diffusely distributed in the cytoplasm, whereas the ΔB mutant was found in large punctate structures in the cytoplasm (Figure 3D, white arrows). Neither mutant was able to revert the aberrant mitochondrial morphology of DNM1L-depleted cells. In the FRAP-based quantification assay, mito-dsRed-labelled mitochondria in the presence of both mutants showed a high degree of fluorescence recovery after bleaching, underpinning their inability to catalyse mitochondrial fission (Figure 3E). Thus, both the GPRP motif and the B-insert in DNM1L are required for mitochondrial fission.
An alternative oligomerization mode of DNM1L
DNM1L dimers in the crystals were stacked next to each other via an additional interface of the stalk (interface-4), opposite of interface-2 (Figure 4A). This interaction was present twice in the asymmetric unit. Interface-4 had a size of 430 A2, which was dominated by ionic interactions and showed only limited sequence similarity to dynamin and MxA (Supplementary Figures 3D and 4). The alternating assembly of DNM1L via interfaces 2 and 4 led to the formation of stacked DNM1L filaments in the crystals (Figure 4A; Supplementary Figure 7A). In this oligomer, all G domains were located on one side of the filament whereas all B-inserts pointed towards the opposing side. The assembly of these filaments in the crystals was different from that of dynamin1 and MxA (Supplementary Figure 7).
Figure 4.

DNM1L oligomerizes via an alternative interface. (A) Left and middle panels: Top and side views on a surface representation of the DNM1L oligomer in the crystal. Interface-4 is indicated (white box). The direction of the oligomer is indicated by arrows. Right panel: close-up view of interface-4. Hydrogen bonds are indicated with dotted lines. (B) Oligomerization experiments and liposome binding assays for DNM1L and interface-4 mutants (E426A, R430D), as in Figure 2B. Lanes are representative of three independent experiments. SN, supernatant; P, pellet fraction. (C) Basal and PS liposome-stimulated GTPase activities of DNM1L and interface-4 mutants were determined, as in Figure 2C. (D) Representative negative-stain electron micrographs of DNM1L interface-4 mutants, as in Figure 2D. Scale bars=2 μm. (E) Cellular localization and mitochondrial morphology studies in COS-7 cells depleted of endogenous DNM1L by siRNA and co-transfected with mito-dsRed and siRNA-resistant interface-4 mutants, as in Figure 2E. Scale bars: 50 μm. (F) Mitochondrial network connectivity quantified by an FRAP assay, as in Figure 2G.
To analyse the functional relevance of interface-4, individual mutations E426A and R430D were introduced in DNM1L. These mutations did not alter the assembly in AUC and RALS experiments compared to DNM1L (Supplementary Figures 2 and 8). Also in oligomerization, liposome co-sedimentation and GTPase assays, the E426A and R430D mutants behaved mostly similarly to DNM1L (Figure 4B and C). Strikingly, both mutants failed to tubulate liposomes, both in the presence and in the absence of nucleotide, and we never observed a regular protein coat on these liposomes (Figure 4D; Supplementary Figure 9). When expressed in COS-7 cells depleted of endogenous DNM1L, none of the mutants localized to mitochondria and both failed to rescue mitochondrial morphology in DNM1L-depleted COS-7 cells (Figure 4E). This was confirmed by FRAP-based quantifications (Figure 4F). These results indicate a role of interface-4 in the functional assembly of DNM1L at membrane surfaces and in mitochondrial division; they are consistent with interface-4 as an additional assembly site.
To obtain further structural insights into DNM1L oligomerization, we fitted our DNM1L model into the EM reconstruction of nucleotide-free yeast Dnm1 (Figure 5; Mears et al, 2011). The resolution of the EM reconstruction was not sufficient to unambiguously determine the orientation of DNM1L in the electron density. We therefore considered the following constraints: (1) as in dynamin and MxA, we assumed that the G domains are located in the outer layer of the oligomer. Since the B-insert is likely unstructured, it is not expected to result in a defined electron density in the EM reconstruction. In agreement with previous suggestions (Mears et al, 2011) and similar to the loop L4 in MxA (Gao et al, 2011) and the PH domain in dynamin, we oriented the B-insert towards the observed gap between protein and liposome surface in the electron density. (2) Based on our mutagenesis data, we assumed that DNM1L forms filaments similar to those of dynamin and MxA, employing stalk interfaces 1, 2 and 3. (3) We hypothesized that the stalks also assemble via interface-4. (4) We employed the helical spacing obtained from the EM reconstruction for our fittings.
Figure 5.

Structural model of the DNM1L oligomer. (A) DNM1L tetramers were manually fitted into the EM reconstruction of yeast Dnm1 (Mears et al, 2007). In this model, the stalks of DNM1L are oriented tangentially to the lipid tubule, with the B-insert pointing towards the tubule. Oligomerization of DNM1L into a filament proceeds via stalk interface-1, -2 and -3, as in dynamin and MxA. Additionally, stalk interface-4 mediates assembly of a double stalk filament. G domains could dimerize across neighbouring double filaments. (B) Schematic illustration of the DNM1L molecules in the oligomeric model. The repeating unit of the reconstruction (boxed) contains eight DNM1L monomers.
Using these assumptions, we constructed an oligomeric model of DNM1L in which DNM1L dimers assemble tangentially to the lipid tubule, as observed for dynamin and MxA. Instead of forming a one start filament, however, the stalks assemble with a neighbouring stalk via interface-4 to form a double layer filament (Figure 5A and B). These filaments further assemble via interface-1 and -3 to form a helix with a pitch of 14.4 nm. Two double filaments, extending next to each other around the lipid tubule, account for the observed two-start helix with a 28.8 nm helical pitch. In this model, G domains of each double stalk filament dimerize across helical turns with G domains of a neighbouring filament allowing nucleotide-dependent rearrangements of adjacent filaments.
Discussion
Structural and functional studies of dynamin and dynamin-like MxA led to the identification of the stalk as the central assembly hub forming filaments via three distinct interfaces (Gao et al, 2010, 2011; Faelber et al, 2011, 2012; Ford et al, 2011). Similarly to dynamin and MxA, the stalks of DNM1L assemble via the central interface-2 to form stable dimers which are the minimal building block of DNM1L oligomers (Ingerman et al, 2005). Charge reversals in interface-2 lead to the formation of stable monomeric DNM1L variants. For dynamin, helix α4S of the stalk and/or α3B of the BSE were proposed to swap to a neighbouring stalk/BSE thereby mediating dimerization (Chappie et al, 2010; Chappie et al, 2011). The existence of monomeric interface-2 mutants in DNM1L is not consistent with such a model for DNM1L. Our liposome co-sedimentation data for interface-2 mutants also indicate that the affinity of a DNM1L monomer for membranes is low; at least two membrane interaction sites in the DNM1L dimer are required for efficient membrane recruitment.
We suggest that DNM1L dimers further oligomerize via interface-3 to form higher order assemblies. Consequently, mutations in the GPRP motif in loop L2S prevent formation of higher order oligomers in DNM1L. Interestingly, the G385E mutation next to loop L1NS of yeast Dnm1 (Sesaki and Jensen, 1999; Ingerman et al, 2005) and the corresponding G392D mutation in MxA (Gao et al, 2010) also prevent higher order assembly leading to stable dimers. We previously suggested a model for interface-3 in dynamin, which includes interactions from loops L1NS and L2S (Faelber et al, 2011). The mutagenesis data for DNM1L indicate a similar architecture of interface-3. Also residues in interface-1 are highly conserved in dynamin, MxA and DNM1L (Supplementary Figure 3D). These observations strongly suggest that DNM1L uses a similar assembly mode as dynamin and MxA to form filaments via interface-1 and -3.
Finally, we uncovered a novel assembly surface in the stalk. Via this interface-4, DNM1L dimers were stacked next to each other in the crystal. This interface has not been observed for other dynamin family members. Mutants in interface-4 can still be recruited to liposomes but fail to induce membrane remodelling, suggesting that assembly via interface-4 is required for the formation of a stable membrane-anchored scaffold with membrane remodelling activity. This observation also points to an intrinsic, adaptor protein-independent functional deficit. Interestingly, the R376E mutation was recently reported to prevent DNM1L recruitment to mitochondria (Strack and Cribbs, 2012). It was suggested that this mutation affects binding to the adaptor protein Mff. However, in pull-down assays and isothermal calorimetry experiments, we did not find evidence for a stable interaction of DNM1L and Mff (CF and OD, unpublished data). Arg376 is located in the direct vicinity of stalk interface-4 (Figure 3A) and its mutation might thus affect the integrity of this interface.
We propose that stalk interface-4 is used to assemble two neighbouring DNM1L filaments (Figures 4 and 5). Such a model is in agreement with EM reconstructions of yeast Dnm1 showing a broader filament size (14.4 nm versus 9.4 nm) and different oligomer architectures compared to dynamin (Mears et al, 2011). Since the basic architecture of such DNM1L and dynamin filaments is similar, this model would allow for a related mechanism for DNM1L in membrane constriction involving GTP-dependent dynamic rearrangements of double filaments across helical turns (Chappie et al, 2010; Chappie et al, 2011). The reduced GTPase activity of DNM1L compared to dynamin, despite the related architectures of the G domains, might indicate that not all G domains are in register to dimerize across helical turns.
The architecture of stalk filaments might be adapted to the relevant lipid template. The neck of a clathrin-coated vesicle has a typical diameter of 40 nm (Roux et al, 2010) and can be encircled by ∼14 dynamin dimers. Electron microscopic studies indicated that mitochondrial constriction sites have diameters of around 110 nm (Ingerman et al, 2005). Our current model predicts that around 48 tetramers are assembled around such a constriction site. To apply force on a membrane template of this size for membrane constriction and scission, a more rigid double filament might be required than for the scission of clathrin-coated vesicles.
Our data indicate that despite their related domain architecture, dynamin superfamily proteins can follow alternative assembly modes, depending on the cellular context and the specific functional requirements at their subcellular sites.
Materials and methods
Protein expression and purification
Human DNM1L isoform 2 (UniProtID: O00429-3, residues 1–710) was cloned into a modified pET28a vector as an N-terminal His6-tag fusion followed by a PreScission cleavage site. Mutations were inserted using standard protocols. The B-Insert (aa 514–613) was deleted as previously described (Hansson et al, 2008). All constructs were expressed in Escherichia coli host strain BL21 DE3 Rosetta2 (Novagen). Bacteria were cultured in TB medium at 37°C to an OD600 of about 0.4 followed by a temperature shift to 18°C. The protein was expressed for 18 h by addition of 40 μM isopropyl β-D-1-thiogalactopyranoside (IPTG). Bacteria were collected by centrifugation and resuspended in buffer A (50 mM HEPES/NaOH (pH 7.5), 400 mM NaCl, 5 mM MgCl2, 40 mM imidazole, 2.5 mM 2-mercaptoethanol, 1 mM DNase (Roche), 100 μM Pefabloc (Roth)), followed by cell disruption in a microfluidizer (Microfluidics). Lysates were cleared by centrifugation at 40 000 g for 30 min at 4°C, and the filtered supernatant applied to a Ni-NTA column pre-equilibrated with buffer B (50 mM HEPES/NaOH (pH 7.5), 400 mM NaCl, 5 mM MgCl2, 40 mM imidazole, 2.5 mM 2-mercaptoethanol). The column was extensively washed with buffer B, followed by buffer C (50 mM HEPES/NaOH (pH 7.5), 800 mM NaCl, 5 mM MgCl2, 40 mM imidazole, 2.5 mM 2-mercaptoethanol, 1 mM ATP, 10 mM KCl) and buffer D (50 mM HEPES/NaOH (pH 7.5), 400 mM NaCl, 5 mM MgCl2, 80 mM imidazole, 2.5 mM 2-mercaptoethanol, 0.5% (w/v) CHAPS). Bound DNM1L was eluted with buffer E (50 mM HEPES/NaOH (pH 7.5), 400 mM NaCl, 5 mM MgCl2, 300 mM imidazole, 2.5 mM 2-mercaptoethanol) and dialysed overnight at 4°C (18 kDa cutoff) against buffer B without imidazole in the presence of PreScission protease to cleave the N-terminal His6-tag. The protein was re-applied to a Ni-NTA column pre-equilibrated with buffer B without imidazole to which it bound under these conditions also in the absence of the His6-tag. Subsequently, the protein was eluted with buffer B. In a final step, DNM1L was purified by size-exclusion chromatography on a Superdex-200 column (GE) in buffer F containing 20 mM HEPES/NaOH (pH 7.5), 300 mM NaCl, 2.5 mM MgCl2 and 2.5 mM dithiothreitol. Fractions containing DNM1L were pooled, concentrated and flash frozen in liquid nitrogen.
Crystallization and structure determination
Crystallization trials by the sitting-drop vapour-diffusion method were performed at 4°C using a Hydra-plus-One pipetting robot (Matrix Technologies Corporation) and Rock Imager storage system (Formulatrix). 300 nl of DNM1L at a concentration of 10 mg/ml was mixed with an equal volume of reservoir solution containing 12% PEG3350, 50 mM K(HCOO). Crystals of the protein appeared after 3 days and had dimensions of 150 μm × 80 μm × 10 μm. For flash cooling of the crystals in liquid nitrogen, a cryo solution containing mother liquor and additionally 27% PEG3350 was used. All data were recorded at BL14.1 at BESSY II, Berlin, using a Rayonics MX-225 CCD detector. A native data set was collected from a single crystal and processed and scaled using the XDS program suite (Kabsch, 2010). The structure was solved by molecular replacement with Phaser (McCoy et al, 2007) employing the structures of the isolated nucleotide-free G domain and the stalks of human dynamin1 (pdb 3SNH) as search models (Faelber et al, 2011). The model was built using COOT (Emsley and Cowtan, 2004) and refined using refmac5 (Murshudov et al, 1997) and finally Phenix (Adams et al, 2010), including the use of a reference model, TLS parameterization and the four-fold non-crystallographic symmetry of the separated G domains, stalks and BSE. In the final model, chain A consists of residues 1–54, 58–71, 85–118, 125–152, 161–231, 234–251, 256–350, 360–401, 408–449, 454–507, 616–703; chain B of residues 1–73, 84–119, 124–152, 160–350, 361–401, 408–449, 455–504, 616–675, 679–701; chain C of residues 1–55, 58–71, 88–118, 124–249, 255–326, 329–350, 360–399, 408–450, 455–503, 617–676, 683–704; and chain D of residues 1–52, 58–71, 88–117, 125–153, 159–323, 329–350, 358–401, 406–449, 454–502, 615–705. In all, 94.3% of all residues are in the favoured regions of the Ramachandran plot and there are nine outliers, as analysed by MolProbity (Chen et al, 2010). Figures were prepared with the PyMOL Molecular Graphics System, Version 1.3r1 (Schrödinger, LLC). Buried surfaces areas (per molecule) were calculated using CNS (Schroder et al, 2010). Domain superpositions were performed with lsqkab (Winn et al, 2011). DNM1L tetramers were manually fitted into the EM reconstruction using Chimera (Pettersen et al, 2004). Sequences were aligned using CLUSTAL W (Thompson et al, 1994) and adjusted by hand.
Isothermal titration calorimetry
ITC experiments were carried out at 8°C in a VP-ITC (Microcal) in a buffer containing 20 mM HEPES, pH 7.5, 300 mM NaCl, 2.5 mM DTT, 5 mM MgCl2 at a protein concentration of 44 μM. Nucleotide concentration in the syringe was 1 mM. Binding isotherms were fitted and equilibrium dissociation constants were calculated using the Microcal ORIGIN software.
Oligomerization and liposome co-sedimentation assays
For liposome co-sedimentation assays, PS liposomes were prepared in PBS as previously described (www.endocytosis.org). In all, 0.5 mg/ml liposomes (non-filtered) were incubated at 37°C with 0.5 mM MgCl2, 2 mM GDP and 10 μM of the indicated DNM1L construct for 10 min in 50 μl reaction volume, followed by a 200 000 g spin for 15 min at 20°C. Supernatant and pellet fractions were analysed on SDS gels and quantified using ImageJ (Sheffield, 2007). For oligomerization assays, the indicated DNM1L constructs were sedimented with or without 2 mM GDP in the absence of PS liposomes. Nucleotide-free spin assays were performed without MgCl2.
Floatation assays
PS liposomes containing 1% (w/w) 1,2-dioleoyl-sn-glycero-3-phospho-L-serine-N-(5-dimethylamino-1-naphthalenesulfonyl) (Dansyl-PS) were prepared in PBS. Liposomes at a lipid concentration of 100 μM (non-filtered) were incubated for 5 min at 37°C with 1.25 μM DNM1L and then mixed with 75% (w/v) sucrose or Accudenz® to a final sucrose/Accudenz concentration of 30%. The batch was then carefully overlayed stepwise with 200 μl 25% sucrose/Accudenz and 50 μl PBS to a total volume of 500 μl. After ultracentrifugation at 200 000 g for 1 h at 4 or 20°C, samples were analysed by UV light and protein contents of equal volumes of top and bottom fractions analysed by SDS–PAGE.
Electron microscopy
In all, 6 μM DNM1L or the indicated mutants were incubated with pre-warmed (37°C) PS liposomes (0.5 mg/ml) in PBS, in the absence or presence of 2 mM nucleotides and 0.5 mM MgCl2. For MgCl2 concentration-dependent PS liposome deformation assay, 0.5 mg/ml PS liposomes in PBS were incubated with increasing concentrations of MgCl2. The volume of all reactions was 25 μl. After 5 min incubation at 37°C, 10 μl of the reaction mix was spotted on carbon-coated copper grids, stained with 2.5% uranyl acetate and inspected using a Zeiss EM910 electron microscope.
GTP hydrolysis assays
GTPase activities of 5 μM of the indicated DNM1L constructs were determined at 37°C in PBS, 2.5 mM DTT, 0.5 mM MgCl2, in the absence and presence of 0.5 mg/ml PS liposomes using saturating concentrations of GTP as substrate (1 mM). Reactions were initiated by the addition of GTP to the batch. At different time points, reaction aliquots were diluted 15-fold in GTPase buffer and quickly transferred to liquid nitrogen. Nucleotides in the samples were separated via a reversed-phase Hypersil ODS-2 C18 column (250 × 4 mm), with 10 mM tetrabutylammonium bromide, 100 mM potassium phosphate (pH 6.5) and 7.5% acetonitrile as running buffer. Denatured proteins were adsorbed at a C18 guard column. Nucleotides were detected by absorption at 254 nm and quantified by integration of the corresponding peaks. Rates were derived from a linear fit to the initial reaction (<40% GTP hydrolysed).
Analytical ultracentrifugation
Sedimentation velocity experiments were carried out at 35 000 r.p.m. in an XLI analytical ultracentrifuge (Beckmann). For all samples 400 μl of DNM1L at a concentration of 1.8 mg/ml and reference buffer (PBS+2.5 mM DTT) were loaded in two-channel centre pieces with an optical path length of 12 mm. Samples were measured at 10°C in an An-50 Ti rotor at a wavelength of 280 nm with radial spacing of 0.003 cm. The program Sednterp (http://bitcwiki.sr.unh.edu/index.php/Main_Page) was used to estimate the partial specific volume from amino-acid composition as well as the density ρ and viscosity η of the buffer. Data were then analysed with the program Sedfit (Schuck, 2000) using a continuous c(s) distribution model. Theoretical sedimentation coefficients for monomeric, dimeric and tetrameric protein species were calculated using the following equation
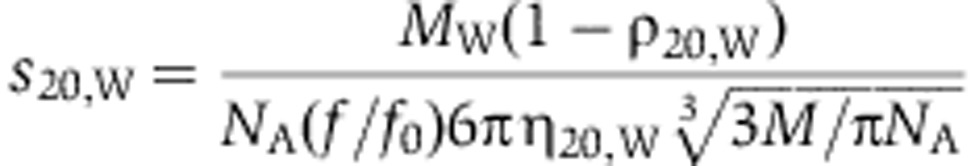 |
with ρ20,w and η20,w being the density and viscosity of water at 20°C, respectively, NA Avogadro’s number and MW and f/f0 the protein’s molecular weight and frictional ratio, respectively. Assuming frictional ratios of 1.5, 1.8 and 2.0 for monomer, dimer and tetramer, sedimentation coefficients of 4.2, 5.6 and 8.0 S were calculated for a protein with a molecular weight of 80 kDa, respectively.
In the case of DNM1L, DNM1L E426A, DNM1L R430D and DNM1L E490A additional broadening of the boundary was observed in the raw data. Furthermore, for all of these four constructs, the best fit was achieved assuming frictional ratios f/f0<1. This indicates fast chemical exchange reactions in the sample and was interpreted as rapid interchange between different oligomeric species.
Analytical gel filtration and RALS
A coupled RALS-refractive index detector (Malvern) was connected in line to an analytical gel filtration column Superdex 200 10/300 to determine absolute molecular masses of the applied proteins. Data were analysed with the provided software Omnisec. The running buffer contained 20 mM HEPES (pH 7.5), 300 mM NaCl, 2.5 mM MgCl2 and 2.5 mM DTT. For each protein sample, 100 μl of a 4 mg/ml solution was applied.
Cell culture, siRNA rescue and live-cell microscopy
COS-7 cells were routinely maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum. For live-cell microscopy, cells were cultured on 35 mm glass bottom dishes (MatTek, Ashland, MA) and transferred to low bicarbonate DMEM without phenol red supplemented with 25 mM HEPES pH 7.4. To replace endogenous DNM1L with siRNA-resistant pmEGFP-DNM1L, the underlined nucleotides were exchanged to introduce silent mutations in the DNM1L siRNA target sequence (DNM1L siRNA: 5′-CCTGCTTTATTTGTGCCTGAGGTT-3′). siRNA oligonucleotides duplexes were made by Invitrogen (5′-CCUGCUUUAUUUGUGCCUGAGGUUU-3′ and 5′-AAACCUCAGGCACAAAUAAAGCAGG-3′). Cells were transiently transfected with 150 pmol DNM1L-targeting siRNA duplexes using Roti-Fect (Roth) according to manufacturer’s instructions. Twenty-four hours post siRNA, cells were co-transfected with 300 ng pDsRed2-Mito (mito-dsRed) and 800 ng of the different DNM1L constructs using Roti-Fect. Live-cell microscopy was performed 48 h post siRNA (24 h post DNA) on a Olympus FluoView FV1000 confocal microscope equipped with a × 60 NA:1.30 silicone UPlanSApo objective. dsRed and GFP were imaged using 559 and 488 nm laser lines, respectively.
To test efficiency of siRNA-mediated DNM1L knockdown, western blots against endogenous DNM1L were performed using a mouse monoclonal anti-DNM1L antibody (#WH0010059M1, Sigma-Aldrich), which was detected by a goat anti-mouse horse-radish peroxidase (HRP)-coupled secondary antibody (Jackson ImmunoResearch). Actin was stained using a rabbit antibody (MAB386, Sigma) and detected via a goat anti-rabbit HRP-coupled secondary antibody (Jackson ImmunoResearch).
Mitochondrial connectivity FRAP assay
For FRAP experiments 48 h post siRNA transfection (24 h post DNA transfection), mito-dsRed images (51 × 51 μm) were acquired and a circular ROI (d=6.21 μm) ∼10 μm distant from the nucleus was defined. The ROI was bleached for 1 s with 100% laser power (559 nm) and mito-dsRed fluorescence recovery was monitored for 90 s (1 frame/second). Mean fluorescence intensities within the ROI were calculated over time, initial values from pre-bleach images were normalized to one (n=18–22 for each construct). Experiments were performed at room temperature.
Accession number
PDB coordinates of nucleotide-free DNM1L have been submitted to the PDB database using accession code 4BEJ.
Supplementary Material
Acknowledgments
We would like to acknowledge A Dick and S Srivastava for experimental help, S Werner and M Papst for technical assistance, F Hezel for help with video processing, B Purfürst for help with the electron microscope, K Oberheide and S Marino for helpful comments to the figures and the manuscript and the BESSY II staff for assistance during data collection. This work was supported by grants of the German Research Foundation (SFB 958/A12 and SFB740/C7) and of the Human Frontier Science Organization to OD.
Author contributions: CF, ER, KF, OR and OD designed research, CF, SG, KF and ER performed experiments, CF, SG, DS, KF, ER, JM, OR and OD analysed data, CF, KF and OD wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard G, Karbowski M (2009) Mitochondrial fusion and division: Regulation and role in cell viability. Semin Cell Dev Biol 20: 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Voth M, Mai S, Jendrach M (2008) Structural implications of mitochondrial dynamics. Biotechnol J 3: 765–780 [DOI] [PubMed] [Google Scholar]
- Bossy B, Petrilli A, Klinglmayr E, Chen J, Lutz-Meindl U, Knott AB, Masliah E, Schwarzenbacher R, Bossy-Wetzel E (2010) S-Nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer’s disease. J Alzheimers Dis 20(Suppl 2): S513–S526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie JS, Acharya S, Leonard M, Schmid SL, Dyda F (2010) G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature 465: 435–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie JS, Acharya S, Liu YW, Leonard M, Pucadyil TJ, Schmid SL (2009) An intramolecular signaling element that modulates dynamin function in vitro and in vivo. Mol Biol Cell 20: 3561–3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie JS, Mears JA, Fang S, Leonard M, Schmid SL, Milligan RA, Hinshaw JE, Dyda F (2011) A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Cell 147: 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC (2009) Mitochondrial dynamics--fusion, fission, movement, and mitophagy in neurodegenerative diseases. Hum Mol Genet 18: R169–R176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Zhang P, Egelman EH, Hinshaw JE (2004) The stalk region of dynamin drives the constriction of dynamin tubes. Nat Struct Mol Biol 11: 574–575 [DOI] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324: 102–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Lipton SA (2010) Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol Life Sci 67: 3435–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Giacomello M, Hudec R, Lopreiato R, Ermak G, Lim D, Malorni W, Davies KJ, Carafoli E, Scorrano L (2010) Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol Med 2: 490–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Faelber K, Held M, Gao S, Posor Y, Haucke V, Noe F, Daumke O (2012) Structural insights into dynamin-mediated membrane fission. Structure 20: 1621–1628 [DOI] [PubMed] [Google Scholar]
- Faelber K, Posor Y, Gao S, Held M, Roske Y, Schulze D, Haucke V, Noe F, Daumke O (2011) Crystal structure of nucleotide-free dynamin. Nature 477: 556–560 [DOI] [PubMed] [Google Scholar]
- Feuerstein J, Goody RS, Wittinghofer A (1987) Preparation and characterization of nucleotide-free and metal ion-free p21 ‘apoprotein’. J Biol Chem 262: 8455–8458 [PubMed] [Google Scholar]
- Ford MG, Jenni S, Nunnari J (2011) The crystal structure of dynamin. Nature 477: 561–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334: 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandre-Babbe S, van der Bliek AM (2008) The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19: 2402–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, von der Malsburg A, Dick A, Faelber K, Schroder GF, Haller O, Kochs G, Daumke O (2011) Structure of myxovirus resistance protein a reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity 35: 514–525 [DOI] [PubMed] [Google Scholar]
- Gao S, von der Malsburg A, Paeschke S, Behlke J, Haller O, Kochs G, Daumke O (2010) Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 465: 502–506 [DOI] [PubMed] [Google Scholar]
- Griffin EE, Graumann J, Chan DC (2005) The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol 170: 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson MD, Rzeznicka K, Rosenback M, Hansson M, Sirijovski N (2008) PCR-mediated deletion of plasmid DNA. Anal Biochem 375: 373–375 [DOI] [PubMed] [Google Scholar]
- Hinshaw JE, Schmid SL (1995) Dynamin self-assembles into rings suggesting a mechanism for coated vesicle budding. Nature 374: 190–192 [DOI] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J (2007) The machines that divide and fuse mitochondria. Annu Rev Biochem 76: 751–780 [DOI] [PubMed] [Google Scholar]
- Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J (2005) Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol 170: 1021–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010) Xds. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E (2008) Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9: 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch A, Thiemann M, Grabenbauer M, Yoon Y, McNiven MA, Schrader M (2003) Dynamin-like protein 1 is involved in peroxisomal fission. J Biol Chem 278: 8597–8605 [DOI] [PubMed] [Google Scholar]
- Koch A, Yoon Y, Bonekamp NA, McNiven MA, Schrader M (2005) A role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 16: 5077–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM (1999) C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4: 815–826 [DOI] [PubMed] [Google Scholar]
- Lackner LL, Horner JS, Nunnari J (2009) Mechanistic analysis of a dynamin effector. Science 325: 874–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443: 787–795 [DOI] [PubMed] [Google Scholar]
- Marks B, Stowell MH, Vallis Y, Mills IG, Gibson A, Hopkins CR, McMahon HT (2001) GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 410: 231–235 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, Hinshaw JE (2011) Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol 18: 20–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears JA, Ray P, Hinshaw JE (2007) A corkscrew model for dynamin constriction. Structure 15: 1190–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, Lippincott-Schwartz J (2010) Analysis of mitochondrial dynamics and functions using imaging approaches. Curr Protoc Cell Biol Chapter 4(Unit 4.25): 21–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148: 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth M, Schapira AH (2001) Mitochondria and degenerative disorders. Am J Med Genet 106: 27–36 [DOI] [PubMed] [Google Scholar]
- Otera H, Mihara K (2011) Molecular mechanisms and physiologic functions of mitochondrial dynamics. J Biochem 149: 241–251 [DOI] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191: 1141–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT (2011) MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep 12: 565–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Roux A, Koster G, Lenz M, Sorre B, Manneville JB, Nassoy P, Bassereau P (2010) Membrane curvature controls dynamin polymerization. Proc Natl Acad Sci USA 107: 4141–4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux A, Uyhazi K, Frost A, De Camilli P (2006) GTP-dependent twisting of dynamin implicates constriction and tension in membrane fission. Nature 441: 528–531 [DOI] [PubMed] [Google Scholar]
- Schapira AH (2008) Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol 7: 97–109 [DOI] [PubMed] [Google Scholar]
- Schon EA, DiMauro S (2003) Medicinal and genetic approaches to the treatment of mitochondrial disease. Curr Med Chem 10: 2523–2533 [DOI] [PubMed] [Google Scholar]
- Schroder GF, Levitt M, Brunger AT (2010) Super-resolution biomolecular crystallography with low-resolution data. Nature 464: 1218–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78: 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE (1999) Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol 147: 699–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield JB (2007) ImageJ, a useful tool for biological image processing and analysis. Microsc Microanal 13: 200–201 [Google Scholar]
- Shirendeb UP, Calkins MJ, Manczak M, Anekonda V, Dufour B, McBride JL, Mao P, Reddy PH (2012) Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington's disease. Hum Mol Genet 21: 406–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12: 2245–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM (1998) A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol 143: 351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell MH, Marks B, Wigge P, McMahon HT (1999) Nucleotide-dependent conformational changes in dynamin: evidence for a mechanochemical molecular spring. Nat Cell Biol 1: 27–32 [DOI] [PubMed] [Google Scholar]
- Strack S, Cribbs JT (2012) Allosteric modulation of drp1 mechanoenzyme assembly and mitochondrial fission by the variable domain. J Biol Chem 287: 10990–11001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweitzer SM, Hinshaw JE (1998) Dynamin undergoes a GTP-dependent conformational change causing vesiculation. Cell 93: 1021–1029 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu Q, Nunnari J (2000) Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol 151: 353–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9: 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Malsburg A, Abutbul-Ionita I, Haller O, Kochs G, Danino D (2011) Stalk domain of the dynamin-like MxA GTPase protein mediates membrane binding and liposome tubulation via the unstructured L4 loop. J Biol Chem 286: 37858–37865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J, Chen Q (2011) Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 286: 11649–11658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Fujioka H, Zhu X (2008) Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol 173: 470–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV (2007) A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 356: 1736–1741 [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y, Krueger EW, Oswald BJ, McNiven MA (2003) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23: 5409–5420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Hinshaw JE (2001) Three-dimensional reconstruction of dynamin in the constricted state. Nat Cell Biol 3: 922–926 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao X, Garavito RM (2011) Biochemical characterization of human dynamin-like protein 1. J Biochem 150: 627–633 [DOI] [PubMed] [Google Scholar]
- Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M (2011) Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J 30: 2762–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.