

Abstract
Despite remarkable advances in human genetics and other genetic model systems, the fruit fly, Drosophila melanogaster, remains a powerful experimental tool to probe with ease the inner workings of a myriad of biological and pathological processes, even when evolutionary forces impart apparent divergences to some of such processes. The understanding of such evolutionary differences provides mechanistic insights into genotype-phenotype correlations underpinning biological processes across metazoans. The pioneering work developed by the William Pak laboratory for the past four decades, and the work of others, epitomize the notion of how the Drosophila system breaks new fertile ground or complements research fields of high scientific and medical relevance. Among the three major genetic complementation groups produced by the Pak's laboratory and impairing distinct facets of photoreceptor neuronal function, the nina group (ninaA….J) selectively affects the biogenesis of G protein-coupled receptors (GPCR) mediating the photoconversion and transduction of light-stimuli. Among the nina genes identified, ninaA arguably assumes heightened significance for several reasons. First, it presents unique physiological selectivity toward the biogenesis of a subset of GPCRs, a standalone biological manifestation yet to be discerned for most mammalian homologues of NinaA. Second, NinaA belongs to a family of proteins, immunophilins, which are the primary targets for immunosuppressive drugs at the therapeutic forefront of a multitude of medical conditions. Third, NinaA closest homologue, cyclophilin-B (CyPB/PPIB), is an immunophilin whose loss-of-function was found recently to cause osteogenesis imperfecta in the human. This report highlights advances made by studies on some members of immunophilins, the cyclophilins. Finally, it re-examines critically data and dogmas derived from past and recent genetic, structural, biological and pathological studies on NinaA and few other cyclophilins that support some of such paradigms to be less than definite and advance our understanding of cyclophilins' roles in cell function, disease and therapeutic interventions.
Keywords: ninaA, cyclophilin, chaperone, proly isomerase, cyclosporine A (CsA), immunophilin, protein biogenesis
Historical overview of ninaA gene
The application of forward genetics screens pioneered by the William Pak laboratory identified three major genetic complementation groups affecting the function of photoreceptor neurons. Each of these groups comprised genes affecting the phototransduction (light-signaling cascade), the biogenesis of the G protein-coupled receptors (GPCRs), opsins, which initiate visual transduction upon photoconversion of light-stimuli, or synaptic transmission (Pak, 1995). The second group was designated nina (nor inactivation nor afterpotential), because electroretinogram (ERG) recordings from the compound eye presented reduced prolonged depolarization afterpotential (PDA) upon a light stimulus (Larrivee et al., 1981; Pak, 1979; Stephenson et al., 1983). The amplitude of PDA is closely proportional to the amount of rhodopsin available for photoconversion (Hamdorf and Razmjoo, 1979; Minke, 1986). Hence, mutants with low rhodopsin content present underdeveloped or absent PDAs. Analogous to the vertebrate visual system, the Drosophila visual system is comprised of four classes of photoreceptors: R1–6, R7, R8 and ocelli (simple eyes) (Zuker, 1996). Each of these classes of photoreceptors expresses evolutionary conserved and homologous opsins, Rh1, Rh2, Rh3, Rh4 Rh5 and Rh6 (Papatsenko et al., 1997). These homologous GPCRs impart distinct spectral tuning properties to rhodopsin photoconversion and activation of the visual transduction cascade (Britt et al., 1993). A member of the nina group, ninaA, was initially found to impair selectively R1–6, but not R7 photoreceptors of Drosophila (Larrivee et al., 1981; Stephenson et al., 1983). Subsequently, work by Stamnes et al. showed that ninaA impairs also the production of functional Rh2 opsin expressed only in photoreceptors of the ocelli (simple eyes) (Stamnes et al., 1991). Collectively, genetic, electrophysiological, molecular and cellular evidences prior and after ninaA identification support that mutations in ninaA causes the selective impairment of the production of Rh1 and Rh2 opsins, but not of other opsins, even though NinaA is expressed across all photoreceptor neurons. Hence, NinaA presents physiological substrate specificity toward a subset of opsins.
The cloning of ninaA showed that it encodes a cyclophilin, comprising an N-terminal cleavable signal sequence, a cyclophilin domain, and a C-terminal transmembrane domain followed by a short cytoplasmic tail (Schneuwly et al., 1989; Shieh et al., 1989). NinaA was shown to be an integral membrane protein associated with the endoplasmic reticulum and secretory vesicles, where it colocalizes and associates stably with Rh1 opsin (Baker et al., 1994; Colley et al., 1991; Stamnes et al., 1991). Mutations in ninaA, such as those leading to the truncation of its transmembrane domain, promote the prominent accumulation of Rh1 opsin in the ER lumen, thus supporting that its localization and anchoring to the ER membrane is crucial to its function (Colley et al., 1991). Finally, NinaA exhibits strong homology to other cyclophilins, which present peptidyl-prolyl cis-trans isomerase (PPIase) activity (Fischer et al., 1984; Fischer et al., 1989; Takahashi et al., 1989), a non-covalent conversion implicated in a rate-limiting step of protein folding (Jakob and Schmid, 2009). All these observations provided strength to the notion that NinaA acts as a chaperone and/or foldase (rotamase) during Rh1/Rh2 biogenesis (e.g. co-translational insertion into the ER, chromophore binding, and/or during its vesicular sorting to the plasma membrane of fly photoreceptors). However, direct evidence that NinaA exhibits PPIase activity is still lacking. As later discussed, this is relevant to understand the mechanisms underpinning the function of NinaA and its homologues, as it will allow determining whether PPIase and chaperone activities are independent and discernible biological properties of NinaA and other cyclophilins. In this regard, the enzymatic proficiency of cyclophilins compared to that of other enzymes is arguably low (Radzicka and Wolfenden, 1995). Further, it was shown recently with model substrates that there was no difference in the acceleration of refolding of carbonic anhydrase II in vitro between wild-type (CyPA\PPIA) and a mutant without PPIase activity, CyPA (R55A) (Moparthi et al., 2009). CyPA also acts as a chaperone in vitro by functioning as a hydrophobic basket toward misfolding-prone regions of model substrates, such as carbonic anhydrase II, creatin kinase and citrate synthase (Moparthi et al., 2010; Ou et al., 2001). This chaperone activity promotes productive folding routes by preventing or delaying the trapping of misfolded conformers and/or unproductive interactions with folding intermediates or unfolded proteins that can result in amyloid-fibril formation (Smajlovic et al., 2009).
Immunophilins' role(s) and beyond
PPIases are grouped in three families of proteins: cyclophilins, FK506-binding proteins (FKBPs) and parvulins (Schiene-Fischer et al., 2011). The primary and tertiary structures are distinct among these families of proteins (Schiene-Fischer et al., 2011). Cyclophilins are also designated as cyclosporin A (CsA)-binding proteins (Handschumacher et al., 1984; Harding et al., 1986), even though not all cyclophilins bind the cyclic peptide, CsA, typically due to subtle non-conservative changes in one or more key residue(s) (Galat and Bua, 2010). Cyclophilins and FKBPs are often called immunophilins, because they are the targets of chemically unrelated and immunosuppressive drugs, CsA and FK506 (a peptide macrolide), which are at the forefront of pharmacological management of a wide variety of clinical conditions, ranging from the suppression of graft rejection to the treatment of a variety of immunological, oncogenic and neurological disorders (Friedman and Weissman, 1991; Liu et al., 1991; Medyouf et al., 2007; Medyouf and Ghysdael, 2008; Schreiber, 1991, 1992; Snyder et al., 1998). CyPA (PPIA) is the primary physiological and molecular target of CsA, a physiological effect reflected by the resistance of Ppia−/− mice to CsA-mediated immunosupression (Colgan et al., 2005). CsA and FK506 inhibit the PPIase activity of cyclophilins and FKBPs, respectively, because the drug binding and PPIase active sites overlap (Fischer et al., 1989; Harding et al., 1989; Kallen and Walkinshaw, 1992; Pflugl et al., 1993). However, the loss of PPIase activity does not underlie immunosuppression by these drugs for several reasons. Among these, CyPA/PPIA and CyPB/PPIB (and other immunophilins) are extremely abundant in cells (up to μM), whereas the efficacy of the immunosuppressants ranges from pico- to nano-molar concentrations (Colgan et al., 2005; Fischer et al., 1989; Galat and Bouet, 1994; Handschumacher et al., 1984; Harding et al., 1989). Immunosuppression is achieved instead by a gain-of-function mechanism, whereby the immunophilin-immunosupressor drug complex associates with and inhibits calcineurin, a protein phosphatase which is crucial for the dephosphorylation of the nuclear factor for activation of T cells (NF-AT) (Liu et al., 1991). The inhibition of NF-AT dephosphorylation prevents its nuclear translocation and the stimulation of cytokine and other genes critical for T-cell proliferation (e.g. IL-2 production) (Rao et al., 1997). On the other hand, subsequent structural analysis of other cyclophilins showed that some cyclophilins might present polymorphic functions (i.e. multimodal structural ways of exerting multiple activities). For example, the binding of CsA to CyPL1 (PPIL1) and SnuCyP-20 (PPIH) does not affect their association to the substrates, SKIP and U4/U6 snRNP-60K, respectively, since they bind to the opposite face of the PPIase active site (Reidt et al., 2003; Xu et al., 2006). A similar scenario likely occurs also with CyP33 (PPIE), because its PPIase activity is stimulated upon specific binding of mRNA (Wang et al., 2008). By contrast, the chaperone activity of PPIase-deficient CyPA is inhibited by CsA in a model substrate (Moparthi et al., 2010; Moparthi et al., 2009).
Cyclophilins are widely conserved from Archea to humans and they occur as single domain proteins or as part of modular proteins with high degree of complexities and functions (Schiene-Fischer et al., 2011). The modular organization of cyclophilins varies greatly between phyla and even classes of organisms. For example, the 358 kDa cyclophilin-related protein, Ran-binding protein 2 (RanBP2)/nucleoporin 358 (Nup358), which was isolated from a screen for transcripts encoding proteins homologous to NinaA as well as by independent approaches by others, comprises a C-terminal cyclophilin domain highly homologous to CyPA among many other unrelated domains present in this protein (Ferreira et al., 1995; Wu et al., 1995; Yokoyama et al., 1995). The functional implications of such structural diversities in multi-modular cyclophilins remain largely elusive. Although none of the eight yeast cyclophilins and four FKBPs are essential to the survival of budding yeast (Dolinski et al., 1997), the knowledge about the roles of most of the 18 annotated single and multi-domain human cyclophilins is still very limited. In fact, the native substrates for most cyclophilins and their exact roles have yet to be discerned. Importantly, genetic evidence support that a considerable degree of functional redundancy or complementarity may exist even among unrelated PPIase or its targets. For example, the PPIase activity of the single domain CyPA counteracts the function and suppresses the loss-of-function of the parvulin, Ess1/Pin1 (Arevalo-Rodriguez et al., 2000), a PPIase with unique specificity toward phosphoserine-prolyl peptides and important for cell cycle progression (Lu et al., 1996; Ranganathan et al., 1997). On the other hand, Pin1 and CyPB (PPIB) share a similar oncoprotein substrate, the signal transducer and activator of transcription 3 (Stat3), apparently via distinct domains of Stat3, and promote its trans-activating potential (Bauer et al., 2009). Regardless, the cyclophilin, NinaA, sets itself apart from all other cyclophilins, because it remains most likely the only cyclophilin for which a physiological substrate (Rh1 and Rh2 opsin) has been ascertained and whose loss is not complemented by other cyclophilins (Baker et al., 1994; Colley et al., 1991; Schneuwly et al., 1989; Shieh et al., 1989; Stamnes et al., 1991).
Cyclophilin B (CyPB/PPIB): a NinaA orthologue, functional redundancy in PPIases or substrates?
CyPB/PPIB and NinaA are the only known cyclophilins that are targeted to and reside in the endoplasmic reticulum (ER) (Colley et al., 1991; Price et al., 1994; Price et al., 1991; Smith et al., 1995; Stamnes et al., 1991). Further, the cyclophilin domain of NinaA exhibits the highest sequence homology to its counterpart domain in human CyPB/PPIB (e.g. 49% vs 43% identity to CyPB and CyPA, respectively) (Figure 1A). The alignment between NinaA, CyPB/PPIB and CyPA/PPIA, show that the known primary and secondary structures are best conserved between NinaA and CyPB/PPIB than with CyPA/PPIA. Of the nine residues implicated in forming the catalytic PPIase site of CyPB/PPIB and CyPA/PPIA (Kallen et al., 1991; Kallen and Walkinshaw, 1992), seven are identical in NinaA, whereas the remaining two, M61 and F113, are conservatively replaced by leucine and tyrosine, respectively (Figure 1B). Similar residues are also replaced in the same exact positions in PPIL4 and SDCCAG-10, or PPIL6 (Davis et al., 2010). We generated a homology model structure of the cyclophilin domain of NinaA based on known structures of CyPB/PPIB and CyPA/PPIA (Figure 1C) (Davis et al., 2010; Kallen et al., 1991; Ke et al., 1991) . The CyPB/PPIB and CyPA/PPIA provide excellent template structures for modeling the homologous NinaA. The structures have eight anti-parallel β-sheets with two sets of four β-sheets orthogonal to each other. The β-sheets are connected by loops of varying sizes, α-helices separate the sheets, β-sheet 2 – β-sheet 3 and β-sheet 7-β-sheet 8, and a short α-helical turn is located in the β6–β7 loop.
FIGURE 1.

Molecular modeling of NinaA to CyPB/PPIB and CyPA/PPIA. (A) Sequence alignment of NinaA, CyPB/PPIB, CyPA/PPIA. Identical and conserved residues are highlighted in green and yellow, respectively. NinaA exhibits 49% and 43% identity to CyPB/PPIB and CyPA/PPIA, respectively. Consensus sequence is noted above the alignment. Consensus of secondary-structure elements are shown below the primary sequence alignment. The secondary structure assignment is taken from the PDB files 2rma and 3ici for CyPA/PPIA and CyPB/PPIB, respectively, and the model of NinaA. Legend: Green arrows and solid green triangles, β-sheet; red cylinders and spheres, α-helices; purple cylinders, non-canonical helices. (B) Conservation of residues comprising the PPIase active site of NinaA, CyPB/PPIB and CyPA/PPIA. Identical residues are highlighted in green. (C) Structural superposition of the NinaA model (green ribbon), the CyPB/PPIB template crystal structure (red ribbon), and the CyPA/PPIA crystal structure (grey ribbon). The residues of the PPIase active site are shown in stick representation and the carbon atoms are colored red and orange, respectively. The main structural differences between the three structures are in three loop regions: β1 to β2 (circled in purple), β4 to β5 (circled in light blue), and β2 to β8 (circled in red). The β1 to β2 loop turn is conserved between the NinaA model and the PPIB template and this turn differs from the turn seen in PPIA structures. A two residue insertion in the β4 to β5 loop in NinaA gives rise to a different loop structure in this region compared to PPIA and PPIB. The α2 to β8 loop is one residue shorter in PPIA and therefore differs to the loops seen in NinaA and PPIB. The modeling indicates that there may be an additional β-sheet in this loop in NinaA compared to PPIA and PPIB.
As shown in figure 1C, the main structural differences between the model structure of NinaA and the known structures of CyPB/PPIB and CyPA/PPIA are in three loop regions connecting β-sheet 1 to β-sheet 2, β-sheet 4 to β-sheet 5, and α-helix 2 to β-sheet 8. The loop turn between β-sheet 1 to β-sheet 2 is conserved between the NinaA and the PPIB template and this turn differs from the turn seen in PPIA structures. A two residue insertion in the loop between β-sheet 4 to β-sheet 5 in NinaA gives rise to a different loop structure in this region compared to CyPA/PPIA and CyPB/PPIB. Notably, the β-sheet 4 to β-sheet 5 loop is implicated in CyPA/PPIA oligomerization upon CsA binding and hence, it may be important to mediate the interaction of NinaA with other proteins (Pflugl et al., 1993). Finally, the loop between α-helix 2 to β-sheet 8 is one residue shorter in CyPA/PPIA and therefore differs from the loops seen in NinaA and CyPB/PPIB. Among these loops, the β-sheet 1 to β-sheet 2 and α-helix 2 to β-sheet 8 loops are located away from and toward the back of the active site and may contribute to substrate selectivity independently or together with the active site. Further, the modeling indicates that there may be an additional anti-parallel β-sheet in this loop in NinaA compared to CyPA/PPIA and CyPB/PPIB. Hence, NinaA and the ubiquitously expressed CyPB/PPIB share the highest sequence homology, subcellular compartmentalization and topology in the ER, and overall structure, even though two out of three loops are unique to NinaA. These features are not shared by any other known cyclophilins of metazoans.
In light of the strong sequence conservation and subcellular compartmentalization between NinaA and CyPB/PPIB, a prediction would be that loss-of-function of CyPB/PPIB will cause phenotypes of similar etiologies to those observed in ninaADrosophila mutants, such as a deficit(s) in the production of one or more members of the opsin family, a deficit that would manifest clinically by an impairment of night or color vision or both. However, lack of expression of CyPB/PPIB in the human caused by mutations in CyPB/PPIB were found to cause osteogenesis imperfecta (OMIM 166200, 166210, 259420, 166220]) in the human, a genetically heterogeneous disease that is manifested by an impairment of the biogenesis of α1 chains of collagen type 1 and clinically by bone fragility (Barnes et al., 2010; Pyott et al., 2011; van Dijk et al., 2009). Loss-of-function of CyPB/PPIB in the human appears to cause the accumulation of pro-collagen in the ER and delay of the assembly of trimer formation (Pyott et al., 2011), however other reports have shown normal or near normal collagen folding and levels of prolyl 3-hydroxylation (Barnes et al., 2010; van Dijk et al., 2009). A mouse model lacking CyPB/PPIB expression was also generated (Choi et al., 2009). Although the mouse model recapitulates clinical features of the human disease, the phenotypes observed seem not to provide additional insights into the disease pathogenesis from what is already known of the disease affecting the human yet. Further, a missense G31R mutation in CyPB/PPIB was found in the horse to cause the degenerative skin disorder, HERDA, without changes in skeletal or collagen being reported (Tryon et al., 2005). Hence, additional studies are needed to understand what physiological substrate(s) are directly impaired by the loss of CyPB/PPIB function and promote disease pathogenesis. For example, CyPB was implicated also in the ER-associated degradation (ERAD) of misfolded glycopolypeptides and triggering the cell adhesion of T-lymphocytes to the extracellular matrix (Allain et al., 2002; Bernasconi et al., 2010a; Bernasconi et al., 2010b). Regardless, the human genetic findings on CyPB/PPIB came as a surprise, because of the properties shared between NinaA and CyPB/PPIB and no visual impairments of the retina are reported to be associated with osteogenesis imperfecta. Collectively, these data raise several important questions. Among these, it will be important to address: i) What is the degree of functional redundancy between the NinaA, CyPB/PPIB and other unrelated PPIases known to reside also in the ER and to associate with collagen [e.g. FKBP65 (Ishikawa et al., 2008)]? ii) What is the nature and degree of conservation of the prolyl and/or chaperone substrates between the NinaA and CyPB/PPIB? iii) In light of the findings that CyPB/PPIB can be secreted to the extracellular milieu upon various inflammatory stimuli, up-taken by cells and translocate into the nuclear compartment, where it modulates transcriptional activity in mammalian cells, are there new and non-conserved biological roles acquired by CypB/PPIB in that may underlie unique pathological outcomes? iv) what is/are the molecular bases of the substrate specificity of NinaA toward a subset of GPCRs and how has the requirement for such PPIase/chaperone activity evolved toward other homologous substrates?
Re-examination of mutations in ninaA: new insights into function(s) of cyclophilins
Recently, the structures of a number of additional cyclophilins were characterized by Davis et al. (Davis et al., 2010) from which the authors propose the existence of two pockets in cyclophilins that contribute to specificity toward substrate binding and turnover (Figure 2A). The first pocket, S1, mediates proline binding to cyclophilin and it is defined by a subset of residues delineating the active PPIase site of cyclophilins (Figures 1 and 2A). These residues are highly conserved between all cyclophilins. The second pocket, S2, mediates the interaction with the two residues (P2 and P3) preceding the proline residue (P1). The base of the S2 pocket is defined by the β5–β6 loop and it forms a deep pocket, which is promiscuous toward binding of a variety of residues. The authors propose that access to the S2 pocket is determined by gatekeeper residues at its surface (Figure 2A) that preferentially discriminate among a diversity of P2 and P3 of peptidyl-prolyl substrates (Davis et al., 2010). However, there are important limitations in these analyses. First, the effect(s) of non-conservative changes in the gatekeeper residues are not tested for the proposed functions. Second, the analyses do not take into account the role of substrate residues further away from the P2 and P3 prolyl peptide that may contribute to significant long-range interactions between the primary structures of native substrates and one or more cyclophilin-interacting domains and conceivable important for the docking of substrates to cyclophilins.
Figure 2.
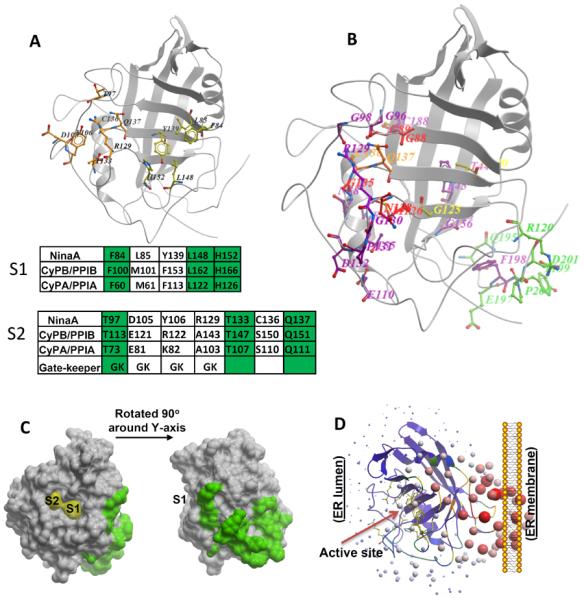
Assignment of physiological relevant residues of NinaA to new cylophilin domains. (A) Location of residues comprising S1 and S2 pockets as defined by Davis et al, 2010, in NinaA 3D-structure. Conservation of residues comprising the S1 and S2 pockets of NinaA, CyPB/PPIB and CyPA/PPIA are shown in the tables below the 3D-structure. The conservation of gatekeeper residues is also depicted. Identical residues are highlighted in green. (B) Mapping of residues caused by mutations in ninaA (Ondek et al., 1992; Table 1) to the 3D-model structure of NinaA. The mutations were grouped in five groups based on their location. Residues nearby the S1 pocket are in yellow, residues in the S2 pocket are in orange, residues in the S2 extended (S2e) region are in red, residues comprising Pm domain facing the membrane surface are in green, all other residues are in magenta. Note F198 points away from the surface. The S2e region comprises the residues, G88, G89, M126, N128 and G135. All residues, but N128, are buried in a shell next to the periphery of S2 binding pocket. (C) Surface representation of residues of Pm domain of NinaA and their relative location to S1 and S2 pockets. (D) Ribbon representation of the relative orientation of the PPIase active site of NinaA to its Pm domain. Active site residues are represented in the purple ribbons by sticks in yellow.
Hence, we examined a large number of mutations generated in ninaA by saturation mutagenesis screens previously reported (Table 1), because the resulting residue changes in NinaA were found to affect physiologically and significantly the biogenesis of Rh1 opsin in photoreceptors of Drosophila (Ondek et al., 1992). Up-to-date predictions of the effects of such mutations are noted and based on the current structural knowledge of multiple cyclophilins (Table 1). The mapping of the residue substitutions to the NinaA structural model generated (Figure 1C) and comparison of these changes to the PPIase active site, S1 and S2 sites, of other cyclophilins (e.g. CyPA/PPIA), will help to validate or repeal the significance of such sites and likely delineate novel domains of physiological relevance to the function and interaction of NinaA and other cyclophilins with native substrates. As shown in figure 2B, the mutations in NinaA affect residues that are clustered in five major domains: a region nearby the S1/PPIase sites (e.g. Del [129–132]), the S2 region, an expanded region encompassing the S2 domain (hereafter named S2e), a region opposite to the PPIase region and likely facing the inner membrane of the ER (hereafter named Pm), and other domains of interest. The majority of the mutations fall in S2e region. This region comprises residues, such as G88, G89, M126, N128 and G135. All residues, but N128, are buried in a shell next to the periphery of S2 binding pocket, and they may have a “knock-on” effect on the packing of the residues of the S2 pocket. Another significant group of mutations (missense and deletions) are clustered in the Pm region, which is made-up of unstructured loops located away from the expanded S2 and canonical PPIase domains (e.g. R120K). Further, since most of the residues forming the previously designated S1/PPIase and S2 pockets are located along one face of the cyclophilin structure, we also examined whether any of the residues affected by mutations were exposed to the surface of NinaA. For example, changes in residues on the surface of NinaA would support that they may impair the docking or recognition of substrate(s) by cyclophilin. Indeed, we found that a significant number of residues were located in the Pm region away from the S1/PPIase and S2 pockets (Figure 2C). It is possible that this region (Pm) defines a novel domain mediating protein-membrane interaction(s) with other ER membrane proteins and ER lipids for the correct positioning of the S1/PPIase and S2/S2e pockets (Figure 2D). Alternatively, the novel Pm region may play an active role on capturing emerging unstructured and elongated polypeptide chains upon co-translation into the ER, where they first interact with and are chaperoned by cyclophilin. This Pm domain may perform similar functions in other cyclophilins, many of which are commonly regarded as being cytosolic (e.g. CyPA/PPIA). However, the subcellular compartmentation of CyPA/PPIA, like CyPB/PPIB, is modulated upon inflammatory stimuli upon which they are secreted to the extracellular by mechanisms not yet understood (Billich et al., 1997; De Ceuninck et al., 2003; Nigro et al., 2011; Satoh et al., 2009; Sherry et al., 1992; Yang et al., 2008). Finally, mutations in the exposed cytoplasmic tail or transmembrane domains of NinaA are very intriguing in the context of their role in opsin biogenesis (Table 1). It is possible that either of these domains cross-talk with the cyclophilin domain via conformational changes with other neighboring proteins cooperating also in opsin biogenesis.
Table 1.
Structure-function effects of changes in residues caused by mutations in ninaA and affecting Rh1opsin production (Ondek et al., 1992).
| Mutation | Location | Predicted Functional Effect |
|---|---|---|
| M1I/L | No template structure | |
| DEL [44–45] | Not included in analysis | |
| G46R/E | Beta 2 | |
| T55M | Alpha 1 | |
| N58K | Alpha 1 | Possible structure stabilization. Makes 2 H-bonds to backbone G135 and I102 |
| R60P | Alpha 1 | Possible structure stabilization. Salt bridge with E190 |
| G88D | Beta 4 | Proximity to S2 pocket; residue located in S2e region |
| G89D/S | Beta 4 to Beta 5 Loop | Proximity to S2 pocket; residue located in S2e region |
| DPL [94–109] | Beta 4 to Beta 5 Loop | Potential oligomerization site (mediates oligomerization of cyclophilin A upon CsA binding) |
| G96E | Beta 4 to Beta 5 Loop | Potential oligomerization site (mediates oligomerization of cyclophilin A upon CsA binding) |
| G98S/D | Beta 4 to Beta 5 Loop | Potential oligomerization site (mediates oligomerization of cyclophilin A upon CsA binding) |
| E110K/V | Beta 4 to Beta 5 Loop | Possible structure stabilization makes two H-bonds with the backbone of T54 and K54 at the N-terminal of alpha 1. |
| R120K | Beta 4 to Beta 5 Loop | Predicted protein or membrane interaction site. |
| G125D | Beta 5 | Proximity to S1 pocket |
| M126I | Beta 5 | Proximity to S2 pocket; residue located in S2e region |
| N128Y | Beta 5 to Beta 6 Loop | Proximity to S2 pocket; residue located in S2e region |
| DEL [129–132] | Beta 5 to Beta 6 Loop | Partial deletion of the Beta 5 to Beta 6 Loop |
| G135D | Beta 5 to Beta 6 Loop | Proximity to S2 pocket; residue located in S2e region |
| C136G | Beta 5 to Beta 6 Loop | S2 Pocket |
| Q137L/H | Beta 5 to Beta 6 Loop | S2 Pocket |
| V140E | Beta 6 | Proximiity to S1 pocket |
| G156D | Beta 7 | Structural instability; Residue is buried without counter-charges in the vicinity |
| P179L | Alpha 1 to Beta 8 Loop | Maybe important for loop conformation |
| C188Y | Beta 8 | Possible disulfide bond with C63 |
| Q195STOP | C-terminal | Predicted protein or membrane interaction site. |
| DEL [197–201] | C-terminal | Predicted protein or membrane interaction site. (Not residue 198 it is buried) |
| P200L | C-terminal | Predicted protein or membrane interaction site. |
| W208STOP | No template structure (truncates ER transmembrane domain) | |
| G213D | No template structure (within ER transmembrane domain) | |
| S219F | No template structure (within ER transmembrane domain) | |
| H227L | No template structure (next to the ER transmembrane domain facing the cytosol) | |
| Q232STOP | No template structure (truncates exposed cytoplasmictail by 5 residues) |
As previously noted, NinaA shows substrate selectivity for Rh1 and Rh2 opsins, but not any other opsins expressed in other photoreceptor cell types, even though NinaA is expressed in all classes of photoreceptor neurons. This observation is highly significant, because it is contrarian to the notion that cyclophilins present low specificity toward model peptidyl-prolyl substrates (REF). Further, the mutation of the conserved proline residue to histidine (P23H) at the N-terminal region of human rhodopsin causes the degeneration of photoreceptors that is clinically manifested as retinitis pigmentosa (Dryja et al., 1991; Hartong et al., 2006). P23 of rhodopsin is targeted to the ER lumen upon its translation, whereas it is localized in the extracellular domain upon rhodopsin targeting to the plasma membrane. In Drosophila, a mutation of such equivalent and conserved proline residue (P37H) in Rh1 opsin was also shown to cause the accumulation of Rh1 opsin in the ER and the degeneration of photoreceptors (Galy et al., 2005). Hence, the N-terminal region of Rh1 opsin comprising P37, and possibly of other opsins subtypes, is a strong candidate to be a direct substrate of NinaA. To test this possibility, we used an in silico docking method to calculate the energetic binding properties of cis- and trans-prolyl peptides comprising the N-terminal P37 and equivalent proline residues of Rh1, Rh2, Rh3 and Rh4 opsins of Drosophila (Figure 3A) to the NinaA structural 3D-model. There was optimal fitting of cis-prolyl peptides of Rh1 (Figure 3B) and Rh2 (Figure 3C) to the PPIase pocket of NinaA, whereas the counterpart trans-prolyl peptides were poorer substrates. By contrast, the cis-prolyl peptides of Rh3 and Rh4 were non-binders to NinaA. In addition to the PPIase pocket, the S2 gatekeepers accommodated well P5 and P4 residues of the Rh1 and Rh2 in the S2 pocket. Finally, Rh1 had a significantly better docking score than Rh2 as a docking substrate of NinaA. Hence, it will be important to probe further by mutagenesis the N-terminal region of Rh1 and Rh2 to validate these in silico structural studies between NinaA and Rh1, Rh2 and other opsins. The outcomes of such studies are likely to provide novel insights into fundamental biological and pathological processes and mechanisms underlying the biogenesis of GPCRs and the role of cyclophilins and other PPIases in mediating such processes. One of such mechanisms may involve unidirectional and long-range effects caused by the cis-trans prolyl isomerization of the N-terminal region of opsin. The cis-trans isomerization causes the rotation of the C-terminal end of the polypeptide chain and thus the propagation of the rotational effects to C-terminal regions of the polypeptide remote from the isomerized peptidyl-prolyl bond (Jakob and Schmid, 2009; Reimer and Fischer, 2002; Wang et al., 2010; Weininger et al., 2009). Such mechanism could act effectively as a prolyl molecular switch with important implications in the modulation of protein-protein interactions, such as those reported first for the interconversion of M-opsin by the cyclophilin and Ran-binding domain-4 of the modular and multifunctional cyclophilin-related protein, RanBP2 (Ferreira et al., 1996; Ferreira et al., 1997). RanBP2 is expressed strongly in cone photoreceptors and associates directly with M-opsin and thus, it is another cyclophilin-related protein candidate to mediate M-opsin biogenesis and/or the survival of cone photoreceptors.
Figure 3.
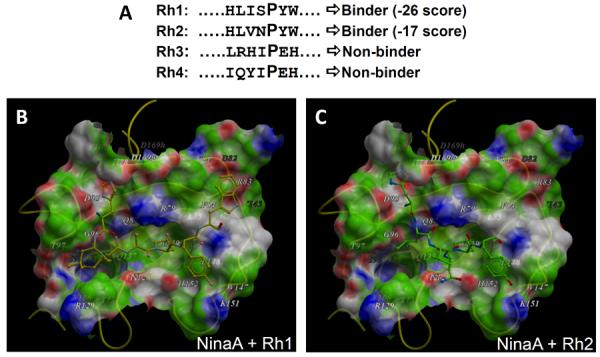
Docking of Rh1, Rh2, Rh3 and Rh4 prolyl-peptide substrates of opsins of Drosophila and equivalent to P23 of human rhodopsin to S1 and S2 pockets of NinaA. (A) Alignment of Rh1, Rh2, Rh3 and Rh4 prolyl-peptide sequences of opsins of Drosophila comprising the equivalent P23 (in bold) of human rhodopsin. Only Rh1 and Rh2 scored as binders according to their fit in the S1 and S2 pockets of NinaA and with the Rh1 being a better binder. (B, C) are surface representations of NinaA upon docking of Rh1 (B) and Rh2 (C) prolyl substrates shown in (A) to its S1 and S2 pockets.
Concluding remarks
The pioneering work spearheaded by the Pak laboratory and continued by colleagues, culminates with the genetic identification of numerous components and biological processes with crucial ramifications in the understanding of biological and disease pathways of complex physiological systems, such as the human. The identification and studies of NinaA are a prime example of how genetic model systems, such as Drosophila, open new ground to test hypothesis about fundamental biological processes and mechanisms mediating protein biogenesis. Further, the outcome of studies like these can be employed to probe pharmacologically the role of a variety of processes in health and disease of the human (Vidal et al., 2005). Cyclophilins are immunophilins and primary targets with a crucial role in the pharmacological modulation of a variety of immune, inflammatory and other responses (Allen et al., 2004; Bauer et al., 2009; Galat and Bua, 2010; Medyouf et al., 2007; Medyouf and Ghysdael, 2008; Nigro et al., 2011). Importantly, cyclophilins present a high degree of pharmacological versatility as reflected by the large number of new and chemically re-designed pharmacological ligands for cyclophilins (and other immunophilins) with novel therapeutic properties (Allen et al., 2004; Galat and Bua, 2010). This is reflected by well over 40,000 PubMed references on CsA and its derivatives (Galat and Bua, 2010). It is likely that Drosophila and other tractable genetic model systems will serve as advantageous experimental tools to explore the vast therapeutic potential of such cyclophilin ligands in a variety of human conditions.
MATERIALS AND METHODS
NinaA modeling
The NinaA model was built using a stochastic global energy optimization procedure in Internal Coordinate Mechanics (ICM) in the ICM-Pro desktop modeling package version 3.7 (MolSoft LLC, San Diego) (Abagyan et al., 2010; Abagyan et al., 1994). A Blast search was performed to identify the cyclophilin structure in the Protein Data Bank (PDB) with the best sequence similarity and coverage compared to the NinaA primary sequence. Based on this analysis the PPIB structure with PDB code 3ici was chosen as the template for modeling. An alignment was generated between NinaA and the template sequence using an adaptation of the Needleman and Wunsch algorithm(Abagyan and Batalov, 1997; Needleman and Wunsch, 1970). The model was refined by globally optimizing the side-chains and annealing the backbone. The iterative refinement procedure contains three major steps: (i) random sampling of the dihedral angles according to the biased probability Monte Carlo method (Abagyan and Totrov, 1994), (ii) a local minimization step, (iii) the Metropolis criterion (Metropolis et al., 1953) is then used to accept or reject a conformation. The lowest energy structure was selected as the final NinaA model.
In silico docking of peptidyl-prolyl substrates of Rh1, Rh2, Rh3 and Rh4 to NinaA structure
Models of four peptidyl-prolyl substrates shown in figure 3A were built and docked to the NinaA model using ICM-Pro 3.7 (MolSoft LLC, San Diego) (Abagyan et al., 2010; Abagyan et al., 1994). During docking, the P1 peptide residue was tethered to the active site in the cis or trans conformations and the ICM biased probability Monte Carlo method (Schapira et al., 1999) was used to sample the energy of the complex. The non-tethered regions of the peptide and the side-chains of the gatekeeper residues in the S2 site were flexible during docking. The docked ligands were then ranked by energy and scored according to their fit in the pocket (Abagyan and Totrov, 1994).
REFERENCES
- Abagyan R, Orry A, Raush E, Totrov M. ICM User Guide 3.7. La Jolla, CA; Molsoft LLC: 2010. [Google Scholar]
- Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- Abagyan R, Totrov M, Kuznetsov D. ICM-a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. [Google Scholar]
- Abagyan RA, Batalov S. Do aligned sequences share the same fold? J Mol Biol. 1997;273:355–368. doi: 10.1006/jmbi.1997.1287. [DOI] [PubMed] [Google Scholar]
- Allain F, Vanpouille C, Carpentier M, Slomianny MC, Durieux S, Spik G. Interaction with glycosaminoglycans is required for cyclophilin B to trigger integrin-mediated adhesion of peripheral blood T lymphocytes to extracellular matrix. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2714–2719. doi: 10.1073/pnas.052284899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen A, Zheng Y, Gardner L, Safford M, Horton MR, Powell JD. The novel cyclophilin binding compound, sanglifehrin A, disassociates G1 cell cycle arrest from tolerance induction. J Immunol. 2004;172:4797–4803. doi: 10.4049/jimmunol.172.8.4797. [DOI] [PubMed] [Google Scholar]
- Arevalo-Rodriguez M, Cardenas ME, Wu X, Hanes SD, Heitman J. Cyclophilin A and Ess1 interact with and regulate silencing by the Sin3-Rpd3 histone deacetylase. The EMBO journal. 2000;19:3739–3749. doi: 10.1093/emboj/19.14.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EK, Colley NJ, Zuker CS. The cyclophilin homolog NinaA functions as a chaperone, forming a stable complex in vivo with its protein target rhodopsin. Embo J. 1994;13:4886–4895. doi: 10.1002/j.1460-2075.1994.tb06816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AM, Carter EM, Cabral WA, Weis M, Chang W, Makareeva E, Leikin S, Rotimi CN, Eyre DR, Raggio CL, et al. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N Engl J Med. 2010;362:521–528. doi: 10.1056/NEJMoa0907705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer K, Kretzschmar AK, Cvijic H, Blumert C, Loffler D, Brocke-Heidrich K, Schiene-Fischer C, Fischer G, Sinz A, Clevenger CV, et al. Cyclophilins contribute to Stat3 signaling and survival of multiple myeloma cells. Oncogene. 2009;28:2784–2795. doi: 10.1038/onc.2009.142. [DOI] [PubMed] [Google Scholar]
- Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. The Journal of cell biology. 2010a;188:223–235. doi: 10.1083/jcb.200910042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi R, Solda T, Galli C, Pertel T, Luban J, Molinari M. Cyclosporine A-sensitive, cyclophilin B-dependent endoplasmic reticulum-associated degradation. PLoS ONE. 2010b;5:e13008. doi: 10.1371/journal.pone.0013008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billich A, Winkler G, Aschauer H, Rot A, Peichl P. Presence of cyclophilin A in synovial fluids of patients with rheumatoid arthritis. The Journal of experimental medicine. 1997;185:975–980. doi: 10.1084/jem.185.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt SG, Feiler R, Kirschfeld K, Zuker CS. Spectral tuning of rhodopsin and metarhodopsin in vivo. Neuron. 1993;11:29–39. doi: 10.1016/0896-6273(93)90268-v. [DOI] [PubMed] [Google Scholar]
- Choi JW, Sutor SL, Lindquist L, Evans GL, Madden BJ, Bergen HR, 3rd, Hefferan TE, Yaszemski MJ, Bram RJ. Severe osteogenesis imperfecta in cyclophilin B-deficient mice. PLoS genetics. 2009;5:e1000750. doi: 10.1371/journal.pgen.1000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan J, Asmal M, Yu B, Luban J. Cyclophilin A-deficient mice are resistant to immunosuppression by cyclosporine. J Immunol. 2005;174:6030–6038. doi: 10.4049/jimmunol.174.10.6030. [DOI] [PubMed] [Google Scholar]
- Colley NJ, Baker EK, Stamnes MA, Zuker CS. The cyclophilin homolog ninaA is required in the secretory pathway. Cell. 1991;67:255–263. doi: 10.1016/0092-8674(91)90177-z. [DOI] [PubMed] [Google Scholar]
- Davis TL, Walker JR, Campagna-Slater V, Finerty PJ, Paramanathan R, Bernstein G, MacKenzie F, Tempel W, Ouyang H, Lee WH, et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS biology. 2010;8:e1000439. doi: 10.1371/journal.pbio.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ceuninck F, Allain F, Caliez A, Spik G, Vanhoutte PM. High binding capacity of cyclophilin B to chondrocyte heparan sulfate proteoglycans and its release from the cell surface by matrix metalloproteinases: possible role as a proinflammatory mediator in arthritis. Arthritis Rheum. 2003;48:2197–2206. doi: 10.1002/art.11099. [DOI] [PubMed] [Google Scholar]
- Dolinski K, Muir S, Cardenas M, Heitman J. All cyclophilins and FK506 binding proteins are, individually and collectively, dispensable for viability in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13093–13098. doi: 10.1073/pnas.94.24.13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, Hahn LB, Cowley GS, McGee TL, Berson EL. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88:9370–9374. doi: 10.1073/pnas.88.20.9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira PA, Hom JT, Pak WL. Retina-specifically expressed novel subtypes of bovine cyclophilin. J Biol Chem. 1995;270:23179–23188. doi: 10.1074/jbc.270.39.23179. [DOI] [PubMed] [Google Scholar]
- Ferreira PA, Nakayama TA, Pak WL, Travis GH. Cyclophilin-related protein RanBP2 acts as chaperone for red/green opsin. Nature. 1996;383:637–640. doi: 10.1038/383637a0. [DOI] [PubMed] [Google Scholar]
- Ferreira PA, Nakayama TA, Travis GH. Interconversion of red opsin isoforms by the cyclophilin-related chaperone protein Ran-binding protein 2. Proc Natl Acad Sci U S A. 1997;94:1556–1561. doi: 10.1073/pnas.94.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, Bang H, Mech C. [Determination of enzymatic catalysis for the cis-transisomerization of peptide binding in proline-containing peptides] Biomed Biochim Acta. 1984;43:1101–1111. [PubMed] [Google Scholar]
- Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature. 1989;337:476–478. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]
- Friedman J, Weissman I. Two cytoplasmic candidates for immunophilin action are revealed by affinity for a new cyclophilin: one in the presence and one in the absence of CsA. Cell. 1991;66:799–806. doi: 10.1016/0092-8674(91)90123-g. [DOI] [PubMed] [Google Scholar]
- Galat A, Bouet F. Cyclophilin-B is an abundant protein whose conformation is similar to cyclophilin-A. FEBS Lett. 1994;347:31–36. doi: 10.1016/0014-5793(94)00501-x. [DOI] [PubMed] [Google Scholar]
- Galat A, Bua J. Molecular aspects of cyclophilins mediating therapeutic actions of their ligands. Cell Mol Life Sci. 2010;67:3467–3488. doi: 10.1007/s00018-010-0437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy A, Roux MJ, Sahel JA, Leveillard T, Giangrande A. Rhodopsin maturation defects induce photoreceptor death by apoptosis: a fly model for RhodopsinPro23His human retinitis pigmentosa. Hum Mol Genet. 2005;14:2547–2557. doi: 10.1093/hmg/ddi258. [DOI] [PubMed] [Google Scholar]
- Hamdorf K, Razmjoo S. Photoconvertible pigment states and excitation in Calliphora; the induction and properties of the prolonged depolarizing afterpotential. Biophys Struct Mech. 1979;5:137–161. doi: 10.1007/BF00535444. [DOI] [PubMed] [Google Scholar]
- Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science. 1984;226:544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- Harding MW, Galat A, Uehling DE, Schreiber SL. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature. 1989;341:758–760. doi: 10.1038/341758a0. [DOI] [PubMed] [Google Scholar]
- Harding MW, Handschumacher RE, Speicher DW. Isolation and amino acid sequence of cyclophilin. J Biol Chem. 1986;261:8547–8555. [PubMed] [Google Scholar]
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Vranka J, Wirz J, Nagata K, Bachinger HP. The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. The Journal of biological chemistry. 2008;283:31584–31590. doi: 10.1074/jbc.M802535200. [DOI] [PubMed] [Google Scholar]
- Jakob RP, Schmid FX. Molecular determinants of a native-state prolyl isomerization. Journal of molecular biology. 2009;387:1017–1031. doi: 10.1016/j.jmb.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Kallen J, Spitzfaden C, Zurini MG, Wider G, Widmer H, Wuthrich K, Walkinshaw MD. Structure of human cyclophilin and its binding site for cyclosporin A determined by X-ray crystallography and NMR spectroscopy. Nature. 1991;353:276–279. doi: 10.1038/353276a0. [DOI] [PubMed] [Google Scholar]
- Kallen J, Walkinshaw MD. The X-ray structure of a tetrapeptide bound to the active site of human cyclophilin A. FEBS Lett. 1992;300:286–290. doi: 10.1016/0014-5793(92)80865-e. [DOI] [PubMed] [Google Scholar]
- Ke HM, Zydowsky LD, Liu J, Walsh CT. Crystal structure of recombinant human T-cell cyclophilin A at 2.5 A resolution. Proc Natl Acad Sci U S A. 1991;88:9483–9487. doi: 10.1073/pnas.88.21.9483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrivee DC, Conrad SK, Stephenson RS, Pak WL. Mutation that selectively affects rhodopsin concentration in the peripheral photoreceptors of Drosophila melanogaster. J Gen Physiol. 1981;78:521–545. doi: 10.1085/jgp.78.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Jr., Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- Medyouf H, Alcalde H, Berthier C, Guillemin MC, dos Santos NR, Janin A, Decaudin D, de The H, Ghysdael J. Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nature medicine. 2007;13:736–741. doi: 10.1038/nm1588. [DOI] [PubMed] [Google Scholar]
- Medyouf H, Ghysdael J. The calcineurin/NFAT signaling pathway: a novel therapeutic target in leukemia and solid tumors. Cell Cycle. 2008;7:297–303. doi: 10.4161/cc.7.3.5357. [DOI] [PubMed] [Google Scholar]
- Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E. Equation of State Calculations by Fast Computing Machines. J Chem Phys. 1953;21:1087. [Google Scholar]
- Minke B. In: The Molecular Mechanisms of Photoreception. Stieve H, editor. Springer; Berlin, Dahlem Konferenzen: 1986. pp. 241–265. [Google Scholar]
- Moparthi SB, Fristedt R, Mishra R, Almstedt K, Karlsson M, Hammarstrom P, Carlsson U. Chaperone activity of Cyp18 through hydrophobic condensation that enables rescue of transient misfolded molten globule intermediates. Biochemistry. 2010;49:1137–1145. doi: 10.1021/bi901997q. [DOI] [PubMed] [Google Scholar]
- Moparthi SB, Hammarstrom P, Carlsson U. A nonessential role for Arg 55 in cyclophilin18 for catalysis of proline isomerization during protein folding. Protein science : a publication of the Protein Society. 2009;18:475–479. doi: 10.1002/pro.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Nigro P, Satoh K, O'Dell MR, Soe NN, Cui Z, Mohan A, Abe J, Alexis JD, Sparks JD, Berk BC. Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E-deficient mice. The Journal of experimental medicine. 2011;208:53–66. doi: 10.1084/jem.20101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondek B, Hardy RW, Baker EK, Stamnes MA, Shieh BH, Zuker CS. Genetic dissection of cyclophilin function. Saturation mutagenesis of the Drosophila cyclophilin homolog ninaA. J Biol Chem. 1992;267:16460–16466. [PubMed] [Google Scholar]
- Ou WB, Luo W, Park YD, Zhou HM. Chaperone-like activity of peptidyl-prolyl cis-trans isomerase during creatine kinase refolding. Protein science : a publication of the Protein Society. 2001;10:2346–2353. doi: 10.1110/ps.23301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak WL. In: Neurogenetics: Genetic Approaches to the Nervous System. Breakfield X, editor. Elsevier; New York: 1979. pp. 67–99. [Google Scholar]
- Pak WL. Drosophila in vision research. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1995;36:2340–2357. [PubMed] [Google Scholar]
- Papatsenko D, Sheng G, Desplan C. A new rhodopsin in R8 photoreceptors of Drosophila: evidence for coordinate expression with Rh3 in R7 cells. Development. 1997;124:1665–1673. doi: 10.1242/dev.124.9.1665. [DOI] [PubMed] [Google Scholar]
- Pflugl G, Kallen J, Schirmer T, Jansonius JN, Zurini MG, Walkinshaw MD. X-ray structure of a decameric cyclophilin-cyclosporin crystal complex. Nature. 1993;361:91–94. doi: 10.1038/361091a0. [DOI] [PubMed] [Google Scholar]
- Price ER, Jin M, Lim D, Pati S, Walsh CT, McKeon FD. Cyclophilin B trafficking through the secretory pathway is altered by binding of cyclosporin A. Proc Natl Acad Sci U S A. 1994;91:3931–3935. doi: 10.1073/pnas.91.9.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price ER, Zydowsky LD, Jin MJ, Baker CH, McKeon FD, Walsh CT. Human cyclophilin B: a second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proc Natl Acad Sci U S A. 1991;88:1903–1907. doi: 10.1073/pnas.88.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyott SM, Schwarze U, Christiansen HE, Pepin MG, Leistritz DF, Dineen R, Harris C, Burton BK, Angle B, Kim K, et al. Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes. Human molecular genetics. 2011;20:1595–1609. doi: 10.1093/hmg/ddr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzicka A, Wolfenden R. A proficient enzyme. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Reidt U, Wahl MC, Fasshauer D, Horowitz DS, Luhrmann R, Ficner R. Crystal structure of a complex between human spliceosomal cyclophilin H and a U4/U6 snRNP-60K peptide. Journal of molecular biology. 2003;331:45–56. doi: 10.1016/s0022-2836(03)00684-3. [DOI] [PubMed] [Google Scholar]
- Reimer U, Fischer G. Local structural changes caused by peptidyl-prolyl cis/trans isomerization in the native state of proteins. Biophys Chem. 2002;96:203–212. doi: 10.1016/s0301-4622(02)00013-3. [DOI] [PubMed] [Google Scholar]
- Satoh K, Nigro P, Matoba T, O'Dell MR, Cui Z, Shi X, Mohan A, Yan C, Abe J, Illig KA, et al. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nature medicine. 2009;15:649–656. doi: 10.1038/nm.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira M, Totrov M, Abagyan R. Prediction of the binding energy for small molecules, peptides and proteins. J Mol Recognit. 1999;12:177–190. doi: 10.1002/(SICI)1099-1352(199905/06)12:3<177::AID-JMR451>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Schiene-Fischer C, Aumuller T, Fischer G. Peptide Bond cis/trans Isomerases: A Biocatalysis Perspective of Conformational Dynamics in Proteins. Top Curr Chem. 2011 doi: 10.1007/128_2011_151. [DOI] [PubMed] [Google Scholar]
- Schneuwly S, Shortridge RD, Larrivee DC, Ono T, Ozaki M, Pak WL. Drosophila ninaA gene encodes an eye-specific cyclophilin (cyclosporine A binding protein) Proc Natl Acad Sci U S A. 1989;86:5390–5394. doi: 10.1073/pnas.86.14.5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science. 1991;251:283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- Schreiber SL. Immunophilin-sensitive protein phosphatase action in cell signaling pathways. Cell. 1992;70:365–368. doi: 10.1016/0092-8674(92)90158-9. [DOI] [PubMed] [Google Scholar]
- Sherry B, Yarlett N, Strupp A, Cerami A. Identification of cyclophilin as a proinflammatory secretory product of lipopolysaccharide-activated macrophages. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:3511–3515. doi: 10.1073/pnas.89.8.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh BH, Stamnes MA, Seavello S, Harris GL, Zuker CS. The ninaA gene required for visual transduction in Drosophila encodes a homologue of cyclosporin A-binding protein. Nature. 1989;338:67–70. doi: 10.1038/338067a0. [DOI] [PubMed] [Google Scholar]
- Smajlovic A, Berbic S, Schiene-Fischer C, Tusek-Znidaric M, Taler A, Jenko-Kokalj S, Turk D, Zerovnik E. Essential role of Pro 74 in stefin B amyloid-fibril formation: dual action of cyclophilin A on the process. FEBS Lett. 2009;583:1114–1120. doi: 10.1016/j.febslet.2009.02.037. [DOI] [PubMed] [Google Scholar]
- Smith T, Ferreira LR, Hebert C, Norris K, Sauk JJ. Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles. A role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum. The Journal of biological chemistry. 1995;270:18323–18328. doi: 10.1074/jbc.270.31.18323. [DOI] [PubMed] [Google Scholar]
- Snyder SH, Lai MM, Burnett PE. Immunophilins in the nervous system. Neuron. 1998;21:283–294. doi: 10.1016/s0896-6273(00)80538-3. [DOI] [PubMed] [Google Scholar]
- Stamnes MA, Shieh BH, Chuman L, Harris GL, Zuker CS. The cyclophilin homolog ninaA is a tissue-specific integral membrane protein required for the proper synthesis of a subset of Drosophila rhodopsins. Cell. 1991;65:219–227. doi: 10.1016/0092-8674(91)90156-s. [DOI] [PubMed] [Google Scholar]
- Stephenson RS, O'Tousa J, Scavarda NJ, Randall LL, Pak WL. In: The Biology of Photoreception. Cosens DJ, Vince-Price D, editors. Cambriddge, U.K.: 1983. pp. 477–501. [Google Scholar]
- Takahashi N, Hayano T, Suzuki M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature. 1989;337:473–475. doi: 10.1038/337473a0. [DOI] [PubMed] [Google Scholar]
- Tryon RC, White SD, Famula TR, Schultheiss PC, Hamar DW, Bannasch DL. Inheritance of hereditary equine regional dermal asthenia in Quarter Horses. Am J Vet Res. 2005;66:437–442. doi: 10.2460/ajvr.2005.66.437. [DOI] [PubMed] [Google Scholar]
- van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PG, Piersma SR, Fratantoni SA, Jimenez CR, Huizer M, Morsman AC, Cobben JM, et al. PPIB mutations cause severe osteogenesis imperfecta. American journal of human genetics. 2009;85:521–527. doi: 10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Wells S, Ryan A, Cagan R. ZD6474 suppresses oncogenic RET isoforms in a Drosophila model for type 2 multiple endocrine neoplasia syndromes and papillary thyroid carcinoma. Cancer research. 2005;65:3538–3541. doi: 10.1158/0008-5472.CAN-04-4561. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang S, Zhang J, Huang X, Xu C, Wang W, Liu Z, Wu J, Shi Y. A large intrinsically disordered region in SKIP and its disorder-order transition induced by PPIL1 binding revealed by NMR. The Journal of biological chemistry. 2010;285:4951–4963. doi: 10.1074/jbc.M109.087528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Han R, Zhang W, Yuan Y, Zhang X, Long Y, Mi H. Human CyP33 binds specifically to mRNA and binding stimulates PPIase activity of hCyP33. FEBS Lett. 2008;582:835–839. doi: 10.1016/j.febslet.2008.01.055. [DOI] [PubMed] [Google Scholar]
- Weininger U, Jakob RP, Eckert B, Schweimer K, Schmid FX, Balbach J. A remote prolyl isomerization controls domain assembly via a hydrogen bonding network. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12335–12340. doi: 10.1073/pnas.0902102106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Matunis MJ, Kraemer D, Blobel G, Coutavas E. Nup358, a cytoplasmically exposed nucleoporin with peptide repeats, Ran-GTP binding sites, zinc fingers, a cyclophilin A homologous domain, and a leucine-rich region. J Biol Chem. 1995;270:14209–14213. doi: 10.1074/jbc.270.23.14209. [DOI] [PubMed] [Google Scholar]
- Xu C, Zhang J, Huang X, Sun J, Xu Y, Tang Y, Wu J, Shi Y, Huang Q, Zhang Q. Solution structure of human peptidyl prolyl isomerase-like protein 1 and insights into its interaction with SKIP. The Journal of biological chemistry. 2006;281:15900–15908. doi: 10.1074/jbc.M511155200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Lu N, Zhou J, Chen ZN, Zhu P. Cyclophilin A up-regulates MMP-9 expression and adhesion of monocytes/macrophages via CD147 signalling pathway in rheumatoid arthritis. Rheumatology (Oxford) 2008;47:1299–1310. doi: 10.1093/rheumatology/ken225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama N, Hayashi N, Seki T, Pante N, Ohba T, Nishii K, Kuma K, Hayashida T, Miyata T, Aebi U, et al. A giant nucleopore protein that binds Ran/TC4. Nature. 1995;376:184–188. doi: 10.1038/376184a0. [DOI] [PubMed] [Google Scholar]
- Zuker CS. The biology of vision of Drosophila. Proc Natl Acad Sci U S A. 1996;93:571–576. doi: 10.1073/pnas.93.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]