

Abstract
Environmentally responsive synthesis of surface proteins represents a hallmark of the infectious cycle of the Lyme disease agent, Borrelia burgdorferi. Here we created and analyzed a B. burgdorferi mutant lacking outer-surface protein C (OspC), an abundant Osp that spirochetes normally synthesize in the tick vector during the blood meal and down-regulate after transmission to the mammal. We demonstrate that B. burgdorferi strictly requires OspC to infect mice but not to localize or migrate appropriately in the tick. The induction of a spirochetal virulence factor preceding the time and host in which it is required demonstrates a developmental sequence for transmission of this arthropod-borne pathogen.
Lyme disease, caused by Borrelia burgdorferi, is the most common vector-borne disease in the United States. B. burgdorferi exists in nature in an enzootic cycle. The spirochetes are transmitted to small mammals, mainly rodents, through the bite of Ixodes ticks (1). During tick feeding, which lasts for several days, bacteria migrate from the tick midgut to the salivary glands, from which they are transmitted through the saliva (2). A switch in the major outer-surface proteins (Osp) of B. burgdorferi from OspA to OspC accompanies this change in location within the tick (3, 4). The switch from OspA to OspC is believed to be essential in releasing B. burgdorferi from the midgut and facilitating migration to the salivary glands. In this model, OspA is proposed to be an adhesin that tethers the spirochetes to the midgut epithelium (5, 6), whereas OspC is thought to be important for movement of the spirochete within the tick (7, 8). Aspects of the midgut environment that change during tick feeding, such as temperature, pH, and nutrients, influence the expression of many B. burgdorferi genes, including ospC (3, 9-11). Although the functional bases for these global changes in B. burgdorferi gene expression are not understood, they are generally considered to constitute an adaptive response that facilitates transition between two distinct niches: the tick and the mammal.
In this study, we investigated the role of OspC in B. burgdorferi in both the tick vector and the mammalian host. We took advantage of recent advances that enable genetic manipulation of an infectious B. burgdorferi clone (12) and efficient artificial infection of ticks (13). This experimental system permits analysis of the requirement for individual genes by the spirochete at each stage of the infectious cycle and allows careful dissection of pathogen-host interactions.
Methods
Bacterial Strains and Growth Conditions. B. burgdorferi B31-A3 is an infectious clone that was derived from B31 MI (12). It contains all plasmids present in B31 MI except cp9. B. burgdorferi was grown at 35°C in Barbour-Stoenner-Kelly (BSK-II) medium with gelatin (14) supplemented with 6% rabbit serum (Cedarlane Laboratories). pH induction of ospC expression was done as described in ref. 15 (see Supporting Methods, which is published as supporting information on the PNAS web site).
Construction of the ospC Inactivation and Complementation Plasmids. The ospC mutant strain was created by allelic exchange in WT B31-A3 by using plasmid pACYCΔtet-ospC::flaBp-kan1, yielding clone ospC7. We complemented the mutation in ospC7 by integration of a WT copy of ospC adjacent to the mutated ospC gene using plasmid pGTEC-Δbla2, yielding clone ospC7/ospC+4. The plasmids used in B. burgdorferi transformations were characterized by restriction enzyme digestion, PCR, and sequencing (see Supporting Methods).
Transformation of B. burgdorferi. Electrocompetent B. burgdorferi cells were prepared and transformed with 10-30 μg of DNA as described in ref. 12. Transformations were plated in solid BSK-II medium containing 200 μg/ml kanamycin (transformation of B31-A3), or 200 μg/ml kanamycin and 40 μg/ml gentamicin (transformation of ospC7). Colonies were screened by PCR with primers amplifying the respective antibiotic resistance cassette or ospC (Table 3, which is published as supporting information on the PNAS web site) as described in ref. 16. Plasmid content of transformants was determined by PCR using unique primer pairs (12, 17).
Experimental Mouse-Tick Infectious Cycle. All animal experiments were performed in accordance with the guidelines of the National Institutes of Health. The protocols were approved by the institution's Animal Care and Use Committee. Rocky Mountain Laboratories (RML) is accredited by the International Association for Assessment and Accreditation of Laboratory Animal Care. B. burgdorferi clones were tested for their proficiency in the mouse-tick infectious cycle by using naïve RML mice and naïve Ixodes scapularis larvae from a colony kept at RML, as described in ref. 18. RML mice represent an outbred strain that has been maintained at RML since 1937. Mice were tested for infection by serology, xenodiagnosis, and culture of different organs (18). Infection of ticks was assessed by immunofluorescence assay (IFA) (5) or culture (13). Immunodeficient B6.CB17-Prkdcscid/SzJ mice (The Jackson Laboratories) were used in one set of experiments.
Serology. Whole-cell lysates from B. burgdorferi and from Escherichia coli expressing recombinant P39 [BmpA (Borrelia membrane protein A)] (19) or recombinant His-tagged OspC (20) were prepared as described in ref. 21. Equal amounts were separated by electrophoresis through 12.5% polyacrylamide gels and subsequently transferred to nitrocellulose membranes. The membranes were incubated with mouse sera obtained 3-5 weeks after inoculation (1:200 dilution), followed by three wash steps for 15 min each and incubation for 1-2 h with peroxidase-conjugated sheep anti-mouse IgG (whole molecule) or goat anti-mouse polyvalent immunoglobulins (1:10,000 dilution) (Sigma-Aldrich). The blots underwent three more 15-min wash steps before incubation with enhanced chemiluminescence reagents (SuperSignal, Pierce).
IFA on Tick Midguts. IFAs were performed on dissected midguts from fed ticks as described in ref. 5. Hyperimmune rabbit anti-B. burgdorferi Sh-2-82 antiserum was used as primary antibody (1:100 dilution) and FITC- or Alexa 488-labeled goat anti-rabbit IgG (1:100 dilution) (Kierkegaard & Perry Laboratories, Gaithersburg, MD) as secondary antibody. Samples were analyzed by epifluorescence microscopy (Nikon Eclipse E800 or Nikon Microphot-FXA). A tick was scored positive if at least ten spirochetes were detected per midgut.
Artificial Tick Feeding. Larval I. scapularis ticks were infected by immersion in exponential phase cultures from B. burgdorferi clones B31-A3, ospC7, and ospC7/ospC+4 as described in ref. 13 by using two independent batches of ticks (≈120 larvae each) per bacterial strain. Ticks from each batch were fed to repletion on separate mice. Subsets of larvae were dissected immediately after immersion and 5 d after repletion, and the isolated midguts were analyzed by IFA for presence of spirochetes. Sera from mice obtained 3 weeks after tick-feeding were tested against recombinant B. burgdorferi antigens and whole-cell B. burgdorferi lysates. Mice were killed, and ear skin, bladders, and joints were cultured for the presence of spirochetes.
IFA on Tick Salivary Glands. Infected nymphs (artificially infected as larvae) were fed on naïve mice and removed at various time points during feeding. Salivary glands and midguts were dissected separately and analyzed by IFA. Spirochetes in the salivary glands were stained with rabbit anti-Borrelia antiserum (1:100 dilution) and subsequently with Alexa 488-labeled anti-rabbit IgG (1:100 dilution). The salivary glands were counterstained with DRAQ5 (1:1,000) (Biostatus Limited, Shepshed, U.K.). These samples were analyzed with a confocal microscope system (Bio-Rad MRC 1024 coupled to a Zeiss Axiovert 135).
Results
Inactivation of ospC. We inactivated the ospC gene in B. burgdorferi B31-A3 by targeted insertion of a kanamycin resistance cassette. We confirmed by PCR that the resultant mutant clone, named ospC7, retained all plasmids present in B31-A3. Southern blot analysis demonstrated that ospC7 contained a single copy of ospC on the 26-kb circular plasmid (cp26) that was interrupted by the kan marker (Figs. 1 and 2). Sequence analysis confirmed the structure of the mutated ospC locus. Western blot analysis of whole-cell lysates demonstrated that ospC7 no longer synthesized OspC (Fig. 3). The ospC mutant and B31-A3 WT had comparable in vitro doubling times.
Fig. 1.

Linear representation of the ospC loci of WT, mutant, and complemented B. burgdorferi clones. The scale indicates the location of the ORFs and restriction sites in base pairs; the gene identification numbers are according to the database designation (40). Arrowheads below the diagram denote the positions of primers (Table 3) used in making the allelic exchange constructs for inactivation of ospC or complementation. Relevant restriction sites used for cloning or for Southern blot analysis are indicated.
Fig. 2.

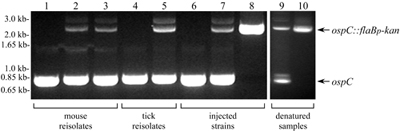
Southern blot analysis of B. burgdorferi clones that are WT, mutant, or complemented at the ospC locus. Genomic DNA of clones B31-A3, ospC7, and ospC7/ospC+4 was separated through agarose gels, transferred to a membrane, and hybridized with a 32P-labeled probe specific for ospC. There is a single AvrII site in cp26, a single SalI site in the integrated pGTEC-Δbla vector, and no SalI sites in cp26. AvaII cuts twice in the cp26 sequences flanking ospC and not in pGTEC-Δbla. The positions of nicked and linear cp26 and of ospC and its derivatives are indicated on the right. Migration positions of molecular size standards are indicated on the left.
Fig. 3.

Western blot analysis of whole-cell lysates of B31-A3 WT, ospC7 mutant, and ospC7/ospC+4 complemented B. burgdorferi clones grown at pH 7 or pH 8 and probed with anti-OspC monoclonal antibody B5 (20). The arrow indicates OspC, which is produced at higher levels in WT and complemented cells grown at pH 7 compared with pH 8. Migration positions of molecular mass standards are indicated on the left.
Defective Phenotype of ospC Mutant in Mice. We conducted preliminary experiments to determine whether inactivation of ospC altered the ability of B. burgdorferi to infect mammals. Mice were inoculated with 5 × 103 B31-A3 WT or ospC mutant bacteria by i.p./s.c. injection. All three mice injected with WT bacteria seroconverted to B. burgdorferi antigens, and spirochetes were cultured from mouse tissues. In contrast, all six mice challenged with ospC7 mutant bacteria remained sero-negative; spirochetes were not acquired by feeding ticks, and spirochetes could not be cultured from mouse tissues. These data suggested that OspC may be required for B. burgdorferi to infect mice.
To determine whether a higher infectious dose or a different route of inoculation would facilitate infection of mice by the ospC mutant, we injected mice by either intracardiac or i.p./s.c. routes with 107 bacteria (one mouse each route for WT, two mice each route for ospC mutant). Although this high dose elicited a seroconversion in all mice injected with either B31-A3 (2/2) or ospC7 mutant bacteria (4/4), only mice inoculated with B31-A3 became infected (2/2), as assessed by xenodiagnosis with larval ticks and reisolation from mouse tissues. These data indicate that OspC may be necessary for B. burgdorferi to infect mice, regardless of the infectious dose and the route of infection.
Complementation of the ospC Mutation. To demonstrate that the defective phenotype of ospC7 was due to the lack of OspC and to determine whether reintroduction of ospC was sufficient to restore mouse infectivity, we complemented the ospC mutation in ospC7 by integrating a WT copy of ospC adjacent to the mutated ospC gene on cp26 (Fig. 1). Southern blot analysis of the complemented clone, named ospC7/ospC+4, demonstrated that it contained both WT and mutated copies of ospC on cp26 as well as the flgBp-aacC1 fusion downstream of the ospC gene (Fig. 2). PCR analysis confirmed that the plasmid content of the complemented clone was identical to that of the progenitor B31-A3 WT and ospC mutant clones. Western blot analysis indicated that the complemented clone ospC7/ospC+4 synthesized OspC at a level comparable to the WT parental strain and was influenced by pH, as described in ref. 9 (Fig. 3). Insertion of the kan marker into ospC is unlikely to alter expression of adjacent genes, because they are well isolated and divergently transcribed (Fig. 1). Consistent with this theoretical consideration, analysis of gene products from adjacent loci (guaA/B and BBB22) demonstrated comparable levels in B31-A3 WT, ospC mutant, and complemented bacteria (data not shown).
Infectious Phenotype of the ospC-Complemented Clone. The ability of the ospC7/ospC+4 clone to infect mice was tested by needle-inoculation with a moderate i.p./s.c. dose of 5 × 103 ospC7/ospC+4 organisms, in parallel with the isogenic B31-A3 WT and ospC7 mutant clones (Table 1). As in previous experiments, the ospC mutant was not infectious for mice and not acquired by feeding ticks. However, the ospC7/ospC+4 clone was able to infect mice at levels comparable to the WT strain, as assessed by seroconversion to B. burgdorferi, acquisition of spirochetes by feeding ticks, and recovery of spirochetes from mouse tissues. Mice infected with B31-A3 and ospC7/ospC+4 strains mounted typical and similar immune responses to OspC, P39 [used as a marker for infection in animals (22)], and additional borrelial proteins (Fig. 4). Ticks that acquired B31-A3 and ospC7/ospC+4 spirochetes by feeding on needle-inoculated mice transmitted B. burgdorferi to naïve mice at the next blood meal (Table 1). The ospC loci of B. burgdorferi B31-A3 and ospC7/ospC+4 cultured from mice and ticks were identical to those of input bacteria (see Fig. 6, which is published as supporting information on the PNAS web site). Restoration of the WT phenotype in mouse infections and ticks by complementation of the ospC mutation in a previously noninfectious clone, ospC7, unambiguously demonstrated that ospC inactivation rendered B. burgdorferi noninfectious for mice.
Table 1. Infectivity and transmission of WT, ospC mutant, and complemented B. burgdorferi clones in mice and ticks.
| Mice infected/analyzed
|
Ticks infected/analyzed by IFA or culture
|
|||
|---|---|---|---|---|
| Clone | Infection route | Seroconversion | Reisolation* | |
| B31-A3 | Needle inoculation† | 7/9 | 4/9 | 3/17‡ |
| Tick bite | 5/6 | 5/6 | 26/28§ | |
| ospC7 | Needle inoculation† | 0/12 | 0/12 | 0/20‡ |
| Tick bite¶ | 0/1 | 0/1 | 0/1§ | |
| ospC7/ospC+4 | Needle inoculation† | 4/6 | 4/6 | 5/27‡ |
| Tick bite | 8/12 | 8/12 | 17/30§ | |
Two to four tissues were analyzed per mouse.
Inoculum of 4 × 103 organisms i.p. and 1 × 103 organisms s.c.
Larval ticks.
Nymphal ticks.
Larvae fed on four mice injected with ospC7 and were negative for spirochetes by IFA. After the molt, a subset of nymphs were fed on one mouse.
Fig. 4.

Immunoblot analysis of recombinant antigens and B. burgdorferi lysates with representative sera from mice inoculated by i.p./s.c. routes with B31-A3 WT, ospC7 mutant, and ospC7/ospC+4 complemented B. burgdorferi clones. Lane 1, whole-cell lysate of E. coli containing vector pBluescript SK (Stratagene) without insert; lane 2, whole-cell lysate of E. coli containing pBluescript SK with bmpA (encoding P39, which is commonly used as a marker for infection in animals) and a gene encoding P28 (an antigen often recognized by sera from infected animals) (21, 44); lane 3, whole-cell lysate of E. coli containing vector pET-29b without insert; lane 4, whole-cell lysate of E. coli containing pET-29b with ospC inserted (20); lane 5, B. burgdorferi whole-cell lysate. • indicates reaction with P39. ○ indicates reaction with OspC (His-tagged). Migration positions of molecular mass standards are indicated on the left.
Phenotype of ospC Mutant in Immunodeficient Mice. To address the possibility that OspC is required for immune evasion, we inoculated severe combined immunodeficient (SCID) mice in parallel with WT, ospC mutant, and complemented clones (Table 4, which is published as supporting information on the PNAS web site). At one week after inoculation, spirochetes were cultured from the blood of SCID mice inoculated with B31-A3 or ospC7/ospC+4 but not from mice injected with the ospC mutant. Larval ticks fed 2-3 weeks after inoculation acquired spirochetes from mice infected with B31-A3 (positive larvae from three of four mice as assessed by IFA and culture) and ospC7/ospC+4 (positive larvae from two of four mice). When subsequently fed as nymphs, infected larvae retained the spirochetes through the molt and transmitted them to naïve mice. In contrast, no spirochetes were cultured from >300 larval ticks that fed on four SCID mice injected with the ospC mutant. Spirochetes were recovered from tissues of mice inoculated with WT and complemented clones but not from mice inoculated with the ospC mutant clone. These results confirmed that the defective phenotype of the ospC mutant clone in mice was independent of the acquired immune response because it was cleared from the host, even in the absence of functional B and T cells.
Artificial Infection of Ticks. To address the role of OspC in tick colonization by B. burgdorferi, we used a recently described method for artificial infection of larval ticks (13) (Table 2). Individual cohorts of larval I. scapularis ticks were immersed in suspensions of B31-A3, ospC7, or ospC7/ospC+4. IFA analysis of midguts from a subset of ticks immediately after immersion demonstrated that a small number of spirochetes from all three strains had been ingested. The remaining ticks were subsequently fed to repletion on mice, and midguts from a subset of ticks were examined by IFA. Larval ticks from all three groups contained increased numbers of spirochetes, suggesting that the ospC mutant, in addition to the WT and complemented clones, could infect and replicate in the tick midgut. However, only mice fed upon by ticks immersed in WT and complemented clones seroconverted to B. burgdorferi antigens and yielded tissue cultures positive for spirochetes. These data demonstrated that OspC was not necessary for spirochetes to colonize the tick midgut but that mice remained refractory to infection with the ospC mutant, even by tick challenge.
Table 2. Artificial infection of larval ticks with WT, ospC mutant, and complemented B. burgdorferi clones and transmission to mice.
| Ticks infected/analyzed
|
Mice infected/analyzed
|
||||
|---|---|---|---|---|---|
| Clone | Feeding-tick stage | IFA midgut | IFA salivary gland* | Seroconversion | Reisolation† |
| B31-A3 | Larvae | 6/6 | NA | 2/2 | 2/2 |
| Nymphs | 21/26‡ | + | 3/3 | 3/3 | |
| ospC7 | Larvae | 5/6 | NA | 0/2 | 0/2 |
| Nymphs | 19/22‡ | + | 0/3 | 0/3 | |
| ospC7/ospC+4 | Larvae | 3/6 | NA | 2/2 | 2/2 |
| Nymphs | 17/23‡ | + | 3/3 | 3/3 | |
Localization of spirochetes within the salivary glands of feeding nymphal ticks as assessed by confocal laser scanning microscopy. NA, not addressed.
Three tissues were analyzed per mouse.
Unfed and fed nymphal ticks.
Following the larval blood meal, the remainder of infected ticks molted to nymphs. Subsets of these ticks were examined by IFA before, during, and after their blood meal on naïve mice (Table 2). Unfed and fed nymphs from all three groups retained midgut infections of spirochetes (Fig. 5A). Midguts and salivary glands of infected nymphs were analyzed during tick feeding (at 48, 67, and 72 h after attachment) to determine whether spirochetes lacking OspC disseminated from the midgut and entered the salivary glands, as is required for transmission. Small numbers of spirochetes were detected in a similar proportion of the salivary glands from all three groups of ticks at all time points. Confocal microscopy confirmed that spirochetes were present within the salivary glands and did not represent surface contamination by midgut contents during dissection (Fig. 5B). These results demonstrated that OspC was not required for migration of B. burgdorferi in the tick. Serology and reisolation from mice on which nymphal ticks fed confirmed that WT and complemented clones infected mice, whereas none of the mice fed upon by nymphs colonized with the ospC mutant became infected (Table 2).
Fig. 5.

(A) IFA of midgut tissues from partially fed nymphal ticks infected with B31-A3 WT, ospC7 mutant, or ospC7/ospC+4 complemented B. burgdorferi clones. Spirochetes were stained with hyperimmune rabbit anti-B. burgdorferi antiserum; binding was detected with Alexa 488-labeled anti-rabbit antibody. (B) Confocal image of IFA of salivary glands from partially fed nymphal ticks. Spirochetes were stained with hyperimmune rabbit anti-B. burgdorferi antiserum (detected with Alexa 488-labeled anti-rabbit antibody); salivary glands were stained with DRAQ5. Spirochetes of all three clones were within the salivary glands. (Scale bar, 10 μm.)
Piesman (23, 24) and Crippa et al. (25) showed that tick-derived spirochetes are physiologically different from culture-derived spirochetes and that infectivity for mice is enhanced with a tick-derived inoculum. We injected four mice with homogenized midguts from partially fed, ospC7-infected nymphs to determine whether the ospC mutant was infectious when obtained directly from a feeding tick. None of the mice seroconverted and spirochetes were not reisolated from mouse tissues. In contrast, an infection was established in a mouse injected with a midgut from a partially fed tick infected with the ospC7/ospC+4 clone. This observation confirmed that OspC is essential for colonization of the mammalian host, independent of the source of the inoculum or the mode of infection.
Discussion
In this study we have analyzed the phenotype of a B. burgdorferi mutant lacking OspC. We demonstrated that the ospC mutant cannot infect mice by needle inoculation or natural tick challenge and that it is cleared from the host, even in the absence of an acquired immune response. In contrast to its defective phenotype in mice, the ospC mutant is competent to colonize the tick vector and migrate from the midgut to the salivary glands during tick feeding, as is required for transmission.
The finding that OspC is an essential virulence factor of B. burgdorferi for infection in mice, but not in ticks, is striking. Spirochetes begin to synthesize OspC during the tick blood meal, while they are still in the tick midgut, and the expression of ospC is subsequently down-regulated after transmission of B. burgdorferi to the mammal (3, 26, 27). It has been suggested that OspC plays a role in directing the dissemination of spirochetes within the tick. Our data argue that this function may be performed by a different spirochetal component. The results obtained by Ohnishi et al. (28), who detected transmission of OspA+/OspC- spirochetes by feeding ticks, support our proposed model that OspC is not required for spirochete migration from the tick midgut to the salivary glands.
The ospC mutant exhibits in vitro growth parameters similar to WT B. burgdorferi. The transient pattern of ospC expression, coupled with the phenotype of the mutant, suggests that OspC does not provide a physiologic function required for growth in the mammalian host, but has a critical function early in host infection, preceding the acquired immune response. The crystal structure of OspC, indicating that it is a predominantly α-helical protein with a putative binding pocket for an unidentified ligand (29, 30), has led to speculations on the role of OspC in invasive disease (30). Wooten et al. (31) recently demonstrated that the innate host defense to B. burgdorferi plays a key role in limiting the number of spirochetes in infected mouse tissues. It is possible that expression of ospC is required to evade innate host defenses during the brief phase of dissemination through the bloodstream. However, ospC mutant bacteria do not establish localized infections in the skin or joints, even when injected directly into these sites (Table 5, which is published as supporting information on the PNAS web site).
The relapsing fever spirochete Borrelia hermsii encodes the large Vsp/Vlp family of surface-exposed proteins that are homologous to OspC (32-34). Analogous to OspC, Vsp33 of B. hermsii is present on relapsing fever spirochetes during tick transmission (35). To evade the acquired immune response of the host while persisting in the blood, B. hermsii subsequently undergoes antigenic variation by expressing different alleles of the vsp/vlp gene family. We suggest that Vsp33 of B. hermsii has a function similar to that of OspC in B. burgdorferi, in accordance with their structural similarities (29, 36), but that B. hermsii undergoes antigenic variation with the vsp/vlp gene family to facilitate persistent bloodstream infection, while maintaining an OspC-like protein on its surface.
The ospC gene is located on a plasmid, cp26, that is ubiquitously present in all examined isolates of B. burgdorferi (37, 38). In contrast to ospC, a number of other genes on cp26 encodes proteins of presumed physiologic significance to the spirochete (39-43). These genes include the single-copy gene encoding the telomere resolvase, which converts the replicated telomeres of linear DNA into the characteristic hairpin ends of the linear chromosome and plasmids of B. burgdorferi (43). Linkage on cp26 of a gene such as ospC, which is needed only during a brief phase of the infectious cycle, with a gene encoding a presumably essential housekeeping function, such as telomere resolution, assures retention of a critical trait during extended periods in which it confers no selective advantage. The ability to persist in both the tick vector and mammalian host and to be transmitted between them are absolute requirements for the maintenance of B. burgdorferi in nature.
We conclude that the OspC protein of B. burgdorferi is a virulence factor required for the initial stage of mammalian infection. Although induction of ospC expression occurs while spirochetes are still in the tick vector, OspC does not play a role in tick colonization or the migration of B. burgdorferi to the salivary glands during tick feeding. The demonstration that induction of an essential virulence factor precedes both temporally and physically the point in the infectious cycle at which it is required reveals a developmental sequence of transmission of this tick-borne pathogen and represents an important feature of Lyme disease pathogenesis.
Supplementary Material
Acknowledgments
We thank D. Dorward and O. Steele-Mortimer for help with confocal microscopy; S. Raffel and G. Sylva for sequencing; T. Hackstadt for assistance with photography; M. Schrumpf for supplying protein lysates and E. coli strains; R. Gilmore, M. L. Mbow, and O. Steele-Mortimer for providing antibodies; K. Caughey, G. Hettrick, and A. Mora for graphic support; and F. DeLeo, T. Hackstadt, and S. Priola for critical review of the manuscript.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Osp, outer-surface protein; RML, Rocky Mountain Laboratories; IFA, immunofluorescence assay.
References
- 1.Burgdorfer, W., Barbour, A. G., Hayes, S. F., Benach, J. L., Grunwaldt, E. & Davis, J. P. (1982) Science 216, 1317-1319. [DOI] [PubMed] [Google Scholar]
- 2.Ribeiro, J. M. C., Mather, T. N., Piesman, J. & Spielman, A. (1987) J. Med. Entomol. 24, 201-205. [DOI] [PubMed] [Google Scholar]
- 3.Schwan, T. G., Piesman, J., Golde, W. T., Dolan, M. C. & Rosa, P. A. (1995) Proc. Natl. Acad. Sci. USA 92, 2909-2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Silva, A. M., Telford, S. R., III, Brunet, L. R., Barthold, S. W. & Fikrig, E. (1996) J. Exp. Med. 183, 271-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwan, T. G. & Piesman, J. (2000) J. Clin. Microbiol. 39, 382-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pal, U., de Silva, A. M., Montgomery, R. R., Fish, D., Anguita, J., Anderson, J. F., Lobet, Y. & Fikrig, E. (2000) J. Clin. Invest. 106, 561-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Silva, A. M., Zeidner, N. S., Zhang, Y., Dolan, M. C., Piesman, J. & Fikrig, E. (1999) Infect. Immun. 67, 30-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilmore, R. D. & Piesman, J. (2000) Infect. Immun. 68, 411-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carroll, J. A., Garon, C. F. & Schwan, T. G. (1999) Infect. Immun. 67, 3181-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Revel, A. T., Talaat, A. M. & Norgard, M. V. (2002) Proc. Natl. Acad. Sci. USA 99, 1562-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ojaimi, C., Brooks, C., Casjens, S., Rosa, P., Elias, A., Barbour, A. G., Jasinskas, A., Benach, J., Katona, L., Radolf, J., et al. (2003) Infect. Immun. 71, 1689-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elias, A. F., Stewart, P. E., Grimm, D., Caimano, M. J., Eggers, C. H., Tilly, K., Bono, J. L., Akins, D. R., Radolf, J. D., Schwan, T. G., et al. (2002) Infect. Immun. 70, 2139-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Policastro, P. F. & Schwan, T. G. (2003) J. Med. Entomol. 40, 364-370. [DOI] [PubMed] [Google Scholar]
- 14.Barbour, A. G. (1984) Yale J. Biol. Med. 57, 521-525. [PMC free article] [PubMed] [Google Scholar]
- 15.Carroll, J. A., Cordova, R. M. & Garon, C. F. (2000) Infect. Immun. 68, 6677-6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elias, A. F., Bono, J. L., Carroll, J. A., Stewart, P., Tilly, K. & Rosa, P. (2000) J. Bacteriol. 182, 2909-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purser, J. E. & Norris, S. J. (2000) Proc. Natl. Acad. Sci. USA 97, 13865-13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimm, D., Elias, A. F., Tilly, K. & Rosa, P. A. (2003) Infect. Immun. 71, 3138-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simpson, W. J., Cieplak, W., Schrumpf, M. E., Barbour, A. G. & Schwan, T. G. (1994) FEMS Microbiol. Lett. 119, 381-388. [DOI] [PubMed] [Google Scholar]
- 20.Mbow, M. L., Gilmore, R. D., Jr., & Titus, R. G. (1999) Infect. Immun. 67, 5470-5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simpson, W. J., Burgdorfer, W., Schrumpf, M. E., Karstens, R. H. & Schwan, T. G. (1991) J. Clin. Microbiol. 29, 236-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simpson, W. J., Schrumpf, M. E., Hayes, S. F. & Schwan, T. G. (1991) J. Clin. Microbiol. 29, 1940-1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piesman, J. (1993) J. Med. Entomol. 30, 199-203. [DOI] [PubMed] [Google Scholar]
- 24.Piesman, J. (1993) J. Infect. Dis. 167, 1082-1085. [DOI] [PubMed] [Google Scholar]
- 25.Crippa, M., Rais, O. & Gern, L. (2002) Vector Borne Zoonotic Dis. 2, 3-9. [DOI] [PubMed] [Google Scholar]
- 26.Montgomery, R. R., Malawista, S. E., Feen, K. J. M. & Bockenstedt, L. K. (1996) J. Exp. Med. 183, 261-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang, F. T., Jacobs, M. B., Bowers, L. C. & Philipp, M. T. (2002) J. Exp. Med. 195, 415-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohnishi, J., Piesman, J. & de Silva, A. M. (2001) Proc. Natl. Acad. Sci. USA 98, 670-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eicken, C., Sharma, V., Klabunde, T., Owens, R. T., Pikas, D. S., Höök, M. & Sacchettini, J. C. (2001) J. Biol. Chem. 276, 10010-10015. [DOI] [PubMed] [Google Scholar]
- 30.Kumaran, D., Eswaramoorthy, S., Luft, B. J., Koide, S., Dunn, J. J., Lawson, C. L. & Swaminathan, S. (2001) EMBO J. 20, 971-978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wooten, R. M., Ma, Y., Yoder, R. A., Brown, J. P., Weis, J. H., Zachary, J. F., Kirschning, C. J. & Weis, J. J. (2002) J. Immunol. 168, 348-355. [DOI] [PubMed] [Google Scholar]
- 32.Margolis, N., Hogan, D., Cieplak, W., Jr., Schwan, T. G. & Rosa, P. A. (1994) Gene 143, 105-110. [DOI] [PubMed] [Google Scholar]
- 33.Carter, C. J., Bergström, S., Norris, S. J. & Barbour, A. G. (1994) Infect. Immun. 62, 2792-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwan, T. G., Burgdorfer, W. & Rosa, P. A. (1999) in Manual of Clinical Microbiology, ed. Murray, P. R. (American Society for Microbiology, Washington, D.C.), pp. 746-758.
- 35.Schwan, T. G. & Hinnebusch, B. J. (1998) Science 280, 1938-1940. [DOI] [PubMed] [Google Scholar]
- 36.Zückert, W. R., Kerentseva, T. A., Lawson, C. L. & Barbour, A. G. (2001) J. Biol. Chem. 276, 457-463. [DOI] [PubMed] [Google Scholar]
- 37.Tilly, K., Casjens, S., Stevenson, B., Bono, J. L., Samuels, D. S., Hogan, D. & Rosa, P. (1997) Mol. Microbiol. 25, 361-373. [DOI] [PubMed] [Google Scholar]
- 38.Casjens, S., Palmer, N., van Vugt, R., Huang, W. M., Stevenson, B., Rosa, P., Lathigra, R., Sutton, G., Peterson, J., Dodson, R. J., et al. (2000) Mol. Microbiol. 35, 490-516. [DOI] [PubMed] [Google Scholar]
- 39.Margolis, N., Hogan, D., Tilly, K. & Rosa, P. A. (1994) J. Bacteriol. 176, 6427-6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraser, C. M., Casjens, S., Huang, W. M., Sutton, G. G., Clayton, R., Lathigra, R., White, O., Ketchum, K. A., Dodson, R., Hickey, E. K., et al. (1997) Nature 390, 580-586. [DOI] [PubMed] [Google Scholar]
- 41.Bono, J. L., Tilly, K., Stevenson, B., Hogan, D. & Rosa, P. (1998) Microbiology (Reading, U.K.) 144, 1033-1044. [DOI] [PubMed] [Google Scholar]
- 42.Tilly, K., Elias, A. F., Errett, J., Fischer, E., Iyer, R., Schwartz, I., Bono, J. L. & Rosa, P. (2001) J. Bacteriol. 183, 5544-5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobryn, K. & Chaconas, G. (2002) Mol. Cell 9, 195-201. [DOI] [PubMed] [Google Scholar]
- 44.Simpson, W. J., Schrumpf, M. E. & Schwan, T. G. (1990) J. Clin. Microbiol. 28, 1329-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}