

Abstract
Loss of the PTEN tumor suppressor enhances metastatic risk in breast cancer, although the underlying mechanisms are poorly defined. We report that homozygous deletion of PTEN in mammary epithelial cells induces tubulin-based microtentacles (McTNs) that facilitate cell reattachment and homotypic aggregation. Treatment with contractility-modulating drugs showed that McTNs in PTEN−/− cells are suppressible by controlling the actin cytoskeleton. Since outward microtubule extension is counteracted by actin cortical contraction, increased activity of actin-severing proteins could release constraints on McTN formation in PTEN−/− cells. One such actin-severing protein, cofilin, is activated in detached PTEN−/− cells which could weaken the actin cortex to promote McTNs. Expression of wild-type cofilin, an activated mutant (S3A), and an inactive mutant (S3E) demonstrated that altering cofilin phosphorylation directly affects McTNs formation. Chemical inhibition of PI3K did not reduce McTNs or inactivate cofilin in PTEN−/− cells. Additionally, knock-in expression of the two most common PI3K-activating mutations observed in human cancer patients did not increase McTNs or activate cofilin. PTEN loss and PI3K activation also caused differential activation of the cofilin regulators, LIM-kinase1 (LIMK) and Slingshot-1L (SSH). Furthermore, McTNs were suppressed and cofilin was inactivated by restoration of PTEN in the PTEN−/− cells, indicating that both the elevation of McTNs and activation of cofilin are specific results arising from PTEN loss. These data identify a novel mechanism by which PTEN loss could remodel the cortical actin network to facilitate McTNs that promote tumor cell reattachment and aggregation. Using isogenic MCF-10A PTEN−/− and PIK3CA mutants, we have further demonstrated that there are clear differences in activation of cofilin, LIMK, and SSH between PTEN loss and PI3K activation, providing new evidence that these mutations yield distinct cytoskeletal phenotypes that could impact tumor biology.
Keywords: PTEN, microtentacle, actin cortex, contractility, cofilin, metastasis, circulating tumor cells
INTRODUCTION
Seeding of solid breast tumors in metastatic sites generally requires release from extracellular matrix (ECM) and periods of suspension in the vasculature or lymphatics (1). This anchorage-independent survival is a major determinant of tumor dormancy, recurrence, and clinical prognosis (2–4). Since nearly 90% of human breast tumors are epithelial carcinomas, it is important to study how mammary epithelial cells (MECs) respond to ECM-detachment to thoroughly understand metastatic progression and therapeutic opportunities. We have previously shown that mammary epithelial cells extend long dynamic microtubule-based protrusions of the plasma membrane, termed microtentacles (McTNs). McTNs are induced by ECM-detachment, are structurally distinct from classical actin-based extensions of adherent cells (5), and persist for days in breast tumor lines that are resistant to anoikis (6, 7). Although traditional studies of tumor dormancy have established the notion that solitary disseminated tumor cells undergo extended periods of inactivity during mitotic quiescence, (8) our findings indicate that detached cells continue to actively respond to the microenvironment via cytoskeletal rearrangement and McTNs without requiring active cell growth.
Metastatic breast tumor cells often display abnormalities in actin cytoskeletal organization compared to their non-tumorigenic counterparts (9–12). Normal cells counteract the expansion of microtubules with inward tension from the actin cortex to control cell morphology. However, altering the balance between microtubules and actin has serious implications for circulating tumor cell (CTC) dissemination, as metastatically efficient CTCs can avoid shear-induced fragmentation by undergoing sphere-to-cylinder shape transformations within capillaries (13). PTEN can influence actin organization through dephosphorylation of its lipid substrate phosphatidylinositol 3,4,5-trisphosphate (PIP3) to yield phosphatidylinositol 4,5-bisphosphate (PIP2). Using homologous recombination, we engineered a homozygous deletion in PTEN in MCF-10A human mammary epithelial cells (14). These PTEN−/− cells have an increase in the PIP3:PIP2 ratio compared to the MCF-10A parental cells, that has been verified by mass spectrometry (15). Both phosphoinositide lipids are important regulators of the actin cytoskeleton. PIP2 directly binds actin-associated proteins to tether the actin cytoskeleton to the plasma membrane, or activates proteins that are involved in the initiation of de novo actin polymerization (e.g. vinculin, profilin, and α-actinin) (16–18). PIP3 can modify Guanine nucleotide Exchange Factors (GEFs) and GTPase Activating Proteins (GAPs), regulators of the Rho-family GTPases that function as modulators of the actin cytoskeleton (19). Interestingly, the activities of virtually all actin-severing proteins are downregulated by association with PIP2, while proteins that promote actin filament nucleation and bundling, and membrane coupling are typically activated by PIP2 (20). Therefore the net effect of reduced PIP2 is actin disassembly.
Cofilin is an actin-severing protein that promotes the dissociation of G-actin from the minus end of F-actin and is activated by dephosphorylation. Cofilin-PIP2 binding is believed to abrogate the affinity of cofilin for actin (21, 22). It is possible that the reduced PIP2 in the PTEN−/− cells could yield more cofilin available to bind actin. By decreasing the ability to counteract PI3K-driven increases in PIP3, PTEN−/− cells could also undergo greater actin rearrangement.
In this study, we have determined that the loss of PTEN expression in MECs enhances the formation of long, dynamic McTNs following ECM detachment. By manipulating cellular contractility, we further established that these tubulin-based McTNs promote cell attachment. The actin-severing protein, cofilin, was more highly activated in the PTEN−/− cells providing a mechanism for greater McTN formation. However, neither the McTN increase nor the cofilin activation were dependent on activation of PI3K or Akt. Patient-derived PI3K mutations were also unable to induce McTNs or cofilin activation. These findings reveal that PTEN loss has a further ability to both induce McTNs and rescue cells from apoptosis (14), producing a unique phenotype which could facilitate tumor cell metastasis by rendering epithelial cells insensitive to extracellular matrix detachment while simultaneously increasing epithelial reattachment responses.
RESULTS
PTEN −/− MECs produce more microtentacles
We have previously evaluated the production of tubulin-based McTNs in a panel of human breast tumor cell lines (23). Detachment of the invasive cell lines SUM-149, Hs578t, HCC1395, and MDA-MB-436 yields McTNs that are longer and more numerous compared with noninvasive ZR-75-1, Bt-20, SkBr3, and MDA-MB-468 lines, which display relatively short and rigid McTNs. Interestingly, most of these breast cancer cell lines have either already lost PTEN expression or harbor the reciprocal PI3K activating mutations and are not suitable for studying PTEN loss in human breast epithelial cells (Supplemental Table T1). PTEN loss can be more clearly addressed in the isogenic MCF-10A background without complication from the numerous other genetic differences in this panel of breast tumor cell lines. To determine whether the absence of PTEN expression is sufficient to alter McTNs, parental MCF-10A and PTEN−/− clones were evaluated for McTN production. Each of the three PTEN−/− clones exhibited double the amount of cells forming McTNs compared to their isogenic PTEN-expressing counterparts (Fig. 1A). Immunofluorescence staining confirmed the presence of α-tubulin within these microtentacle protrusions (Fig. 1B).
Figure 1. Tubulin-based McTNs are increased on PTEN−/− MECs.
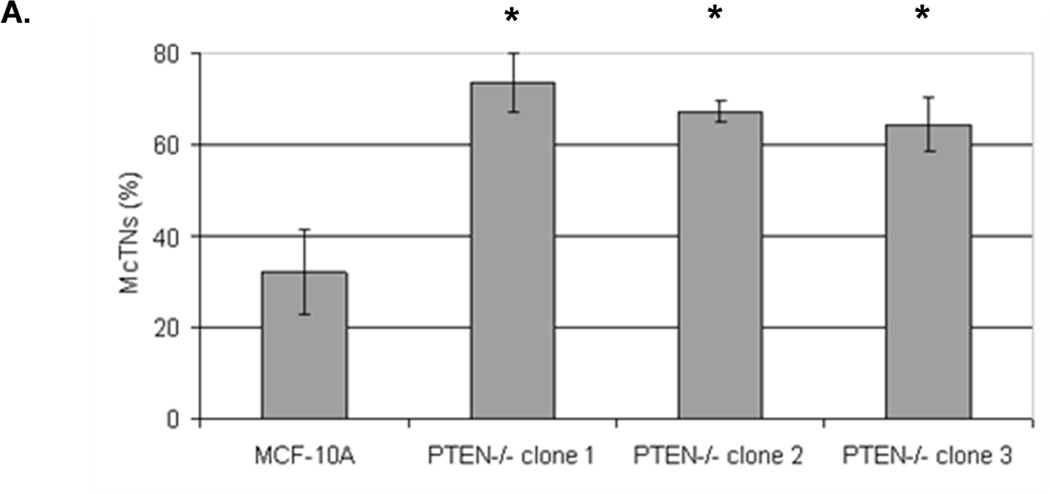
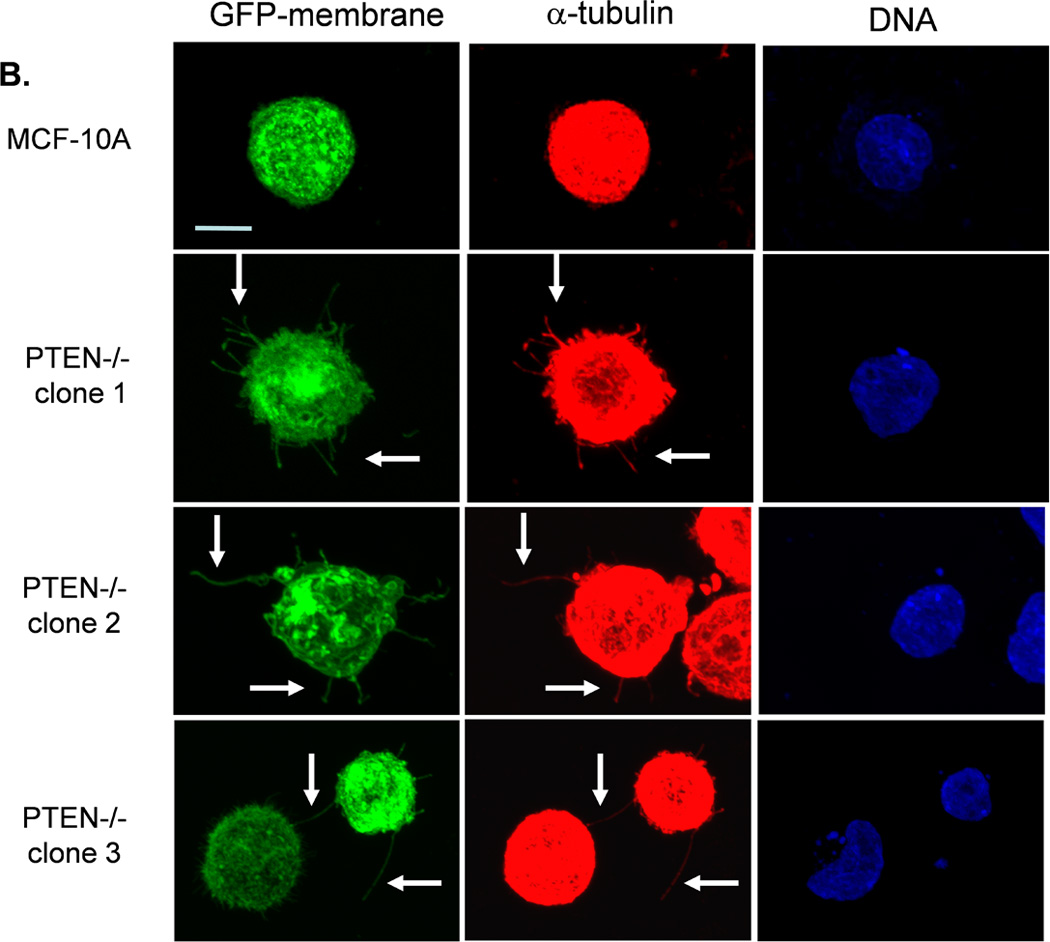
A. MCF-10A cells and three PTEN−/− clones were fluorescently stained with CellMask Orange and suspended in an ultra-low attachment plate in complete media. Blinded counting showed a consistent McTN increase in the PTEN−/− cells (n=6, *p<0.002). B. Suspended cell immunofluorescence reveals α-tubulin within the membrane protrusions (green - GFP-mem, red - α-tubulin, blue - Hoechst) (bar = 10µM).
Cell attachment and homotypic aggregation are increased by PTEN deletion
To assess the effect of McTN induction in PTEN−/− cells, cell adherence was assessed by real-time changes in electrical impedance as cells attached to microwells with integrated electrodes. This impedance measurement was then normalized to parental MCF-10A cells to calculate relative attachment curves. Within the first hour, initial adhesion was elevated in each of the PTEN−/− cells clones, which attached over 2-fold more rapidly than the MCF-10A parental cells (Fig. 2A; raw data, Supplemental S1).
Figure 2. Attachment and homotypic aggregation are enhanced in PTEN−/− cells.
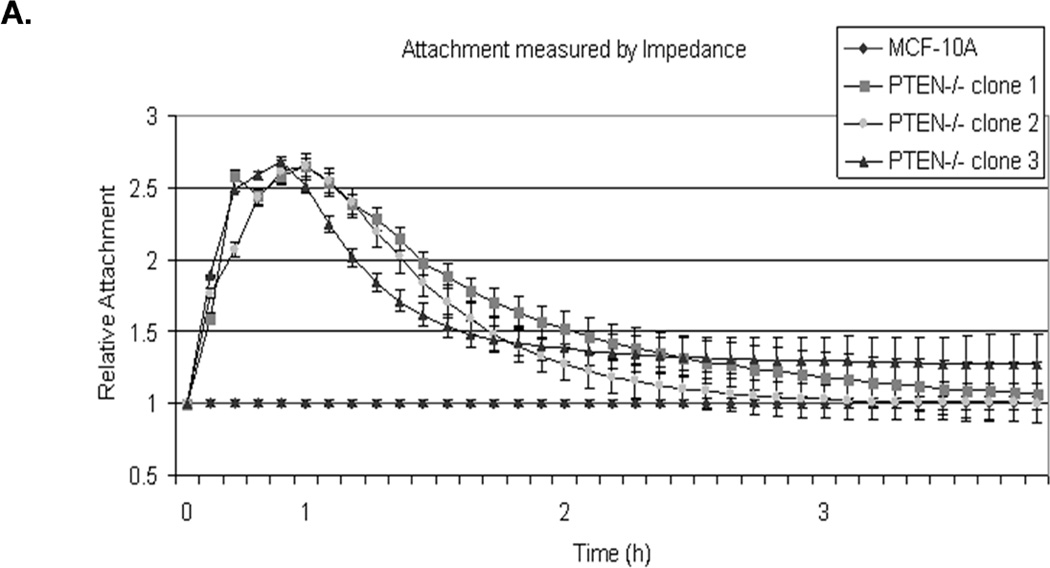
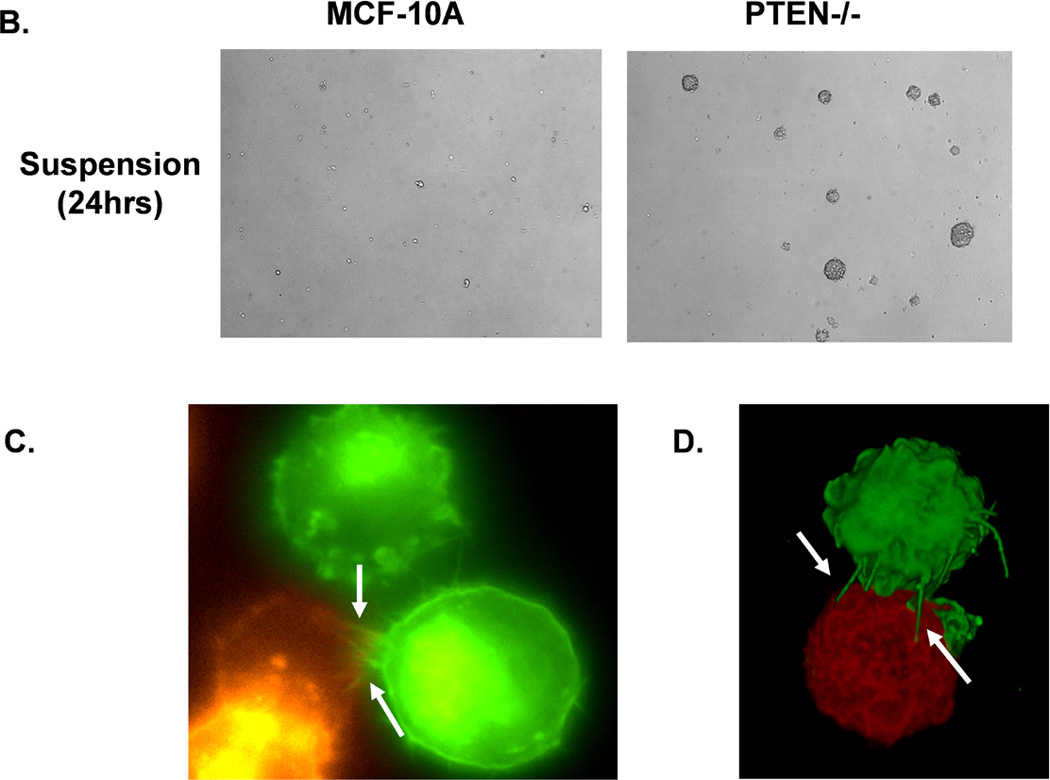
A. A greater change in impedance of the PTEN−/− cells illustrates an increase in attachment compared to the MCF-10A cells. Impedance values are normalized to MCF-10A (n=4, representative results from triplicate experiments). B. An equal number of cells were suspended in normal growth media in ultra-low attachment plates. After 48h, only the PTEN−/− cells aggregate into tight, spheroid structures. C. Live cell imaging of two cell populations of the same PTEN clone, one GFP-mem transfected (green) and the other CellMask stained (red), were combined and suspended over a BSA coated glass surface to prevent attachment. Arrows indicate McTNs extending from the GFP-mem cell attaching to a CellMask stained cell. D. 3D computer rendered image of PTEN−/− cells from separate populations (GFP-mem transfected and CellMask stained) associating. Arrows indicate McTNs from the GFP-mem cell contacting the CellMask stained cell.
Increased cellular aggregation correlates with an elevated metastatic potential in vivo (24), so we sought to examine the influence of PTEN−/− induced McTNs on homotypic cell-cell binding. When suspended, PTEN−/− cells start aggregating within 6 hours, while the MCF-10A cells remained as individual cells. By 24 hours, the PTEN−/− cells formed distinct spheroid clusters (Fig. 2B). Flow cytometry confirmed that, after 24 hours, the MCF-10A and PTEN−/− cells are viable and arrested in G1 (Supplemental S2), indicating that the spheroid clusters of PTEN−/− cells arise from cell aggregation rather than division. Time-lapse fluorescence microscopy of live cells expressing membrane-targeted GFP has illustrated that McTNs promote aggregation by encircling neighboring cells (7, 25). Mixing PTEN−/− cells differentially labeled with red and green fluorescent membrane probes further demonstrated this cell-wrapping phenomenon with epifluorescence (Fig. 2C) and confocal microscopy (Fig. 2D).
Attenuating actomyosin contractility increases McTNs production and cell reattachment
Current evidence supports a model where the balance between the expansive force of polymerizing microtubules and inward force of the actin cortex regulates McTN protrusion from the cell surface (25). Accordingly, microtubule-stabilization enhances the prevalence of McTNs on detached epithelial cells (7, 23, 26, 27). McTNs are supported by coordination of the intermediate filament protein, vimentin, and stable posttranslational modified forms of α-tubulin (Glu-tubulin) (23). However neither vimentin nor Glu-tubulin levels were affected by deletion of PTEN (Supplemental S3). Therefore, we investigated the potential role of the actin cytoskeleton on McTN formation in response to PTEN deletion. Small molecule inhibitors were used to examine the role of non-muscle myosin II (NM II), which plays a fundamental role in cell shape, movement, and contractile activity by regulating the actin cytoskeleton (28). Blebbistatin binds to the myosin-ADP-Pi complex to interfere with phosphate release, thus blocking myosin in a functionally inactive, actin-detached state, paralyzing the motor activity of NM II (29). The myosin head is unable to contact actin and perform its “power stroke,” resulting in a weakened actin cortex. The decrease in cellular contractility induced by blebbistatin elevated McTNs 3-fold in MCF-10A parental cells and 2-fold in PTEN−/− cells (Fig. 3A). The presence of tubulin within the protrusions confirmed their McTN identity (Fig. 3B). This result suggests that decreasing tension in the actin cortex results in the outward expansion of microtubules during McTN extension (30).
Figure 3. Decreased contractility enhances McTNs.
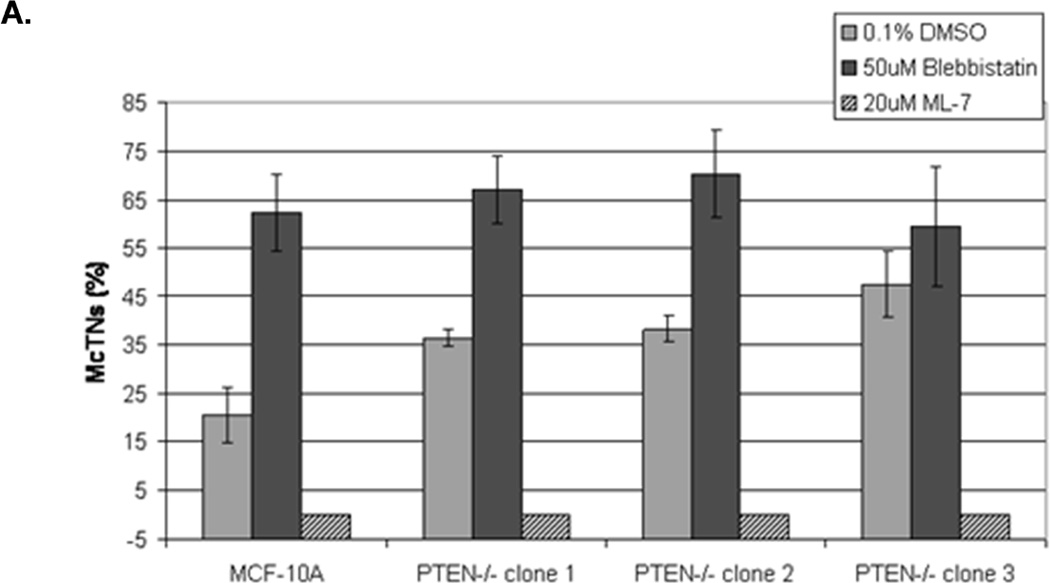
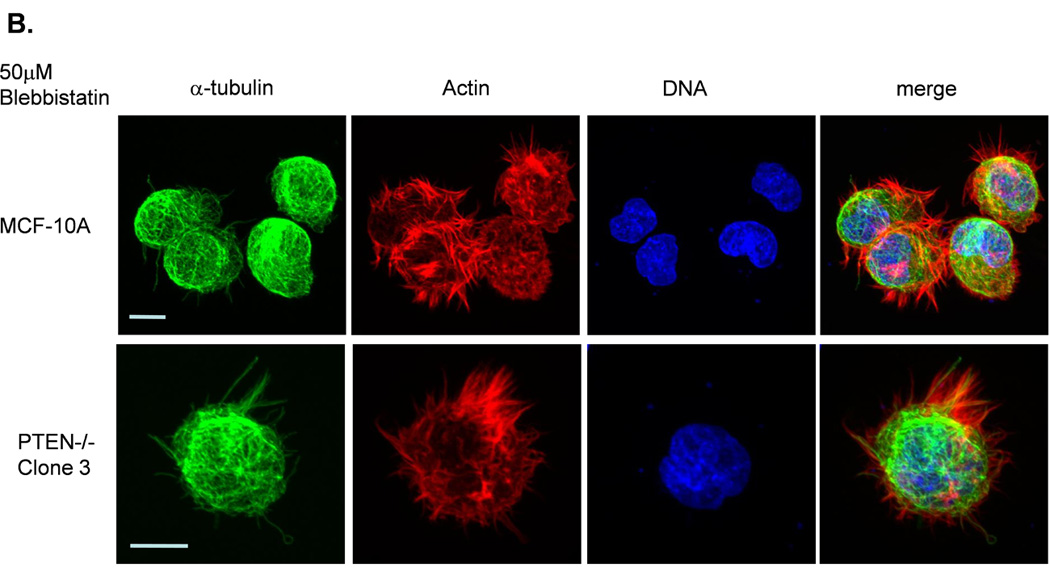
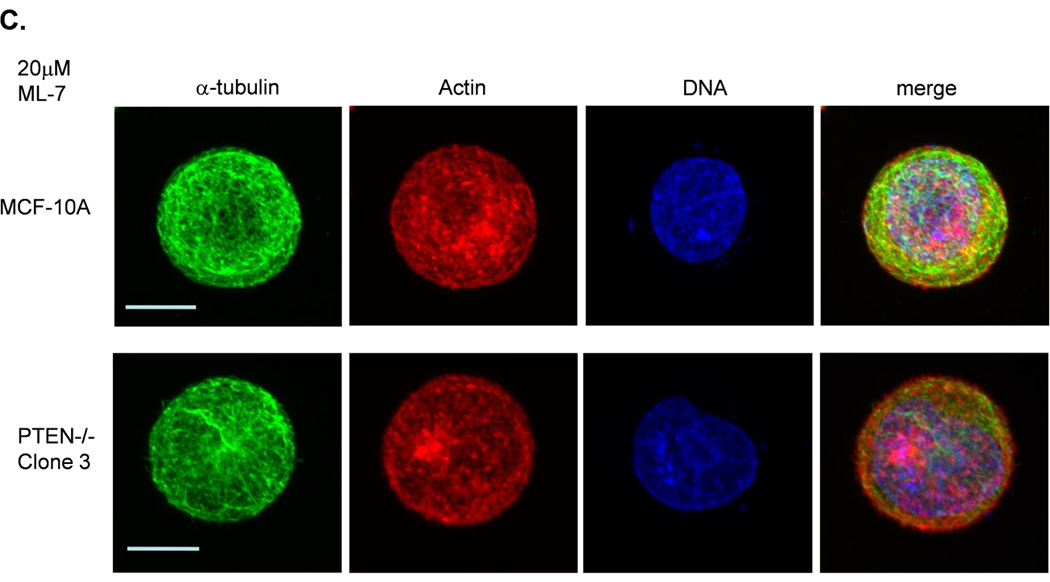
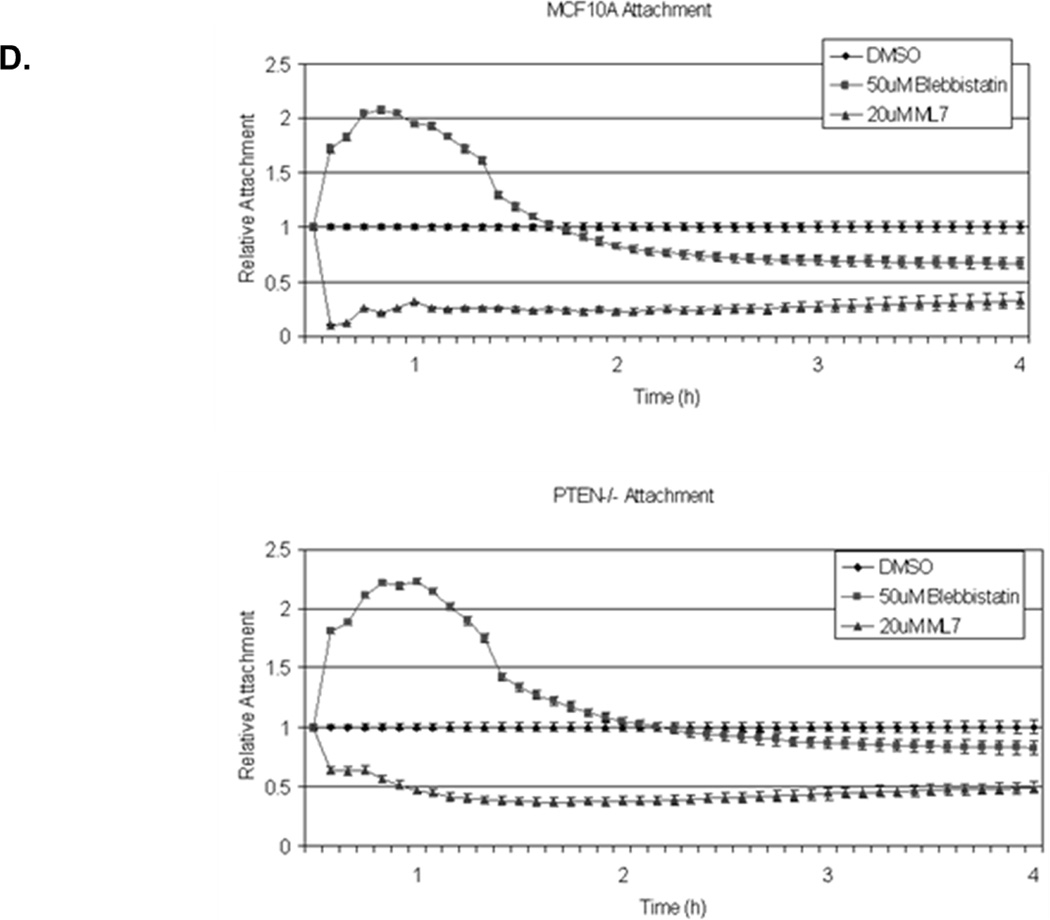
A. McTNs increase in cells suspended in 50µM blebbistatin (p<0.05), but are suppressed in cells suspended in 20µM ML-7 (p<0.001). B. Suspended cell immunofluorescence of MCF-10A cells and a representative PTEN−/− clone in the presence of 50µM blebbistatin or C. 20 µM ML-7. Induced McTNs contain α-tubulin (green) extending under the actin (red) cortex. Cells treated with ML-7 show no outwardly extending protrusions (bar = 10µM). D. Real time attachment measurements of blebbistatin treated MCF-10A and PTEN−/− cells attach approximately 2-fold quicker at 1 hour compared to the DMSO control cells. At all time points, ML-7 treated cells attached approximately 50% less than that of the controls. Impedance values are normalized to DMSO controls (n=4, representative results from triplicate experiments).
ML-7 is a myosin light chain kinase (MLCK) inhibitor that prevents MLCK from phosphorylating myosin light chain (MLC), thus decreasing NMII’s ATPase activity and inhibiting contraction (31). ML-7 was also expected to greatly reduce cortical tension and contractility, thereby increasing McTN formation. Surprisingly, McTN production was completely abolished by ML-7 treatment (Fig. 3A and C). MCF-10A and PTEN−/− cells treated with 20µM ML-7, showed only a minimal reduction of pMLC (Supplemental S4) consistent with others who have reported that ML-7 does not decrease pMLC (31–33). MLCK has also been shown to play a role in disrupting the cortex (34) and have a kinase-independent activity that stimulates the dissociation of actin and myosin (35). If cortical disruption and dissociation of actin and myosin is inhibited by ML-7, the actin cortex would essentially be “locked,” a prediction consistent with our observations that ML-7 induces a rounded, smooth shape that suppresses McTNs. Despite their inhibitory effects on myosin, blebbistatin treatment results in a “loosening,” while ML-7 treatment results in a “tightening” of actin cortex (30).
Our previous studies have shown McTNs promote the reattachment of cells to tissue culture substrates and endothelial monolayers (7, 23, 27). However, when McTNs were induced by actin depolymerization, cell attachment was not significantly increased (7). While actin depolymerization can decrease cellular contractility to increase McTNs, it may be also impair cytoskeletal functions necessary for firm adhesion. Blebbistatin induced McTNs, but to verify that these induced McTNs maintained functionality, we assessed cell-substratum reattachment. Change in impedance was measured for PTEN−/− and parental cells during initial adhesion (the first hour after initial cell seeding). In the presence of blebbistatin, cells adhered 2-fold quicker than that of the control (Fig. 3D; raw data, Supplemental S5). Conversely, cells plated in ML-7 containing media attached 2-fold slower and never regained the ability to spread (Fig. 3D; raw data, Supplemental S5). While broadly depolymerizing actin generates McTNs that cannot increase attachment, specifically altering actomyosin contractility can either suppress McTNs (ML-7) or induce McTNs that remain functional for cell-substratum attachment (blebbistatin).
Activated cofilin is elevated in the suspended PTEN−/− MECs and regulates McTNs
We were able to manipulate cellular contractility, the actin cytoskeleton, and McTN production by modulating the function of NM II. Therefore, we examined the phosphorylation status of MLC, but no consistent differences between suspended MCF-10A and PTEN−/− clones were observed (Supplemental S6). Alternatively, we found that the MCF-10A parental cells maintained a higher level of phosphorylated (inactive) cofilin (Fig. 4A). Densitometry analysis of four separate experiments shows that pCofilin levels only decrease 20% in MCF-10A cells after detachment. By comparison, pCofilin levels are reduced 55–75% in PTEN−/− cells, indicating a more robust detachment-induced activation of cofilin in PTEN−/− cells (Fig. 4B). We constructed a non-phosphorylatable mutant cofilin (S3A) which remains active (36) upstream of the IRES-GFP marker and scored GFP-positive transfected cells for McTNs production. There is a significant difference in cofilin phosphorylation and McTN generation between vector control and cofilin WT expressing cells and between the cofilin WT and S3A expressing cells (Fig. 4C). MCF-10A cells were able to highly phosphorylate exogenous WT cofilin and suppress McTNs. Although the S3A mutant did not induce McTNs to the levels observed in PTEN−/− cells, likely due to the fact that it was unable to suppress phosphorylation of endogenous cofilin as the levels of pCofilin remain similar to that of the non-transfected and vector transfected controls (Fig. 4C), it was able to relieve suppression of McTNs observed in the WT overexpressing cells.
Figure 4. Cofilin is activated in suspended cells, but to a much higher degree in cells without PTEN expression.
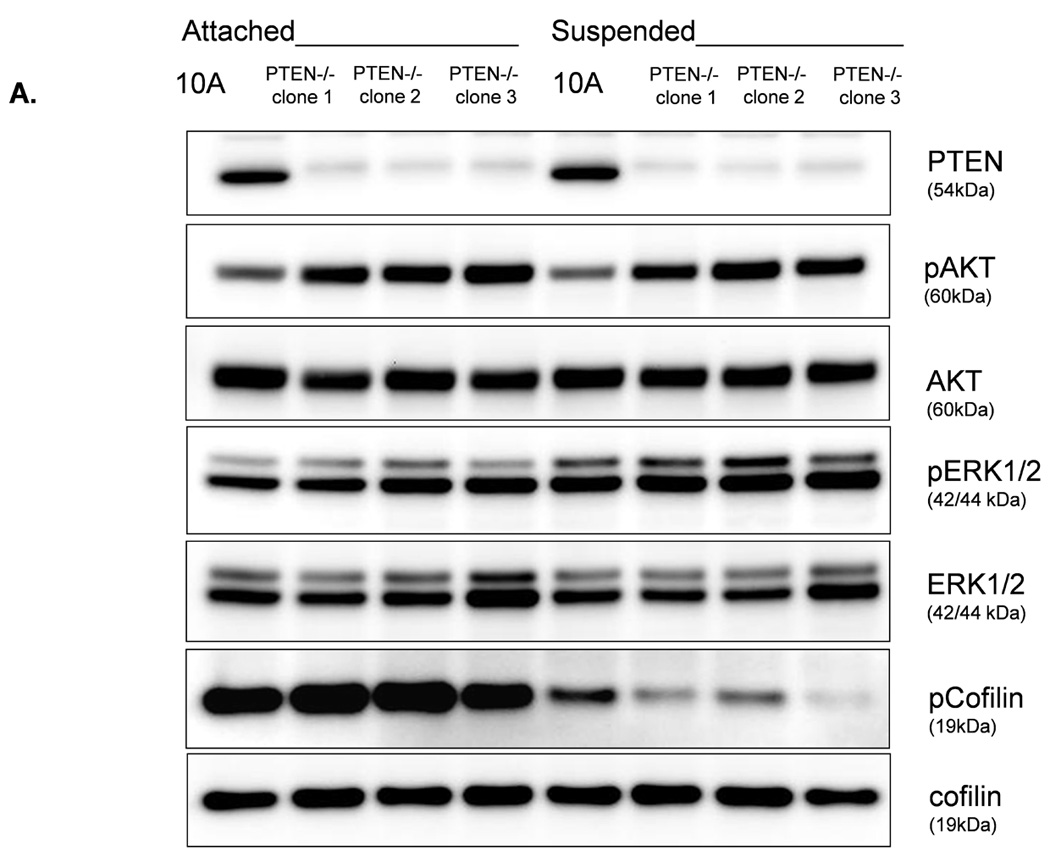
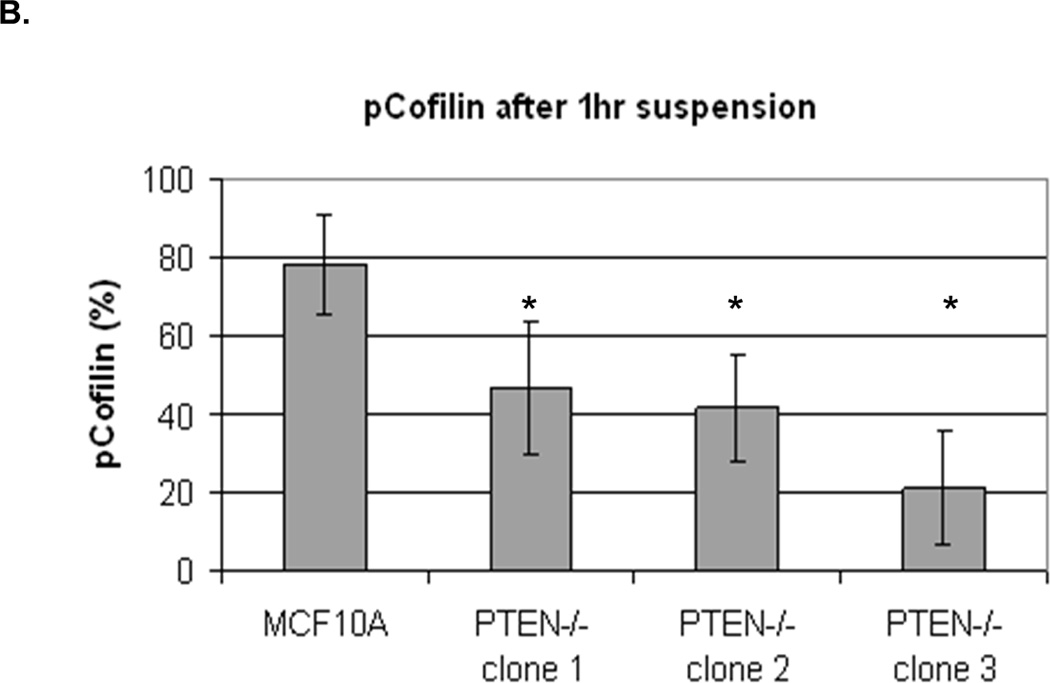
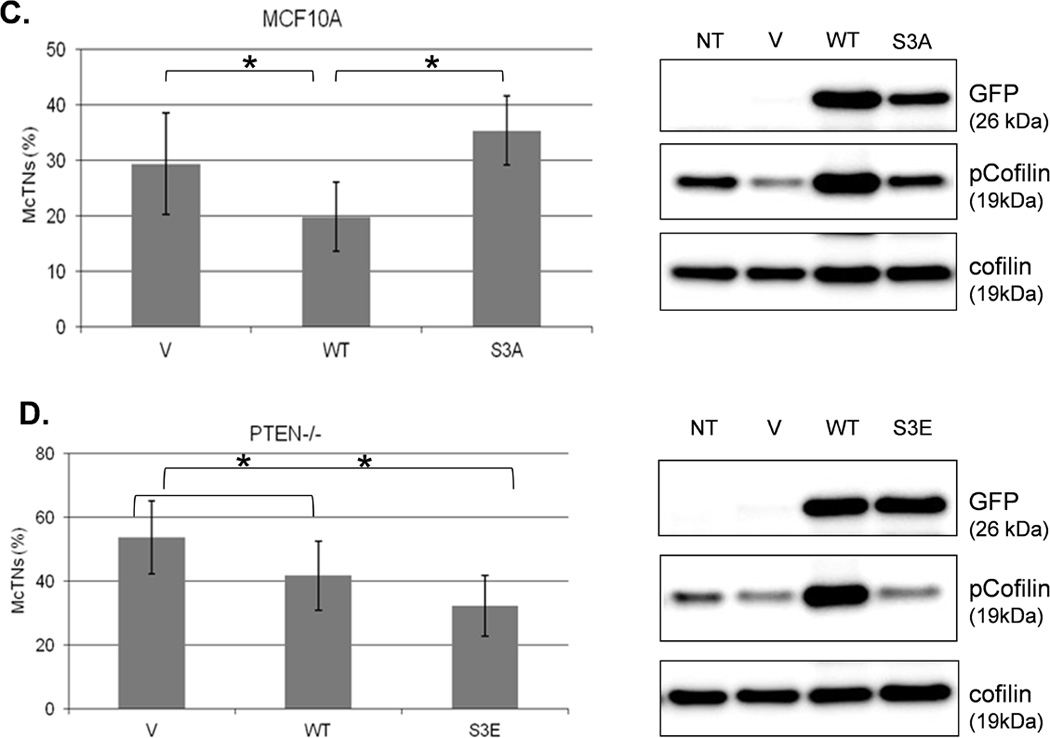
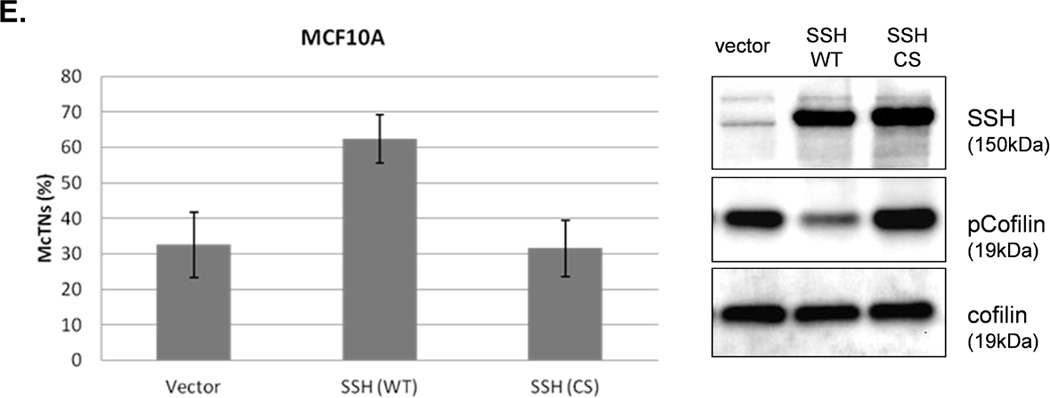
A. Representative Western blot analysis shows that cofilin is highly phosphorylated (pCofilin) and inactive when cells are attached and dephosphorylated to an active form when cells are detached. B. Densitometry analysis shows that pCofilin levels only decrease 20% in MCF-10A cells after detachment. By comparison, pCofilin levels are reduced 55–75% in PTEN−/− cells, indicating a more robust detachment-induced activation of cofilin in PTEN−/− cells. Levels of pCofilin were normalized to total cofilin in each sample and the ratio of pCofilin in suspended cells compared to attached cells was determined (n=4, *p<0.01). C. MCF-10A cells transfected with vector only (V), cofilin (WT), or mutant cofilin (S3A) and D. PTEN−/−cells transfected with vector only (V), cofilin (WT), or mutant cofilin (S3E) for 24h. Untransfected (NT) and transfected cells were blindly counted for McTNs (n=6, *p<0.01) and analyzed by Western blotting. E. MCF-10A cells were transfected with vector only (V), wild-type Slingshot-1L (SSH WT), or catalytically inactive Slingshot-1L (SSH CS) for 24hr and scored blindly for McTNs (n=6, *p<0.01) or analyzed by Western blotting.
The PTEN−/− cells were similarly able to highly phosphorylate exogenous WT cofilin which resulted in suppression of McTNs (Fig. 4D). To further confirm an increase in inactivated cofilin could decrease McTNs production, a cofilin S3E phospho-mimic was also constructed for expression in the PTEN−/− cells (36) and able to suppress McTNs (Fig. 4D). Beyond exogenous expression of wild-type and mutant cofilin, we were able to manipulate endogenous cofilin phosphorylation levels via expression of the cofilin phosphatase, Slingshot-1L (SSH). SSH expression dephosphorylates cofilin (activates) leading to an increase in McTN production (Fig. 4E). Both the vector control and the catalytically inactive SSH (SSH CS) are unable to dephosphorylate cofilin or produce any increase in McTNs (Fig. 4E).
Increased activated cofilin in the PTEN−/− MECs is not due to elevated PI3K or AKT activity
The absence of PTEN expression promotes the activation of the PI3K and MAPK pathways (14), and PTEN−/− cells maintain higher levels of pAKT than parental controls in suspension (Fig. 4A). To determine the importance of the PI3K pathway in the production of McTNs, a small molecule inhibitor (LY294002) was used to suppress PI3K activity, which was confirmed by a dose-dependent decrease in pAKT in all three PTEN−/− clones (Fig. 5A). If the increase in PI3K activity or pAKT in the PTEN−/− cells were responsible for the decrease in pCofilin, the pCofilin levels should increase upon LY294002 treatment. However, this was not the case. Despite the presence of the PI3K inhibitor, the pCofilin levels remained much lower in all three PTEN−/− clones than the MCF-10A controls (Fig. 5A) and addition of LY294002 did not alter McTN production (Fig. 5B). Therefore, PI3K activity and/or pAKT are not responsible for the cofilin activation and increased McTNs in the suspended PTEN−/− cells. In support of this conclusion, the MCF-10A cells with the “knock-in” PIK3CA mutations in exon 9 or exon 20, which render PI3K constitutively active (37), do not exhibit increased McTNs over the MCF-10A cells (Fig. 5C). Additionally, although the clones with the activating PI3K maintain elevated pAKT levels in suspension, their pCofilin levels do not decrease past that of the suspended MCF-10A cells (Fig. 5D and E). Future investigations will be required to determine if PTEN’s role as a broad-spectrum protein phosphatase or cytoskeletal docking protein are responsible for this cofilin regulation, but our results establish that PTEN loss activates cofilin without requiring PI3K or Akt activity.
Figure 5. Increased cofilin activity in the PTEN−/− MECs is not due to elevated PI3K or AKT activity.
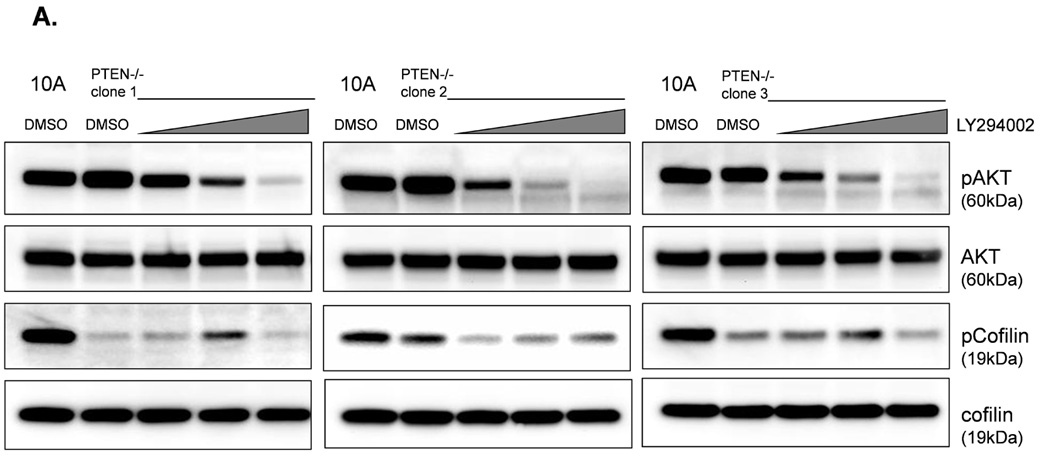
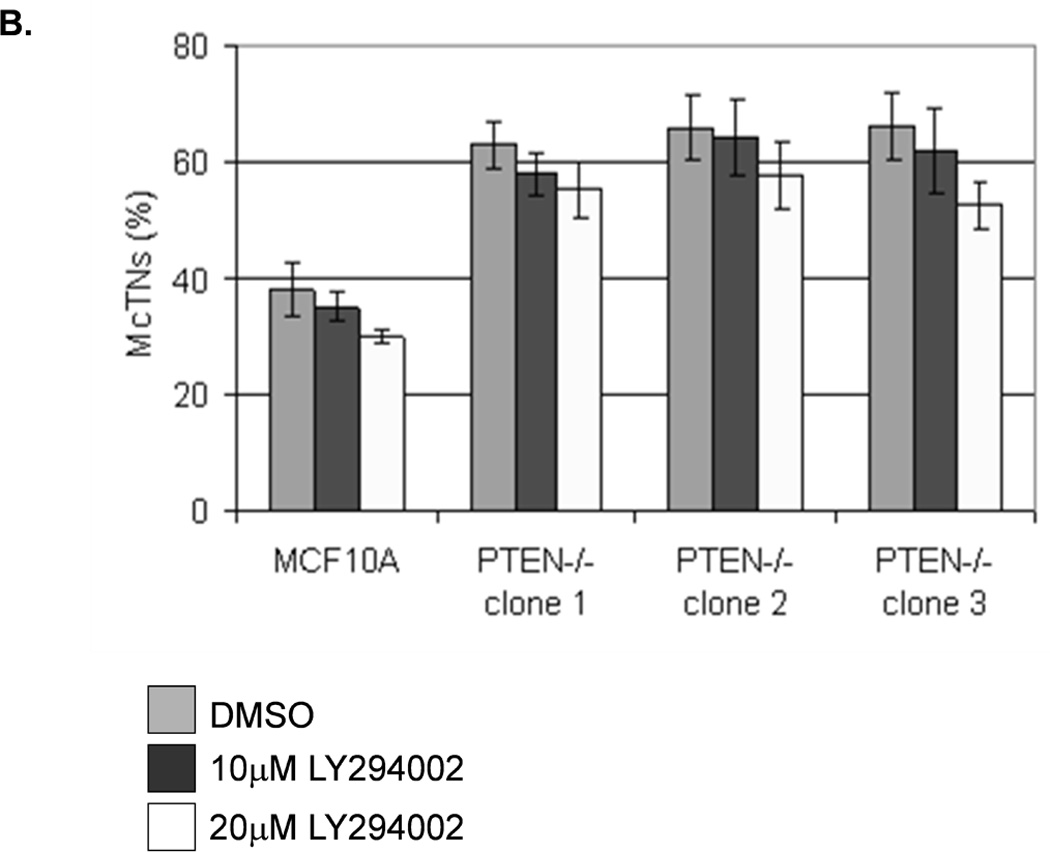
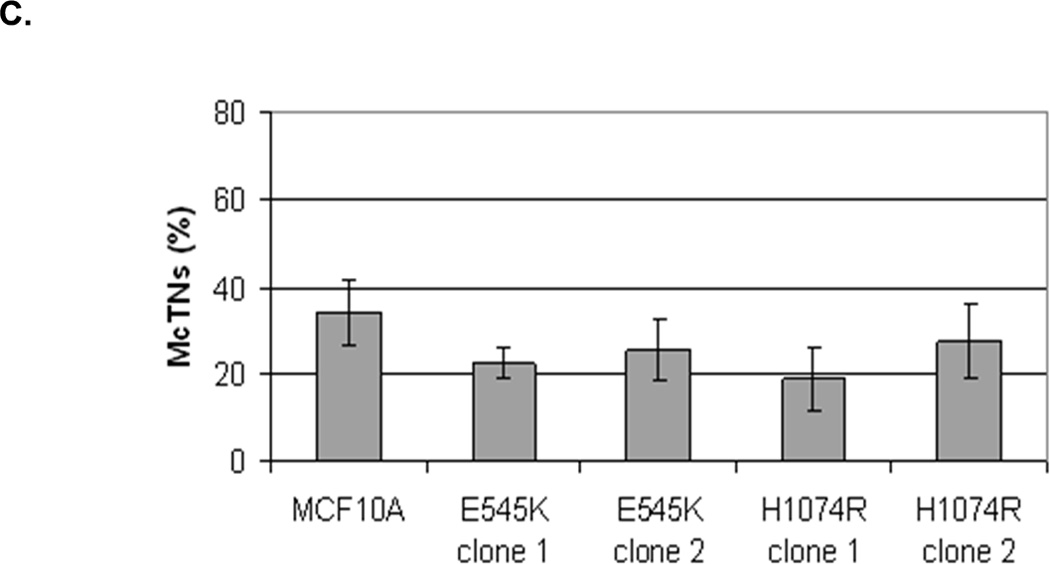
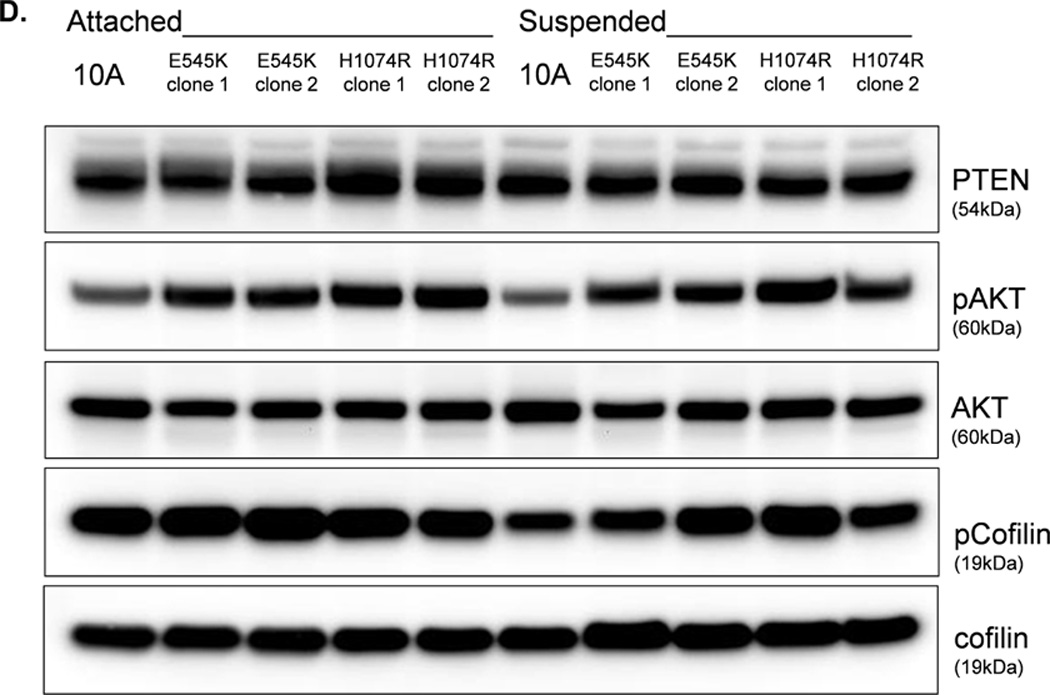
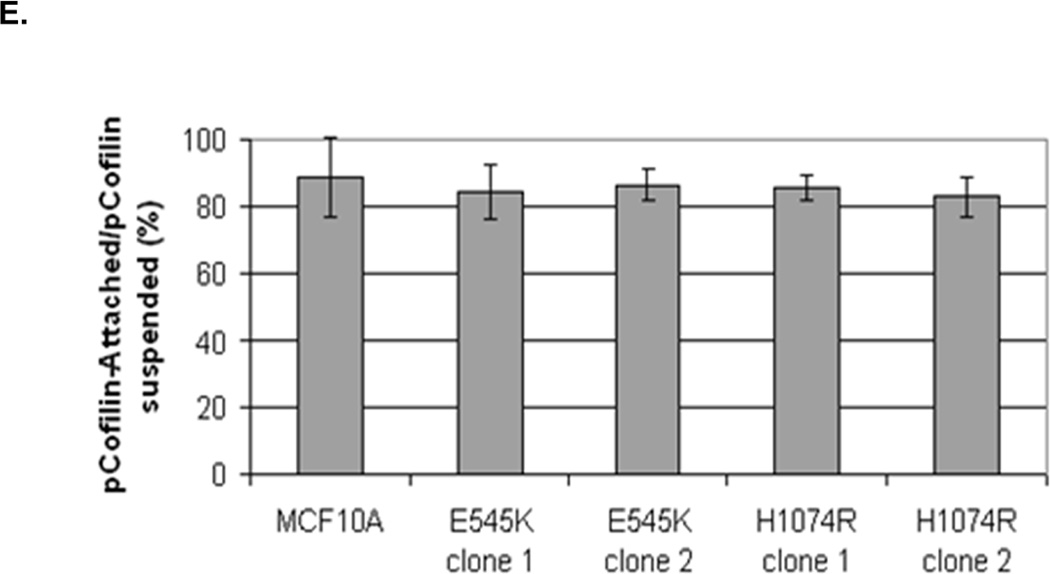
A. Western blot analysis of suspended MCF-10A and PTEN−/− clones in the presence of DMSO vehicle, or treatment with LY294002 (5µM, 10µM, and 20µM). B. Blinded McTNs counts performed on suspended cells in the presence and absence of LY294002 (n=6). C. Blinded McTNs counts performed on suspended MCF-10A and two clones of MCF-10A PIK3CA with the patient-derived E545K activating mutation in exon 9 and two clones of MCF-10A PIK3CA with the patient-derived H1074R mutation in exon 20 (n=6). D. Representative Western blot analysis of attached and suspended MCF-10A cells compared to those bearing the E545K and H1074R PIK3CA mutations. E. Densitometry results show the ratio of suspended normalized pCofilin levels to attached normalized cofilin levels is similar in the MCF-10A and PIK3CA mutant cells (n=3).
PI3K mutation and PTEN loss differentially regulate LIMK and SSH
LIM-kinase-1 (LIMK) and Slingshot-1L (SSH-1L) are known to specifically regulate cofilin phosphorylation. LIMK can be activated through phosphorylation of T508 by downstream kinases in the Rho family (38–40), but SSH-1L can be inactivated by phosphorylation (S978) (41, 42). Western blot analysis of PTEN−/− cells reveals an absence of phosphorylated pLIMK-T508 and pSSH-1L-S978 (Fig. 6A), and there is no difference in pLIMK-T508 and pSSH-1L-S978 levels in attached and detached PTEN−/− cells. Therefore, the phosphorylation states of LIMK and SSH-1L at these sites are likely not responsible for the robust decrease in cofilin phosphorylation in the suspended PTEN−/− cells. This finding lends additional support to our conclusion that the change in pCofilin in the PTEN−/− cells is a PI3K-independent event as SSH-1L has been shown to modulate cofilin through PI3K (43). In contrast; cells expressing mutant PI3K do maintain higher levels of pLIMK-T508 and pSSH-1L-S978 (Fig. 6B). Therefore, the presence of phosphorylated pLIMK-T508 and pSSH-1L-S978 in the mutant PI3K expressing cells and the absence of pLIMK-T508 and pSSH-1L-S978 in the PTEN−/− cells provides a mechanistic difference that could account for the different levels of pCofilin observed between these 2 genotypes. Defining the complete signaling mechanisms by which PTEN loss and PI3K mutation cause this differential regulation of pLIMK-T508 and pSSH-1L-S978 will be an important focus for future studies.
Figure 6. LIMK and SSH are differentially regulated by PTEN loss and PIK3CA activation in detached MECs.
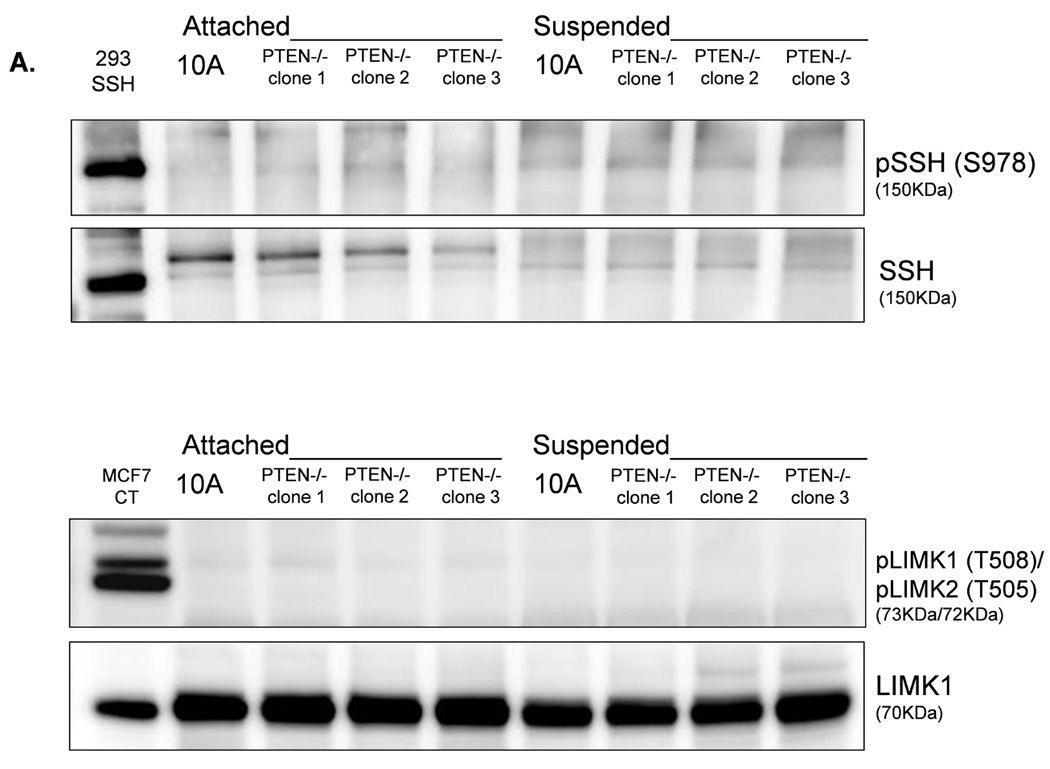
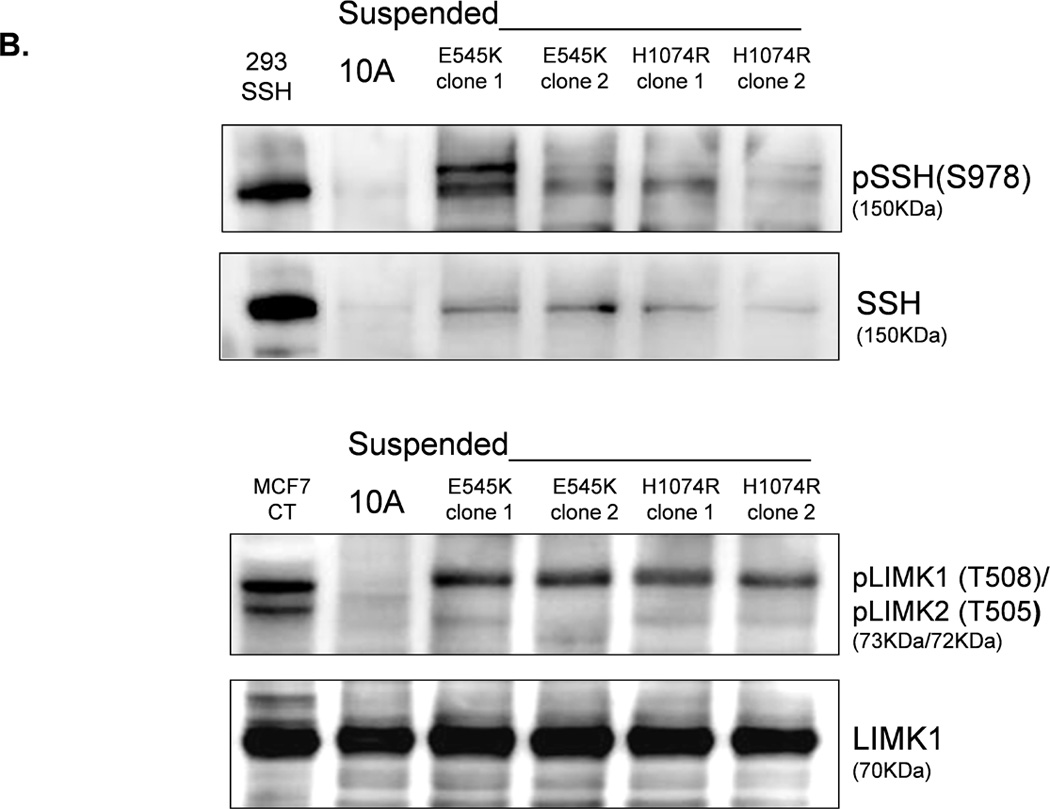
A. Western blot analysis of attached and suspended MCF-10A and PTEN−/− clones. 293T cells overexpressing SSH and MCF7 cells are used as positive controls for the SSH and LIMK antibodies, respectively. B. Western blot analysis of suspended MCF-10A cells and mutant PI3K clones.
Cofilin activation is directly due to PTEN loss
To determine whether the increase in activated cofilin is a direct effect of PTEN loss, we restored PTEN expression in the PTEN−/− cells. When both the PTEN or GFP proteins were expressed in adherent PTEN−/− cells, there was no difference in the pCofilin levels (Fig. 7A). However, an increase in pCofilin was observed in the suspended PTEN−/− cells when PTEN was re-expressed as compared to GFP-expressing control cells (Fig. 7A). Multiple cell samples were fixed in suspension, and subjected to blinded McTN scoring (Fig. 7B). The results of quantitative McTN analysis indicate that re-expression of PTEN in PTEN−/− cells suppresses McTNs. Therefore, PTEN has a direct effect on McTNs formation and cofilin phosphorylation.
Figure 7. Expression of PTEN decreases activated cofilin and McTNs in PTEN−/− cells.
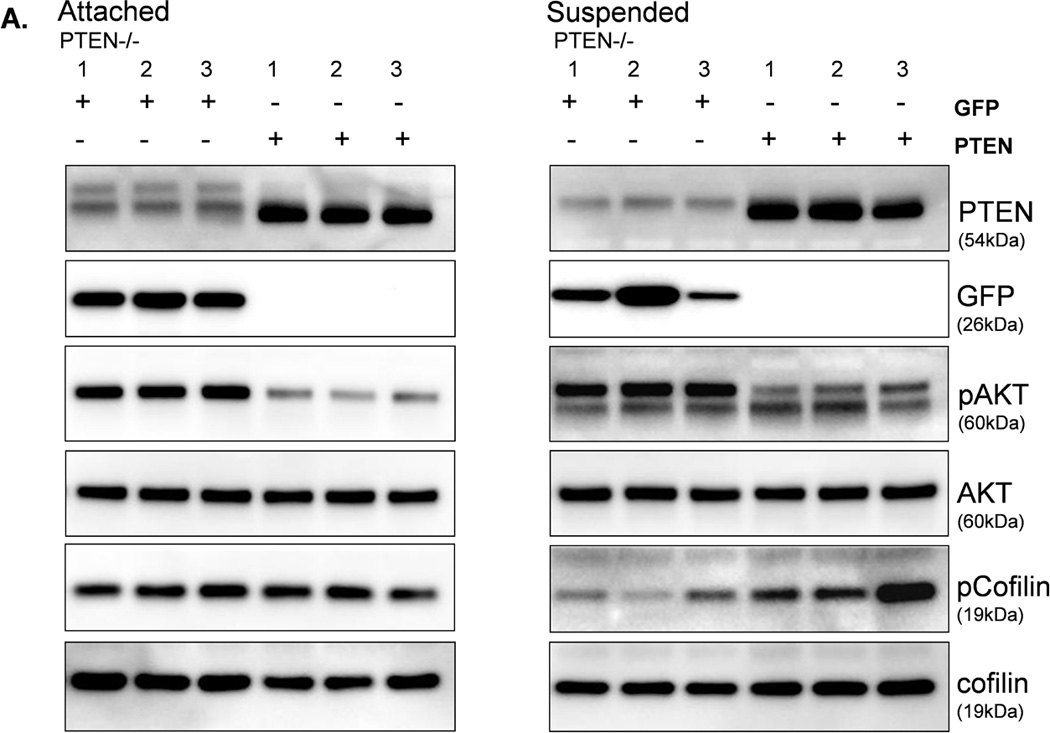
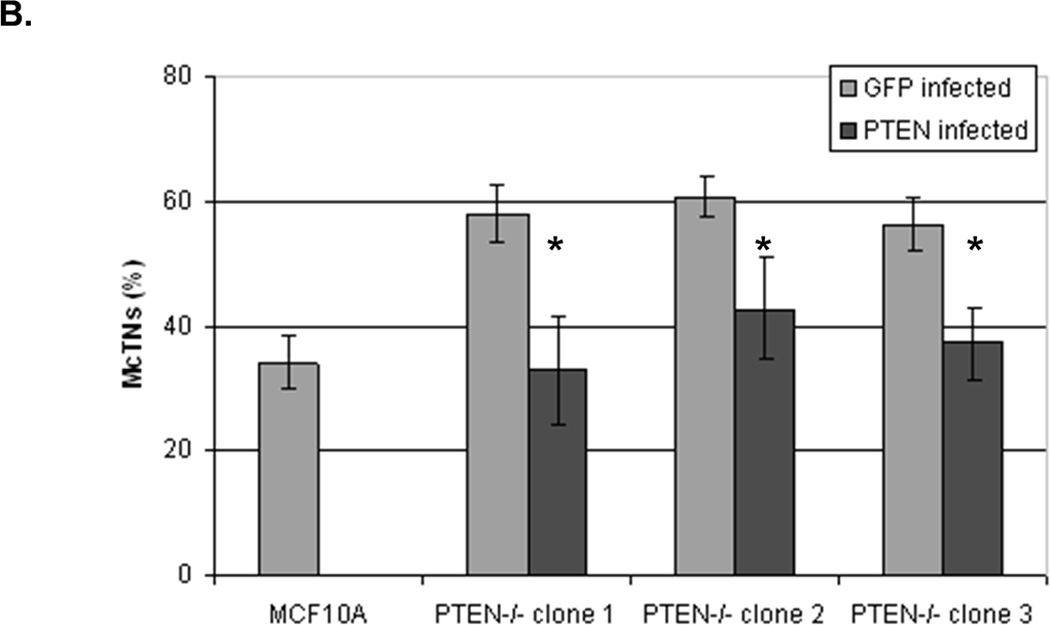
MCF-10A PTEN−/− clones (1, 2, and 3) were infected with either PTEN or GFP (CT) adenovirus once a day for two days. A. Both attached and suspended (1h) cells were analyzed by Western blot for pCofilin (n=3, representative blot) and B. McTNs formation (n=6, *p<0.002).
DISCUSSION
According to the cellular tensegrity model, epithelial cells counteract the outward force of microtubules, originating from the microtubule organizing center, through contraction of the cortical actin cytoskeleton (44). Imbalances in cortical regulation can weaken the actin cortex to allow McTN formation. Cofilin is a ubiquitously expressed actin-binding protein required for the reorganization of actin filaments, and its phopho-regulation has been directly linked to cortical actin integrity where depleted or inactivated cofilin results in elevated actomyosin contractility (45). Our study demonstrates that suspended MECs devoid of PTEN exhibit an increase in activated cofilin compared to their isogenic PTEN-expressing controls (Fig. 4), leading to increased McTNs formation, and that the phosphorylation state of cofilin serine-3 is sufficient to regulate McTNs (Fig.4C,D,E).
The low levels of pCofilin in suspended PTEN−/− cells is not due to activation of the PI3K pathway. While pAKT diminishes in the PTEN−/− cells with increasing doses of the PI3K inhibitor, LY294002, the pCofilin levels remain low and the McTNs counts do not change. Additionally, the PIK3CA cells containing activating PI3K mutations do not exhibit an increased McTNs or decreased pCofilin, supporting the conclusion that it is not PI3K pathway activation which causes increased McTNs production and cofilin activation in the PTEN−/− cells. Although in another cell system, growth factor induced dephosphorylation of pCofilin was due to PI3K-mediated SSH-1L activation (43), the dephosphorylation of pCofilin we observe with homozygous deletion of PTEN in detached mammary epithelial cells does not seem to operate via a similar mechanism since both PI3K inhibition with LY294002 and PI3K-activating mutants did not affect cofilin activation. The differences in cell type, ECM attachment state, and growth factor stimulation could explain these apparent differences in signaling mechanism.
The cofilin regulating molecules LIMK1 and Slingshot-1L also differ between suspended PTEN−/− and PIK3CA cells. The PTEN−/− cells maintain lower levels of T508 phosphorylated LIMK1 (pLIMK) and S978 phosphorylated SSH-1L (pSSH), which would both favor reduced pCofilin. In contrast to PTEN−/− cells, the PIK3CA cells exhibit higher levels of pLIMK and pSSH than the PTEN−/− cells, events which would both favor increased pCofilin. Further studies are needed to assess whether LIMK is activated via another mechanism or whether other known cofilin-specific phosphatases (SSH2, SSH3, chronophin) are more active in the PTEN−/− cells (46, 47). Nonetheless, differential activation of LIMK, SSH, cofilin, and McTNs between the PI3K mutant and PTEN−/− cells defines a series of important mechanistic differences in cytoskeletal signaling between PTEN loss and PI3K activation.
In addition to regulating the PI3K pathway and phosphoinositide production, PTEN may have other catalytic, binding, or dephosphorylating properties affecting cofilin activation. As a protein phosphatase, PTEN can dephosphorylate tyrosine-, serine-, and threonine-phosphorylated peptides (48), though it is not clear how or if PTEN and cofilin associate. PTEN contains a conserved catalytic domain shared with other protein-tyrosine-phosphatases (PTPs), flanked by noncatalytic, regulatory sequences (PTPs reviewed in ref. (49)). In addition to its catalytic domain, PTEN has a potential binding site for PDZ domain-containing proteins at its C-terminus (50). PTEN also contains a stretch of sequence, overlaping the catalytic domain, that is similar to a domain within the cytoskeletal proteins tensin and auxilin, suggesting that PTEN may participate in regulating cytoskeletal phosphorylation events (51).
PTEN loss/mutation and the reciprocal oncogenic PIK3CA are common targets of mutation in human cancers, being the second most commonly mutated target next to p53. While some have shown these mutations to be mutually exclusive (52), others argue PTEN loss and PIK3CA are concordant. It is believed that PTEN protein loss and PIK3CA mutations have different functional effects on activation of the PI3K signaling pathway which could lead to different outcomes (53, 54). A recent study of 125 patients under the age of 55 with lymph-node negative invasive breast cancers treated with adjuvant chemotherapy, identified PTEN positivity correlated with an excellent prognosis, while the PI3K/AKT/mTOR pathway had no prognostic value (55). Another study determined that PTEN loss is strongly associated with both mouse and human basal-like breast cancer (BBC), which is among the breast cancer subtypes with the worst prognosis (56). A third study analyzing gene profiles from 351 stage II breast cancer (BC) tumors determined 86% of tumor samples had PTEN mRNA levels below the median and a signature PTEN loss expression profile, which included 11 (3%) samples with PIK3CA mutation along with the low PTEN mRNA levels (52). The same group used another set of 295 BC samples for gene profiling and the cases with signature PTEN loss expression profile had significantly worse disease-free survival by Kaplan-Meier survival curves.
Recent clinical studies highlight differences in patient outcomes depending on whether a PTEN loss/mutation or PIK3CA mutation is harbored, and PTEN loss leads to a worse prognosis (52, 55, 56). In this study, we have observed that dynamic, tubulin-based McTNs increase in MCF-10A PTEN−/− clones compared to their isogenic, parental MCF-10A or the PIK3CA mutated MCF-10A cells. These results establish novel and important distinctions in cytoskeletal pathway activation that occur with PTEN loss as compared to PIK3CA activation. Significant differences in the activation state of cofilin, LIMK and SSH were all noted between cells with PTEN loss and those with activation of PI3KCA, which could each contribute to the differential elevation of McTNs in PTEN−/− cells. Data indicates that McTNs regulate adhesive properties of detached cells and can increase the efficiency of CTCs reattachment to metastatic sites (26, 27). This is consistent with the in vivo finding that CTC adhesion is tubulin-dependent and enhanced by actin depolymerization (57). Our studies demonstrate that PTEN deletion increases both tumor cell survival (14) and McTN formation in detached cells. These cellular effects provide two possible explanations for why PTEN loss is thought to specifically increase the risk of hematogenous metastasis in breast cancer (52). Elucidating the critical mechanisms by which PTEN loss enhances these metastatic risk factors may aid the design of novel therapeutic strategies that reduce metastasis by selectively targeting the survival and reattachment of CTCs. Given the numerous differences in cytoskeletal structure and signaling noted between cells with PTEN loss and PIK3CA activation, our data reveal that the prognostic implications and treatment strategies for these two genotypes may ultimately be very distinct.
MATERIALS AND METHODS
Cell Culture
MCF-10A cells were purchased from the American Type Culture Collection. The maintenance of the MCF-10A, MCF-10A/PTEN−/−, and MCF-10A/PIK3CA “knock in” cell lines has been previously described (14, 37) . Cells were maintained in a 37°C incubator with 5% CO2.
Membrane microtentacle scoring
Cells were trypsinized and suspended in complete media without phenol red for 30 minutes. Cells were either counted live as previously described (23) or fixed. Briefly, a 50°C prewarmed solution of PBS-buffered paraformaldehyde was added to the suspended cells for a final concentration of 4%. Cells were incubated for 10 minutes at RT. An equal volume of Fluoromount plus CellMask Orange plasma membrane stain (1:10,000, Invitrogen, Carlsbad, CA) and Hoechst 33342 (1:5000, Sigma, St. Louis, MO) was added. A portion of the fixed and stained cell solution was transferred to a slide and protected with a coverslip. Single cells were scored blindly for microtentacles. Cells with two or more microtentacles extending greater than the radius of the cell body were scored as positive.
Plasmids and Transfections
Cofilin, S3A, and S3E were PCR amplified using the following primers and ligated into the XhoI/EcoRI sites of the pIRES2-AcGFP1 plasmid (Clontech, Mountain View, CA). Sense primers: (WT) CTCGAGATGGCCTCCGGTGTGGCTGTCTCT, (S3A) CTCGAGATGGCCGCCGGTGTGGCTGTCTCT, (S3E) CTCGAGATGGCCGAGGGTGTGGCTGTCTCT, antisense primer: GAATTCTCACAAAGGCTTGCCCTCCAGG The SSH and SSH(CS) plasmids were very generous gifts from Dr. Alyson Fournier (58). pAcGFP1-Mem (GFP-Mem) was purchased from Clontech. Transfections were performed using Fugene-HD (Promega, Madison, WI) following manufacturer’s instructions.
Immunofluorescence
Cells were fixed in suspension with 0.3% glutaraldehyde in 80 mM Pipes, pH 6.8, 1 mM MgCl2, 5 mM EGTA, 0.5% NP40 for 20 minutes at room temperature then centrifuged (300 rpm, 5 min) onto poly-L-lysine (Sigma-Aldrich, St Louis, MO) -coated coverslips. After washing with PBS, cells were incubated with 10 mg/ml NaBH4 for 7 minutes, washed with PBS, then incubated 10 mg/ml BSA and 0.1% Tween 20 in PBS prior to antibody staining. Microtubules were stained indirectly using an anti-α-tubulin (DM1A, 1:1000; eBioscience, San Diego, CA) followed by AlexaFluor568- or AlexaFluor488-conjugated secondary antibody (1:1000; Invitrogen, Carlsbad, CA). Hoechst 33342 (1:5000, Sigma-Aldrich, St Louis, MO) was added during secondary antibody incubation. Z-stacks were obtained using an Olympus (Center Valley, PA) FV1000 confocal microscope. Stacks were imaged as maximum intensity projections using ImageJ (NIH, Bethesda, MD).
Cell Association Assay
The same PTEN−/− clone was plated in duplicate, one stained with CellMask Orange, the other transfected with GFP-mem. Cells were trypsinized, mixed in a single well, and allowed to associate. Live-cell images were captured using an Olympus IX81 inverted microscope. 3D-surface rendering was done using the Volocity software (ImproVision Inc., Waltham, MA).
Attachment assay
The impedance assay used an xCelligence RTCA SP real-time cell sensing device (Roche Applied Science, Indianapolis, IN) to assess attachment (27). Impedance values were normalized to the parental MCF-10A control line for each time point. Test cell lines were represented as the fold change of the control cell line attachment.
Western blot analysis
Western blots were performed as previously described (14). Primary antibodies for PTEN, pAKT (S473), AKT, phosphorylated extracellular signal-regulated kinase 1/2 (pERK1/2), ERK1/2, pCofilin (Ser3), and cofilin were purchased from Cell Signaling; a second cofilin antibody recognizing the C-terminal end, LIM-kinase (LIMK), and pLIMK were purchased from Abcam; Slingshot (SSH) and pSSH were purchased from ECM biosciences; anti-GFP was purchased from Santa Cruz. All antibodies were used at the manufacturers' recommended dilutions. Densitometry was performed using ImageJ.
Adenoviral infection
Equal numbers of PTEN−/− cells were infected daily for two days with either a PTEN or GFP control adenovirus (Vector Biolabs, Philadelphia, PA). Cell lysates were harvested while the cells were attached or after 1h suspension. Lysates were analyzed by Western blotting. Duplicate PTEN or GFP infected cells; both attached and suspended, were fixed, stained, and blindly scored for McTNs production.
Statistical Analysis
Significance was measured by t-test (Excel, Microsoft, Redmond, WA, USA).
Supplementary Material
Raw data from the impedance attachment assays before control normalization. Cell index is a dimensionless parameter derived as a relative change in measured electrical impedance to represent cell status. Examples of MCF-10A and PTEN−/− attachment (Data normalized and presented in Fig. 2A)
Flow cytometry analysis (20,000 counts) of (A.) MCF-10A and (B.) a representative PTEN−/− clone after suspension in complete media for 24hrs. Less than 1% of the cells are in sub-G1, while 82–83% of the cells are arrested in G1.
Western blot analysis of attached and suspended MCF-10A cells and three PTEN−/− clones for vimentin, Glu-tubulin, and acetylated-tubulin expression.
A. Western blot analysis of suspended MCF-10A cells or a representative PTEN−/− clone in the presence of 0.1% DMSO (DMSO), 50µM blebbistatin (Bleb), and 20µM ML-7. B. Densitometry results after normalizing pMLC to total MLC and setting the pMLC:MLC ratio of the MCF-10A cells to 1.
Western blot analysis of suspended MCF-10A cells and PTEN−/− clones for pMLC and MLC expression.
Acknowledgements
This work was supported by the Ruth L. Kirschstein-National Service Research Award, T32-HL07698 (MIV), National Cancer Institute, R01-CA124704 (SSM), Susan G. Komen Foundation, KG100240 (SSM) and an Era of Hope Scholar award from the Department of Defense, BC100675 (SSM).
Footnotes
Conflict of Interest: The PTEN−/− cells are licensed by Horizon Discovery Ltd. (Cambridge, UK). Dr. Vitolo receives compensation from the sale of these cells. All other authors declare no conflict of interest.
REFERENCES
- 1.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006 Jun;6(6):449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 2.Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, Wilson SM, et al. Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res. 2002 Apr 1;62(7):2162–2168. [PubMed] [Google Scholar]
- 3.Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A. 2003 Jun 24;100(13):7737–7742. doi: 10.1073/pnas.1331931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002 Aug;2(8):563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr Opin Cell Biol. 2005 Oct;17(5):559–564. doi: 10.1016/j.ceb.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Martin SS, Vuori K. Regulation of Bcl-2 proteins during anoikis and amorphosis. Biochim Biophys Acta. 2004 Jul 5;1692(2–3):145–157. doi: 10.1016/j.bbamcr.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 7.Whipple RA, Cheung AM, Martin SS. Detyrosinated microtubule protrusions in suspended mammary epithelial cells promote reattachment. Exp Cell Res. 2007 Apr 15;313(7):1326–1336. doi: 10.1016/j.yexcr.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007 Nov;7(11):834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asch HL, Head K, Dong Y, Natoli F, Winston JS, Connolly JL, et al. Widespread loss of gelsolin in breast cancers of humans, mice, and rats. Cancer Res. 1996 Nov 1;56(21):4841–4845. [PubMed] [Google Scholar]
- 10.Guck J, Schinkinger S, Lincoln B, Wottawah F, Ebert S, Romeyke M, et al. Optical deformability as an inherent cell marker for testing malignant transformation and metastatic competence. Biophys J. 2005 May;88(5):3689–3698. doi: 10.1529/biophysj.104.045476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu CR, Ma CS, Ning JY, You JF, Liao SL, Zheng J. Differential thymosin beta 10 expression levels and actin filament organization in tumor cell lines with different metastatic potential. Chin Med J (Engl) 2004 Feb;117(2):213–218. [PubMed] [Google Scholar]
- 12.Zschiesche W, Schonborn I, Behrens J, Herrenknecht K, Hartveit F, Lilleng P, et al. Expression of E-cadherin and catenins in invasive mammary carcinomas. Anticancer Res. 1997 Jan-Feb;17(1B):561–567. [PubMed] [Google Scholar]
- 13.Weiss L. Biomechanical interactions of cancer cells with the microvasculature during hematogenous metastasis. Cancer Metastasis Rev. 1992 Nov;11(3–4):227–235. doi: 10.1007/BF01307179. [DOI] [PubMed] [Google Scholar]
- 14.Vitolo MI, Weiss MB, Szmacinski M, Tahir K, Waldman T, Park BH, et al. Deletion of PTEN promotes tumorigenic signaling, resistance to anoikis, and altered response to chemotherapeutic agents in human mammary epithelial cells. Cancer Res. 2009 Nov 1;69(21):8275–8283. doi: 10.1158/0008-5472.CAN-09-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark J, Anderson KE, Juvin V, Smith TS, Karpe F, Wakelam MJ, et al. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat Methods. 2011 Mar;8(3):267–272. doi: 10.1038/nmeth.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilmore AP, Burridge K. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature. 1996 Jun 6;381(6582):531–535. doi: 10.1038/381531a0. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto M, Hilgemann DH, Feng S, Bito H, Ishihara H, Shibasaki Y, et al. Phosphatidylinositol 4,5-bisphosphate induces actin stress-fiber formation and inhibits membrane ruffling in CV1 cells. J Cell Biol. 2001 Mar 5;152(5):867–876. doi: 10.1083/jcb.152.5.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraley TS, Pereira CB, Tran TC, Singleton C, Greenwood JA. Phosphoinositide binding regulates alpha-actinin dynamics: mechanism for modulating cytoskeletal remodeling. J Biol Chem. 2005 Apr 15;280(15):15479–15482. doi: 10.1074/jbc.M500631200. [DOI] [PubMed] [Google Scholar]
- 19.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006 Oct 12;443(7112):651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 20.Janmey PA, Lindberg U. Cytoskeletal regulation: rich in lipids. Nat Rev Mol Cell Biol. 2004 Aug;5(8):658–666. doi: 10.1038/nrm1434. [DOI] [PubMed] [Google Scholar]
- 21.Kusano K, Abe H, Obinata T. Detection of a sequence involved in actin-binding and phosphoinositide-binding in the N-terminal side of cofilin. Mol Cell Biochem. 1999 Jan;190(1–2):133–141. [PubMed] [Google Scholar]
- 22.Yonezawa N, Nishida E, Iida K, Yahara I, Sakai H. Inhibition of the interactions of cofilin, destrin, and deoxyribonuclease I with actin by phosphoinositides. J Biol Chem. 1990 May 25;265(15):8382–8386. [PubMed] [Google Scholar]
- 23.Whipple RA, Balzer EM, Cho EH, Matrone MA, Yoon JR, Martin SS. Vimentin filaments support extension of tubulin-based microtentacles in detached breast tumor cells. Cancer Res. 2008 Jul 15;68(14):5678–5688. doi: 10.1158/0008-5472.CAN-07-6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glinsky VV, Glinsky GV, Glinskii OV, Huxley VH, Turk JR, Mossine VV, et al. Intravascular metastatic cancer cell homotypic aggregation at the sites of primary attachment to the endothelium. Cancer Res. 2003 Jul 1;63(13):3805–3811. [PubMed] [Google Scholar]
- 25.Matrone MA, Whipple RA, Balzer EM, Martin SS. Microtentacles tip the balance of cytoskeletal forces in circulating tumor cells. Cancer Res. 2010 Oct 15;70(20):7737–7741. doi: 10.1158/0008-5472.CAN-10-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balzer EM, Whipple RA, Cho EH, Matrone MA, Martin SS. Antimitotic chemotherapeutics promote adhesive responses in detached and circulating tumor cells. Breast Cancer Res Treat. 2010 May;121(1):65–78. doi: 10.1007/s10549-009-0457-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matrone MA, Whipple RA, Thompson K, Cho EH, Vitolo MI, Balzer EM, et al. Metastatic breast tumors express increased tau, which promotes microtentacle formation and the reattachment of detached breast tumor cells. Oncogene. 2010 Jun 3;29(22):3217–3227. doi: 10.1038/onc.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009 Nov;10(11):778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hale CM, Sun SX, Wirtz D. Resolving the role of actoymyosin contractility in cell microrheology. PLoS One. 2009;4(9):e7054. doi: 10.1371/journal.pone.0007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Even-Ram S, Doyle AD, Conti MA, Matsumoto K, Adelstein RS, Yamada KM. Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat Cell Biol. 2007 Mar;9(3):299–309. doi: 10.1038/ncb1540. [DOI] [PubMed] [Google Scholar]
- 31.Tohtong R, Phattarasakul K, Jiraviriyakul A, Sutthiphongchai T. Dependence of metastatic cancer cell invasion on MLCK-catalyzed phosphorylation of myosin regulatory light chain. Prostate Cancer Prostatic Dis. 2003;6(3):212–216. doi: 10.1038/sj.pcan.4500663. [DOI] [PubMed] [Google Scholar]
- 32.Betapudi V, Licate LS, Egelhoff TT. Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res. 2006 May 1;66(9):4725–4733. doi: 10.1158/0008-5472.CAN-05-4236. [DOI] [PubMed] [Google Scholar]
- 33.Shin DH, Chun YS, Lee KH, Shin HW, Park JW. Arrest defective-1 controls tumor cell behavior by acetylating myosin light chain kinase. PLoS One. 2009;4(10):e7451. doi: 10.1371/journal.pone.0007451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doreian BW, Fulop TG, Meklemburg RL, Smith CB. Cortical F-actin, the exocytic mode, and neuropeptide release in mouse chromaffin cells is regulated by myristoylated alanine-rich C-kinase substrate and myosin II. Mol Biol Cell. 2009 Jul;20(13):3142–3154. doi: 10.1091/mbc.E09-03-0197. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Okagaki T, Hayakawa K, Samizo K, Kohama K. Inhibition of the ATP-dependent interaction of actin and myosin by the catalytic domain of the myosin light chain kinase of smooth muscle: possible involvement in smooth muscle relaxation. J Biochem. 1999 Mar;125(3):619–626. doi: 10.1093/oxfordjournals.jbchem.a022328. [DOI] [PubMed] [Google Scholar]
- 36.Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004 Apr 30;304(5671):743–746. doi: 10.1126/science.1094561. [DOI] [PubMed] [Google Scholar]
- 37.Gustin JP, Karakas B, Weiss MB, Abukhdeir AM, Lauring J, Garay JP, et al. Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci U S A. 2009 Feb 24;106(8):2835–2840. doi: 10.1073/pnas.0813351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem. 2000 Feb 4;275(5):3577–3582. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- 39.Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999 Sep;1(5):253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- 40.Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999 Aug 6;285(5429):895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 41.Eiseler T, Doppler H, Yan IK, Kitatani K, Mizuno K, Storz P. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat Cell Biol. 2009 May;11(5):545–556. doi: 10.1038/ncb1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagata-Ohashi K, Ohta Y, Goto K, Chiba S, Mori R, Nishita M, et al. A pathway of neuregulin-induced activation of cofilin-phosphatase Slingshot and cofilin in lamellipodia. J Cell Biol. 2004 May 24;165(4):465–471. doi: 10.1083/jcb.200401136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishita M, Wang Y, Tomizawa C, Suzuki A, Niwa R, Uemura T, et al. Phosphoinositide 3-kinase-mediated activation of cofilin phosphatase Slingshot and its role for insulin-induced membrane protrusion. J Biol Chem. 2004 Feb 20;279(8):7193–7198. doi: 10.1074/jbc.M312591200. [DOI] [PubMed] [Google Scholar]
- 44.Ingber DE. Tensegrity I. Cell structure and hierarchical systems biology. J Cell Sci. 2003 Apr 1;16(Pt 7):1157–1173. doi: 10.1242/jcs.00359. [DOI] [PubMed] [Google Scholar]
- 45.Wiggan O, Shaw AE, DeLuca JG, Bamburg JR. ADF/cofilin regulates actomyosin assembly through competitive inhibition of myosin II binding to F-actin. Dev Cell. 2012 Mar 13;22(3):530–543. doi: 10.1016/j.devcel.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang TY, DerMardirossian C, Bokoch GM. Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol. 2006 Feb;18(1):26–31. doi: 10.1016/j.ceb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 47.Kobayashi M, Nishita M, Mishima T, Ohashi K, Mizuno K. MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 2006 Feb 22;25(4):713–726. doi: 10.1038/sj.emboj.7600973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1997 Aug 19;94(17):9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Denu JM, Stuckey JA, Saper MA, Dixon JE. Form and function in protein dephosphorylation. Cell. 1996 Nov 1;87(3):361–364. doi: 10.1016/s0092-8674(00)81356-2. [DOI] [PubMed] [Google Scholar]
- 50.Craven SE, Bredt DS. PDZ proteins organize synaptic signaling pathways. Cell. 1998 May 15;93(4):495–498. doi: 10.1016/s0092-8674(00)81179-4. [DOI] [PubMed] [Google Scholar]
- 51.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999 Apr 13;96(8):4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A. 2007 May 1;104(18):7564–7569. doi: 10.1073/pnas.0702507104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008 Aug 1;68(15):6084–6091. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perez-Tenorio G, Alkhori L, Olsson B, Waltersson MA, Nordenskjold B, Rutqvist LE, et al. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin Cancer Res. 2007 Jun 15;13(12):3577–3584. doi: 10.1158/1078-0432.CCR-06-1609. [DOI] [PubMed] [Google Scholar]
- 55.Janssen EA, Soiland H, Skaland I, Gudlaugson E, Kjellevold KH, Nysted A, et al. Comparing the prognostic value of PTEN and Akt expression with the Mitotic Activity Index in adjuvant chemotherapy-treated node-negative breast cancer patients aged<55 years. Cell Oncol. 2007;29(1):25–35. doi: 10.1155/2007/569246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saal LH, Gruvberger-Saal SK, Persson C, Lovgren K, Jumppanen M, Staaf J, et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet. 2008 Jan;40(1):102–107. doi: 10.1038/ng.2007.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Korb T, Schluter K, Enns A, Spiegel HU, Senninger N, Nicolson GL, et al. Integrity of actin fibers and microtubules influences metastatic tumor cell adhesion. Exp Cell Res. 2004 Sep 10;299(1):236–247. doi: 10.1016/j.yexcr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 58.Hsieh SH, Ferraro GB, Fournier AE. Myelin-associated inhibitors regulate cofilin phosphorylation and neuronal inhibition through LIM kinase and Slingshot phosphatase. J Neurosci. 2006 Jan 18;26(3):1006–1015. doi: 10.1523/JNEUROSCI.2806-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Raw data from the impedance attachment assays before control normalization. Cell index is a dimensionless parameter derived as a relative change in measured electrical impedance to represent cell status. Examples of MCF-10A and PTEN−/− attachment (Data normalized and presented in Fig. 2A)
Flow cytometry analysis (20,000 counts) of (A.) MCF-10A and (B.) a representative PTEN−/− clone after suspension in complete media for 24hrs. Less than 1% of the cells are in sub-G1, while 82–83% of the cells are arrested in G1.
Western blot analysis of attached and suspended MCF-10A cells and three PTEN−/− clones for vimentin, Glu-tubulin, and acetylated-tubulin expression.
A. Western blot analysis of suspended MCF-10A cells or a representative PTEN−/− clone in the presence of 0.1% DMSO (DMSO), 50µM blebbistatin (Bleb), and 20µM ML-7. B. Densitometry results after normalizing pMLC to total MLC and setting the pMLC:MLC ratio of the MCF-10A cells to 1.
Western blot analysis of suspended MCF-10A cells and PTEN−/− clones for pMLC and MLC expression.