

Abstract
The genes encoding the ribonucleases RNase J1 and RNase Y have long been considered essential for Bacillus subtilis cell viability, even before there was concrete knowledge of their function as two of the most important enzymes for RNA turnover in this organism. Here we show that this characterization is incorrect and that ΔrnjA and Δrny mutants are both viable. As expected, both strains grow relatively slowly, with doubling times in the hour range in rich medium. Knockout mutants have major defects in their sporulation and competence development programs. Both mutants are hypersensitive to a wide range of antibiotics and have dramatic alterations to their cell morphologies, suggestive of cell envelope defects. Indeed, RNase Y mutants are significantly smaller in diameter than wild-type strains and have a very disordered peptidoglycan layer. Strains lacking RNase J1 form long filaments in tight spirals, reminiscent of mutants of the actin-like proteins (Mre) involved in cell shape determination. Finally, we combined the rnjA and rny mutations with mutations in other components of the degradation machinery and show that many of these strains are also viable. The implications for the two known RNA degradation pathways of B. subtilis are discussed.
INTRODUCTION
Defining the full set of essential genes in any organism is of considerable interest to the identification of the minimal assortment of genes required to sustain life and the identification of potential targets for new antimicrobial compounds. In a previous study, a total of 271 genes were deemed essential for growth of Bacillus subtilis in rich medium at 37°C (1). The criteria for classifying about 150 of these genes as essential were based on (i) the inability to interrupt the coding sequence by Campbell recombination of a plasmid bearing homology to a short (200- to 400-bp) internal portion of the gene and (ii) the strain becoming IPTG (isopropyl-β-d-thiogalactopyranoside) dependent for growth when a Pspac promoter fusion was made to the intact gene by Campbell insertion of a similar plasmid bearing homology to the N-terminal portion of the coding sequence. Strains containing Pspac-dependent fusions to essential genes generally show significantly reduced growth in the absence of IPTG and essentially no growth if a plasmid expressing additional copies of the LacI repressor (e.g., pMAP65) is added to the strain. Residual growth, if observed, is usually attributed to leakiness of the Pspac promoter.
In a follow-up study of 11 of these 150 putative essential genes, four genes of unknown function (ydiB, yloQ, yqeI, and ywlC) were deemed nonessential because they were successfully inactivated in a second attempt (2). However, at least three attempts to recover Campbell insertions for the 7 remaining genes (yacA, ydiC, ykqC, ylaN, ymdA, yneS, and yqjK) failed, while IPTG-dependent strains were successfully made. The essential nature of these genes was thus considered to be confirmed. Three of these genes, yqjK, ykqC, and ymdA, have since been shown to encode the ribonucleases RNase Z, RNase J1, and RNase Y, and their genes were renamed rnz, rnjA, and rny, respectively (3–7). The yacA and yneS genes have been renamed tilS and plsY, encoding a tRNAIle-lysidine synthetase and an acylphosphate:glycerol-phosphate acyltransferase, respectively (8–10), while the functions of ydiC and ylaN remain unknown.
Based on the above-mentioned observations, we and others (4, 6, 7) have made IPTG-dependent and xylose-dependent derivatives of the rnjA and rny genes and confirmed a lack of growth, or very slow growth, in the absence of an inducer, both on plates and in liquid culture, depending on the “tightness” of the promoter in question and on the presence or absence of pMAP65. These depletion strains have played a fundamental role in demonstrating the 5′-to-3′ exoribonuclease function of RNase J1 in the maturation of the 5′ end of 16S rRNA and in mRNA decay (4, 5, 11, 12) and that of RNase Y in endonucleolytic cleavage of mRNA and rRNA degradation in B. subtilis spores (6, 7, 13–15). In a recent tiling array study, we used these depletion strains to show that RNase J1 and RNase Y together account for the degradation of almost half of B. subtilis mRNAs (16).
B. subtilis laboratory strains typically contain multiple prophages in their genomes that contain several type 1 toxin/antitoxin modules, where expression of the toxin is regulated by an antisense RNA expressed from the complementary strand. These toxins may play a role in maintenance of the prophages on the genome (by postsegregational killing of daughter cells that do not inherit the antitoxin gene) or in linking phage biology to the physiology of the cell. We have recently shown that the essential role of RNase III in B. subtilis is to silence the expression of two such toxin genes, txpA and yonT, encoded by the prophages Skin and SPβ, respectively (17). RNase III is therefore essential in B. subtilis only in the presence of these two prophages. In our tiling array study of RNase Y-depleted strains, we observed that 92% of the genes of the prophage PBSX were overexpressed (16). To determine whether the presence of prophages and their encoded toxin genes could also account for the essential nature of RNase Y and/or J1, we performed an experiment in which we attempted to delete the rny and rnjA genes from strains lacking up to three prophages (SPβ, Skin, and PBSX). As a control, we included the 168 trpC2 parental strain. To our surprise (and embarrassment), we were able to delete both rny and rnjA from the parental B. subtilis chromosome. The resulting mutants grew very poorly, as expected, but were clearly viable.
MATERIALS AND METHODS
Bacterial strains and plasmids used.
Bacterial strains used in this study are shown in Table 1. CCB084 (= RB530) was a kind gift from R. Britton. It contains a deletion of the rnjA gene extending from 164 nucleotides (nt) upstream to 147 nt downstream of the coding sequence, replaced by a Spcr cassette (from 283 nt upstream to 116 nt downstream of the coding sequence) from plasmid pDR111. It also contains a copy of the rnjA gene under the control of a xylose-dependent promoter integrated at amyE [amyE::pCT1 (Pxyl-ykqC) Cm].
Table 1.
Strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| W168 | trp+ | Laboratory strain |
| JH642 | trpC2 pheA1 | M. Nakano |
| PY79 | ΔSPβ | P. Stragier |
| 168 | trpC2 | J. M. van Dijl |
| 168 ΔSPβ | 168 trpC2 ΔSPβ | 22 |
| 7TFC7a | 168 trpC2 ΔSPβ skin | 22 |
| TF8a | 168 trpC2 ΔSPβ skin PBSX | 22 |
| CCB078 | W168 rnjB::spc | 5 |
| CCB084 | trpC2 pheA1 amyE::pCT1(Pxyl-rnjA) Cm rnjA::spc | 11 |
| CCB294 | amyE::Pspac-ymdA lacI Cm rny::spc | 16 |
| CCB304 | W168 skin amyE::pX(Pxyl-rny) Cm rny::spc | This study |
| CCB411 | W168 amyE::pCT1(Pxyl-rnjA) Cm rnjA::spc | This study |
| CCB423 | 168 trpC2 rny::spc | This study |
| CCB432 | 168 trpC2 pnp::kan | This study |
| CCB433 | 168 trpC2 rnjA::spc | This study |
| CCB444 | 168 trpC2 rnjB::spc | This study |
| CCB434 | W168 rnjA::spc | This study |
| CCB435 | PY79 rnjA::spc | This study |
| CCB436 | JH642 rnjA::spc | This study |
| CCB438 | 168 trpC2 ΔSPβ rny::spc | This study |
| CCB439 | 168 trpC2 ΔSPβ Δskin rny::spc | This study |
| CCB440 | 168 trpC2 ΔSPβ Δskin ΔPBSX rny::spc | This study |
| CCB441 | W168 rny::spc | This study |
| CCB442 | JH642 rny::spc | This study |
| CCB443 | PY79 rny::spc | This study |
| CCB445 | 168 trpC2 rppH::pMUTIN ery | This study |
| CCB448 | 168 trpC2 rnjA::kan | This study |
| CCB449 | 168 trpC2 rnjB::spc rnjA::kan | This study |
| CCB451 | 168 trpC2 rppH::pMUTIN ery rnjA::spc | This study |
| CCB453 | 168 trpC2 rppH::pMUTIN ery rny::spc | This study |
| CCB455 | 168 trpC2 pnp::kan rny::spc | This study |
| CCB481 | 168 trpC2 amyE::pX(Pxyl-rny) Cm rny::pMUTIN Ery | This study |
| CCB482 | 168 trpC2 rny::pMUTIN Ery | This study |
| CCB483 | 168 trpC2 amyE::pCT1(Pxyl-rnjA) Cm rnjA::pMUTIN Ery | This study |
| CCB484 | 168 trpC2 rnjA::pMUTIN Ery | This study |
Strain CCB304 was made by transforming plasmid pX-rny into the W168 trp+ skin strain, a kind gift from P. Stragier, followed by transfer of the rny::spc construct from CCB294 (16). In strain CCB294, the coding sequence of the rny gene has been replaced by the coding sequence of the Spcr gene. Plasmid pX-rny was made by amplifying the rny gene by PCR using oligonucleotides CC772 and CC773, cleaving with SpeI and BamHI, and cloning the resulting fragment into plasmid pX (18) cut with the same enzymes.
Strain CCB411 was made by first transferring the amyE::pCT1(Pxyl-rnjA) Cm construct from strain CCB084 into W168, creating strain CCB374. The native rnjA coding sequence of CCB374 was then replaced by the coding sequence of the Spcr cassette by transformation with a DNA fragment generated by overlapping PCR using oligonucleotide pairs CC1091/CC1092, CC1093/CC1095, and CC1094/CC1096.
Strain CCB448 was constructed similarly to CCB374, with the native rnjA coding sequence being replaced by a Kanr coding sequence. The overlapping PCR fragment to create the rnjA::kan mutation was generated by using oligonucleotides CC1091/CC1114, CC1115/CC1117, and CC1116/CC1096.
Strain CCB449 was constructed by transferring the rnjA::kan construct from strain CCB448 into CCB444 (168 trpC2 rnjB::spc). The rnjB::spc construct (originally from strain CCB078) was described previously (5).
Strain CCB445 contains the rppH::pMUTIN Ery mutation in the 168 trpC2 background. The rppH::pMUTIN construct originally came from strain BFA627, a kind gift from P. Stragier.
Strains CCB451 (rppH::pMUTIN rnjA::spc) and CCB453 (rppH::pMUTIN rny::spc) were made by transferring the rnjA::spc and rny::spc constructs from CCB411 and CCB304, respectively, into CCB445.
Strain CCB455 (pnp::kan rny::spc) was made by transforming CCB432 with CCB304 chromosomal DNA. CCB432 contains the pnp::kan mutation in the 168 trpC2 background. This construct originally came from strain BG517, a kind gift from D. Bechhofer.
Strain CCB481 was made by transforming the 168 trpC2 amyE::Pxyl-rny strain (CCB479) with plasmid pMUTIN-rny. This plasmid was made by first amplifying an ∼250-nt internal fragment of the rny gene by PCR using oligonucleotide pair CC1178/CC1179 and cutting with EcoRI and BamHI and then cloning into pMUTIN-4M (19) digested with the same enzymes. Chromosomal DNA from CCB481 was then used to transform the 168 trpC2 strain to obtain strain CCB482.
Strain CCB483 was made by transforming the 168 trpC2 amyE::Pxyl-rnjA strain (CCB478) with plasmid pMUTIN-rnjA. This plasmid was made by first amplifying an ∼250-nt internal fragment of the rnjA gene by PCR using oligonucleotide pair CC014/CC1177 and cutting with EcoRI and BamHI and then cloning into pMUTIN-4M (19) digested with the same enzymes. Chromosomal DNA from CCB483 was then used to transform the 168 trpC2 strain to obtain strain CCB484.
Gene deletions were confirmed by PCR using the following oligonucleotide pairs (see Table S1 in the supplemental material): CC1090/CC1096 for rnjA::spc, CC768/CC770 for rny::spc, CC132/CC133 for pnp::kan, CC175/CC1010 for rnjB::spc, HP279/CC546 for rppH::pMUTIN, CC1090/HP279 for rnjA::pMUTIN, and CC768/HP279 for rny::pMUTIN.
Competence and sporulation assays.
Competence assays were performed by growing bacteria for 1 h after the transition into stationary phase in MD medium (20) supplemented with 0.1% hydrolyzed casein (Oxoid). Cells were concentrated 10-fold in spent medium containing 10% glycerol and stored frozen at −80°C. For transformation, 100 μl of competent cells was incubated with 2.5 μg of the integrative plasmid pX (18) at 37°C for 20 min in 400 μl MD medium. Two hundred microliters of 2× YT (yeast extract, tryptone) medium was added, and cells were allowed to grow for 90 min at 37°C, before spreading onto selective LB plates. Viable cells were counted by plating dilutions of the competent cell preparation onto nonselective plates.
Sporulation assays were performed by growing cells in Difco nutrient broth for 48 h, followed by heating at 80°C for 10 min and spreading the appropriate dilutions onto LB plates. Viable cells were counted by plating dilutions before heat treatment.
Microscopy.
Light microscopy images were generated on a Zeiss Axio Observer Z1 microscope. For transmission electron microscopy, bacteria were pelleted and resuspended in a small amount of LB medium. The concentrated suspension was taken up in a cellulose capillary tube (Leica Microsystems, Vienna, Austria). The tube was cut into closed pieces not longer than 2 mm with a modified scalpel (21) and placed into type A planchettes (Agar Scientific, Stansted, United Kingdom) of a 0.2-mm depth filled with 1-hexadecene and closed with a type B planchette. Cryoimmobilization was done by using an HPM 010 high-pressure freezer (Abra Fluid AG [formerly BalTec], Widnau, Switzerland). 1-Hexadecene is not soluble at the starting temperature of the freeze substitution. To allow access of the substitution fluid, small cracks were introduced into frozen 1-hexadecene with a precooled forceps (no. 5; Dumont, Switzerland), and samples were transferred under liquid nitrogen for freeze substitution in 2% osmium tetroxide (Merck, Darmstadt, Germany) in dry acetone in an automatic freeze substitution instrument (AFS; Leica Microsystems, Vienna, Austria). Substitution was performed for 24 h at −90°C, and samples were warmed (2°C/h) to −60°C (8 h) and then to −30°C (8 h). Samples were then put on ice for 1 h and washed three times with dry acetone on ice before stepwise embedding in epoxy resin. After heat polymerization, thin sections were cut with an Ultracut UCT microtome (Leica Microsystems, Vienna, Austria).
Sections were collected onto 200-mesh carbon-Formvar-coated copper grids. After poststaining with 4% uranyl acetate and Reynolds' lead citrate, the sections were observed with a JEOL 1010 instrument at 80 kV equipped with a KeenView camera (Soft Imaging System; Olympus, Münster, Germany).
RESULTS
Deletion of the rny and rnjA genes is possible in B. subtilis.
To attempt to delete the rny and rnjA genes from B. subtilis strains lacking different numbers of prophages, we used chromosomal DNA isolated from strains CCB304 and CCB411, respectively (see Materials and Methods). Each strain has a copy of the RNase gene expressed under the control of a xylose-dependent promoter integrated at the amyE locus, selectable by chloramphenicol resistance (Cmr), and the native copy of the gene completely replaced by a spectinomycin resistance (Spcr) cassette. Healthy growth of these strains is ensured by the addition of xylose to the growth medium. Five micrograms of chromosomal DNA from CCB304 or CCB411 was transformed into B. subtilis strains lacking one (SPβ), two (SPβ and Skin), or three (SPβ, Skin, and PBSX) prophages (22) and selected either for the benign amyE marker (Cmr) or for deletion of the RNase gene (Spcr). The B. subtilis 168 trpC2 parental strain was transformed as a negative “wild-type” control. Surprisingly, very similar numbers of colonies were obtained on all plates (data not shown), including those corresponding to the control strain. Transformation of the wild-type strain with the rny::spc construct yielded hundreds of pinpoint Spcr colonies at 37°C overnight (Fig. 1A), while transformation with the rnjA::spc construct yielded a similar number of slightly larger Spcr colonies (Fig. 1B). Despite their small size, we were able to purify the Spcr colonies on fresh antibiotic-containing plates, where they grew to their original size (Fig. 1). None of 100 small Spcr colonies tested were Cmr (data not shown), ruling out the possibility that the amyE::Pxyl-rny or amyE::Pxyl-rnjA constructs were cotransferred during the transformation (congression). The replacement of the rny and rnjA genes by the Spcr cassette was confirmed by colony PCR using oligonucleotides flanking the rny and rnjA genes (see Materials and Methods). An example is shown in Fig. 2. We further confirmed that the rnjA and rny genes were not duplicated elsewhere on the genome by colony PCR using oligonucleotides internal to the coding sequence (data not shown). Together, these experiments show that it is indeed possible to completely inactivate the rny and rnjA genes in B. subtilis and that the presence of prophages has no impact on this observation.
Fig 1.
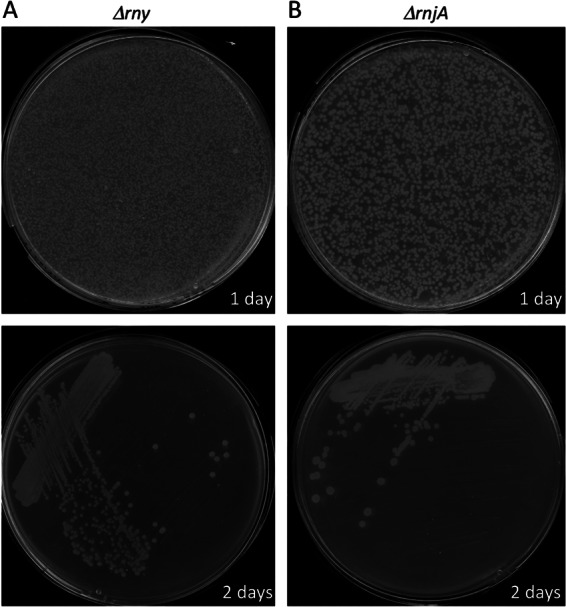
RNase Y and RNase J1 deletion mutants are viable. Colonies of B. subtilis 168 trpC2 cells after transformation with rny::spc (A)- or rnjA::spc (B)-containing DNA and selection on plates containing spectinomycin. (Top) Transformation plates; (bottom) purification plates.
Fig 2.
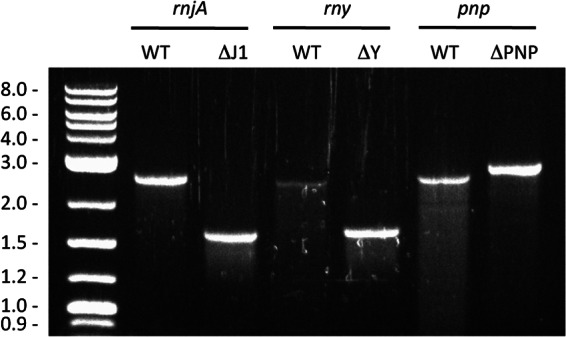
Confirmation of rny::spc and rnjA::spc deletions. Deletions were verified by colony PCR using flanking DNA oligonucleotides for each gene. The PCR products change in size for wild-type and mutant copies of the RNase gene (oligonucleotide pairs CC1090/CC1096 and CC768/CC770 for rnjA and rny, respectively). A pnp::kan mutant strain which was used throughout the study was also included (oligonucleotides CC132 and CC133). A DNA marker is shown at the left of the gel. Expected sizes are as follows: rnjA, 2,571 bp for the wild type (WT) versus 1,656 bp for the mutant; rny, 2,541 bp for the wild type versus 1,731 bp for the mutant; pnp, 2,591 kb for the wild type versus 2,829 kb for the mutant.
We confirmed that this observation was not unique to the B. subtilis 168 trpC2 strain by transforming other commonly used B. subtilis laboratory strains, W168, JH642, and PY79, with CCB304 and CCB411 chromosomal DNA. Very similar transformation efficiencies and colony sizes were observed for each of the four backgrounds for the rny::spc deletion; the rnjA::spc colonies were more variable in size in the different backgrounds (see Fig. S1 in the supplemental material). Colonies of strains lacking RNase Y were consistently more translucent than either wild-type or ΔrnjA colonies, suggesting a possible cell surface defect. This translucent phenotype does not appear to be caused by any of the major prophages, since the Δrny mutation also causes a translucent colony phenotype in strains lacking Skin, SPβ, and PBSX (data not shown).
We measured the growth rate of 168 trpC2 strains lacking RNase Y or RNase J1 in 2× YT medium at 37°C. The wild-type strain had an average doubling time of 26 min in this medium, while the rny mutant doubled every 56 min and the rnjA mutant doubled every 76 min (Table 2). In comparison, a PNPase (polynucleotide phosphorylase) mutant lacking the major 3′-to-5′ exoribonuclease in B. subtilis had a doubling time of 33 min. We used this PNPase mutant for comparison throughout the paper, as it is known to be sensitive to some of the phenotypic tests that we wished to perform. The rny mutant shows a characteristic growth curve, whereby the optical density rises steadily to 1.0 to 1.2 before falling to around 0.8 (see Fig. S2 in the supplemental material), without observable cell lysis, and then rises again to reach a maximum of around 1.2 in cultures grown overnight. This phenomenon was also independent of the presence of the three prophages SPβ, Skin, and PBSX (data not shown).
Table 2.
Doubling times of RNase mutants
| Strain | Avg doubling time (min) ± SDa |
|---|---|
| Wild-type 168 trpC2 | 26 ± 4 |
| Single mutants | |
| 168 trpC2 rnjA::spc | 76 ± 4 |
| 168 trpC2 rny::spc | 56 ± 5 |
| 168 trpC2 pnp::kan | 33 ± 1 |
| 168 trpC2 rnjB::spc | 23 ± 1 |
| 168 trpC2 rppH::pMUTIN ery | 24 ± 1 |
| Double mutants | |
| 168 trpC2 rppH::pMUTIN ery rnjA::spc | 71 ± 11 |
| 168 trpC2 rppH::pMUTIN ery rny::spc | 68 ± 4 |
| 168 trpC2 rnjB::spc rnjA::kan | 66 ± 13 |
| 168 trpC2 pnp::kan rny::spc | 165 ± 20 |
Doubling times are the averages of data from between three and five experiments.
We also examined the ability of the rny and rnjA deletion mutants to grow on more defined media. Both mutants could form colonies on minimal medium supplemented with glucose and Casamino Acids (MD medium) or, indeed, on minimal medium (M9) with glucose as the sole carbon source. The RNase Y mutant maintained its translucent phenotype on these media (data not shown).
Strains lacking RNase Y or RNase J1 are highly defective in competence and sporulation.
We tested the ability of the rnjA and rny deletion strains to become naturally competent and to sporulate, two key developmental pathways in B. subtilis. The results are shown in Table 3. Under the experimental conditions used (integration of the vector pX at the amyE locus), the PNPase mutant was about 3 orders of magnitude less competent than the wild type, about 10-fold more deficient than observed previously (23), while both the RNase J1 and RNase Y mutants were not measurably competent. We were also unable to transform the RNase J1 and RNase Y mutants with a replicative plasmid (data not shown).
Table 3.
Competence and sporulation efficiencies of RNase mutantsa
| Strain | Competence (CFU/μg/109 cells) | Sporulation (CFU/108 cells) (%) |
|---|---|---|
| 168 trpC2 | 776 | 9.6 × 106 (10) |
| 168 trpC2 pnp::kan | 0.9 | 1.0 × 107 (10) |
| 168 trpC2 rny::spc | 0 | 780 (0.0008) |
| 168 trpC2 rnjA::spc | 0 | 0 |
Values are the averages of data from at least two experiments.
In sporulation assays, the PNPase mutant had a sporulation efficiency similar to that of the wild-type strain, while the RNase Y and RNase J1 mutants were reduced by 4 and 7 orders of magnitude, respectively. This suggests that these two RNases play important roles in both developmental pathways. A previous tiling array analysis that we performed on strains depleted for RNase Y and RNase J1 (16) showed defects in many mRNAs of these pathways that could explain these two phenotypes (see Discussion).
Strains lacking RNase J1 are cold sensitive.
We next examined the effect of temperature on the growth phenotype of the RNase mutants. At 42°C and 45°C, all strains showed the same relative growth as they did at 37°C (see Fig. S3 in the supplemental material). The strain lacking RNase J1 showed relatively poor growth at 30°C and did not grow at 25°C or 18°C. This cryosensitive phenotype is consistent with the role of RNase J1 in 16S rRNA processing (5); mutations leading to ribosome assembly defects are typically cold sensitive. The RNase Y deletion strain grew at all temperatures, while the PNPase strain was cold sensitive at 18°C, a phenotype that was already known and which B. subtilis shares with Escherichia coli (23).
Strains lacking RNase Y or RNase J1 are hypersensitive to a wide range of antibiotics.
We noticed that, in addition to the translucent phenotype of the RNase Y deletion strain, the strain lacking RNase J1 does not form a tight pellet when centrifuged. These observations suggested to us that both mutants might in fact have altered cell envelopes. We therefore decided to test their levels of resistance to some commonly used antibiotics in a simple disk assay. We plated wild-type and mutant cells to form lawns of bacteria and placed a filter paper disk impregnated with different concentrations of antibiotic in the center. The diameters of the zones of inhibition were measured for each RNase mutant strain to compare its antibiotic sensitivity to that of the wild type. The PNPase mutant was included as a control because it is known to be hypersensitive to tetracycline (24). Strains lacking RNase J1 or Y showed increased sensitivity to a wide range of antibiotics, including the translational inhibitors tetracycline, streptomycin, erythromycin, and chloramphenicol; the transcriptional inhibitor rifampin; the DNA replication inhibitor trimethoprim; and the inhibitors of cell wall biosynthesis ampicillin and vancomycin (Fig. 3). The greater sensitivity of the ΔrnjA and Δrny strains to a broad spectrum of antibiotics suggests an increased permeability of the cell envelope. The RNase J1 mutant also showed increased sensitivity to nalidixic acid, while the RNase Y mutant was more resistant to this topoisomerase/gyrase inhibitor. The PNPase mutant, in contrast, primarily showed increased sensitivity to translation inhibitors, with the exception of streptomycin and nalidixic acid, to which it was more resistant.
Fig 3.
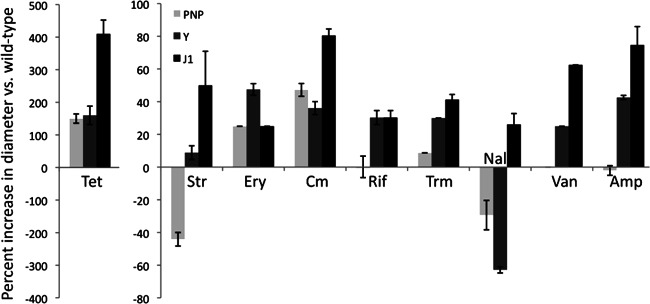
RNase Y and RNase J1 mutants are hypersensitive to antibiotics. Histograms show the percent increase in the diameter of the zone of inhibition compared to the wild type in filter disk assays (see the text). The PNPase mutant was included as a control. Disks (5 mm) were impregnated with 3 μl of antibiotic solutions at the following concentrations: tetracycline (Tet) at 2 mg/ml, streptomycin (Str) at 40 mg/ml, erythromycin (Ery) at 0.5 mg/ml, chloramphenicol (Cm) at 4 mg/ml, rifampin (Rif) at 4 mg/ml, trimethoprim (Trm) at 0.6 mg/ml, nalidixic acid (Nal) at 4 mg/ml, vancomycin (Van) at 2 mg/ml, and ampicillin (Amp) at 4 mg/ml. Values are the averages of data from 2 experiments with standard deviations as shown. Curiously, the zones of inhibition of the RNase mutant strains caused by tetracycline are significantly larger than those for other antibiotics, hence the different scale.
Strains lacking RNase Y or RNase J1 have major defects in cell morphology.
Since various lines of evidence were suggestive of cell surface defects (see above), we examined the ΔrnjA and Δrny mutants under a microscope. Under a light microscope, wild-type cells were of regular shape and size, while the PNPase mutant formed long filaments, as seen previously (24), indicative of a problem with septum formation. The cell morphologies of the ΔrnjA and Δrny mutants were remarkably different from those of either the wild type or the Δpnp mutant or from each other. Representative images are shown in Fig. 4. Cells lacking RNase J1 formed clumps of densely packed tight spirals interspersed with long chains, with few obvious septa and very few individual cells. In contrast, those lacking RNase Y formed thin curved structures consisting of 2 to 3 cells.
Fig 4.
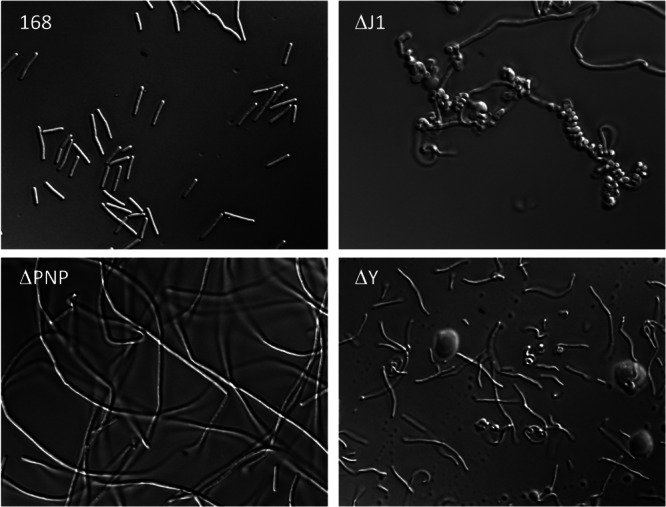
RNase Y and RNase J1 mutants have altered cell morphology. Shown are representative light microscopy images of the wild type (WT) and the pnp (ΔPNP), rnjA (ΔJ1), and rny (ΔY) mutants.
When analyzed by transmission electron microscopy, all three RNase mutants showed a disordered peptidoglycan layer compared to the well-organized peptidoglycan of the wild-type strain (Fig. 5). This defect was most dramatic in the RNase Y mutant, resulting in a “solar flare” appearance, with loosely attached pieces of peptidoglycan protruding from the cell surface. As seen by light microscopy, Δrny cells were much thinner, with a diameter of only about 0.5 μm, compared to 0.8 μm for wild-type or Δpnp cells. RNase J1 and PNPase mutants also had clear defects in their peptidoglycan layers, with Δrny ≫ ΔrnjA > Δpnp in terms of their phenotypes. Consistent with observations by light microscopy, the shapes of the RNase J1 mutant cells were highly irregular. The morphological defects observed for the RNase J1 and RNase Y mutants are likely explained by a subset of mRNAs involved in cell shape determination previously shown to be affected in cells depleted for these enzymes (see Discussion).
Fig 5.

RNase Y and RNase J1 mutants have altered cell walls. Shown are representative transmission electron microscopy images of the wild type (WT) and the pnp (ΔPNP), rnjA (ΔJ1), and rny (ΔY) mutants. The peptidoglycan (pg) layer, an electron-dense layer representing the base (b) of the peptidoglycan layer, the thin (white) cellular membrane (m), and ribosomes (r) are indicated where visible.
Knockout of rny and rnjA genes in strains lacking other components of the degradation machinery.
RNase J1 and RNase Y are both sensitive to the 5′ phosphorylation status of their RNA substrates, preferring 5′-monophosphate to the 5′-triphosphate of primary transcripts (7, 11). We have recently shown that the enzyme RppH can perform this mRNA deprotection reaction in B. subtilis. We therefore asked whether it was possible to inactivate the rnjA or rny gene in strains already lacking RppH. Chromosomal DNA from strains CCB304 and CCB411 were used to transform strain CCB445, bearing an inactive copy of the rppH gene. We readily recovered rppH rny and rppH rnjA double mutants, which were confirmed by PCR (see Fig. S4 in the supplemental material). The doubling time of the rppH rnjA mutant in 2× YT medium was similar to that of strains lacking rnjA alone (71 versus 76 min), while growth of the rppH rny mutant was slower than that of the rny single mutant (68 versus 56 min) (Table 2).
We have previously shown that RNase J1 forms a complex with its paralogue RNase J2 and that RNase J2 has about 100-fold-lower 5′-to-3′ exoribonuclease activity than RNase J1 (25). We therefore asked whether it was possible that the RNase J1 deletion mutant survives because of the residual 5′-to-3′ exoribonuclease activity of RNase J2. Chromosomal DNA from strain CCB447 (similar to CCB411 but bearing an rnjA::kan deletion) (Table 2) was transformed into the CCB444 rnjB::spc strain. In this case also, we readily recovered rnjA rnjB double mutants with a slightly shorter doubling time than that of the strain lacking RNase J1 alone (66 versus 76 min) (Table 2). Thus, strains lacking both RNase J activities are viable, and the survival of strains lacking RNase J1 cannot be attributed to RNase J2.
Finally, we asked whether it was possible to delete the rny or rnjA gene in strains lacking the major 3′-to-5′ exonuclease PNPase. Since Δrny and ΔrnjA cells could not be made competent, but the Δpnp mutant was slightly better, we tried transforming the PNPase mutant with larger amounts of chromosomal DNA (20 μg) from strains CCB304 and CCB411. A few very-slow-growing transformants were obtained in each case, which we screened by colony PCR (see Fig. S4 in the supplemental material). We successfully obtained pnp rny double mutants that grew extremely slowly (doubling time, 165 min), but we failed to recover strains lacking both PNPase and RNase J1. We also failed to simultaneously transfer two markers to wild-type strains by transformation with a mixture of chromosomal DNAs (either Δpnp::kan plus Δrny::spc or Δpnp::kan plus ΔrnjA::spc). A similar double-transformation strategy also failed to generate Δrny ΔrnjA double mutants.
DISCUSSION
In this paper, we have shown that it is possible to inactivate the genes encoding the two key ribonucleases RNase J1 and RNase Y in B. subtilis, contrary to previous data from both the literature and our own experience that suggested that they were essential. The homogenous colony sizes and high transformation efficiencies in four different genetic backgrounds make it relatively unlikely that these deletion strains require second-site suppressor mutations for growth. This observation should serve as a cautionary tale for the choice of method used to declare genes to be essential in bacteria. Without further genetic analysis of the type performed here, there is necessarily some doubt about all 150 genes labeled as being essential based on the failure to obtain viable clones using the plasmid-borne replacement technique in the original paper by Kobayashi et al. (1).
The difference in previous attempts to inactivate the rnjA and rny genes seems to be explained in large part by the much higher recombination efficiency of long chromosomal DNA fragments than that of the short homology regions offered on plasmid replacement systems. We managed, with some perseverance, to inactivate both the rnjA and rny genes using a plasmid-borne replacement system in strains containing a second copy of the genes expressed from a xylose-dependent promoter at the amyE locus. Once this construct was established on the chromosome, it was readily transferable into the 168 wild-type background by transformation with chromosomal DNA and grew similarly to the rnjA::spc or rny::spc deletion strain (data not shown). Thus, the major problem is not with the plasmid-borne construct per se, i.e., there are no polar effects on the expression of downstream genes, nor are the truncated peptides synthesized by the Campbell-recombined plasmid a source of toxicity, but rather with the efficiency of recombination. It cannot be solely a question of recombination efficiency, however. A plasmid-borne gene replacement system would be predicted to yield similar numbers of colonies for a gene that is unnecessary for growth on rich medium as for a gene that makes a major contribution to cell growth under these conditions; only colony size should vary. Clearly, the fact that deletion of these genes has a major effect on cell doubling time has an additional negative impact on the ability to recover viable colonies with the plasmid-borne constructs compared to transformation with chromosomal DNA. The reasons for this are unclear.
The RNase J1 and RNase Y deletion strains have major defects in cell growth and morphology. Given their globally important roles, it was surprising to learn that the cell can live without them. This is in contrast, for example, to the major enzyme of E. coli mRNA degradation, RNase E. While the RNase J1 and RNase Y deletion strains have understandably much longer doubling times than wild-type strains in rich medium, they are not impossible to work with and will be a major asset in future experiments. Although the RNase Y deletion strain has a shorter doubling time than the strain lacking RNase J1 (Table 2), it forms smaller colonies (Fig. 1), presumably due to its smaller cell size (Fig. 5). Both deletion stains grow reasonably well in minimal medium and at high temperatures. The RNase J1 mutant is cryosensitive, even at room temperature, presumably explained by its role in rRNA maturation (5). The increased sensitivity of the RNase J1 and RNase Y mutants to a wide range of antibiotics suggested that they might have cell permeability defects, and we confirmed by electron microscopy that the peptidoglycan layer is disordered. Curiously, however, the RNase Y mutant was more resistant to the topoisomerase/gyrase inhibitor nalidixic acid (as was the PNPase mutant). It will be interesting to explore the basis for this behavior in the future. RNase J1-depleted cells were previously shown by Hunt et al. to be more resistant to the dihydrofolate reductase inhibitor trimethoprim (2); it is not clear to us why the null mutant is more sensitive to this antibiotic in our hands.
It would not be surprising if the defects in the cell envelope contributed to the sporulation and competence deficiencies of these strains. However, in a previous tiling array experiment (16), a number of key sporulation genes showed increased expression levels in RNase J1 and RNase Y depletion mutants, including those of the master regulator Spo0A (RNases J1 and Y) and its modulators Spo0B and Spo0E (RNase J1) (see Table S2 in the supplemental material). Phosphorylated SpoOA also plays a key role in competence by first indirectly increasing the expression level of ComK, the master competence regulator, and then directly inhibiting its transcription by binding to its promoter (26). Interestingly, levels of the comK mRNA were also significantly increased in RNase J1-depleted strains (16), while those of other important regulators were decreased in either RNase J1 (ComS and ComQ) or RNase Y (ComN) mutants. It is likely that the perturbation of the optimal balance between these different regulators accounts at least in part for the sporulation and competence deficiencies observed for the ΔrnjA and Δrny strains.
Under a light microscope, the RNase J1 mutant forms tight spirals interspersed by long chains, while the RNase Y mutant forms thin curvy chains of 2 to 3 cells. Defects in cell morphology were observed previously in cells depleted for RNases J1 and Y (2), but they appear to be less severe than those observed here with the null mutants. The spiraled phenotype of the RNase J1 deletion mutant is very similar to that of cells lacking the actin-like proteins of the MreB family that are part of the bacterial cytoskeleton (27, 28). We looked at the mRNA levels of genes involved in cell morphology in strains depleted for RNase J1 or Y in the tiling array data set (16). The most dramatic effect on the RNase J1 mutant was an 11-fold stabilization of the mreBH operon mRNA (Table 4). Overexpression of MreBH (or MreB) has been shown to lead to a similar spiraled phenotype in B. subtilis (29), and this may therefore be a good candidate for a contributor to the morphology phenotype of the RNase J1 mutant. The greatest effect of RNase Y depletion on mRNAs involved in cell morphology was a 12-fold increase in the expression level of the rodA gene. Depletion of this protein leads to the production of spherical cells in B. subtilis (30), but the effect of its overproduction is not known. Mutations in many other genes involved in cell wall biosynthesis also have effects on cell shape, and the expression of a great many of these genes was affected in strains depleted for RNase J1 or RNase Y (see Table S3 in the supplemental material). Indeed, this functional category was overrepresented in cells depleted for RNase Y, with about half of the mRNAs involved in cell wall biosynthesis and cell envelope stress having increased levels in cells deficient for this enzyme (16). The effects on cell morphology observed in the knockout mutants may thus be far more complicated than just the effects on mreBH or rodA alone. Consistent with this idea, the RNase J1 morphology defect was not corrected by the addition of 25 mM magnesium to cultures (data not shown), which was previously shown to rescue growth and morphology of a B. subtilis mreB mutant (31).
Table 4.
Effect of RNase J1 and RNase Y depletion on mRNAs involved in cell shape determination
| Genea | Fold expression level change relative to wild typeb |
|
|---|---|---|
| RNase J1↓ | RNase Y↓ | |
| mreBCD | 1.9 | 2.8 |
| mreBH | 11.3 | 4.3 |
| mbl | 0.8 | 0.8 |
| rodA | 3.2 | 12.1 |
| rodZ (ymfM) | 1.1 | 1.1 |
| yvcK | 1.5 | 1.6 |
Genes were taken from the Subtiwiki website (http://subtiwiki.uni-goettingen.de/wiki/index.php/Categories).
Values are expression levels relative to those of the wild type and are taken from data reported previously (16).
We tested whether it was possible to knock out the RNase J1 and RNase Y genes in cells already lacking other components of the degradation machinery. There are currently two known pathways of RNA turnover in B. subtilis: (i) endonucleolytic cleavage by RNase Y followed by degradation of the downstream fragment by the 5′-to-3′ exoribonuclease activity of RNase J1 and degradation of the upstream fragment by 3′-to-5′ exonucleases, principally PNPase, and (ii) removal of the protecting 5′-triphosphate group of primary transcripts by RppH, followed by degradation from the 5′ end by RNase J1. We were particularly interested in asking whether inhibiting the second pathway, by inactivating RppH, would be synthetic lethal in strains lacking the key endonuclease of the first pathway, RNase Y. Although the rppH rny double mutant was viable, it did show significantly slower growth than strains lacking RNase Y alone. We suspect that there is a redundant RppH-like activity in B. subtilis (see reference 32) that provides sufficient RNA pyrophosphohydrolase activity to maintain the second pathway in the absence of RppH. Strains lacking both RppH and RNase J1 had doubling times similar to those of the single rnjA mutants. Together, these data are consistent with the idea that RppH acts in the same degradation pathway as RNase J1 but in a different pathway from RNase Y.
We were also able to inactivate RNase J1 in strains already lacking its paralog RNase J2. We have previously shown that these two enzymes form a complex and that RNase J2 has 5′-to-3′ exoribonuclease activity that is about 2 orders of magnitude lower than that of RNase J1, but our ability to make rnjA rnjB double mutants shows that the viability of RNase J1 deletion strains is not ensured by the residual activity of RNase J2. Indeed, if anything, the rnjA rnjB double mutant grows slightly faster than the rnjA mutant alone. We have previously seen that mutation of the RNase J2 catalytic histidine motif (HGHDEN) to match that of RNase J1 (HGHEDH) did not increase exoribonuclease activity as expected but rather abolished it completely (33), raising questions as to the role of RNase J2. We are currently leaning toward the idea that its principal role is as a scaffold to regulate or stabilize the RNase J1 subunit of the complex. In the absence of RNase J1, the continued presence of RNase J2 may become slightly inhibitory.
We managed to make a pnp rny double mutant that grew much slower than the strain lacking RNase Y alone. This also suggests that PNPase plays an important role in the cell that is independent of its role in the RNase Y-dependent degradation pathway. Although there is no evidence so far of direct degradation from the 3′ end in B. subtilis (similar to that performed by the exosome in eukaryotes), this remains a possibility to be considered. Furthermore, PNPase has also been shown to play a role in B. subtilis DNA damage repair by degrading single-stranded DNA ends (34, 35).
We failed to make either pnp rnjA or rnjA rny double mutants in multiple attempts to either transform pnp mutants or simultaneously transform two mutations into wild-type cells. However, given the extremely low competence levels of the PNPase mutant or the low probability of successfully transforming two markers into the same cell, these results are suggestive only, and it is not possible to state definitively that these combinations are synthetic lethal.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funds from the CNRS (UPR 9073), Université Paris VII-Denis Diderot, and the Agence Nationale de la Recherche (subtilRNA2 and Labex Dynamo).
We thank laboratory members Mathias Springer and Rut Carballido-Lopez for helpful discussion. We thank Naima Belgareh-Touzé, Flore Sinturel, and Lionel Bénard for help with light microscopy and Sandra Duperrier and Patrick Stragier for advice on sporulation assays and providing strains.
Footnotes
Published ahead of print 15 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00164-13.
REFERENCES
- 1. Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, Boland F, Brignell SC, Bron S, Bunai K, Chapuis J, Christiansen LC, Danchin A, Debarbouille M, Dervyn E, Deuerling E, Devine K, Devine SK, Dreesen O, Errington J, Fillinger S, Foster SJ, Fujita Y, Galizzi A, Gardan R, Eschevins C, Fukushima T, Haga K, Harwood CR, Hecker M, Hosoya D, Hullo MF, Kakeshita H, Karamata D, Kasahara Y, Kawamura F, Koga K, Koski P, Kuwana R, Imamura D, Ishimaru M, Ishikawa S, Ishio I, Le Coq D, Masson A, Mauel C, et al. 2003. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U. S. A. 100:4678–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hunt A, Rawlins JP, Thomaides HB, Errington J. 2006. Functional analysis of 11 putative essential genes in Bacillus subtilis. Microbiology 152:2895–2907 [DOI] [PubMed] [Google Scholar]
- 3. Pellegrini O, Nezzar J, Marchfelder A, Putzer H, Condon C. 2003. Endonucleolytic processing of CCA-less tRNA precursors by RNase Z in Bacillus subtilis. EMBO J. 22:4534–4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Even S, Pellegrini O, Zig L, Labas V, Vinh J, Brechemmier-Baey D, Putzer H. 2005. Ribonucleases J1 and J2: two novel endoribonucleases in B. subtilis with functional homology to E. coli RNase E. Nucleic Acids Res. 33:2141–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Britton RA, Wen T, Schaefer L, Pellegrini O, Uicker WC, Mathy N, Tobin C, Daou R, Szyk J, Condon C. 2007. Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol. Microbiol. 63:127–138 [DOI] [PubMed] [Google Scholar]
- 6. Commichau FM, Rothe FM, Herzberg C, Wagner E, Hellwig D, Lehnik-Habrink M, Hammer E, Volker U, Stulke J. 2009. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol. Cell. Proteomics 8:1350–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shahbabian K, Jamalli A, Zig L, Putzer H. 2009. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 28:3523–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soma A, Ikeuchi Y, Kanemasa S, Kobayashi K, Ogasawara N, Ote T, Kato J, Watanabe K, Sekine Y, Suzuki T. 2003. An RNA-modifying enzyme that governs both the codon and amino acid specificities of isoleucine tRNA. Mol. Cell 12:689–698 [DOI] [PubMed] [Google Scholar]
- 9. Paoletti L, Lu YJ, Schujman GE, de Mendoza D, Rock CO. 2007. Coupling of fatty acid and phospholipid synthesis in Bacillus subtilis. J. Bacteriol. 189:5816–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoshimura M, Oshima T, Ogasawara N. 2007. Involvement of the YneS/YgiH and PlsX proteins in phospholipid biosynthesis in both Bacillus subtilis and Escherichia coli. BMC Microbiol. 7:69 doi:10.1186/1471-2180-7-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mathy N, Benard L, Pellegrini O, Daou R, Wen T, Condon C. 2007. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell 129:681–692 [DOI] [PubMed] [Google Scholar]
- 12. Deikus G, Condon C, Bechhofer DH. 2008. Role of Bacillus subtilis RNase J1 endonuclease and 5′-exonuclease activities in trp leader RNA turnover. J. Biol. Chem. 283:17158–17167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yao S, Bechhofer DH. 2010. Initiation of decay of Bacillus subtilis rpsO mRNA by endoribonuclease RNase Y. J. Bacteriol. 192:3279–3286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lehnik-Habrink M, Schaffer M, Mader U, Diethmaier C, Herzberg C, Stulke J. 2011. RNA processing in Bacillus subtilis: identification of targets of the essential RNase Y. Mol. Microbiol. 81:1459–1473 [DOI] [PubMed] [Google Scholar]
- 15. Segev E, Smith Y, Ben-Yehuda S. 2012. RNA dynamics in aging bacterial spores. Cell 148:139–149 [DOI] [PubMed] [Google Scholar]
- 16. Durand S, Gilet L, Bessieres P, Nicolas P, Condon C. 2012. Three essential ribonucleases—RNase Y, J1, and III—control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet. 8:e1002520 doi:10.1371/journal.pgen.1002520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Durand S, Gilet L, Condon C. 2012. The essential function of B. subtilis RNase III is to silence foreign toxin genes. PLoS Genet. 8:e1003181 doi:10.1371/journal.pgen.1003181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim L, Mogk A, Schumann W. 1996. A xylose-inducible Bacillus subtilis integration vector and its application. Gene 181:71–76 [DOI] [PubMed] [Google Scholar]
- 19. Vagner V, Dervyn E, Ehrlich SD. 1998. A vector for systematic gene inactivation in Bacillus subtilis. Microbiology 144:3097–3104 [DOI] [PubMed] [Google Scholar]
- 20. Hardy KG. 1985. B. subtilis cloning methods, p 1–17 In Glover DM. (ed), DNA cloning, a practical approach, vol II IRL Press, Oxford, United Kingdom [Google Scholar]
- 21. McDonald K, Schwarz H, Muller-Reichert T, Webb R, Buser C, Morphew M. 2010. “Tips and tricks” for high-pressure freezing of model systems. Methods Cell Biol. 96:671–693 [DOI] [PubMed] [Google Scholar]
- 22. Westers H, Dorenbos R, van Dijl JM, Kabel J, Flanagan T, Devine KM, Jude F, Seror SJ, Beekman AC, Darmon E, Eschevins C, de Jong A, Bron S, Kuipers OP, Albertini AM, Antelmann H, Hecker M, Zamboni N, Sauer U, Bruand C, Ehrlich DS, Alonso JC, Salas M, Quax WJ. 2003. Genome engineering reveals large dispensable regions in Bacillus subtilis. Mol. Biol. Evol. 20:2076–2090 [DOI] [PubMed] [Google Scholar]
- 23. Luttinger A, Hahn J, Dubnau D. 1996. Polynucleotide phosphorylase is necessary for competence development in Bacillus subtilis. Mol. Microbiol. 19:343–356 [DOI] [PubMed] [Google Scholar]
- 24. Wang W, Bechhofer DH. 1996. Properties of a Bacillus subtilis polynucleotide phosphorylase deletion strain. J. Bacteriol. 178:2375–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mathy N, Hebert A, Mervelet P, Benard L, Dorleans A, de la Sierra-Gallay IL, Noirot P, Putzer H, Condon C. 2010. Bacillus subtilis ribonucleases J1 and J2 form a complex with altered enzyme behaviour. Mol. Microbiol. 75:489–498 [DOI] [PubMed] [Google Scholar]
- 26. Mirouze N, Desai Y, Raj A, Dubnau D. 2012. Spo0A∼P imposes a temporal gate for the bimodal expression of competence in Bacillus subtilis. PLoS Genet. 8:e1002586 doi:10.1371/journal.pgen.1002586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsuoka S, Chiba M, Tanimura Y, Hashimoto M, Hara H, Matsumoto K. 2011. Abnormal morphology of Bacillus subtilis ugtP mutant cells lacking glucolipids. Genes Genet. Syst. 86:295–304 [DOI] [PubMed] [Google Scholar]
- 28. Schirner K, Errington J. 2009. Influence of heterologous MreB proteins on cell morphology of Bacillus subtilis. Microbiology 155:3611–3621 [DOI] [PubMed] [Google Scholar]
- 29. Kawai Y, Asai K, Errington J. 2009. Partial functional redundancy of MreB isoforms, MreB, Mbl and MreBH, in cell morphogenesis of Bacillus subtilis. Mol. Microbiol. 73:719–731 [DOI] [PubMed] [Google Scholar]
- 30. Henriques AO, Glaser P, Piggot PJ, Moran CP., Jr 1998. Control of cell shape and elongation by the rodA gene in Bacillus subtilis. Mol. Microbiol. 28:235–247 [DOI] [PubMed] [Google Scholar]
- 31. Formstone A, Errington J. 2005. A magnesium-dependent mreB null mutant: implications for the role of mreB in Bacillus subtilis. Mol. Microbiol. 55:1646–1657 [DOI] [PubMed] [Google Scholar]
- 32. Richards J, Liu Q, Pellegrini O, Celesnik H, Yao S, Bechhofer DH, Condon C, Belasco JG. 2011. An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol. Cell 43:940–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Condon C, Gilet L. 2011. The metallo-beta-lactamase family of ribonucleases, p 245–267 In Nicholson AW. (ed), Ribonucleases, vol 26 Springer-Verlag, Berlin, Germany [Google Scholar]
- 34. Cardenas PP, Carrasco B, Sanchez H, Deikus G, Bechhofer DH, Alonso JC. 2009. Bacillus subtilis polynucleotide phosphorylase 3′-to-5′ DNase activity is involved in DNA repair. Nucleic Acids Res. 37:4157–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cardenas PP, Carzaniga T, Zangrossi S, Briani F, Garcia-Tirado E, Deho G, Alonso JC. 2011. Polynucleotide phosphorylase exonuclease and polymerase activities on single-stranded DNA ends are modulated by RecN, SsbA and RecA proteins. Nucleic Acids Res. 39:9250–9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.