

ABSTRACT
Propionibacterium acnes constitutes a major part of the skin microbiome and contributes to human health. However, it has also been implicated as a pathogenic factor in several diseases, including acne, one of the most common skin diseases. Its pathogenic role, however, remains elusive. To better understand the genetic landscape and diversity of the organism and its role in human health and disease, we performed a comparative genome analysis of 82 P. acnes strains, 69 of which were sequenced by our group. This collection covers all known P. acnes lineages, including types IA, IB, II, and III. Our analysis demonstrated that although the P. acnes pan-genome is open, it is relatively small and expands slowly. The core regions, shared by all the sequenced genomes, accounted for 88% of the average genome. Comparative genome analysis showed that within each lineage, the strains isolated from the same individuals were more closely related than the ones isolated from different individuals, suggesting that clonal expansions occurred within each individual microbiome. We also identified the genetic elements specific to each lineage. Differences in harboring these elements may explain the phenotypic and functional differences of P. acnes in functioning as a commensal in healthy skin and as a pathogen in diseases. Our findings of the differences among P. acnes strains at the genome level underscore the importance of identifying the human microbiome variations at the strain level in understanding its association with diseases and provide insight into novel and personalized therapeutic approaches for P. acnes-related diseases.
IMPORTANCE
Propionibacterium acnes is a major human skin bacterium. It plays an important role in maintaining skin health. However, it has also been hypothesized to be a pathogenic factor in several diseases, including acne, a common skin disease affecting 85% of teenagers. To understand whether different strains have different virulent properties and thus play different roles in health and diseases, we compared the genomes of 82 P. acnes strains, most of which were isolated from acne or healthy skin. We identified lineage-specific genetic elements that may explain the phenotypic and functional differences of P. acnes as a commensal in health and as a pathogen in diseases. By analyzing a large number of sequenced strains, we provided an improved understanding of the genetic landscape and diversity of the organism at the strain level and at the molecular level that can be further applied in the development of new and personalized therapies.
Introduction
Propionibacterium acnes is a major commensal of the human skin. It contributes to maintaining skin health by inhibiting the invasion of common pathogens, such as Staphylococcus aureus and Streptococcus pyogenes. It does so by hydrolyzing triglycerides and releasing free fatty acid that contributes to the acidic pH of the skin surface (1). On the other hand, P. acnes has been historically linked to acne vulgaris, a chronic inflammatory disease of the pilosebaceous unit affecting more than 85% of adolescents and young adults (2). Our metagenomic study previously demonstrated that P. acnes was a dominant bacterium in the pilosebaceous unit in both healthy individuals and acne patients (3, 4). At the strain level, however, the population structures of P. acnes were different between the two groups. Our findings suggested that microbe-related human diseases are often caused by certain strains of a species rather than the entire species, in line with the studies of other diseases (5, 6).
P. acnes has been classified into three major genetic lineages. Studies by Johnson and Cummins (7) first revealed two distinct phenotypes of P. acnes, known as types I and II, that were able to be distinguished based on serological agglutination tests and cell wall sugar analysis. McDowell et al. (8) differentiated types I and II P. acnes by monoclonal antibody typing. Furthermore, their phylogenetic analysis of P. acnes strains based on the nucleotide sequences of the recA gene and a more-variable hemolysin/cytotoxin gene (tly) demonstrated that types I and II represent distinct lineages. Their investigations also revealed that strains within the type I lineage can be further split into two clades, known as types IA and IB (8, 9). An additional phylogenetic group of P. acnes, known as type III, was described later (10). Recent studies based on multilocus sequence typing (MLST) further subdivided P. acnes into closely related clusters—IA1, IA2, IB, IC, II, and III or I-1a, I-1b, I-2, II, and III—some of which were associated with various diseases, including acne (11–13).
The first complete genome sequence of P. acnes, KPA171202, a type IB strain, provided insight into the pathogenic potential of this Gram-positive bacterium (14). The genome is 2.56 Mb with 60% GC content. It encodes 2,333 open reading frames (ORFs), including multiple gene products, such as sialidases, neuraminidases, endoglycoceramidases, lipases, and pore-forming factors, that are involved in degrading host molecules. However, the sequence of a single genome does not reflect the genetic landscape of the organism and how genetic variations among strains determine their various phenotypes and pathogenic properties.
To better understand the human microbiome variations at the strain level, as part of the Human Microbiome Project (HMP) (15, 16), we previously generated the reference genome sequences of 66 P. acnes strains selected from a collection of over 1,000 strains isolated from a cohort of healthy subjects and acne patients (4). These 66 strains represent the major lineages of P. acnes found on the human skin, including types IA, IB, and II. To cover all the main P. acnes lineages in the analysis, we newly sequenced three additional P. acnes strains, including the first available type III P. acnes genome. Thirteen P. acnes genomes sequenced by other research groups (14, 17–22) were also available at the time of analysis. With a total of 82 genomes, we performed a comparative genome analysis to characterize the pan-genome of P. acnes, the phylogenetic relationships among different lineages, the microevolution of the strains in the same individual microbiome, and the genetic elements specific to each lineage and their associations with health and disease.
RESULTS
P. acnes strains and general genome features.
To understand the genomic diversity of this important skin commensal at the strain level, we analyzed the genomes of 69 P. acnes strains that we sequenced. Among them, 67 P. acnes strains were isolated from the skin of healthy individuals and acne patients (4) and two P. acnes strains, HL201PA1 and HL202PA1, were isolated from refractory endodontic lesions (23) (Table 1). These 69 strains cover all the known P. acnes lineages isolated to date. We classified the strains based on their 16S rRNA sequences. Each unique 16S rRNA sequence was defined as a ribotype (RT). All the sequenced P. acnes genomes had three identical copies of 16S rRNA. Based on our metagenomic study of the skin microbiome associated with acne (4), among the top ten major ribotypes, RT1, RT2, and RT3 were the most abundant and found in both healthy individuals and acne patients with no significant differences. RT4, RT5, and RT8, however, were enriched in acne patients, while RT6 was found mostly in healthy individuals. Our 69 strains included 19 RT1 strains, five RT2 strains, 15 RT3 strains, eight RT4 strains, seven RT5 strains, four RT6 strains, six RT8 strains, four strains of minor ribotypes, and one type III strain. The sequence types of all the strains were assigned based on two published MLST schemes (11–13) and are shown in Table S1 in the supplemental material. The average genome size was 2.50 Mb (ranging from 2.46 to 2.58 Mb), and the GC content was 60%. On average, each genome encoded 2,626 ORFs (ranging from 2,393 to 2,806) (Table 1).
TABLE 1 .
General features of the 82 P. acnes genomes
| Genome no. | Strain name | Origin | Ribotype | recA type | Sequencing method |
Genome size (Mb) |
GC (%) | No. of ORFs |
|---|---|---|---|---|---|---|---|---|
| 1 | HL001PA1 | Skin | 2 | II | Illumina | 2.49 | 60.0 | 2,661 |
| 2 | HL002PA1 | Skin | 3 | IB | Illumina | 2.48 | 60.1 | 2,549 |
| 3 | HL002PA2 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,594 |
| 4 | HL002PA3 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,565 |
| 5 | HL005PA1 | Skin | 4 | IA | Illumina | 2.53 | 60.2 | 2,724 |
| 6 | HL005PA2 | Skin | 1 | IA | Illumina | 2.48 | 60.0 | 2,645 |
| 7 | HL005PA3 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,579 |
| 8 | HL005PA4 | Skin | 3 | IB | Illumina | 2.47 | 60.0 | 2,607 |
| 9 | HL007PA1 | Skin | 4 | IA | Illumina | 2.53 | 60.2 | 2,691 |
| 10 | HL013PA1 | Skin | 3 | IB | Illumina | 2.48 | 60.0 | 2,618 |
| 11 | HL013PA2 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,588 |
| 12 | HL020PA1 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,554 |
| 13 | HL025PA1 | Skin | 1 | IB | Illumina | 2.54 | 60.1 | 2,581 |
| 14 | HL025PA2 | Skin | 3 | IB | Illumina | 2.48 | 60.0 | 2,616 |
| 15 | HL027PA1 | Skin | 3 | IB | Illumina | 2.53 | 60.1 | 2,711 |
| 16 | HL027PA2 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,629 |
| 17 | HL030PA1 | Skin | 1 | IB | Illumina | 2.54 | 60.0 | 2,662 |
| 18 | HL030PA2 | Skin | 3 | IB | Illumina | 2.51 | 60.1 | 2,647 |
| 19 | HL036PA1 | Skin | 532 | IA | Illumina | 2.48 | 60.1 | 2,575 |
| 20 | HL036PA2 | Skin | 532 | IA | Illumina | 2.48 | 60.1 | 2,565 |
| 21 | HL036PA3 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,601 |
| 22 | HL037PA1 | Skin | 3 | IB | Illumina | 2.48 | 60.1 | 2,617 |
| 23 | HL038PA1 | Skin | 4 | IA | Illumina | 2.54 | 60.2 | 2,663 |
| 24 | HL042PA3 | Skin | 6 | II | Roche/454 | 2.53 | 60.1 | 2,610 |
| 25 | HL043PA1 | Skin | 5 | IA | Illumina | 2.53 | 60.2 | 2,698 |
| 26 | HL043PA2 | Skin | 5 | IA | Illumina | 2.53 | 60.2 | 2,688 |
| 27 | HL045PA1 | Skin | 4 | IA | Illumina | 2.53 | 60.2 | 2,692 |
| 28 | HL046PA1 | Skin | 3 | IB | Illumina | 2.48 | 60.0 | 2,599 |
| 29 | HL046PA2 | Skin | 1 | IA | Illumina | 2.53 | 60.1 | 2,692 |
| 30 | HL050PA1 | Skin | 3 | IB | Illumina | 2.48 | 60.0 | 2,652 |
| 31 | HL050PA2 | Skin | 1 | II | Illumina | 2.46 | 60.1 | 2,581 |
| 32 | HL050PA3 | Skin | 3 | IB | Illumina | 2.48 | 60.0 | 2,558 |
| 33 | HL053PA1 | Skin | 4 | IA | Illumina | 2.53 | 60.2 | 2,632 |
| 34 | HL053PA2 | Skin | 8 | IB | Illumina | 2.51 | 60.1 | 2,664 |
| 35 | HL056PA1 | Skin | 4 | IA | Illumina | 2.48 | 60.1 | 2,581 |
| 36 | HL059PA1 | Skin | 16 | IB | Illumina | 2.48 | 60.1 | 2,570 |
| 37 | HL059PA2 | Skin | 16 | IB | Illumina | 2.48 | 60.0 | 2,535 |
| 38 | HL060PA1 | Skin | 2 | II | Illumina | 2.48 | 60.1 | 2,601 |
| 39 | HL063PA1 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,520 |
| 40 | HL063PA2 | Skin | 3 | IB | Illumina | 2.53 | 60.0 | 2,669 |
| 41 | HL067PA1 | Skin | 3 | IB | Illumina | 2.53 | 60.1 | 2,633 |
| 42 | HL072PA1 | Skin | 5 | IA | Illumina | 2.53 | 60.1 | 2,594 |
| 43 | HL072PA2 | Skin | 5 | IA | Illumina | 2.53 | 60.1 | 2,672 |
| 44 | HL074PA1 | Skin | 4 | IA | Illumina | 2.53 | 60.2 | 2,723 |
| 45 | HL078PA1 | Skin | 1 | IA | Illumina | 2.58 | 60.1 | 2,785 |
| 46 | HL082PA1 | Skin | 8 | IB | Illumina | 2.50 | 60.1 | 2,648 |
| 47 | HL082PA2 | Skin | 2 | II | Illumina | 2.51 | 60.0 | 2,644 |
| 48 | HL083PA1 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,575 |
| 49 | HL083PA2 | Skin | 3 | IB | Illumina | 2.48 | 60.0 | 2,633 |
| 50 | HL086PA1 | Skin | 8 | IB | Illumina | 2.53 | 60.1 | 2,610 |
| 51 | HL087PA1 | Skin | 3 | IB | Illumina | 2.48 | 60.1 | 2,584 |
| 52 | HL087PA2 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,572 |
| 53 | HL087PA3 | Skin | 3 | IB | Illumina | 2.52 | 60.1 | 2,619 |
| 54 | HL092PA1 | Skin | 8 | IB | Illumina | 2.50 | 60.1 | 2,590 |
| 55 | HL096PA1 | Skin | 5 | IA | Roche/454 | 2.49 | 60.0 | 2,393 |
| 56 | HL096PA2 | Skin | 5 | IA | Illumina | 2.56 | 60.1 | 2,638 |
| 57 | HL096PA3 | Skin | 1 | IA | Illumina | 2.56 | 60.0 | 2,651 |
| 58 | HL097PA1 | Skin | 5 | IC | Illumina | 2.52 | 60.2 | 2,617 |
| 59 | HL099PA1 | Skin | 4 | IA | Illumina | 2.58 | 60.1 | 2,735 |
| 60 | HL100PA1 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,562 |
| 61 | HL103PA1 | Skin | 2 | II | Illumina | 2.48 | 60.1 | 2,546 |
| 62 | HL106PA1 | Skin | 2 | II | Illumina | 2.48 | 60.1 | 2,533 |
| 63 | HL106PA2 | Skin | 1 | IA | Illumina | 2.48 | 60.1 | 2,567 |
| 64 | HL110PA1 | Skin | 8 | IB | Illumina | 2.51 | 60.1 | 2,667 |
| 65 | HL110PA2 | Skin | 8 | IB | Illumina | 2.50 | 60.1 | 2,614 |
| 66 | HL110PA3 | Skin | 6 | II | Illumina | 2.54 | 60.1 | 2,806 |
| 67 | HL110PA4 | Skin | 6 | II | Illumina | 2.54 | 60.1 | 2,724 |
| 68 | HL201PA1 | Refractory endodontic lesion |
Not assigned | III | Illumina | 2.48 | 60.1 | 2,629 |
| 69 | HL202PA1 | Refractory endodontic lesion |
6 | II | Illumina | 2.56 | 60.0 | 2,821 |
| 70 | KPA171202 | Plate | 1 | IB | Sanger | 2.56 | 60.0 | 2,297 |
| 71 | J139 | Skin | 2 | II | Roche/454 | 2.48 | 60.0 | 2,364 |
| 72 | J165 | Skin | 1 | IA | Roche/454 | 2.50 | 60.0 | 2,403 |
| 73 | SK137 | Skin | 1 | IA | Roche/454 | 2.50 | 60.0 | 2,352 |
| 74 | SK187 | Skin | 3 | IB | Roche/454 | 2.51 | 59.0 | 2,381 |
| 75 | SK182 | Skin | 1 | IA | Roche/454 | 2.48 | 60.0 | 2,338 |
| 76 | 266 | Pleuropulmonary infection |
1 | IA | Roche/454 | 2.50 | 60.0 | 2,412 |
| 77 | 6609 | Skin | 1 | IB | Solid | 2.56 | 60.0 | 2,358 |
| 78 | ATCC 11828 | Abscess | 2 | II | Solid | 2.49 | 60.0 | 2,260 |
| 79 | P.acn17 | Corneal scrape | 3 | IB | Solid | 2.52 | 60.0 | 2,266 |
| 80 | P.acn31 | Aqueous humor | 3 | IB | Solid | 2.50 | 60.0 | 2,247 |
| 81 | P.acn33 | Aqueous humor | 3 | IB | Solid | 2.49 | 60.0 | 2,236 |
| 82 | PRP-38 | Skin | 5 | IC | Solid | 2.51 | 60.0 | 2,233 |
Our analysis included 13 additional P. acnes genomes that were publicly available (14, 17–22) (Table 1). The average genome size of these 13 P. acnes strains was 2.51 Mb (ranging from 2.48 to 2.56 Mb), and the GC content was 60%, encoding 2,319 ORFs on average (ranging from 2,233 to 2,412). These 13 genomes include six RT1 strains, two RT2 strains, four RT3 strains, and one RT5 strain; however, no genomes of RT4, RT6, RT8, and type III strains were available. Our sequencing effort significantly increased the number of genomes for each P. acnes lineage as well as the number of lineages covered.
P. acnes pan-genome.
To determine the genetic landscape of P. acnes, we estimated the pan-genome based on the 82 P. acnes genomes. We first estimated the number of new genes that would be discovered by sequencing additional P. acnes genomes by using a power law regression analysis, n = κN−α (24) (Fig. 1A). Our analysis identified α as 0.788. The average number of new genes added by a novel genome was three when the 82nd genome was added. We then estimated the number of P. acnes pan-genes that would be accumulated by sequencing additional P. acnes genomes by using a power law regression analysis, n = κNγ (Fig. 1B). The exponent γ was 0.067, and P. acnes had 3,136 pan-genes (n = 82). Based on these results, the pan-genome of P. acnes is defined as open, as the exponent α was less than one and γ was greater than zero (24). However, since α was close to one and γ was close to zero, we speculate that this organism evolved tightly without large expansions.
FIG 1 .

P. acnes pan-genome. (A) A power law regression for new genes (n) discovered with the addition of new genome sequences (N). (B) A power law regression for total genes (n) accumulated with the addition of new genome sequences (N). Circles are the medians of n for 200 simulations. Error bars indicate the standard deviations for the 200 simulations.
Phylogenetic relationships among the P. acnes genomes.
Genome comparison of the 82 P. acnes strains revealed that 2.20 Mb (88% of the average genome) were shared by all the P. acnes genomes, a section which we refer to as the “core regions.” Within the core regions, 123,223 unique single nucleotide polymorphisms (SNPs) were detected among the strains. Twenty-seven percent of the SNPs were unique to type I, 22% were unique to type II, and 22% were unique to type III (see Fig. S1 in the supplemental material). We constructed a phylogenetic tree based on the 123,223 SNPs in the core regions (Fig. 2). The tree showed that the recA type classification of the strains was consistent with the major clades based on the genomes, which we named clades IA-1, IA-2, IB-1, IB-2, IB-3, IC, II, and III (4). The recA type IA, IB, and II strains, except HL097PA1 and PRP-38, were all clustered together within each type. The only recA type III strain, HL201PA1, formed a separate branch from type I and type II strains. The tree also showed that the 16S rRNA ribotypes of the strains were consistent with the phylogenetic relationships inferred from the genome sequences. Most of the RT1 strains were clustered in clade IA-1, while all the RT4 and most of the RT5 strains were clustered in clade IA-2. All six RT8 strains were clustered together in clade IB-1. All the RT3 and RT16 strains except SK187 were clustered together in clade IB-2. HL030PA1 and KPA171202 were clustered together with 6609 as a distinct IB-3 clade which has different phenotypes from strains in clades IB-1 and IB-2, including the lack of expression of dermatan sulfate binding adhesions (13). HL097PA1 and PRP-38, which are antibiotic-resistant strains, were clustered together and were classified as a novel type IC recently named by McDowell et al. (22). All the RT2 strains were clustered in clade II, distant from clade I, together with RT6 strains. HL202PA1, which is an RT6 strain and was isolated from an oral site, was not much different from the skin RT6 isolates and was clustered together with them. The phylogenetic tree based on the core genome regions demonstrated that 16S ribotyping can be used for P. acnes strain identification and classification. The 16S ribotypes of the strains are also consistent with the clonal complexes assigned based on the two MLST schemes (11, 13) with a similar resolution (see Table S1 in the supplemental material). 16S ribotyping provides a much higher resolution than recA typing, and in the meantime, it is much simpler and faster, with only one gene required, than MLST, which is a laborious process generally requiring 7 to 9 genes.
FIG 2 .

A phylogenetic tree of 82 P. acnes strains and the distance matrix among the strains. A phylogenetic tree of 82 P. acnes strains was constructed based on the 123,223 SNPs in the core regions (2.20 Mb). The distances between strains were calculated as nucleotide substitution rates at all SNP sites, colored according to the scale bar. The strains from the same individuals (SSIs) belonging to the same lineages are marked with “+.”
The large number of genome sequences that we generated allowed us to analyze the P. acnes pan-genome at the clade level. Clades IA, IB, and II had 36, 33, and 12 genomes, respectively. Based on the power law regression analyses described above, we determined that at the clade level, P. acnes also has an open pan-genome for recA type IA clade, type IB clade, and type II clade with limited expansions (see Fig. S2 in the supplemental material). The expansion rates were not significantly different among the clades and were similar to the one at the species level. This suggests that all the major lineages of P. acnes evolved at a similar rate.
SNP distribution in the core genome regions.
To understand whether there are “hot spots” for mutation and/or recombination in the P. acnes genomes, we determined whether the SNPs were randomly distributed throughout the genomes or enriched in particular regions. We calculated the frequency of SNPs in each protein coding gene in the core regions. The average rate of polymorphic sites in the core regions was 5.3%, i.e., 5.3 unique SNPs in every 100 bp. This rate is comparable to the ones found in multiple gut bacterial genomes (25). Among the 1,888 genes encoded in the core regions, 55 genes had higher SNP frequencies with more than two standard deviations (SD) and 47 genes had higher frequencies with more than three SD (Fig. 3A). Using the Kolmogorov-Smirnov (K-S) test, we demonstrated that these 102 highly mutated genes were not randomly distributed throughout the genome (P < 0.01) (Fig. 3B). This suggests that P. acnes has an evolutionary risk management strategy. Based on the clusters of orthologous groups (COG) categories, the functions of these 102 genes showed a similar distribution as those of all 1,888 genes in the core regions. There was no enrichment of a particular functional category in these frequently mutated genes.
FIG 3 .
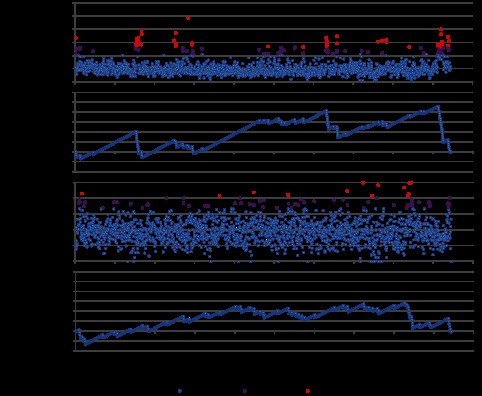
SNP distribution in the core regions. (A) SNP frequencies (percentages of polymorphic sites) of the genes in the core regions. (B) K-S statistics for genes that had higher SNP frequencies with more than two standard deviations (SD). (C) Nonsynonymous mutation frequencies of the genes in the core regions. (D) K-S statistics for genes that had higher nonsynonymous mutation frequencies with more than 2 standard deviations.
We further determined whether the mutations in the core regions were under selection by calculating the ratio of nonsynonymous (NS) to synonymous (S) SNPs for the 1,888 genes. The average rate of NS mutations was 38%. Among the 1,888 genes, 54 genes had higher NS mutation rates with more than two SD and 13 genes had higher NS mutation rates with more than three SD (Fig. 3C). These 67 genes were randomly distributed in the genome and not particularly enriched in certain regions (P > 0.05 with the K-S test) (Fig. 3D). Most of the 102 genes with higher SNP frequencies did not overlap with these 67 genes, suggesting that independent evolutionary events might lead to these gene alternations. Only ten genes had both high SNP frequencies and high NS mutation rates, all annotated as hypothetical proteins.
Evolutionary relationships of the strains isolated from the same individuals.
The large number of P. acnes strains isolated from the cohort of acne patients and healthy individuals allowed us to investigate whether the P. acnes strains in hair follicles from the same individual were clonal. Based on our previous metagenomic analysis, we demonstrated that most individuals harbored multiple P. acnes strains from different lineages (4). However, it is not known whether the strains of the same lineage in the same individual were derived from the same ancestor. Genome sequences of the strains isolated from the same samples make it possible to examine whether the strains from the same individuals (SSIs) evolved from the same origin via clonal expansion. Our 69 sequenced P. acnes strains included 49 SSIs: 13 duets (i.e., 13 pairs of strains isolated from 13 individuals), five trios, and two quartets. Twenty-three SSIs were clustered in the same clades, with nine in clade IA-1, four in clade IA-2, two in clade IB-1, six in clade IB-2, and two in clade II. We calculated the distance (substitution rate at the 123,223 SNP sites in the core regions) between each pair of SSIs (Fig. 2). The average distance of the SSIs in clade IA-1 was 0.0014, while that of strains from different individuals in clade IA-1 was 0.0064 (P < 0.001). Consistent results were observed in other clades, including IA-2, IB-1, IB-2, and II (see Fig. S3A in the supplemental material). This demonstrated that the SSIs in the same lineage were significantly more similar to each other than the strains isolated from different individuals, suggesting that they were clonal in each individual. Among the RT4 and RT5 strains within clade IA-2, however, the average distance between SSIs (0.0004) was not significantly different from the average distance between strains from different individuals (0.0017) (P = 0.072). Moreover, the average distance between RT4/RT5 strains from different individuals (0.0017) was similar to the average distance between the SSIs in clade IA-1 (0.0014) and even shorter than the average distances between the SSIs in clades IB-1 (0.0059), IB-2 (0.0019), and II (0.0022) (Fig. S3A). This suggests that although isolated from different individuals, these RT4 and RT5 strains seemed to be clonal and had evolved from the same recent ancestor. We observed a similar relationship between the two RT5 strains in clade IC, in which HL097PA1 and PRP-38, isolated from different individuals, were closely related to each other with a distance of 0.0012. Our metagenomic study has demonstrated a strong association of strains of RT4 and RT5 with acne (4). The clonality of these strains isolated from different individuals suggests that RT4 and RT5 strains may be transmitted among individuals. This finding is consistent with the previous clinical report that antibiotic-resistant propionibacteria were transmissible between acne-prone individuals, including dermatologists (26), as most of the antibiotic-resistant P. acnes strains belong to RT4 and RT5 (4). Our analysis of SSIs further supports the theory that RT4 and RT5 strains may be a pathogenic factor in acne.
To determine whether the distances between strain pairs from the same individuals but belonging to different lineages were different from those between random strain pairs, we calculated the distances of any pair of SSIs from different clades. The average distance of the SSIs between clades IA-1 and IA-2 (i.e., HL005PA3 versus HL005PA1, HL005PA2 versus HL005PA1, HL096PA3 versus HL096PA1, and HL096PA3 versus HL096PA2) was 0.039, similar to that of the isolates from different individuals (0.040). Similar results were obtained for all other clade pair comparisons (see Fig. S3B in the supplemental material). These results demonstrated that the SSIs from different clades were similarly different from each other as from the strains from different individuals. Our analysis suggests that in each individual microbiome, P. acnes strains undergo clonal expansion in the same population, while multiple strain populations can often coexist in the same community with little recombination.
Noncore genome regions in type I strains.
By comparing the genome sequences of the 82 P. acnes strains, we identified noncore genome regions that were not shared by all 82 strains. The total length of the noncore regions was approximately 0.90 Mb. The average GC content of the noncore regions was slightly lower than that of the core regions, 58% ± 6.9%, suggesting that part of the noncore regions might be originated from other species via horizontal gene transfer.
Different lineages of P. acnes strains have distinct noncore regions. Using hierarchical clustering of the noncore regions, we showed that the strains of the same ribotypes were clustered together, with distinct separations among the clades (Fig. 4). Among the noncore regions, we identified the genetic elements specific to each lineage, which may explain the phenotypic and functional differences of the strains in health and disease. In clade IA-2, we previously identified genomic loci 1, 2, and 3, which were unique to mainly the RT4 and RT5 strains (4). These loci appear to be originated from mobile genetic elements, encode several virulent genes, and may contribute to the virulence of these strains. In the meantime, the genomic island-like cluster 2 (GI2) (18) was uniquely absent in most of the strains in this clade. Clade IB-1 consisted of all RT8 strains, which were also highly associated with acne based on our metagenomic study (4). They all have a unique genomic island (locus 4) which is 20 kb long and encodes a series of nonribosomal peptide synthetases (NRPS) which may contribute to increased virulence of these strains. Most RT3 and RT16 strains belong to clade IB-2 and have fewer noncore regions than the strains in other clades. This may be explained by the lack of entire rearrangement hot spot (RHS) family proteins, which function in genomic rearrangements as previously implicated in Escherichia coli (27). Clade IB-3 consisted of three strains, including KPA171202. Three of the four genomic islands described previously (18), GI1, GI3, and GI4, were unique to this clade and were absent in all other strains. Our analysis suggests that KPA171202, although it was the first sequenced complete genome of P. acnes, did not seem to be a common skin P. acnes strain representing one of the major lineages. This result is consistent with previous studies using MLST (11–13). Strains of clade IC belong to RT5. They also contain locus 3, a linear plasmid, which is highly homologous to the locus 3 in the RT4 and RT5 strains of clade IA-2 and encodes a tight adhesion locus highly homologous to the one in Clostridium leptum (4). In general, although strains in different lineages had a similar genome size with similar gain and loss of genetic materials, they harbor distinct genetic elements which may give rise to their different virulent properties.
FIG 4 .
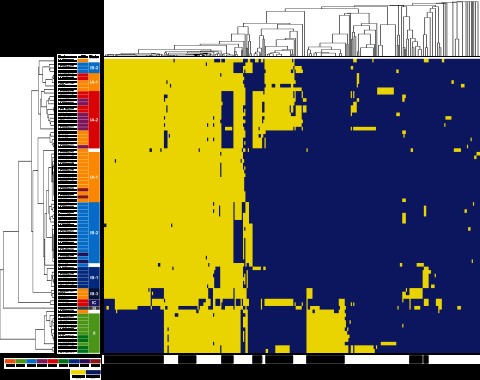
P. acnes strains within each lineage share unique noncore genomic regions. Rows represent 82 P. acnes genomes, and columns represent 314 noncore regions that are longer than 500 bp. The genomes and the noncore regions were clustered based on similarity. The width of each block plotted is not proportional to the genomic length of each noncore region. The presence of a noncore region is colored in yellow, and the absence is colored in blue. The color schemes used for RT and clades are the same as in Fig. 2.
Noncore genome regions in type II strains.
Strains in clade II, mainly RT2 and RT6, were more distantly related to the strains in clade I. Based on our metagenomic study and previous studies, strains in clade II were not associated with acne (4, 11–13), as RT2 was evenly distributed between acne patients and healthy individuals while RT6 was significantly enriched in healthy individuals (4). Compared to type I strains, the genomes of RT2 and RT6 strains lack several regions which are approximately 92 kb long in total and encode 107 ORFs. On the other hand, RT2 and RT6 genomes have additional genomic regions with a similar size encoding 93 ORFs (Fig. 4). Based on the COG classification, there were no significant differences in the distribution of the functional categories between the 107 type I-specific ORFs and the 93 type II-specific ORFs.
The most unique genomic feature of RT2 and RT6 strains is represented by the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas locus (4). The CRISPR/Cas system provides acquired bacterial immunity against viruses and plasmids by targeting nucleic acids in a sequence-specific manner (28). All the sequenced strains of RT2 and RT6 encoded a complete set of CRISPR/Cas genes and at least one repeat-spacer sequence, while none of the other ribotype strains did. Based on its complete genome sequence (20), strain ATCC 11828 appeared to be an exception, having only a terminal sequence but no spacer sequence. However, using PCR and sequencing, we determined that ATCC 11828 has one repeat-spacer sequence (see Table S2 in the supplemental material). A total of 48 spacer sequences were found in the 11 RT2 and RT6 strains, 29 of which were unique. In other bacterial species, it has been established experimentally and computationally that the spacers at the leader-proximal end are more diversified while the spacers at the leader-distal end are more conserved among strains. We analyzed the evolutionary relationships among the RT2 and RT6 strains based on their shared spacer sequences. HL060PA1 and HL082PA2, which were clustered tightly in clade II, shared the same spacer S2 (Fig. S4). J139, ATCC 11828, HL110PA4, HL110PA3, and HL202PA1 shared the same spacers S17 and S18 (Fig. S4). These results suggest that these groups of strains probably evolved from the same ancestors before having acquired additional spacers. The relationships among the strains based on shared CRISPR spacers are consistent with the phylogenetic relationships calculated based on the SNPs in the core regions. In addition, multiple type II strains harbored spacer sequences that match the sequences in loci 2 and 3, which were unique to mainly acne-associated RT4 and RT5 strains (4). The sequences in loci 2 and 3 appeared to be originated from C. leptum and encode potential virulence factors (Fig. S4). As we hypothesized previously, these loci may have been acquired by RT4 and RT5 strains, while the genomes of RT2 and RT6 that encode these spacers may be capable of eliminating the invasion of foreign DNA through the CRISPR mechanism (4).
The large number of high-quality draft genome sequences enabled us to detect not only large genomic variations but also small but essential genomic alterations. It was previously reported that type II strains showed decreased lipase activity (10). Lipase functions in hydrolyzing triglycerides and releasing free fatty acids, which are thought to be essential in P. acnes virulence. Based on the genome annotation, we identified 13 genes with a potential function of lipase (see Fig. S5A in the supplemental material). Among them, we detected insertions/deletions ranging from one nucleotide to 13 nucleotides that may explain the decreased lipase activity in type II strains. Two triacylglycerol lipases were encoded in tandem in P. acnes genomes, HMPREF0675-4855 and HMPREF0675-4856 (according to the annotation of SK137). All the type II strains and IB-3 strains had a deletion of the TATA box 20 bp upstream of the start codon of the second lipase gene, HMPREF0675-4856 (Fig. S5B). In addition, there was a one-nucleotide deletion at the position of G124 of the second lipase gene, leading to a frameshift and the introduction of a premature stop codon. These two deletions may potentially explain the decreased lipase activity and, hence, decreased virulence in acne observed in type II strains in previous studies (4, 10).
Noncore genome regions in the type III strain.
Type III strains are rarely found on the skin surface. We sequenced a type III P. acnes strain, HL201PA1, isolated from a refractory endodontic lesion. This first available type III genome allowed us to identify the genetic elements specific to this lineage. Compared to type I and type II strains, the genome of HL201PA1 lacks a few regions with a total length of 43 kb (Fig. 4). There were 42 ORFs encoded in these regions, including anaerobic dimethyl sulfoxide reductase (PPA0516-PPA0517), iron(III) dicitrate transport system permease (PPA0792-PPA0793), 3-isopropylmalate dehydratase (PPA1361-PPA1363), and maltose transport system permease (PPA1553-PPA1554). Additional genomes of type III strains will provide further insight into the specific genome properties and evolution of this lineage.
DISCUSSION
High-throughput genome sequencing and comparative analysis of a large number of related strains have been used to study the spread and microevolution of several pathogens at the strain level, including methicillin-resistant Staphylococcus aureus (29), Streptococcus pneumoniae (30), and Vibrio cholerae in a Haiti outbreak (31), demonstrating the power of comparative genome analysis of multiple strains in improving our understanding of the bacterial pathogens. However, this approach has been rarely applied to study commensal species to understand their varied virulence potentials among different strains and their roles in both health and diseases.
Our study presents a comparative genome analysis of a major skin commensal, P. acnes, based on a large number of sequenced strains. This collection of strains includes not only strains associated with either healthy skin or acne but also a large number of strain pairs that were isolated from the same individuals. This allowed us to compare the phylogenetic relationships and microevolution of the P. acnes strains associated with health versus disease as well as of the strains in the same individual microbiome.
By comparing 82 P. acnes genomes, we showed that all P. acnes strains had a similar genome size with a similar GC content, encoding 2,577 ORFs on average (Table 1). Although P. acnes has an open pan-genome, unlike many other open-genome species (24), it has limited genome expansion, with only a few new genes added per genome (Fig. 1). The rates of genome expansion are similar within the major lineages (see Fig. S2 in the supplemental material). There was limited recombination among different P. acnes strains, and thus, 16S rRNA ribotypes can be used as a proxy for P. acnes strain identification and classification (Fig. 2). Compared to other typing methods, 16S ribotyping has a much higher resolution than recA typing and is much easier and faster than the traditional MLST method. This method can be applied in a high-throughput manner by combining it with next-generation sequencing and thus allows us to detect the microbiome variations at the strain level (4). This is advantageous and important, as identifying and understanding the strain-level variations of the human microbiome is medically important.
We compared the genomes of the sets of strains isolated from the same individual samples (Fig. 2; see also Fig. S3 in the supplemental material). To our knowledge, this collection of genome data is unique and no such kind of studies to investigate the microevolution of a human commensal within an individual microbiome has been performed. We found that while multiple P. acnes strain populations coexisted in the same individual microbiome, clonal expansions occurred in each population with little recombination among different populations. Within each lineage, the strains isolated from the same individuals were significantly more similar to each other than strains from different individuals except the disease-associated strains, RT4 and RT5 strains (Fig. S3). Although isolated from different individuals, they appeared to be clonal and have evolved from the same ancestor strain (Fig. 2). This supports the observation that these strains were transmissible (26) and that they may play a role in acne pathogenesis (4). This finding is important and will help us control the spread of antibiotic-resistant strains and develop new targeted therapy for acne.
By analyzing the noncore regions, we identified the genomic elements and alterations specific to each lineage (Fig. 4). These lineage-specific elements may render the strains different physiological and functional properties and thus lead to their different roles as a commensal in health or as a pathogen in diseases. Among the acne-associated strains, RT4 and RT5 strains encode three distinct loci originated from mobile genetic elements and RT8 strains encode a distinct region containing a set of NRPS. The virulent genes carried in these strain-specific regions may explain the associations of these strains with acne and help the development of new drugs targeting against these strains. On the other hand, RT2 and RT6 strains, which were not associated with acne and were enriched in healthy skin, respectively, all encode CRISPR/Cas elements. The CRISPR mechanism may prevent these strains from acquiring virulent genes from invading foreign mobile elements. In addition, these strains contain genomic variations in lipases that may alter lipid metabolism and reduce their virulence (see Fig. S5 in the supplemental material).
In conclusion, by characterizing the genetic landscape and diversity of P. acnes with a large number of genomes, we provide genomic evidence that may explain the diverse phenotypes of P. acnes strains and a new insight into the dual role of this commensal in human skin health and disease. The findings from this comparative genome analysis provide new perspectives on the strain diversity and evolution of commensals in the human microbiome. As many current microbiome studies focus on the associations of microbial communities with health and diseases, our study underscores the importance of understanding the commensal microbiome at the strain level (25). Our findings from this study also shed light on new strain-specific therapeutics for acne and other P. acnes-associated diseases.
MATERIALS AND METHODS
P. acnes strains.
Among the 69 P. acnes strains that we sequenced, 67 were isolated from the skin pore samples from acne patients and healthy individuals (4). The other two strains, HL201PA1 and HL202PA1, were isolated from refractory endodontic lesions (23) and provided by David Beighton at King’s College, London, United Kingdom.
Whole-genome shotgun sequencing, assembly, and annotation.
The genome of HL042PA3 was sequenced using Roche/454 FLX and assembled using a combination of Phrap/Consed (32) and GSMAPPER (Roche). HL201PA1 and HL202PA1 were sequenced using Illumina MiSeq (250 bp, paired end) and assembled using Velvet (33). The remaining 66 genomes were sequenced as described previously (4). Coding sequences were predicted using GeneMark (34) and GLIMMER (35).
Computation of the core regions, noncore regions, and pan-genome.
The core regions were defined as genome sequences that were present in all 82 genomes, while the noncore regions were defined as genome sequences that were not present in all the genomes. KPA171202 was used as the reference genome. Each of the other 81 genome sequences (a series of contigs in most of the genomes and ten complete genomes) was mapped to the reference genome using Nucmer (36). All the 81 “.coords” output files of the Nucmer program were analyzed to identify overlap regions based on the KPA171202 coordinates using a Perl script. Core sequences were then extracted based on the genome sequence of KPA171202 with the coordinates calculated above.
The unique regions from each genome were added to the reference genome to make a “revised” reference genome, which contained the original sequence plus the unique genome sequences. This process was repeated for all the genomes until all the unique regions from all genomes were included in the pan-genome.
Lastly, core regions were subtracted from the pan-genome. The remaining regions were defined as noncore regions, which are not shared by all the strains. Protein coding sequences were predicted by GeneMark.hmm using KPA171202 as a reference.
Identification of SNPs in the core regions.
Single nucleotide polymorphisms (SNPs) were identified by using the “show-snps” utility option of the Nucmer program with the default settings (36). The genome sequence of KPA171202 was used as the reference genome. All the 81 “.snps” output files of the Nucmer program were analyzed to identify unique SNP positions based on the KPA171202 coordinates using a Perl script.
Phylogenetic tree construction.
The 82 concatenated sequences of the 123,223 SNP nucleotides in the core regions were used to construct a phylogenetic tree of the P. acnes genomes. MEGA5 (37) was used to calculate the distance based on the SNPs in the core regions using the neighbor-joining method and the p-distance method. The bootstrap tree inferred from 200 replicates was taken.
Sequence type analysis based on MLST schemes.
The sequence types of the 82 isolates were determined based on the MLST schemes published previously (11–13). The MLST gene sequences were aligned using BLAST against all the alleles used in the two MLST schemes.
Identification of CRISPR/Cas.
CRISPRFinder (38) was used to identify the CRISPR repeat-spacer sequences. The annotation of HL110PA3 was used for BLAST alignment in order to identify the presence of CRISPR/Cas structure and CRISPR repeat-spacer sequences in strains HL001PA1, HL060PA1, HL042PA3, HL082PA2, HL103PA1, HL106PA1, HL110PA4, HL202PA1, J139, and ATCC 11828. Each spacer sequence was annotated by BLAST (39) against NCBI’s nonredundant nucleotide database and the reference genomic sequence database (refseq_genomic).
Hierarchical clustering analysis of the noncore regions.
Among the 1,685 noncore fragments (895,905 bp in total), 314 noncore fragments with a length of >500 bp (747,189 bp in total, corresponding to 83% of all the noncore regions) were extracted and used in hierarchical clustering of the noncore regions. The Cluster 3.0 program (40) and average linkage method were used. The clustering matrix was composed of 314 rows and 82 columns, in which “1” denotes presence of the noncore region and “0” denotes absence of the noncore region. The Java TreeView program (41) was used to display the clustering result.
Nucleotide sequence accession numbers.
The accession numbers of the genomes in the described analysis are ADWB00000000, ADWC00000000, ADWF00000000, ADWH00000000, ADWI00000000, ADXP00000000, ADXQ00000000, ADXR00000000, ADXS00000000, ADXT00000000, ADXU00000000, ADXW00000000, ADXX00000000, ADXY00000000, ADXZ00000000, ADYA00000000, ADYB00000000, ADYC00000000, ADYD00000000, ADYE00000000, ADYF00000000, ADYG00000000, ADYI00000000, ADYJ00000000, ADYK00000000, ADYL00000000, ADYM00000000, ADYN00000000, ADYO00000000, ADYP00000000, ADYQ00000000, ADYR00000000, ADYS00000000, ADYT00000000, ADYU00000000, ADYV00000000, ADYW00000000, ADYX00000000, ADYY00000000, ADYZ00000000, ADZA00000000, ADZB00000000, ADZC00000000, ADZD00000000, ADZE00000000, ADZF00000000, ADZG00000000, ADZH00000000, ADZI00000000, ADZJ00000000, ADZK00000000, ADZL00000000, ADZM00000000, ADZN00000000, ADZO00000000, ADZP00000000, ADZQ00000000, ADZR00000000, ADZS00000000, ADZT00000000, ADZV00000000, ADZW00000000, ADFS00000000, CP003293, CP003294, AE017283.1, ADFS00000000, ADJL00000000, CP001977.1, ADJM00000000, AFUM00000000, CP002409.1, CP002815.1, CP003084.1, CP003195.1, CP003196.1, CP003197.1, and AIJP00000000.
SUPPLEMENTAL MATERIAL
Proportion of the 123,223 SNPs in the core regions specific to recA types (1) II and III. Download
Pan-genomes of types IA (A), IB (B), and II (C) strains. Circles are the medians of n for 200 simulations. Error bars are standard deviations for the 200 simulations. Download
Distances between P. acnes strains in the same lineage (A) and in different lineages (B). Download
CRISPR spacer sequences in RT2 and RT6 strains. A total of 48 CRISPR spacer sequences were found in 11 P. acnes genomes, 29 of which were unique. Some CRISPR spacers were found in multiple strains. For example, spacer 2 (S2) was shared by HL060PA1 and HL082PA2. Spacer 17 (S17) was shared by J139, ATCC 11828, HL110PA3, HL110PA4, HL042PA3, and HL202PA1. Spacer 18 (S18) was shared by J139, ATCC 11828, HL110PA3, HL110PA4, and HL202PA1. The tree was from Fig. 2, constructed based on the 123,223 SNPs in the core regions. Download
Genes with putative lipase activity in the P. acnes genomes. (A) Summary of 13 genes with putative lipase activity based on the annotations of KPA171202 and SK137 genomes. (B) Insertions/deletions and frameshift observed in ORF HMPREF0675-4856. Download
Sequence types of the 82 P. acnes strains based on two MLST schemes.
CRISPR spacer sequences found in the genomes of ATCC 11828, HL042PA3, and HL202PA1.
ACKNOWLEDGMENTS
We thank E. Curd and M. Wong for technical support. We thank Sadia Niazi and David Beighton at the Dental Institute, King’s College, London, United Kingdom, for providing strains HL201PA1 and HL202PA1.
This research was partly funded by one of the Demonstration Projects by the NIH Human Microbiome Project (HMP). It was supported by grants UH2AR057503 and R01GM099530 from the NIH to H.L. and grant U54HG004968 from the NIH to G.M.W.
Footnotes
Citation Tomida S, Nguyen L, Chiu B, Liu J, Sodergren E, Weinstock GM, Li H. 2013. Pan-genome and comparative genome analyses of Propionibacterium acnes reveal its genomic diversity in the healthy and diseased human skin microbiome. mBio 4(3):e00003-13. doi:10.1128/mBio.00003-13.
REFERENCES
- 1. Grice EA, Segre JA. 2011. The skin microbiome. Nat. Rev. Microbiol. 9:244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. White GM. 1998. Recent findings in the epidemiologic evidence, classification, and subtypes of acne vulgaris. J. Am. Acad. Dermatol. 39:S34–S37 [DOI] [PubMed] [Google Scholar]
- 3. Li H. 2010. Metagenomic study of the human skin microbiome associated with acne. Nat. Precedings http://dx.doi.org/10.1038/npre.2010.5305.1 [Google Scholar]
- 4. Fitz-Gibbon S, Tomida S, Chiu BH, Nguyen L, Du C, Liu M, Elashoff D, Erfe MC, Loncaric A, Kim J, Modlin RL, Miller JF, Sodergren E, Craft N, Weinstock GM, Li H. 2013. Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J. Invest. Dermatol. http://dx.doi.org/10.1038/jid.2013.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chambers HF, Deleo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7:629–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chase-Topping M, Gally D, Low C, Matthews L, Woolhouse M. 2008. Super-shedding and the link between human infection and livestock carriage of Escherichia coli O157. Nat. Rev. Microbiol. 6:904–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson JL, Cummins CS. 1972. Cell wall composition and deoxyribonucleic acid similarities among the anaerobic coryneforms, classical propionibacteria, and strains of Arachnia propionica. J. Bacteriol. 109:1047–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McDowell A, Valanne S, Ramage G, Tunney MM, Glenn JV, McLorinan GC, Bhatia A, Maisonneuve JF, Lodes M, Persing DH, Patrick S. 2005. Propionibacterium acnes types I and II represent phylogenetically distinct groups. J. Clin. Microbiol. 43:326–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Valanne S, McDowell A, Ramage G, Tunney MM, Einarsson GG, O’Hagan S, Wisdom GB, Fairley D, Bhatia A, Maisonneuve JF, Lodes M, Persing DH, Patrick S. 2005. CAMP factor homologues in Propionibacterium acnes: a new protein family differentially expressed by types I and II. Microbiology 151:1369–1379 [DOI] [PubMed] [Google Scholar]
- 10. McDowell A, Perry AL, Lambert PA, Patrick S. 2008. A new phylogenetic group of Propionibacterium acnes. J. Med. Microbiol. 57:218–224 [DOI] [PubMed] [Google Scholar]
- 11. Lomholt HB, Kilian M. 2010. Population genetic analysis of Propionibacterium acnes identifies a subpopulation and epidemic clones associated with acne. PLoS One 5:e12277 http://dx.doi.org/10.1371/journal.pone.0012277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McDowell A, Gao A, Barnard E, Fink C, Murray PI, Dowson CG, Nagy I, Lambert PA, Patrick S. 2011. A novel multilocus sequence typing scheme for the opportunistic pathogen Propionibacterium acnes and characterization of type I cell surface-associated antigens. Microbiology 157:1990–2003 [DOI] [PubMed] [Google Scholar]
- 13. McDowell A, Barnard E, Nagy I, Gao A, Tomida S, Li H, Eady A, Cove J, Nord CE, Patrick S. 2012. An expanded multilocus sequence typing scheme for Propionibacterium acnes: investigation of “pathogenic," “commensal” and antibiotic resistant strains. PLoS One 7:e41480 http://dx.doi.org/10.1371/journal.pone.0041480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brüggemann H, Henne A, Hoster F, Liesegang H, Wiezer A, Strittmatter A, Hujer S, Dürre P, Gottschalk G. 2004. The complete genome sequence of Propionibacterium acnes, a commensal of human skin. Science 305:671–673 [DOI] [PubMed] [Google Scholar]
- 15. The Human Microbiome Project Consortium 2012. A framework for human microbiome research. Nature 486:215–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. The Human Microbiome Project Consortium 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Human Microbiome Jumpstart Reference Strains Consortium. Nelson KE, Weinstock GM, Highlander SK, Worley KC, Creasy HH, Wortman JR, Rusch DB, Mitreva M, Sodergren E, Chinwalla AT, Feldgarden M, Gevers D, Haas BJ, Madupu R, Ward DV, Birren BW, Gibbs RA, Methe B, Petrosino JF, Strausberg RL, Sutton GG, White OR, Wilson RK, Durkin S, Giglio MG, Gujja S, Howarth C, Kodira CD, Kyrpides N, Mehta T, Muzny DM, Pearson M, Pepin K, Pati A, Qin X, Yandava C, Zeng Q, Zhang L, Berlin AM, Chen L, Hepburn TA, Johnson J, McCorrison J, Miller J, Minx P, Nusbaum C, Russ C, Sykes SM, Tomlinson CM, Young S, Warren WC, Badger J, Crabtree J, Markowitz VM, Orvis J, Cree A, Ferriera S, Fulton LL, Fulton RS, Gillis M, Hemphill LD, Joshi V, Kovar C, Torralba M, Wetterstrand KA, Abouellleil A, Wollam AM, Buhay CJ, Ding Y, Dugan S, FitzGerald MG, Holder M, Hostetler J, Clifton SW, Allen-Vercoe E, Earl AM, Farmer CN, Liolios K, Surette MG, Xu Q, Pohl C, Wilczek-Boney K, Zhu D, Zhu D. 2010. A catalog of reference genomes from the human microbiome. Science 328:994–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brzuszkiewicz E, Weiner J, Wollherr A, Thürmer A, Hüpeden J, Lomholt HB, Kilian M, Gottschalk G, Daniel R, Mollenkopf HJ, Meyer TF, Brüggemann H. 2011. Comparative genomics and transcriptomics of Propionibacterium acnes. PLoS One 6:e21581 http://dx.doi.org/10.1371/journal.pone.0021581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hunyadkürti J, Feltóti Z, Horváth B, Nagymihály M, Vörös A, McDowell A, Patrick S, Urbán E, Nagy I. 2011. Complete genome sequence of Propionibacterium acnes type IB strain 6609. J. Bacteriol. 193:4561–4562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Horváth B, Hunyadkürti J, Vörös A, Fekete C, Urbán E, Kemény L, Nagy I. 2012. Genome sequence of Propionibacterium acnes type II strain ATCC 11828. J. Bacteriol. 194:202–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vörös A, Horváth B, Hunyadkürti J, McDowell A, Barnard E, Patrick S, Nagy I. 2012. Complete genome sequences of three Propionibacterium acnes isolates from the type IA(2) cluster. J. Bacteriol. 194:1621–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McDowell A, Hunyadkürti J, Horváth B, Vörös A, Barnard E, Patrick S, Nagy I. 2012. Draft genome sequence of an antibiotic-resistant Propionibacterium acnes strain, PRP-38, from the novel type IC cluster. J. Bacteriol. 194:3260–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Niazi SA, Clarke D, Do T, Gilbert SC, Mannocci F, Beighton D. 2010. Propionibacterium acnes and Staphylococcus epidermidis isolated from refractory endodontic lesions are opportunistic pathogens. J. Clin. Microbiol. 48:3859–3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tettelin H, Riley D, Cattuto C, Medini D. 2008. Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11:472–477 [DOI] [PubMed] [Google Scholar]
- 25. Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J, Kota K, Sunyaev SR, Weinstock GM, Bork P. 2013. Genomic variation landscape of the human gut microbiome. Nature 493:45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ross JI, Snelling AM, Carnegie E, Coates P, Cunliffe WJ, Bettoli V, Tosti G, Katsambas A, Del Galvan P, Pulgar JI, Rollman O, Torok L, Eady EA, Cove JH. 2003. Antibiotic-resistant acne: lessons from Europe. Br. J. Dermatol. 148:467–478 [DOI] [PubMed] [Google Scholar]
- 27. Jackson AP, Thomas GH, Parkhill J, Thomson NR. 2009. Evolutionary diversification of an ancient gene family (rhs) through C-terminal displacement. BMC Genomics 10:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Horvath P, Barrangou R. 2010. CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170 [DOI] [PubMed] [Google Scholar]
- 29. Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, Edgeworth JD, de Lencastre H, Parkhill J, Peacock SJ, Bentley SD. 2010. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327:469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, Bentley SD. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chin CS, Sorenson J, Harris JB, Robins WP, Charles RC, Jean-Charles RR, Bullard J, Webster DR, Kasarskis A, Peluso P, Paxinos EE, Yamaichi Y, Calderwood SB, Mekalanos JJ, Schadt EE, Waldor MK. 2011. The origin of the Haitian cholera outbreak strain. N Engl J. Med. 364:33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gordon D, Abajian C, Green P. 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8:195–202 [DOI] [PubMed] [Google Scholar]
- 33. Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Borodovsky M, McIninch J. 1993. Recognition of genes in DNA sequence with ambiguities. Biosystems 30:161–171 [DOI] [PubMed] [Google Scholar]
- 35. Salzberg SL, Delcher AL, Kasif S, White O. 1998. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 26:544–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35:W52–W57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 40. de Hoon MJ, Imoto S, Nolan J, Miyano S. 2004. Open source clustering software. Bioinformatics 20:1453–1454 [DOI] [PubMed] [Google Scholar]
- 41. Saldanha AJ. 2004. Java TreeView—extensible visualization of microarray data. Bioinformatics 20:3246–3248 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Proportion of the 123,223 SNPs in the core regions specific to recA types (1) II and III. Download
Pan-genomes of types IA (A), IB (B), and II (C) strains. Circles are the medians of n for 200 simulations. Error bars are standard deviations for the 200 simulations. Download
Distances between P. acnes strains in the same lineage (A) and in different lineages (B). Download
CRISPR spacer sequences in RT2 and RT6 strains. A total of 48 CRISPR spacer sequences were found in 11 P. acnes genomes, 29 of which were unique. Some CRISPR spacers were found in multiple strains. For example, spacer 2 (S2) was shared by HL060PA1 and HL082PA2. Spacer 17 (S17) was shared by J139, ATCC 11828, HL110PA3, HL110PA4, HL042PA3, and HL202PA1. Spacer 18 (S18) was shared by J139, ATCC 11828, HL110PA3, HL110PA4, and HL202PA1. The tree was from Fig. 2, constructed based on the 123,223 SNPs in the core regions. Download
Genes with putative lipase activity in the P. acnes genomes. (A) Summary of 13 genes with putative lipase activity based on the annotations of KPA171202 and SK137 genomes. (B) Insertions/deletions and frameshift observed in ORF HMPREF0675-4856. Download
Sequence types of the 82 P. acnes strains based on two MLST schemes.
CRISPR spacer sequences found in the genomes of ATCC 11828, HL042PA3, and HL202PA1.