

Abstract
Tissue transglutaminase (TG2) is a multifunctional protein that binds to fibronectin and exerts protein transamidating activity in the presence of Ca2+. We previously reported that TG2 is upregulated in ovarian tumors and enhances intraperitoneal (i.p.) metastasis. TG2 is secreted abundantly in ovarian cancer (OC) ascites as an active enzyme, yet its function in the extracellular compartment remains unknown. To study the distinct functions of secreted TG2, we used recombinant His6-tagged TG2 and catalytically inactive enzyme in vitro and in vivo. By using i.p. and orthotopic ovarian xenografts, we show that extracellular transglutaminase promoted OC peritoneal metastasis. The main pathway activated by extracellular TG2 was noncanonical nuclear factor-kappa B (NF-κB) signaling, and the enzymatic function of the protein was required to induce phosphorylation of IκB kinase α and processing of the precursor protein p100 into the active p52 subunit. A specific target of TG2-activated p52/RelB complex is the hyaluronan receptor, CD44. Noncanonical NF-κB activation by extracellular TG2 induced CD44 up-regulation and epithelial-to-mesenchymal transition, contributing to increased cancer cell invasiveness and OC peritoneal dissemination. Taken together, our data support that noncanonical NF-κB activation is the pathway through which extracellular TG2 promotes OC metastasis.
Introduction
Tissue transglutaminase (TG2) is a multifunctional protein with enzymatic and nonenzymatic functions. As an enzyme, it regulates Ca2+-dependent protein posttranslational modifications and cross-linking by transferring acyl groups between glutamine and lysine residues [1]. Its nonenzymatic functions include facilitation of cellular adhesion to the extracellular matrix (ECM), particularly to fibronectin (FN) [2] and GTPase activity, involved in transduction of α-adrenergic responses [3,4]. The functions of the protein are modulated by Ca2+ and guanosine-5′-triphosphate (GTP) levels; high extracellular Ca2+ concentrations enhance the enzymatic function, while high intracellular GTP levels inhibit TGase activity [5]. This reciprocal activation of TG2 by Ca2+ and GTP causes structural allosteric changes affecting the accessibility of the active site [6].
TG2 is overexpressed in breast, ovarian, brain, and pancreatic tumors [7–9]; however, its roles in cancer are not fully elucidated. Several reports, including ours, described TG2 as a protumorigenic protein that enhances cellular invasion, metastasis, and chemoresistance [7–12]. These functions have been attributed to activation of oncogenic signaling, governed in part by the transcription factors nuclear factor-kappa B (NF-κB) [11,13] and cyclic adenosine monophosphate response element-binding protein (CREB) [14] and by the protein kinase B (Akt) [15]. Direct activation of the NF-κB complex by intracellular TG2 following the canonical pathway dependent on IκBα degradation was demonstrated in pancreatic and ovarian cancer (OC) cells [11,13]. Indirect activation of serine/threonine kinase Akt and CREB in cancer cells was attributed to TG2-mediated targeting of the phosphatases phosphatase and tensin homolog and protein phosphatase 2A [14,15]. By regulating β-integrin-dependent cell adhesion to the matrix, TG2 also engages outside-in cellular signaling through activation of the focal adhesion kinase [9]. These processes contribute to increased cell survival under stress and increased cell invasiveness [14,16,17]. However, TG2 is also secreted [18] and its role in the tumor milieu remains unknown, being the subject of the current study.
In OC, the peritoneal space possesses unique characteristics that protect cancer cells, facilitating proliferation and dissemination. Our previous studies established that intracellular TG2 promotes OC metastasis through enhancement of cell adhesion to the matrix [8] and promotion of epithelial-to-mesenchymal transition (EMT) [17]. We have also shown that TG2 is secreted in malignant ascites [8]; however, the function of TG2 in the extracellular compartment was never investigated. By using an OC cell and xenograft model devoid of endogenous intracellular TG2 and recombinant catalytically active enzyme, we now show that TG2 functioning in the extracellular milieu enhances intraperitoneal (i.p.) metastasis. This was facilitated through activation of NF-κB by TG2 through an IκBα-independent mechanism. Activation of the NF-κB complex by extracellular TG2 contributed to loss of E-cadherin, increased cancer cell invasiveness, and up-regulation of the hyaluronan receptor CD44. Taken together, our findings point to a unique pathway, distinct from the mechanisms engaged by intracellular TG2, through which the extracellular compartment of the enzyme promotes OC peritoneal dissemination.
Materials and Methods
Cells
SKOV3 and OV90 cells were from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in growth media containing 1:1 MCDB 105 (Sigma, St Louis, MO) and M199 (Cellgro, Manassas, VA). A2780 cells (Sigma) were grown in Dulbecco's modified Eagle's medium (Sigma). All treatments with recombinant protein were carried out in the absence of FBS.
Expression and Purification of Human TG2
Full-length and catalytically inactive (C277A) TG2 were expressed in Escherichia coli and purified as described [6], with minor modifications (see Supplementary Materials). Purity of recombinant TG2 (rTG2) was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transamidation activity was assayed using a colorimetric kit (Sigma; No. CS1070). Purified proteins were endotoxin free, as measured by ToxinSensor Chromogenic LAL Endotoxin Assay Kit (GeneScript, Piscataway, NJ).
Immunohistochemistry
Ninety-three de-identified paraffin-embedded OC specimens from Pantomics Inc (Richmond, CA; tissue microarrays, n = 51) and from the Indiana University (IU) Tissue Bank (Indianapolis, IN; n = 42) were immunostained. Among the 42 specimens from the IU Tissue Bank, 14 specimens included paired primary tumor and metastatic peritoneal implants, with staining of the primary tumors being included in the primary analysis. A secondary analysis compared TG2 staining in primary versus metastatic implants among the 14 pairs available. Immunohistochemistry (IHC) used a TG2 monoclonal antibody (Neomarkers, Fremont, CA), at a dilution of 1:200 after sodium citrate antigen retrieval, as previously described [8]. Secondary labeling used Avidin/Biotin System (Dako, Carpinteria, CA). Negative controls omitting the primary antibody were run in parallel. Staining was graded from 0 (no staining) to 3+ (strong staining) by a board-certified pathologist (R.E.). Immunoreactivity was recorded if noted in more than 15% of tumor cells. The IU Institutional Review Board approved the use of de-identified human tissue specimens.
Malignant Ascites
Ten samples of OC ascites fluid cytologically positive and five samples of nonmalignant ascites (cirrhosis) were used.
Cell Transfections
Full-length TG2 subcloned in the retroviral vector pQCXIP was transduced into OV90 cells, as described [17]. Pooled, stable clones overexpressing TG2 were collected after puromycin selection. To stably knock down TG2, the antisense construct AS-TG2 subcloned into pcDNA3.1 was transfected into SKOV3 cells, as described [8]. Transient transfection of siRNA targeted RelB (Santa Cruz Biotechnology, Inc, Santa Cruz, CA) and CD44 (Dharmacon, Chicago, IL). Scrambled siRNA was used as control.
I.p. and Orthotopic Ovarian Xenograft Model
Female nu/nu mice (7 weeks old) were from Harlan (Indianapolis, IN). Studies were approved by the IU Institutional Animal Care and Use Committee (IACUC) being in compliance with federal regulations. For the i.p. xenograft model, 5 x 106 OV90 cells were injected i.p. Two experiments were carried out, n = 8 per group and n = 6 per group, with similar results. For the orthotopic ovarian model, ∼5 x 105 OV90 cells diluted in 5 µl of media were injected under the ovarian bursa (n = 4 per group), as described [17]. rTG2 or buffer (control) was delivered three times per week i.p. at a dose of 2.5 µg with the intent of reproducing concentrations of TG2 in the peritoneal space of ∼1 µg/ml. An orthotopic ovarian xenograft experiment was carried out using OV90 cells stably transduced with TG2 (n =8) or pQCXIP vector (n = 9). Five weeks after the i.p. or sub-bursal injection, mice were killed, tumors were measured, and tumor volume was calculated as length x width2/2. Miliary (disseminated) seeding was defined as presence of >10 nodules (yes/no), and peritoneal metastatic implants were counted.
Solid-phase Adhesion Assays
Cells were labeled with calcein (Molecular Probes, Eugene, OR) and seeded at a density of 4 x 104 cells into 96-well plates precoated with FN (5 µg/ml; Sigma), vitronectin (VN, 5 µg/ml; Sigma), collagen type 1 (10 µg/ml; Sigma), or BSA (1% wt/vol; Sigma). rTG2 was either added to serum-free culture media or immobilized directly to the FN matrix. rTG2 (20 µg/ml) immobilization to FN was performed as previously described [19]. All matrices were blocked with BSA (1%) for 1 hour before cell seeding. After 60 minutes, adherent cells were quantified in a fluorescence plate reader (Applied Biosystems, Grand Island, NY). Experiments were performed in quadruplicate and repeated at least twice. The neutralizing α5β1 antibody was from Millipore (Billerica, MA).
Matrigel Invasion Assay
Invasion assay was performed by using two-dimensional (2D) cell culture in a Matrigel matrix (BD Biosciences, Palo Alto, CA). Briefly, 2.5 x 104 cells treated or not treated with rTG2 (1 µg/ml) suspended in 50 µl of cell culture media were seeded onto solidified Matrigel in 24-well plates as a monolayer (2D model). After incubation at 37°C, invasive cells adhered to the surface of the gel and spread to form networks (2D model). These were observed under an inverted microscope and photographed live. The experiments were performed in duplicates and repeated in independent conditions.
Western Blot Analysis
Cells were lysed in RIPA buffer containing protease inhibitors. Nuclear and cytosolic fractions were separated by using NE-PER Nuclear and Cytoplasmic Extraction Reagents (NE-PER Kit; Pierce, Rockford, IL) as per the manufacturer's instructions. Equal amounts of protein were separated by SDS-PAGE and blotted onto polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were probed with primary antibodies followed by HRP-conjugated secondary antibodies. Primary antibodies identified TG2 (Neomarkers), glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Meridian Life Science, Cincinnati, OH), β-catenin (ECM Biosciences LLC, Versailles, KY), IκBα (Santa Cruz Biotechnology, Inc), phosphorylated IκB kinase α (pIKKα; Abcam, Cambridge, MA), E-cadherin, pP100, pP65 (Ser536), p100/p52, Skp2, CD44, and RelB (Cell Signaling Technology Inc, Danvers, MA). The antigen-antibody complexes were visualized using enhanced chemiluminescence substrate (ECL; Amersham Biosciences Corp, Piscataway, NJ). Densitometry analysis used Gel-Pro Analyzer 3.1. Western blots were repeated at least twice.
Immunofluorescence
OV90 cells treated with rTG2, mutant rTG2, or control were plated on FN-coated chamber slides (BD Biosciences). After fixation in 4% paraformaldehyde, cells were permeabilized using 0.2% Triton X-100 in phosphate-buffered saline (15 minutes) and blocked for 1 hour with 3% goat serum in phosphate-buffered saline. Subsequently, cells were incubated for 1 hour with E-cadherin antibody (1:200; Cell Signaling Technology) followed by a 1-hour incubation with Alexa Fluor 488 anti-rabbit secondary antibody (Molecular Probes). Isotype-specific IgG was negative control. Analysis used a Zeiss LSM 510 META confocal multiphoton microscope system under UV excitation.
Quantitative Reverse Transcription-Polymerase Chain Reaction
RNA extracted with RNA STAT-60 reagent (Tel-Test, Inc, Friends-wood, TX) from cells or tumors was reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). FastStart TaqMan Probe Master (Roche, Indianapolis, IN) was used for quantitative reverse transcription-polymerase chain reaction (qRT-PCR) on an ABI Prism 7900 platform (Applied Biosystems), using specific primers and probes (Table 1). Relative mRNA expression levels were normalized to GAPDH. Data analysis used the comparative Ct method [20].
Table 1.
qRT-PCR Primers and TaqMan Probes.
| Gene | 5′ Primer | 3′ Primer | Probe |
| E-cadherin | GCCGAGAGCTACACGTTCA | GACCGGTGCAATCTTCAAA | 80 |
| GAPDH | AGCCACATCGCTGAGACA | GCCCAATACGACCAAATCC | 60 |
| Zeb-1 | GCCAACAGACCAGACAGTGTT | TCTTGCCCTTCCTTTCCTG | 52 |
| Zeb-2 | AAGCCAGGGACAGATCAGC | CCACACTCTGTGCATTTGAACT | 68 |
| Snail | GCTGCAGGACTCTAATCCAGA | ATCTCCGGAGGTGGGATG | 11 |
| CD44 | CAACAACACAAATGGCTGGT | CTGAGGTGTCTGTCTCTTTCATCT | 40 |
Statistical Analysis
All values presented are means ± SE from duplicate (qRT-PCR, Western blot analysis) or triplicate (solid-phase adhesion) experiments. Comparisons between groups used the Student's t test, with P values < .05 being significant. IHC data were not normally distributed; therefore, correlation with histologic subtype, grade, and stage used the nonparametric Kruskal-Wallis test. Pairwise tests were done using exact Wilcoxon rank sum tests with P values adjusted by the Hochberg step-up method.
Results
TG2 Expression in OC
Previously, we demonstrated that TG2 is expressed in a cancer-restricted manner in ovarian tumors compared to normal surface ovarian epithelium [8]. Here, we expanded those initial findings to a larger cohort of ovarian tumors (n = 93) and analyzed TG2 expression relative to tumor stage, histologic grade, and type. The majority of samples in this cohort were serous papillary (n = 47) and of high grade (n = 67; Table 2). Both early (stages I–II, n = 50) and advanced stages (stages III–IV; n = 43) were analyzed. Most tumors displayed at least weak TG2 immunostaining (78 of 93), of which 68 (∼73%) stained 2+ or 3+ (Figure 1A). Four specimens labeled as undifferentiated were excluded from the correlation with histology. TG2 expression was significantly correlated with clear cell subtype versus others (P < .005) and with high-grade histology versus others (P < .005) but not with Federation of Obstetricians and Gynaecologists (FIGO) stage (Table 2).
Table 2.
TG2 Expression by IHC in Ovarian Tumor Specimens.
| Sample (n) | TG2 Expression | P Values | |||
| 0–1+ | 2+ | 3+ | |||
| Histologic type | |||||
| Clear Cell | 16 | 0 | 2 | 14 | P < .005 |
| Endometrioid | 18 | 5 | 7 | 6 | P = .43 |
| Mucinous | 8 | 2 | 6 | 0 | P = .43 |
| Serous | 47 | 15 | 9 | 23 | P = .43 |
| Poorly differentiated | 4 | 3 | 0 | 1 | Excluded |
| Surgical stage | |||||
| Stage I or II | 50 | 11 | 12 | 27 | P = .24 |
| Stage III or IV | 43 | 14 | 13 | 16 | P = .24 |
| Grade | |||||
| Grades 1 and 2 | 22 | 7 | 14 | 1 | P < .005 |
| Grade 3 | 67 | 16 | 10 | 41 | P < .005 |
| Not available | 4 | 2 | 0 | 2 | Excluded |
Figure 1.

TG2 is overexpressed in OC and secreted in malignant ascites. (A) Representative IHC for TG2; panel 1 (x100); panel 2 (x400); panels 3 to 6 are matched primary ovarian tumors (left panels) and peritoneal implants (right panels, x200, selected from n = 14 pairs). (B) TG2 in OC and cirrhosis ascites. Western blot analysis for TG2 used titration standards with purified TG2 (10 to 1000 ng per lane). Ten OC and five benign ascites specimens were loaded in equal volumes (30 µl). (C) Western blot analysis for TG2 in OC cell lines (OV90, A2780, SKOV3, SKOV3-pcDNA3.1, and SKOV3-AS-TG2).
TG2 Is Secreted in the Peritoneal Milieu
As OC disseminates in the peritoneal cavity, ascites containing proteins secreted by tumor and mesothelial cells is generated [21–23]. Previously, we detected abundant TG2 in malignant OC ascites compared to benign effusions [8]. Here, we estimated the concentration of secreted TG2 by immunoblot analysis of ascites specimens compared to known concentrations of purified protein. TG2 was detectable in all malignant samples (n = 10), at concentrations estimated between 0.1 and 10 µg/ml (Figure 1B and Table W1), but not in fluid from nonmalignant disease (n =5).
The objective of the subsequent studies was to investigate the role of secreted TG2 in the peritoneal (extracellular) space. For this, His6-tagged wild-type rTG2 and the mutant TG2 lacking enzymatic activity (C277A, mTG2) were induced and purified on talon and anion exchange columns, as previously described [24]. Protein purity was >90% (Figure W1A) and enzymatic activity was comparable to commercially available purified TG2, with the mutant TG2 lacking cross-linking activity (Figure W1B). A2780 and OV90 cells not expressing endogenously TG2 and SKOV3 cells stably transfected with an antisense TG2 construct or control vector [8] were used. TG2 expression level in these cells was measured by Western blot analysis (Figure 1C).
Extracellular TG2 Promotes Peritoneal Dissemination
To determine the role of secreted TG2 in metastasis, we used i.p. and orthotopic ovarian xenografts derived from OV90 cells. OV90 cells were injected i.p. in nude mice and rTG2 was administered i.p. three times per week at 2.5 µg per dose. Mice treated with rTG2 developed disseminated metastatic implants on the omentum, mesentery, and abdominal flanks compared to controls (Figure 2A). The average number of peritoneal implants in rTG2 and control-treated mice was 27.7 ± 5.2 versus 11.4 ± 3.4 (P < .017, n = 8 per group), but tumor volume was not different (412 mm3 vs 578 mm3, P = .38; Figure 2, B and C).
Figure 2.

rTG2 promotes i.p. metastasis of OC xenografts. (A) OV90 cells injected i.p. generated tumors studding the mesentery. I.p. administration of rTG2 increased peritoneal dissemination of OV90 cells in nude mice (right panel) compared to control-injected mice (left panel). Arrows point to tumor deposits on the mesentery. Average number of tumor implants in the peritoneal space 5 weeks after (B) i.p. (n = 8 mice per group) or (D) orthotopic ovarian (n = 4 mice per group) cancer cell implantation (P < .01). Average tumor volume (mm3) ± SE 5 weeks after (C) i.p. or (E) orthotopic ovarian cancer cell implantation. (F) Western blot analysis for TG2 in OV90 cells stably transduced with empty vector and wild-type TG2 (upper panel) or in media conditioned by these cells (lower panel). (G) Mean tumor volume ± SE generated from OV90-TG2 or control cells after orthotopic ovarian tumor implantation (n = 9 for control and n = 8 for OV90-TG2). (H) Number of mice with military (disseminated) metastases (>10 peritoneal implants; P = .02).
We next used orthotopic ovarian xenografts, with OV90 cells injected under the ovarian bursa and rTG2 administered i.p. The number of peritoneal implants was increased in rTG2-treated mice versus controls (31.7 vs 10.7, P = .002, n = 4 per group; Figure 2D), supporting that extra-cellular TG2 promotes tumor dissemination. The mean volume of primary tumors was not significantly different, although there was a trend in favor of rTG2-treated animals (730 mm3 vs 218.6 mm3, P = .07; Figure 2E).
These data are consistent with effects observed when TG2 was stably overexpressed in OV90 cells leading to its increased cellular expression and secretion in conditioned medium (CM; Figure 2F). TG2 and vector-transduced OV90 cells implanted orthotopically (n = 8 and 9, respectively) induced primary tumors of similar sizes (1825 mm3 vs 1609 mm3, P = .6; Figure 2G) but caused distinct patterns of peritoneal dissemination. Specifically, eight of eight mice injected with OV90-TG2 cells developed disseminated metastases (>10 implants) compared to two of nine mice injected with control cells (P = .002; Figure 2H), supporting the concept that increased cellular TG2 expression level determines metastatic dissemination.
Interestingly, overexpression of TG2 did not affect cellular proliferation as measured by bromodeoxyuridine (BrdU; Figure W2, A and B) or clonogenic assays (Figure W2C) under normal (10%) or low (1%) serum conditions, consistent with the observed lack of association between TG2 expression and primary tumor size. Likewise, extracellular TG2 did not affect OC cell proliferation. rTG2 added to low (1%) or normal (10%) serum containing culture media failed to increase OV90 cell proliferation (Figure W2, D and E).
Effects of Extracellular TG2 on Cell Adhesion to the Matrix
A necessary step in the establishment of peritoneal metastases is adhesion to the ECM. We measured the effects of rTG2 to cell adhesion to collagen, FN, and VN by using solid-phase adhesion assays. TG2 overexpression in OV90 cells increased adhesion to FN compared to control cells (Figure 3A; P < .05), consistent with previous findings that support TG2's role stabilizing the β1-integrin/FN complex [2,8,25]. However, rTG2 added to the culture media (extracellular) did not alter adhesion to VN, collagen, or FN (Figure 3, B-D). When cells were plated on FN surfaces pretreated with rTG2 (FN-rTG2), cell adhesion was increased by ∼20% compared to untreated FN (Figure 3E). Interestingly, addition of C277A mutant rTG2 to culture media or pretreatment of FN with enzymatically inactive rTG2 did not affect cell adhesion (Figure 3E), suggesting that catalytically active rTG2 is required for cell adhesion to FN-rTG2 matrix. Adhesion to TG2-coated FN matrix was blocked by preincubation of cells with an anti-α5β1-integrin antibody (Figure 3F), suggesting that it was mediated by β1-integrins. Collectively, these data suggest that rTG2 added to the CM does not measurably alter cell adhesion to the ECM but support the concept that cell adhesion is increased when cells are plated on an FN matrix premodified by enzymatically active TG2.
Figure 3.

Effects of TG2 on cell adhesion to ECM proteins. (A) Adhesion to FN (5 µg/ml) of OV90-TG2 or pQCXIP-transduced cells. (B) Adhesion of OV90 cells to VN-coated plates (5 µg/ml) in the presence of buffer or rTG2 (2.5 µg/ml) diluted in media. (C) Adhesion of OV90 cells to collagen type 1-coated plates (10 µg/ml) in the presence of buffer or rTG2 (2.5 µg/ml) diluted in media. (D) Adhesion of OV90 cells to FN-coated plates (5 µg/ml) in the presence of buffer or rTG2 (2.5 µg/ml) diluted in media. (E) Adhesion of OV90 cells to FN-coated plates (5 µg/ml) in the presence of rTG2 (1 µg/ml) or the enzymatically inactive C277A recombinant protein (1 µg/ml) either added to media or used to pretreat the FN matrix. (F) Adhesion of OV90 cells to FN-coated plates (5 µg/ml) pretreated or not with rTG2 (20 µg/ml) in the presence of α5β1-integrin neutralizing antibody (nAB, 1:100 dilution) or nonspecific IgG (control). Data represent average of four replicates ±SEM (*P < .05).
Extracellular TG2 Induced OC Cell Invasiveness and E-Cadherin Down-Regulation
To understand how extracellular TG2 affects metastasis, we measured its effects on cellular invasion, a necessary step for tumor dissemination. For this, OC cells were cultured in Matrigel and treated with rTG2 (72 hours). OV90 and A2780 that do not express TG2 grew in cohesive, noninvading clumps. Treatment with rTG2 induced formation of networks, indicative of an invasive phenotype (Figure 4A, lower panels). SKOV3 cells that express TG2 and stably transfected with vector or AS-TG2 [8,14] were used as a second model. Control cells formed networks, whereas AS-TG2 cells remained clumped. Treatment of AS-TG2 cells with rTG2 promoted invasion in Matrigel similar to the phenotype of control cells (Figure 4A). The invasive behavior corresponded to a mesenchymal phenotype appreciated by phase-contrast microscopy. A2780 and OV90 cells treated with rTG2 transitioned from epithelial, cobblestone to spindle-like morphology consistent with rTG2-induced EMT (Figure 4A, right panels). EMT was confirmed by quantifying epithelial and mesenchymal markers, E-cadherin, β-catenin, Snail-1, Zeb-1, and Zeb-2 at protein and mRNA levels (Figure 4B). Cells treated with rTG2 expressed decreased levels of E-cadherin and increased levels of β-catenin and transcriptional repressor for E-cadherin (Zeb-1, Zeb-2, Snail). Treatment with rTG2 diminished E-cadherin expression in OV90 cells, as visualized by immunofluorescence (Figure 4C). However, treatment with the mutant C277A recombinant protein did not affect E-cadherin expression (Figure 4C), suggesting that the enzymatic activity of TG2 is necessary for the induction of the mesenchymal phenotype.
Figure 4.

Extracellular TG2 induces cell invasion and EMT. (A) Cell invasion in Matrigel using 2D cultures with A2780, OV90, and SKOV3/pcDNA3.1 (control) or SKOV3/ASTG2 cells treated with buffer (upper panels, left) or rTG2 (1 µg/ml; lower panels, left) for 72 hours; original magnification, x100. Phase-contrast microscopy demonstrates cell morphology for A2780 and OV90 cells treated with control or with rTG2 (1 µg/ml) for 7 days (original magnification, x200; right panels). (B) Western blot analysis for E-cadherin and β-catenin for control and rTG2 (1 µg/ml)-treated OV90 cells (72-hour treatment; left panel). Densitometry quantifies protein expression relative to GAPDH. qRT-PCR quantifies expression levels for E-cadherin, Snail-1, Zeb-1, and Zeb-2 in OV90 cells treated with control and rTG2 (1 µg/ml) for 72 hours (right panel). Y axis represents fold change in gene expression levels normalized to GAPDH of rTG2-treated OV90 cells compared with control (*P < .05). (C) Immunofluorescence staining for E-cadherin of OV90 cells treated with control, rTG2, or mutant rTG2 (1 µg/ml) for 72 hours (x600).
Extracellular TG2 Activates the NF-κB Complex through a Noncanonical Pathway
As NF-κB activation is a known inducer of EMT in TG2-expressing cells [17,26], we measured whether rTG2 also activates the complex. It is known that intracellular TG2 promotes degradation of IκBα, causing activation of NF-κB through a canonical pathway [7]. In contrast, extracellular TG2 did not measurably alter IκBα levels (Figure 5A). However, rTG2 stimulation of OV90 cells caused phosphorylation of IKKα and p100 (Figure 5B). This corresponded to an increase in p52, the transcriptionally active degradation fragment of p100 (Figure 5B). To understand whether the TG2 enzymatic function is important, active and catalytically inactive rTG2 were used. Induction of p52 expression and phosphorylation of p65 and p100 were induced by wild-type TG2 but not by the C277A mutant (Figure 5C), suggesting that TG2 enzymatic activity in the extracellular space is necessary. Similar processing to p52 and phosphorylation of p65 were observed in TG2-expressing SKOV3 cells and TG2-null A2780 cells stimulated with rTG2 (Figure 5D), suggesting that the observed effects are not cell type specific.
Figure 5.

Extracellular TG2 activates noncanonical NF-κB signaling. (A) Western blot analysis for IκBα (upper panel) in OV90 cells treated with rTG2 (1 µg/ml) for 15 to 45 minutes, under serum starvation. Stimulation with tumor necrosis factor-α (10 ng/ml) was used as a positive control. (B) Western blot analysis for phospho-IκKα, phospho-p100, p100, and p52 in OV90 cells treated with rTG2 (1 µg/ml) or BSA (control, 1 µg/ml). (C) Western blot analysis for phospho-p100, phospho-p65, p100, and p52 in OV90 cells treated with rTG2 (1 µg/ml) or catalytically inactive C277A rTG2 (1 µg/ml); line 4, untreated control. (D) Western blot analysis for phospho-p100 and phosphop65 and for p100 and p52 in SKOV3 (left panel) and A2780 (right panel) cells treated with rTG2 (1 µg/ml) for up to 6 hours. (E) Western blot analysis for RelB in whole-cell lysates from OV90 (left panel) and SKOV3 cells (middle panel) or in nuclear extracts from OV90 cells (right panel) treated with rTG2 (1 µg/ml) or control. Densitometry quantifies protein expression relative to GAPDH.
The final step in noncanonical NF-κB signaling is the formation of the p52/RelB complex that translocates to the nucleus and activates gene transcription. Treatment of OV90 and SKOV3 cells with rTG2 caused increased RelB protein expression (Figure 5E, left and middle panels), as well as RelB translocation to the nucleus (Figure 5E, right panel).
Extracellular TG2 Downregulates E-cadherin through RelB
To assess the effects of TG2-induced NF-κB activation, we focused on two transcripts, CD44 and Skp-2, known to be inducible by p52/RelB [27,28]. Both Skp-2 (Figure 6A) and CD44 (Figure 6B, left panel) were induced by rTG2 in OV90 cells. CD44 induction by rTG2 was also demonstrated in SKOV3 and A2780 cells (Figure 6B, middle and right panels).
Figure 6.

Extracellular TG2 activates noncanonical NF-κB signaling in OC cells. Western blot analysis for Skp2 (A) or CD44 (B) in OV90, SKOV3, and A2780 cells treated with rTG2 (1 µg/ml). Densitometry quantifies protein expression relative to GAPDH. (C) Western blot analysis for RelB in SKOV3 cells untransfected (UT) and transiently transfected with siRNA control or targeting RelB (left panel). qRT-PCR quantifies E-cadherin (middle panel) or CD44 expression (right panel) in SKOV3 cells transiently transfected with siRNA control (si Ctl) or targeting RelB and treated with rTG2 (1 µg/ml) or control for 18 and 72 hours. Y axis represents fold change in E-cadherin (middle panel) or CD44 levels normalized to GAPDH in rTG2-treated cells compared with controls (right panel); *P < .05. (D) Western blot analysis for CD44 in SKOV3 cells transiently transfected with siRNA control or targeting CD44 (left panel). qRT-PCR quantifies E-cadherin expression in SKOV3 cells transiently transfected with siRNA control or targeting CD44 (right panel) and treated with rTG2 for 72 hours. Y axis represents fold change in E-cadherin level normalized to GAPDH; *P < .05; NS, not significant. (E) qRT-PCR quantifies expression levels for CD44 normalized to GAPDH in OV90 xenografts from animals treated with control and rTG2 (n = 3 per group, right panel). Y axis represents fold change in CD44 expression; *P < .05.
To demonstrate that RelB is involved in rTG2-induced E-cadherin repression and CD44 up-regulation, RelB was knocked down by siRNA (Figure 6C, left panel). R-TG2 inhibited E-cadherin expression in SKOV3 cells transfected with scrambled siRNA (control) but not in cells transfected with RelB siRNA (Figure 6C, middle panel). RelB knockdown also repressed basal levels of E-cadherin (Figure 6C, middle panel). R-TG2 augmented CD44 expression in SKOV3 cells transfected with scrambled siRNA (control) but not in cells in which RelB was knocked down (Figure 6C, right panel). Collectively, the data demonstrate that regulation of E-cadherin and CD44 expression by extracellular TG2 depends on RelB signaling.
As CD44 functions have been linked to EMT in other models [29,30], we measured whether CD44 knockdown in OC cells altered E-cadherin expression. CD44 was knocked down by siRNA (Figure 6D, left panel), and E-cadherin expression in response to rTG2 was measured by qRT-PCR. We observed increased E-cadherin expression in cells in which CD44 was silenced compared to siRNA control-transfected cells (Figure 6D, right). Furthermore, rTG2-induced E-cadherin down-regulation was prevented by CD44-targeted siRNA, supporting that CD44 is a necessary element in this process.
To verify the functional significance of rTG2-induced CD44 in vivo, the expression of the receptor was quantified by qRT-PCR in xenografts. More than two-fold increased CD44 mRNA levels were observed in rTG2-treated tumors compared to controls, suggesting that the pathway is relevant to induction of metastasis (Figure 6E). A model illustrating the distinct pathways activated by extracellular (shown here) and intracellular TG2 (described previously and converging on Zeb-1, [17]) to promote metastasis is presented in Figure 7.
Figure 7.
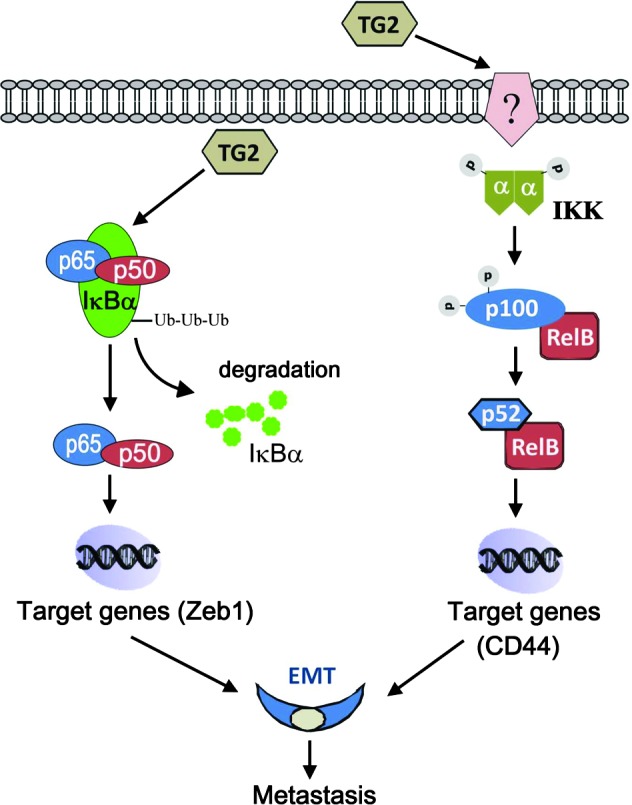
Proposed model for the mechanisms by which TG2 activates NF-κB in the extracellular and intracellular space.
Discussion
This is the first study analyzing the effects of extracellular TG2 on the phenotype of OC cells in vitro and tumor formation and metastasis in vivo. On the basis of clinical specimen-derived observations that TG2 is secreted in the peritoneal milieu, our study demonstrates that extracellular TG2 induces EMT and cancer cell invasiveness and promotes peritoneal metastasis through a distinct pathway leading to noncanonical activation of NF-κB.
Our findings that TG2 is expressed in more than 70% of ovarian tumors are consistent with previous reports [8,31]. We also noted that TG2 expression was increased in highly aggressive cancers (e.g., grade 3 and clear cell tumors), a novel observation. Although TG2 expression did not correlate with surgical stage, in some cases we observed increased TG2 expression in metastatic implants compared to primary tumors. Specifically, for 5 of 14 matched primary serous ovarian tumors and omental metastases, we observed increased TG2 staining in the metastatic implants compared to the primary tumors. In these implants, TG2 immunostaining was more intense at the interface with stroma, suggesting that the enzyme may be overexpressed at the invasive front of tumors.
TG2 secretion in the ECM has remained an enigma, as the enzyme lacks a signal recognition peptide that would target it to the endoplasmic reticulum. A recent study demonstrated that TG2 secretion follows an unconventional pathway through recycling endosomes where TG2 is packaged with internalized β1-integrins [32]. Indeed, in this and in previous studies we detected abundant TG2 in CM and in malignant ascites [8,14]. To demonstrate the distinct effects of extracellular TG2, we used an OC cell model devoid of endogenous TG2 and recombinant wild-type or mutant TG2. This model allowed eliminating undue effects caused by the cellular compartment of the enzyme. We targeted a concentration of extracellular TG2 of ∼1 µg/ml, based on estimated TG2 concentrations in malignant ascites of ∼0.1 to 10 µg/ml, but we recognize high variability of TG2 secretion in the peritoneal milieu. In a previous study, direct intratumoral injection of TG2 at high concentration (200 µg) inhibited tumor growth [33]. Those effects were attributed to TG2-induced alterations in the ECM and, specifically, to increased deposition of collagen-1 [33]. Here, we did not observe a significant effect of extracellular TG2 on tumor size or OC cell proliferation but recorded increased number of metastases, an end point previously not measured. We attribute the pro-metastatic effects to the lower dose of TG2 used in our model. TG2 dose dependence has been previously demonstrated in a wound healing model where angiogenesis was stimulated by low-dose TG2 (10–20 µg) [34] and inhibited by higher concentrations (200 µg) [33].
The use of both i.p. and orthotopic ovarian xenograft models strongly supports the effects of secreted TG2 on peritoneal dissemination. The orthotopic approach better mimics the phenomena of primary tumor formation and tumor spreading initiated from the primary site compared to the i.p. model [35–37]. The demonstration that in vitro rTG2 facilitates EMT and promotes cell invasion provides further support to this concept. A limitation of our in vivo model is that it does not completely eliminate the influence of host TG2, which is present in small amounts in mesothelial cells lining the peritoneal space. Interestingly, a previous study suggested that subcutaneous tumor growth is enhanced in a TG2-/- background [33]; however, the mechanism involved remained unclear. It is known that inflammatory responses are altered in TG2-/- animals, which display deficient transforming growth factor-β, interleukin-12 (IL-12), and macrophage response [38]. That deregulated immune response could contribute to altered immune tolerance, which may promote tumor formation.
We previously showed that cellular TG2 enhances metastasis and induces EMT [17,39]. The current observations highlight a new pathway through which extracellular TG2 activates NF-κB, induces CD44, and promotes EMT and metastasis. We demonstrate that the two TG2 compartments have distinct effects on the NF-κB complex. While intracellular TG2 activates NF-κB through an IκBα degradation-dependent mechanism [7], extracellular TG2 activates IKKα leading to induction of gene transcription through the p52/RelB complex [40]. Furthermore, by demonstrating that the catalytically inactive enzyme fails to induce phosphorylation of IKKα, p65, or p100, we show that the enzymatic function of TG2 is important in this process. This is particularly important as TGase activity is enhanced in the extracellular milieu, where Ca2+ is abundant and GTP concentrations are low.
Remaining yet undefined is how extracellular TG2 initiates non-canonical NF-κB signaling. The engagement of a limited subset of tumor necrosis factor superfamily receptors has been implicated in stabilization of NF-κB inducing kinase (NIK), which activates the non-canonical pathway [41]. These receptors include CD40, RANK, Fn14, B-cell activating factor receptor, and lymphotoxin β receptor and are relevant to B- and dendritic cells' development [40]. Recent reports suggest that this pathway is also activated in osteoclasts, mammary glands, and epithelial cancer cells. Overexpression of Fn14, the receptor for TNF-related weak inducer of apoptosis (TWEAK), has been reported in aggressive breast cancer, glioblastoma, and other cancers [42,43]. Future work will attempt to dissect whether extracellular TG2 engages noncanonical NF-κB signaling directly or indirectly.
Although we had first hypothesized that extracellular TG2 might facilitate cell adhesion to the ECM, based on the knowledge that membrane-bound TG2 stabilizes the β1-integrin/FN complex [2,8,25], our current findings support only a limited role in this process. OC cell adhesion to FN, VN, or collagen was not enhanced by rTG2 in the media. However, OC cells' adhesion to a TG2-primed FN matrix was increased through an α5β1-integrin-dependent mechanism, an observation consistent with previous findings implicating syndecan 4 and β1-integrin co-signaling in adhesion to TG2-bound FN [42,43]. Therefore, our study suggests that in the peritoneal milieu, TG2 crosslinked matrix may provide a more suitable substrate for OC cells. Additionally, we found that extracellular TG2 robustly induces CD44 expression in OC cells and xenografts through an RelB-dependent mechanism. As CD44 mediates cell adhesion to the mesothelium [44], it is possible that by upregulating this receptor, extracellular TG2 could also positively alter cancer cells' interaction with the peritoneal surface. By interacting with cytoskeletal proteins, CD44 has also been linked to EMT [29,30] and here we show that CD44 induction by rTG2 is involved in NF-κB-mediated EMT.
In summary, our in vitro and in vivo data support that extracellular TG2 promotes tumor metastasis by increasing adhesion to a TG2-primed matrix and by activating noncanonical NF-κB, which in turn promotes EMT and cell invasiveness. A model supporting the functions of TG2 in the intracellular and extracellular space is presented in Figure 7. Taken together, our data support that TG2 acts as a metastasis enhancer at the cancer cell-matrix interface.
Supplementary Materials
Materials and Methods
Expression and purification of human TG2. Full-length and catalytically inactive (C277A) TG2 were expressed in E. coli and purified as described [6], with minor modifications. Briefly, overnight cultures from E. coli BL21 cells (Invitrogen, Grand Island, NY) transformed with pET28-TG2 expression vector (gift from Dr E. A. Cerione) were grown at 37°C. Protein expression was induced with 300 µM isopropylthio-β-galactoside (IPTG) for 24 hours at 25°C. Cell pellets were lysed by sonication at 0°C in lysis buffer [50 mM Na2HPO4 (pH 7.5), 400 mM NaCl, 5 mM benzamidine, and 5 mM 2-mercaptoethanol] containing 50 µM GTP, 50 µMadenosine-5′-triphosphate (ATP), and 50 µg/ml phenylmethanesulfonyl fluoride (PMSF). After sonication, NP-40 was added to a concentration of 0.5% (vol/vol). Cell debris was removed by high-speed centrifugation and the supernatant was mixed with Talon metal affinity resins (Clontech, Mountain View, CA). The His6-TG2 fusion protein was eluted with 50 mM Hepes (pH 7.0), 50 mM NaCl, 5 mM 2-mercaptoethanol, 20 µM guanosine diphosphate (GDP), and 160 mM imidazole. The eluted protein was dialyzed against 50 mM MES (pH 6.5), 50 mM NaCl, 10% (vol/vol) glycerol, 1 mM EDTA, and 5 mM DTT and loaded onto a MonoQ anion exchange column (Clontech). rTG2 was eluted by using a gradient of 50 to 450 mM NaCl in MES buffer. The fractions containing rTG2 were pooled and concentrated. Purity of rTG2 was confirmed by SDS-PAGE, and transamidation activity was assayed using a colorimetric kit (Sigma; No. CS1070), according to the manufacturer's instructions.
Cell proliferation. Cells were seeded in 96-well plates at a density of 2 x 104 cells/well and allowed to proliferate from 1 to 7 days in medium containing 1% or 10% FBS. Cell proliferation was measured by using the BrdU proliferation assay (Roche Molecular Biochemicals, Mannheim, Germany) following the manufacturer's directions. Absorbance values were read using the Ultra Multifunctional Microplate Reader (Tecan, Durham, NC). Data shown are means ± SE of four replicates.
Clonogenic assay. Cells were seeded at a concentration of 100 cells/six-well plate and allowed to proliferate in medium containing 1% or 10% FBS over a 2-week period. Colonies were fixed with methanol-acetic acid (3:1), stained with Hema3 (Fisher Diagnostics, Middle-town, VA), and counted. Data represent means ± SE of three replicates.
Results
Acknowledgments
We thank Susan Perkins for assistance with statistical analyses and Kenneth Nephew, Harikrishna Nakshatri, and Mircea Ivan for helpful comments.
Footnotes
This work was supported by the American Cancer Society through a Research Scholar grant to D.M. and by the US Department of Veterans Affairs through a Merit Award to D.M. Conflict of interest: None.
This article refers to supplementary materials, which are designated by Table W1 and Figures W1 and W2 and are available online at www.neoplasia.com.
References
- 1.Pincus JH, Waelsch H. The specificity of transglutaminase. II. Structural requirements of the amine substrate. Arch Biochem Biophys. 1968;126:44–52. doi: 10.1016/0003-9861(68)90557-2. [DOI] [PubMed] [Google Scholar]
- 2.Akimov SS, Belkin AM. Cell surface tissue transglutaminase is involved in adhesion and migration of monocytic cells on fibronectin. Blood. 2001;98:1567–1576. doi: 10.1182/blood.v98.5.1567. [DOI] [PubMed] [Google Scholar]
- 3.Chen S, Lin F, Iismaa S, Lee KN, Birckbichler PJ, Graham RM. α1-Adrenergic receptor signaling via Gh is subtype specific and independent of its transglutaminase activity. J Biol Chem. 1996;271:32385–32391. doi: 10.1074/jbc.271.50.32385. [DOI] [PubMed] [Google Scholar]
- 4.Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001;276:20673–20678. doi: 10.1074/jbc.M010846200. [DOI] [PubMed] [Google Scholar]
- 5.Fesus L, Piacentini M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci. 2002;27:534–539. doi: 10.1016/s0968-0004(02)02182-5. [DOI] [PubMed] [Google Scholar]
- 6.Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci USA. 2002;99:2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mann AP, Verma A, Sethi G, Manavathi B, Wang H, Fok JY, Kunnumakkara AB, Kumar R, Aggarwal BB, Mehta K. Overexpression of tissue transglutaminase leads to constitutive activation of nuclear factor-κB in cancer cells: delineation of a novel pathway. Cancer Res. 2006;66:8788–8795. doi: 10.1158/0008-5472.CAN-06-1457. [DOI] [PubMed] [Google Scholar]
- 8.Satpathy M, Cao L, Pincheira R, Emerson R, Bigsby R, Nakshatri H, Matei D. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res. 2007;67:7194–7202. doi: 10.1158/0008-5472.CAN-07-0307. [DOI] [PubMed] [Google Scholar]
- 9.Verma A, Wang H, Manavathi B, Fok JY, Mann AP, Kumar R, Mehta K. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res. 2006;66:10525–10533. doi: 10.1158/0008-5472.CAN-06-2387. [DOI] [PubMed] [Google Scholar]
- 10.Antonyak MA, Miller AM, Jansen JM, Boehm JE, Balkman CE, Wakshlag JJ, Page RL, Cerione RA. Augmentation of tissue transglutaminase expression and activation by epidermal growth factor inhibit doxorubicin-induced apoptosis in human breast cancer cells. J Biol Chem. 2004;279:41461–41467. doi: 10.1074/jbc.M404976200. [DOI] [PubMed] [Google Scholar]
- 11.Cao L, Petrusca DN, Satpathy M, Nakshatri H, Petrache I, Matei D. Tissue transglutaminase protects epithelial ovarian cancer cells from cisplatin-induced apoptosis by promoting cell survival signaling. Carcinogenesis. 2008;29:1893–1900. doi: 10.1093/carcin/bgn158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao L, Shao M, Schilder J, Guise T, Mohammad KS, Matei D. Tissue transglutaminase links TGF-β, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene. 2012;31:2521–2534. doi: 10.1038/onc.2011.429. [DOI] [PubMed] [Google Scholar]
- 13.Verma A, Mehta K. Transglutaminase-mediated activation of nuclear transcription factor-κB in cancer cells: a new therapeutic opportunity. Curr Cancer Drug Targets. 2007;7:559–565. doi: 10.2174/156800907781662275. [DOI] [PubMed] [Google Scholar]
- 14.Satpathy M, Shao M, Emerson R, Donner DB, Matei D. Tissue transglutaminase regulates matrix metalloproteinase-2 in ovarian cancer by modulating cAMP-response element-binding protein activity. J Biol Chem. 2009;284:15390–15399. doi: 10.1074/jbc.M808331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verma A, Guha S, Wang H, Fok JY, Koul D, Abbruzzese J, Mehta K. Tissue transglutaminase regulates focal adhesion kinase/AKT activation by modulating PTEN expression in pancreatic cancer cells. Clin Cancer Res. 2008;14:1997–2005. doi: 10.1158/1078-0432.CCR-07-1533. [DOI] [PubMed] [Google Scholar]
- 16.Mehta K, Fok J, Miller FR, Koul D, Sahin AA. Prognostic significance of tissue transglutaminase in drug resistant and metastatic breast cancer. Clin Cancer Res. 2004;10:8068–8076. doi: 10.1158/1078-0432.CCR-04-1107. [DOI] [PubMed] [Google Scholar]
- 17.Shao M, Cao L, Shen C, Satpathy M, Chelladurai B, Bigsby RM, Nakshatri H, Matei D. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009;69:9192–9201. doi: 10.1158/0008-5472.CAN-09-1257. [DOI] [PubMed] [Google Scholar]
- 18.Kotsakis P, Griffin M. Tissue transglutaminase in tumour progression: friend or foe? Amino Acids. 2007;33:373–384. doi: 10.1007/s00726-007-0516-1. [DOI] [PubMed] [Google Scholar]
- 19.Verderio EA, Telci D, Okoye A, Melino G, Griffin M. A novel RGD-independent cell adhesion pathway mediated by fibronectin-bound tissue transglutaminase rescues cells from anoikis. J Biol Chem. 2003;278:42604–42614. doi: 10.1074/jbc.M303303200. [DOI] [PubMed] [Google Scholar]
- 20.Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270:41–49. doi: 10.1006/abio.1999.4085. [DOI] [PubMed] [Google Scholar]
- 21.Graves LE, Ariztia EV, Navari JR, Matzel HJ, Stack MS, Fishman DA. Proinvasive properties of ovarian cancer ascites-derived membrane vesicles. Cancer Res. 2004;64:7045–7049. doi: 10.1158/0008-5472.CAN-04-1800. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Gaudette DC, Boynton JD, Frankel A, Fang XJ, Sharma A, Hurteau J, Casey G, Goodbody A, Mellors A, et al. Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin Cancer Res. 1995;1:1223–1232. [PubMed] [Google Scholar]
- 23.Xu Y, Shen Z, Wiper DW, Wu M, Morton RE, Elson P, Kennedy AW, Belinson J, Markman M, Casey G. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA. 1998;280:719–723. doi: 10.1001/jama.280.8.719. [DOI] [PubMed] [Google Scholar]
- 24.Antonyak MA, Jansen JM, Miller AM, Ly TK, Endo M, Cerione RA. Two isoforms of tissue transglutaminase mediate opposing cellular fates. Proc Natl Acad Sci USA. 2006;103:18609–18614. doi: 10.1073/pnas.0604844103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol. 2000;148:825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar A, Xu J, Brady S, Gao H, Yu D, Reuben J, Mehta K. Tissue transglutaminase promotes drug resistance and invasion by inducing mesenchymal transition in mammary epithelial cells. PLoS One. 2010;5:e13390. doi: 10.1371/journal.pone.0013390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barre B, Perkins ND. The Skp2 promoter integrates signaling through the NF-κB, p53, and Akt/GSK3β pathways to regulate autophagy and apoptosis. Mol Cell. 2010;38:524–538. doi: 10.1016/j.molcel.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 28.Damm S, Koefinger P, Stefan M, Wels C, Mehes G, Richtig E, Kerl H, Otte M, Schaider H. HGF-promoted motility in primary human melanocytes depends on CD44v6 regulated via NF-kappa B, Egr-1, and C/EBP-beta. J Invest Dermatol. 2010;130:1893–1903. doi: 10.1038/jid.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, Cheng C. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121:1064–1074. doi: 10.1172/JCI44540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi E, Nagano O, Ishimoto T, Yae T, Suzuki Y, Shinoda T, Nakamura S, Niwa S, Ikeda S, Koga H, et al. Tumor necrosis factor-α regulates transforming growth factor-β-dependent epithelial-mesenchymal transition by promoting hyaluronan-CD44-moesin interaction. J Biol Chem. 2010;285:4060–4073. doi: 10.1074/jbc.M109.056523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang JY, Mangala LS, Fok JY, Lin YG, Merritt WM, Spannuth WA, Nick AM, Fiterman DJ, Vivas-Mejia PE, Deavers MT, et al. Clinical and biological significance of tissue transglutaminase in ovarian carcinoma. Cancer Res. 2008;68:5849–5858. doi: 10.1158/0008-5472.CAN-07-6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zemskov EA, Mikhailenko I, Hsia RC, Zaritskaya L, Belkin AM. Unconventional secretion of tissue transglutaminase involves phospholipid-dependent delivery into recycling endosomes. PLoS One. 2011;6:e19414. doi: 10.1371/journal.pone.0019414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones RA, Kotsakis P, Johnson TS, Chau DY, Ali S, Melino G, Griffin M. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006;13:1442–1453. doi: 10.1038/sj.cdd.4401816. [DOI] [PubMed] [Google Scholar]
- 34.Haroon ZA, Hettasch JM, Lai TS, Dewhirst MW, Greenberg CS. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999;13:1787–1795. doi: 10.1096/fasebj.13.13.1787. [DOI] [PubMed] [Google Scholar]
- 35.Sengupta S, Kim KS, Berk MP, Oates R, Escobar P, Belinson J, Li W, Lindner DJ, Williams B, Xu Y. Lysophosphatidic acid downregulates tissue inhibitor of metalloproteinases, which are negatively involved in lysophosphatidic acid-induced cell invasion. Oncogene. 2007;26:2894–2901. doi: 10.1038/sj.onc.1210093. [DOI] [PubMed] [Google Scholar]
- 36.Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther. 2004;10:1032–1042. doi: 10.1016/j.ymthe.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 37.Fu X, Hoffman RM. Human ovarian carcinoma metastatic models constructed in nude mice by orthotopic transplantation of histologically-intact patient specimens. Anticancer Res. 1993;13:283–286. [PubMed] [Google Scholar]
- 38.Falasca L, Iadevaia V, Ciccosanti F, Melino G, Serafino A, Piacentini M. Transglutaminase type II is a key element in the regulation of the anti-inflammatory response elicited by apoptotic cell engulfment. J Immunol. 2005;174:7330–7340. doi: 10.4049/jimmunol.174.11.7330. [DOI] [PubMed] [Google Scholar]
- 39.Kumar A, Gao H, Xu J, Reuben J, Yu D, Mehta K. Evidence that aberrant expression of tissue transglutaminase promotes stem cell characteristics in mammary epithelial cells. PLoS One. 2011;6:e20701. doi: 10.1371/journal.pone.0020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Razani B, Reichardt AD, Cheng G. Non-canonical NF-κB signaling activation and regulation: principles and perspectives. Immunol Rev. 2011;244:44–54. doi: 10.1111/j.1600-065X.2011.01059.x. [DOI] [PubMed] [Google Scholar]
- 41.Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 42.Huang M, Narita S, Tsuchiya N, Ma Z, Numakura K, Obara T, Tsuruta H, Saito M, Inoue T, Horikawa Y, et al. Overexpression of Fn14 promotes androgen-independent prostate cancer progression through MMP-9 and correlates with poor treatment outcome. Carcinogenesis. 2011;32:1589–1596. doi: 10.1093/carcin/bgr182. [DOI] [PubMed] [Google Scholar]
- 43.Zhou H, Marks JW, Hittelman WN, Yagita H, Cheung LH, Rosenblum MG, Winkles JA. Development and characterization of a potent immunoconjugate targeting the Fn14 receptor on solid tumor cells. Mol Cancer Ther. 2011;10:1276–1288. doi: 10.1158/1535-7163.MCT-11-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cannistra SA, Kansas GS, Niloff J, DeFranzo B, Kim Y, Ottensmeier C. Binding of ovarian cancer cells to peritoneal mesothelium in vitro is partly mediated by CD44H. Cancer Res. 1993;53:3830–3838. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.