

Abstract
Toxoplasma gondii transmission between intermediate hosts is dependent on the ingestion of walled cysts formed during the chronic phase of infection. Immediately following consumption, the parasite must ensure survival of the host by preventing adverse inflammatory responses and/or by limiting its own replication. Since the Toxoplasma secreted effectors rhoptry 16 kinase (ROP16) and dense granule 15 (GRA15) activate the JAK-STAT3/6 and NF-κB signaling pathways, respectively, we explored whether a particular combination of these effectors impacted intestinal inflammation and parasite survival in vivo. Here we report that expression of the STAT-activating version of ROP16 in the type II strain (strain II+ROP16I) promotes host resistance to oral infection only in the context of endogenous GRA15 expression. Protection was characterized by a lower intestinal parasite burden and dampened inflammation. Host resistance to the II+ROP16I strain occurred independently of STAT6 and the T cell coinhibitory receptors B7-DC and B7-H1, two receptors that are upregulated by ROP16. In addition, coexpression of ROP16 and GRA15 enhanced parasite susceptibility within tumor necrosis factor alpha/gamma interferon-stimulated macrophages in a STAT3/6-independent manner. Transcriptional profiling of infected STAT3- and STAT6-deficient macrophages and parasitized Peyer's patches from mice orally challenged with strain II+ROP16I suggested that ROP16 activated STAT5 to modulate host gene expression. Consistent with this supposition, the ROP16 kinase induced the sustained phosphorylation and nuclear localization of STAT5 in Toxoplasma-infected cells. In summary, only the combined expression of both GRA15 and ROP16 promoted host resistance to acute oral infection, and Toxoplasma may possibly target the STAT5 signaling pathway to generate protective immunity in the gut.
INTRODUCTION
The obligate intracellular parasite Toxoplasma gondii is a widespread protozoan pathogen capable of infecting any nucleated cell. It is highly transmissible and able to achieve chronic infection in a wide variety of birds and sea and land mammals, including an estimated third of the human population. Infection is typically asymptomatic, and though most parasites are cleared by the immune system, others escape and form cysts within the brain, heart, and skeletal muscle. In immunocompromised individuals, such as in those with AIDS or in the developing fetus, new infections or rupture of preexisting cysts can lead to inflammation-related pathologies and even death. However, some immunocompetent hosts are also susceptible to acute infection. For example, following oral infection, intestinal inflammation has been observed in deer, pigs, rabbits, cats, and certain strains of laboratory mice (1). Severe toxoplasmosis has also been documented in immunocompetent humans exposed to South American strains (2). Since Toxoplasma spreads when an animal consumes meat containing tissue cysts or food contaminated with oocytes shed from a cat, we hypothesize that Toxoplasma has evolved ways to limit intestinal pathology to promote host survival.
One strategy used by Toxoplasma to achieve chronic infection is through the modulation of the T helper 1 (Th1) response (3), which culminates in the production of gamma interferon (IFN-γ). IFN-γ induces several host mechanisms for intracellular elimination of Toxoplasma, including the induction of reactive nitrogen intermediates (4) and the activation of two families of interferon-regulated GTPases, the immunity-related GTPases (IRGs) (5, 6) and guanylate-binding proteins (GBPs) (7). In the mouse model of toxoplasmosis, the Th1 response must be tightly regulated; otherwise, a lethal inflammatory response develops. For example, mice lacking the Th1-regulatory cytokine interleukin-10 (IL-10) succumb to sublethal oral challenge with Toxoplasma tissue cysts and exhibit severe immune pathology in the small intestine and liver (8). Furthermore, in naturally susceptible mouse strains, the intestinal pathology that develops following oral infection resembles many aspects of Crohn's disease (9). Thus, understanding how Toxoplasma manipulates mucosal immunity may yield novel strategies for treating human inflammatory bowel diseases, like Crohn's disease.
Importantly, not all strains of Toxoplasma cause similar disease. Of the three strain types predominantly isolated from humans in North America and Europe, types I, II, and III, it is the type II strain that is associated with reactive encephalitis in HIV-infected patients (10) and tissue inflammation in susceptible C57BL/6 (B6) mice (11, 12). Recently, we have identified polymorphisms in key parasite effector proteins that are injected into macrophages and activate signaling pathways that regulate inflammation. The Toxoplasma rhoptry 16 kinase (ROP16), which is secreted into the host cytosol upon invasion, directly phosphorylates the STAT3 and STAT6 transcription factors. The versions encoded by the type I/III strains, but not the type II strain, maintain STAT3/6 activation for 24 h (13–15). In contrast, the type II strain, but not the type I/III strains, encodes a version of the Toxoplasma dense granule 15 (GRA15), a protein with no homology to other proteins, that activates the nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) (16). Due to polymorphisms in these effectors, the type I and III strains promote the alternative activation of macrophages (M2), while the type II strain induces the classical activation of macrophages (M1) (17). M1 cells require NF-κB activation, promote Th1 responses, and elicit many of the host's toxoplasmacidal mechanisms but are also associated with pathological inflammation. In contrast, M2 cells driven by STAT6 activation develop in a Th2 environment and inhibit Th1-type inflammatory responses (18). Remarkably, following oral infection with a type II strain engineered to express the STAT3/6-activating version of ROP16 from the type I strain, there was significantly less inflammatory damage in the small intestine compared to that following infection with the parental type II strain (17). Although the mechanism by which ROP16 mediates this effect is unknown, we hypothesized that it was due to its ability to activate the STAT3/6 signaling pathways and inhibit NF-κB activation. Furthermore, the role of GRA15 in intestinal inflammation has not been explored, but since it activates NF-κB and promotes M1 activation, it is possible that by removing GRA15 from Toxoplasma, parasite-induced ileitis may be alleviated.
Finally, death of the infected host can result from failure to control parasite replication or failure to survive tissue damage following infection, in what has been described as host resistance and infectious tolerance, respectively (19). Following oral infection, it is apparent that the intestinal parasite burden often, but not always, correlates with pathology. For example, antibody neutralization of tumor necrosis factor alpha (TNF-α) or IFN-γ on days 6 and 8 following infection or chemical inhibition of inducible nitric oxide synthase (iNOS) reduces ileitis, but only TNF-α neutralization leads to an increase in parasite numbers (12, 20). Other notable examples include oral infection in IL-23 gene-knockout (Il23−/−) and Il10−/− mice. In these studies, intestinal parasite numbers were similar between knockout and wild-type mice; however, Il10−/− mice were highly susceptible and developed severe ileitis, whereas Il23−/− mice survived infection and did not develop ileitis (8, 21). Thus, the Toxoplasma effectors GRA15 and ROP16, which modulate the expression of many inflammatory mediators, including IL-10 and IL-23 (17), could possibly affect both host tolerance and resistance mechanisms to promote chronic infection and subsequent spread in nature.
Here we describe that ROP16 and GRA15 have an immediate impact on host resistance following oral infection. Through the use of a variety of gene-knockout and transgenic parasites, we demonstrate that GRA15 and ROP16 are required to limit parasite replication in the intestines of orally infected mice and enhance parasite susceptibility within TNF-α/IFN-γ-stimulated macrophages. These phenotypes were independent of STAT3/6, and we report here that Toxoplasma ROP16 induced the sustained phosphorylation and nuclear translocation of STAT5. This interaction may play a significant role in modulating host gene expression in parasite-infected macrophages and in generating protective immunity in the gut of infected animals.
MATERIALS AND METHODS
Mice, cells, and Toxoplasma strains.
Eight- to 12-week-old female C57BL/6J (B6), B6.129S2(C)-Stat6tm1Gru/J (Stat6−/−), B6.129P2-Lyzstm1(cre)Ifo/J (LysM-cre), and B6.129S1-Stat3tm1Xyfu/J (Stat3fl/fl) mice were purchased from Jackson Laboratories. Stat3fl/fl and LysM-cre+/+ mice were bred to generate Stat3fl/fl LysM-cre+/− and Stat3fl/fl LysM-cre−/− mice (University of Pennsylvania). Cd274−/− mice were obtained from Jianzhu Chen (MIT) with permission from Leiping Chen (Yale); Stat6−/− and Cd274−/− mice were bred as homozygotes (MIT). All mouse work was performed in accordance with the guidelines set by MIT's Committee for Animal Care (CAC) or the University of Pennsylvania's Institutional Animal Care and Use Committee (IACUC).
Isogenic green fluorescent protein (GFP)- and firefly luciferase (fLUC)-expressing type II Pru (PA7) strains PA7 +HPT (5-8B+, 50-5A+), PA7 +ROP16IHA +HPT (2C4, 1F5), PA7 Δgra15 +HPT (50-5C, 5-8A), and PA7 Δgra15 +ROP16IHA +HPT +BLE (a phleomycin resistance cassette) (1B7) have been previously generated and described before (17). Transgenic protein expression of ROP16IHA in the II Δgra15 +ROP16I and II+ROP16I strains was comparable between strains by Western blotting (see Fig. S1 in the supplemental material). A (Pru) II+ROP16IHA +HPT strain (22) and a type III CEP hxgprt-GFP-cLUC (click beetle luciferase) strain were also used for oral infections. A GFP-cLUC-expressing type I (RH) (1-1) strain and a I Δrop16 (1A2) strain (17) were used in immune fluorescence assays (IFAs). Parasite strains were passaged in monolayers of human foreskin fibroblasts (HFFs) in Dulbecco modified Eagle medium (DMEM) with 1% fetal bovine serum (FBS) and antibiotics. HFFs and the DC2.4 dendritic cell line were cultured as described previously (17). Bone marrow-derived macrophages (BMDMs) were obtained by culturing murine bone marrow cells in DMEM supplemented with 10% FBS, 1× minimal essential medium nonessential amino acids, 1 mM sodium pyruvate (Gibco, Invitrogen), antibiotics, and 20% L929 conditioned medium (final concentration of granulocyte colony-stimulating factor [G-CSF], 8.8 ng/ml) for 7 to 8 days, which yielded a highly pure population of CD11b+ F4/80+ macrophages by fluorescence-activated cell sorter (FACS) analysis.
Cyst isolation, oral infections, and bioluminescence imaging.
Brains from chronically infected mice (>30 days) were harvested, emulsified in 1 ml phosphate-buffered saline (PBS) per brain, and stored at 4°C before use. One hundred microliters of the emulsion was fixed in 900 μl of ice-cold methanol, washed, stained with fluorescein isothiocyanate-labeled Dolichos Biflorus agglutinin (Vector) overnight at 4°C, washed, and resuspended in 1 ml PBS. Cysts were enumerated by counting 3 to 5 50-μl aliquots with a fluorescence microscope (×20 objective); most cysts were typically 7 to 12 μm in diameter. Mice were administered 1,000 or 250 cysts with a feeding needle. In some experiments, mice were injected intraperitoneally (i.p.) with 300 μg neutralizing antibody (BioXcell) against B7-DC (TY25) or rat IgG2a isotype control (2A3) on days 4, 6, and 9 after oral infection. For experiments comparing wild-type to Cd274−/− or Stat6−/− mice, intestinal floras were normalized between mouse strains (23) by first swapping bedding between cages and then placing mice from each genetic background in the same cage for 2 weeks prior to oral infections. Littermates were used for infections comparing Stat3fl/fl LysM-cre+/− and Stat3fl/fl LysM-cre−/− mice.
For in vivo bioluminescence imaging of luciferase-expressing parasites, mice were injected i.p. with 300 μg of d-luciferin (Gold Biotechnology) and anesthetized, and 10 min later, light emission from individual mice was detected with an IVIS Spectrum bioluminescent and fluorescent imaging system (Xenogen Corporation). For subsequent bioluminescence imaging of the small intestine and other organs, mice were injected a second time with luciferin, and 8 min later, organs were harvested, placed on a petri dish, and imaged. Image analysis was performed with Living Image software.
Tissue sectioning and scoring matrix for inflammatory assessment.
After 8 to 9 days of infection, small intestines were fixed in 10% buffered formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin (H&E) or stained with eosin and an anti-Ly6G antibody (BD 550291), followed by development with horseradish peroxidase. A scoring matrix used to assess rat colitis (24) was modified for this study to score murine small intestinal inflammation and takes into account inflammatory cell infiltrate into the lamina propria, the degree of Gr-1 (Ly6G) staining, submucosa thickening and cellular infiltrate, deformation of the villus architecture, the number of goblet cells per villus, and the number of ulcerations that extend through the mucosa and smooth muscle wall. The scoring matrix is described in detail in Table S1 in the supplemental material.
In vitro Toxoplasma infection assays.
BMDMs were plated at 2 × 105 cells per well (96-well black plate, clear bottom; Costar) and stimulated overnight with 25 ng/ml of TNF-α and 10 ng/ml of IFN-γ (Peprotech) in the same 20% L929 conditioned medium used to generate BMDMs. Parasites were obtained by sequentially syringe lysing heavily vacuolated HFF monolayers through 25-gauge and 27-gauge needles and spinning the flowthrough at 32 × g to pellet large debris, followed by a faster spin (582 × g) to pellet parasites, and the parasites were washed in PBS. Prior to infection, the BMDM stimulation medium was removed, and infections were performed at several different multiplicities of infection (MOIs) ranging from 0.5 to 0.1 in DMEM supplemented with 1% FBS and antibiotics. Twenty hours later, luciferase activity within the cellular lysate was detected following automated addition of the luciferin substrate (luciferase assay system; Promega) in a Varioskan Flash plate reader (Thermo Scientific). Parasite viability was inferred by a plaque assay; MOIs that returned a similar number of viable parasites between strains were compared. Infected BMDMs or DC2.4 cells were also analyzed by FACS for the surface expression of B7-H1 by staining with an anti-CD274 antibody (MIH5; BD Pharmingen).
Immunofluorescence assay.
BMDMs or HFFs were plated on coverslips overnight and infected with parasites on the following day. After 20 h of infection, cells were fixed with 3% formaldehyde, permeabilized with ice -old methanol, washed with PBS, blocked, and stained in 3% bovine serum albumin, 5% goat serum, 0.2% Triton X-100 in PBS with antibodies that recognize phospho-Tyr694 STAT5A or STAT5B (C71E5; 1:400; Cell Signaling Technology) or NF-κB p65 (sc-8008; 1:1,000; Santa Cruz Biotechnology). Alexa Fluor 488- or Alexa Fluor 594-coupled secondary antibodies (Molecular Probes) and Hoechst dye were used for antigen and DNA visualization with a fluorescence microscope.
Microarray analysis of infected BMDMs and Peyer's patches.
BMDMs were plated at 3 × 106 cells per well (6-well plate) and infected (MOI, 3) for 18 h, and RNA was isolated using the TRIzol reagent (Invitrogen). cDNA was generated, labeled, and hybridized to a mouse Affymetrix array (Mouse 430 2.0 or Mouse 430A 2), probe intensities were measured and normalized with the MAS5 algorithm, and gene expression was processed as described before (16). Microarray experiments of wild-type C57BL/6 mouse BMDMs infected with the type II, II Δgra15, II+ROP16I, and II Δgra15 +ROP16I strains, as previously reported (17), were originally performed side by side with infections of Stat3fl/fl LysM-cre (B6) mouse BMDMs reported here. Microarray experiments of II+ROP16I-infected Stat6−/− and Stat6+/+ (B6) mouse BMDMs were performed similarly. Our criteria to determine Toxoplasma ROP16-regulated host genes that were modulated through STAT3 or STAT6 are as follows. First, a host gene was considered regulated by ROP16 if its expression changed ±1.7-fold when comparing infections with the type II and II+ROP16I strains or strains II Δgra15 and II Δgra15 +ROP16I. This produced a list of 938 uniquely annotated macrophage genes that were regulated by Toxoplasma ROP16. Second, a ROP16-regulated host gene was considered to be modulated through STAT3 if, for example, the fold change between the type II and II+ROP16I strains no longer occurred in STAT3-deficient macrophages or deviated ±1.7-fold from the value obtained in wild-type macrophages when comparing the same infections. Finally, a ROP16-regulated gene was considered modulated through STAT6 if its expression changed ±1.7-fold when comparing strain II+ROP16I infections in Stat6−/− and wild-type mouse BMDMs.
Peyer's patches from several mice infected with the type II or II+ROP16I strain after 5 days of oral infection and from a naive mouse were dissected, individually analyzed by bioluminescence imaging, and snap-frozen in liquid nitrogen. Peyer's patches with equivalent parasite burdens were processed by using a needle to crunch the frozen Peyer's patch inside an Eppendorf tube placed on dry ice, followed by suspension in TRIzol. Additionally, small intestines from these infected mice, with the Peyer's patches removed, were further dissected into three parts, the upper 1/3 (approximately consisting of the duodenum), middle 1/3 (approximately consisting of the jejunum), and lower 1/3 (approximately consisting of the ileum), and subjected to bioluminescence imaging before snap-freezing. The middle 1/3 intestinal sections that had similar parasite burdens between infections were processed as described above. RNA was isolated, and microarray analysis was performed. Gene expression was normalized using Robust Multiarray Averaging (RMA).
Microarray data accession numbers.
The microarray data generated for infections in the STAT-deficient BMDMs with all strains are accessible through NCBI's Gene Expression Omnibus (GEO) database under accession number GSE45309 and can be compared to data generated for infections in wild-type BMDMs previously published and accessible under accession number GSE29404. Microarray data for parasitized intestines and Peyer's patch samples can be found under the GEO accession number GSE45310.
RESULTS
GRA15 and ROP16 promote host resistance to acute oral infection.
To determine the role of GRA15 and ROP16 in altering host survival, we orally gavaged susceptible C57BL/6 mice with tissue cysts of the luciferase- and GFP-expressing (Pru A7) type II strain or with Pru A7 type II strains that transgenically express the type I copy of ROP16 (strain II+ROP16I) or have the endogenous GRA15II gene deleted (strain II Δgra15). The type II Pru strain is less virulent than the type II ME49 strain used in most oral infection studies; therefore, in our hands, we found that a dose of 1,000 cysts is required to achieve a nearly 100% lethal dose (LD100) in C57BL/6 mice; in comparison, an LD100 of 100 cysts has been reported for the ME49 strain (12, 25). As reported before, transgenic expression of type I ROP16 in a type II strain makes this strain significantly less virulent than the parental type II strain (Fig. 1A). Mice that did succumb to infection with strain II+ROP16I did so 3 to 4 days later than mice infected with the type II strain. In contrast, oral infection with the II Δgra15 strain led to rapid death with kinetics similar to those observed following infection with the wild-type type II strain. A similar trend was observed with a lower-dose infection of 250 cysts (see Fig. S2 in the supplemental material). To test whether transgenic expression of type I ROP16 alone or a combination of GRA15II and ROP16I is required for host protection, we orally challenged mice with a type II Δgra15 strain that expresses type I ROP16 (strain II Δgra15 +ROP16I) (see Fig. S1 in the supplemental material). Interestingly, transgenic expression of type I ROP16 in the II Δgra15 strain did not confer host protection, as this strain had virulence similar to that of the type II and II Δgra15 strains (Fig. 1A), demonstrating that endogenous GRA15 promotes host survival in this setting. Furthermore, these results highlight that both GRA15 and ROP16 are required for optimal host survival following type II challenge.
Fig 1.

Host resistance to oral infection requires expression of both Toxoplasma rhoptry kinase ROP16 and dense granule GRA15. C57BL/6 mice were orally gavaged with 1,000 cysts of the luciferase- and GFP-expressing type II Pru +HXGPRT strains Pru A7 (5-8B+, 50-5A), type II Δgra15 (50-5C, 5-8A), type II+ROP16I (1F5, 2C4), and type II Δgra15 +ROP16I (1B7). A non-GFP- and non-luciferase- expressing Pru +ROP16I strain was also analyzed. (A) Cumulative survival from 3 to 15 separate experiments of mice orally challenged with the indicated parasite strains. The total number (n) of mice orally challenged with each strain is listed. A Mantel-Cox log-rank test was performed to assess significance; only mouse survival following challenge with the II+ROP16I strain was statistically significant (P < 0.05) compared to that following challenge with the type II strain. (B) Bioluminescence images of small intestines from C57BL/6 mice on days 5 and 9 following oral infection with 1,000 cysts of the indicated luciferase-expressing Toxoplasma type II strains. The relative parasite burden (number of photons/s/r2, where r2 = cm2/steradian) between Toxoplasma strains is depicted as a heat map. (C) Representative H&E stains of intestines from C57BL/6 mice after 8 days of infection with the indicated parasite strains or from A/J mice infected with the type II strain. Pictured are two layers of small intestine from a single mouse seen with a ×10 objective. (D) Average inflammatory score + SD of small intestines from C57BL/6 mice 8 days after oral infection with the indicated Toxoplasma strains (n = 3 to 7 mice per group). Inflammatory scores for noninfected C57BL/6 mice and A/J mice infected with the type II strain are also given. Significant differences between the means of each group were calculated by analysis of variance testing; a significant P value was determined for the II+ROP16I strain. A two-tailed Student t test was performed between the type II strain-infected A/J mouse and II+ROP16I strain-infected B6 mouse groups (n.s., not significant). (E) Quantification (n = 2 to 4 intestines per group) of the relative parasite burdens (number of photons/s/r2) in the small intestines (from the experiment shown in panel B) 9 days after oral infection; the P value was calculated by analysis of variance testing. (F) Bioluminescence images of small intestines from C57BL/6 mice orally infected with either the type II or the II+ROP16I strain or A/J mice infected with the type II strain 8 days after oral infection with 1,000 cysts.
NF-κB plays a central role in immune function and inflammatory induction in the small intestine. Since the Toxoplasma strains that lack GRA15II or express ROP16I/III exhibit significantly reduced NF-κB p65 nuclear translocation in infected macrophages (see Fig. S3 in the supplemental material), we investigated whether there was a separation of host tolerance and resistance mechanisms following oral infection with these strains. To first address this issue, parasite burden in the small intestine was directly measured ex vivo by bioluminescence imaging. Host susceptibility to the engineered Toxoplasma strains directly correlated with intestinal parasite burden on days 8 to 9 following oral challenge (Fig. 1B). Notably, the II+ROP16I strain exhibited a 300-fold decrease in luciferase activity compared to that of the other strains, and this reduction required endogenous GRA15II expression (Fig. 1E). The role of GRA15 in promoting host resistance could also be observed at earlier time points of infection (day 5), as there were significantly higher strain II Δgra15 parasite numbers than either parental or II+ROP16I strain numbers in the small intestine (see Fig. S4A and B in the supplemental material). A similar defect in host resistance was reported on day 5 following i.p. infection with the II Δgra15 strain (16). Since NF-κB null mice are highly susceptible to Toxoplasma infection due to diminished IFN-γ responses (26), an organ culture assay was used to measure ex vivo cytokine production of intestines from mice infected with the II Δgra15, II+ROP16I, and wild-type strains. However, no significant difference in either IFN-γ or IL-12p70 production on day 5 following oral infection was observed, although there was considerable variability with these assays (see Fig. S4C in the supplemental material). Whether differences in cytokine production occur at earlier time points of infection remain to be determined.
Importantly, infectious tolerance was not separable from host resistance in this system. A scoring matrix (see Table S1 in the supplemental material) was used to quantify the degree of small intestinal inflammation observed following 8 days of infection. Virulent strains that exhibited high intestinal parasite burdens (Fig. 1B) induced severe intestinal pathology with high inflammatory sores (Fig. 1C and D). Virulent strains induced significant destruction of the villi, infiltration of inflammatory cells, submucosa swelling, and at least one ulceration of the smooth muscle wall and basement membrane (see Table S2 in the supplemental material), presumably exposing the host's peritoneal cavity to the luminal contents of the intestine. In contrast, infection with the II+ROP16I strain induced a very mild inflammation characterized by a few regions (<10%) with moderate cellular infiltrate in the lamina propria and submucosa/villus swelling. The intestinal parasite burden and inflammation following II+ROP16I strain infection in C57BL/6 mice mirrored type II strain infection in A/J mice (Fig. 1C and F; see Table S2 in the supplemental material), a mouse strain known to be resistant to oral Toxoplasma infection (27), demonstrating that synergistic expression of GRA15 and ROP16 can compensate for resistance defects in susceptible hosts. In conclusion, disease outcome is tightly linked to the exact combination of Toxoplasma ROP16 and GRA15 effector proteins, which together function as negative regulators of virulence during oral infection in susceptible mice.
Stat6−/− and B7-H1−/− mice are resistant to oral Toxoplasma strain II+ROP16I infection.
Given the established role of the NF-κB pathway in generating protective immunity during Toxoplasma infection, we decided instead to explore how the STAT3/6 signaling pathways and/or targets of ROP16 influence host resistance during oral infection. Two of the ROP16 targets include the T cell coinhibitory receptors B7-DC (Pdcdl2 or PD-L2) and B7-H1 (Cd274 or PD-L1) (17). Both receptors bind PD-1 on T cells with different affinities and inhibit T cell cytokine secretion (28). Previously, we observed that surface expression of B7-DC on infected macrophages is entirely dependent on Toxoplasma ROP16 and host STAT6 (17). In contrast, B7-H1 upregulation by ROP16 is independent of STAT6, and parasite infection in the absence of ROP16 (i.e., with a type I Δrop16 strain) induces intermediate surface expression on infected macrophages (see Fig. S5 in the supplemental material). The B7-H1–PD1 pathway can have both positive and negative outcomes for the host, depending on the tissue and stage of Toxoplasma infection (29, 30). Hence, we explored oral infection with strain II+ROP16I in Stat6−/− and Cd274−/− mice and in mice treated with neutralizing antibodies against B7-DC (31). Finally, we screened LysM-cre Stat3fl/fl mice, which lack Stat3 in macrophages and neutrophils. We reasoned that mice lacking the relevant ROP16 target should be highly susceptible to oral infection with the II+ROP16I strain with elevated parasite numbers.
However, Stat6−/− mice were even less susceptible to II+ROP16I oral challenge than wild-type mice (Fig. 2A). In addition, Cd274−/− and Stat6−/− mice exhibited lower parasite burdens than wild-type mice, as revealed by whole-body bioluminescence imaging of mice orally challenged with strain II+ROP16I (Fig. 2B and C). In contrast, neutralization of the B7-DC receptor with blocking antibodies or infection in LysM-cre Stat3fl/fl mice produced infections that were not significantly different from those in controls (Fig. 2D to F). Thus, although some of the ROP16 host targets analyzed here modulated resistance to oral infection, in the case of B7-H1 and STAT6, they inhibited rather than promoted parasite clearance.
Fig 2.

Host resistance to the II+ROP16I strain occurs in the absence of B7-H1 and STAT6. (A) Cumulative survival data for Cd274−/−, Stat6−/−, or wild-type C57BL/6 mice orally challenged with the II+ROP16I strain. The total number (n) of mice orally challenged is listed. The Mantel-Cox test was used to address significant differences (P < 0.05) in survival between knockout and wild-type mice (n.s., not significant). (B) Representative whole-body bioluminescence images of individual knockout or wild-type (wt) mice on day 8 following oral infection with strain II+ROP16I. (C) Average bioluminescence (number of photons/s/r2) detected by whole-body imaging of wild-type and knockout mice on various days following oral infection with strain II+ROP16I. Bars depict the mean bioluminescence of the cohort + SD (n = 5 per group). Asterisks indicate statistically significant differences (two-tailed Student t test, P < 0.05) in luminescence between knockout and wild-type mice on that particular day. (D) C57BL/6 mice were injected i.p. with a neutralizing antibody against B7-DC or with isotype control antibody on days 4, 6, and 9 following oral challenge with the II+ROP16I strain. The cumulative results from two experiments are plotted; the number of mice for each treatment is indicated (Mantel-Cox, P = 0.2; n.s., not significant). (E) As in panel D, but the average bioluminescence obtained from whole-body imaging on day 7 following oral challenge with the II+ROP16I strain is plotted (Student t test, P = 0.3). (F) Survival curves of Stat3fl/fl (wild type) or Stat3fl/fl LysM-cre mice following oral infection with the II+ROP16I strain. The combined results of two individual experiments are plotted, and the number of mice challenged is indicated (Mantel-Cox test, P = 0.3).
Synergistic expression of ROP16 and GRA15 increases parasite susceptibility within IFN-γ- and TNF-α-stimulated macrophages.
Inflammatory macrophages that are recruited to the intestine are critically important for the host to limit Toxoplasma replication and promote host resistance at the site of infection (32, 33). Since ROP16 and GRA15 had a drastic effect on parasite burden in vivo, we tested whether the type II engineered strains differed in their ability to evade macrophage toxoplasmacidal mechanisms elicited by stimulation with IFN-γ and TNF-α, cytokines that are expressed in the intestine following oral infection (20). When infections were performed in bone marrow-derived macrophages (BMDMs) previously stimulated with TNF-α (25 ng/ml) and IFN-γ (10 to 1 ng/ml), survival differences between the engineered strains could be observed. In particular, the II+ROP16I strain exhibited the lowest parasite burden in stimulated macrophages (Fig. 3A). Importantly, differences in parasite numbers between the type II and II+ROP16I strains were still evident in IFN-γ- and TNF-α-stimulated Stat6−/− and LysM-cre Stat3fl/fl BMDMs (Fig. 3B) or in these STAT-deficient macrophages pretreated with 0.5 μM the STAT3 inhibitor StatticV (not shown). Thus, Toxoplasma GRA15 and ROP16 play a fundamental role in modulating macrophage resistance mechanisms in vitro, which correlates with the oral infectivity of these strains in vivo.
Fig 3.
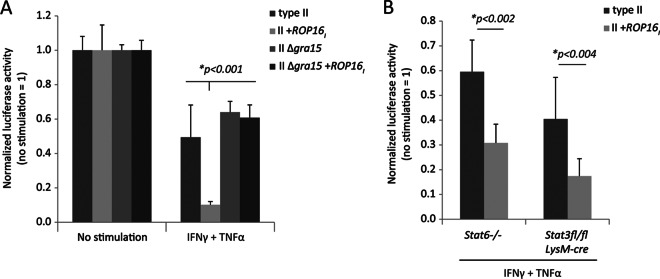
Coexpression of ROP16 and GRA15 enhances parasite susceptibility within IFN-γ- and TNF-α-stimulated macrophages. (A) C57BL/6 BMDMs were stimulated with IFN-γ (10 ng/ml) and TNF-α (25 ng/ml) for 20 h in 20% L929 medium (final concentration of G-CSF, 9 ng/ml). Before infection, the medium was replaced and infection with the indicated luciferase-expressing strains (MOI, ∼0.2) occurred in medium without cytokine or L929 in triplicate; 24 h later, luciferase activity was measured in the cell lysate. The luciferase activity obtained in stimulated macrophages is normalized to the mean luciferase activity observed in nonstimulated macrophages (no stimulation = 1). The mean + SD of the triplicates for each parasite strain is plotted. Significance was addressed by analysis of variance testing; indicated is the P value obtained for the II+ROP16I strain in stimulated macrophages. (B) As in panel A, but Stat6−/− (B6) and LysM-cre Stat3fl/fl (B6) mice were assayed. P values were calculated with a two-tailed Student t test comparing differences between parasite strains.
Evidence for STAT5 activation by Toxoplasma ROP16.
Although the II+ROP16I strain produced an infection characterized by reduced parasite replication and improved host survival (Fig. 1), Stat6 deletion enhanced rather than reversed these phenotypes in vivo (Fig. 2A to C) and Stat3- or Stat6-deficient BMDMs readily killed the II+ROP16I strain (Fig. 3). Since many of the ROP16-regulated host genes lack consensus STAT3/6 transcription factor binding sites (TFBSs) (13), we utilized a microarray strategy to determine ROP16-regulated host genes that were modulated independently of either STAT6 or STAT3 transcription factors. To this end, we infected Stat6−/−, LysM-cre Stat3fl/fl, and Stat3fl/fl control mouse BMDMs with the engineered Toxoplasma strains. We noticed that Stat3 gene expression in LysM-cre Stat3fl/fl mouse BMDMs was reduced by only 50%, suggesting incomplete knockout during in vitro differentiation, which has been observed by others (34). Thus, we chose a lower cutoff value of a 1.7-fold difference for macrophage gene expression that was defined to be influenced by ROP16, STAT3, or STAT6 (see Materials and Methods). Using these criteria, the expression of 938 host genes with unique gene annotations were regulated by Toxoplasma ROP16, and of these, 51% were not modulated through either host STAT3 or STAT6 (Fig. 4A). TFBS analysis of the remaining ROP16-regulated genes revealed significant enrichment in STAT5A/B TFBSs (determined by gene set enrichment analysis [GSEA]). Indeed, type I ROP16 induced the prolonged phosphorylation and nuclear translocation of STAT5 observed in infected HFFs and BMDMs (Fig. 4B), implicating ROP16 as a broadly promiscuous Toxoplasma kinase for the STATs. A shortened list of Toxoplasma ROP16-regulated genes that are modulated through host STAT3 and STAT6, as well as host genes regulated by ROP16 but independent of either host STAT, can be found in Table S3 in the supplemental material.
Fig 4.

Evidence for STAT5 activation by Toxoplasma ROP16. (A) A Venn diagram depicting ROP16-regulated host genes (1.7-fold) that are influenced by STAT3 and/or STAT6 in infected BMDMs. BMDMs from Stat6−/−, Stat3fl/fl LysM-cre, or Stat3fl/fl control mice were infected with the type II engineered Toxoplasma strains to determine ROP16-regulated genes whose expression was regulated by STAT3 and/or STAT6 (see Materials and Methods). Gene symbols and the total numbers of genes (in red) that fall within each category are indicated within the diagram. TFBS analysis of ROP16-regulated genes that were not influenced by STAT3/6 was performed by GSEA, and the most significant TFBSs are listed. (B) At 20 h after infection with the indicated parasite strains, cells were analyzed by immunofluorescence for phosphorylated STAT5. Representative immunofluorescence images of infected HFFs and BMDMs are shown. (C) On day 5 following oral challenge with the indicated parasite strains, individual Peyer's patches from C57BL/6 mice were dissected and analyzed by bioluminescence imagining. Peyer's patches with similar parasite burdens (see Fig. S6 in the supplemental material) and a Peyer's patch from a naive mouse were analyzed by microarray analysis. The normalized microarray chip intensities for the indicated genes are plotted; all genes are within the top 50 genes induced following infection compared to their expression in the naive control. TFBS enrichment analysis (DiRE) was performed on all Peyer's patch genes that were upregulated, on average, 1.7-fold following infection compared to the level of regulation in the uninfected sample [0.8 < log2(average gene expression of type II and II+ROP16I strain infections)/gene expression in the naive sample]. The top-scoring TFBS (rank = 1) and its importance value are given. (D) As in panel C, but the fold change in gene expression between II+ROP16I strain- versus type II strain-infected Peyer's patches and small intestines with Peyer's patches removed (approximately the jejunum) is plotted. The top-scoring TFBS and its importance value (DiRE) are indicated for II+ROP16I strain-upregulated Peyer's patch and intestinal genes. Genes bearing putative STAT5 or RORA2 TFBSs in their promoters are indicated with blue and red asterisks, respectively. (E) Profile of the running enrichment score and positions of members of gene sets for M2 versus M1 activation and hypoxia (determined by GSEA) on a rank-ordered list of II+ROP16I strain- versus type II strain-infected Peyer's patches. The nominal P value is indicated. The GSEA diagrams show the enrichment score (green line), which reflects the degree to which that particular gene set is overrepresented in the differentially expressed genes between II+ROP16I strain- and type II strain-infected Peyer's patches (ranked by their differential expression values, where 1 is the gene most highly induced by the II+ROP16I strain). The middle portion of the diagram shows where the members of the particular gene set (black bars) appear in the ranked gene list.
Since synergistic expression of Toxoplasma GRA15 and ROP16 was required for host protection from oral infection, we searched for host genes that were particularly sensitive to the combined expression of both ROP16 and GRA15. Ninety macrophage genes were at least 1.5-fold different in expression in response to the II+ROP16I strain than to the other engineered strains, many of which contain putative STAT5 and NF-κB TFBSs (TRANSFAC; Genomatix) within conserved promoters regions (Table 1). Interestingly, Ccl1 and Ccl3 chemokine gene expression was highly induced by the II+ROP16I strain, while coagulation factors (F5 and F13a1) were downregulated. CCL3 and its receptor, CCR1, were recently shown to be required for inflammatory monocyte recruitment to the intestine of Toxoplasma-infected mice, which in turn limited the intestinal parasite burden (32). Considering that a putative STAT5 TFBS sequence is 500 bp upstream of the transcriptional start site of CCL3 (not shown), it remains to be determined whether gene expression of Ccl3 by the II+ROP16I strain requires host STAT5 and/or NF-κB.
Table 1.
Macrophage genes that are maximally regulated in response to the II+ROP16I Toxoplasma straina
| Level of expression and MGI gene symbol | Gene title; alternative name(s)b | BMDM gene expression following infection with the indicated strains |
STAT-dependent regulation in infected BMDMsc |
No. of promoter region STAT5 TFBSsd | TFBS sequence in promoter regione |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Uninfected | Type II | II+ROP16I | II Δgra15 | II Δgra15 +ROP16I | STAT6 | STAT3 | STAT | NF-κB | |||
| Expression 1.5-fold higher | |||||||||||
| Retnla | Resistin-like alpha; RELM-alpha, FIZZ | 50 | 168 | 2,154 | 50 | 1,349 | Y | Y | Y | ||
| Slamf1 | Signaling lymphocytic activation molecule family member 1 | 50 | 55 | 316 | 50 | 117 | Y | Y | 7 | Y | Y |
| Aqp9 | Aquaporin 9 | 50 | 178 | 581 | 128 | 306 | Y | ||||
| Ccl1 | Chemokine (C-C motif) ligand 1 | 50 | 253 | 500 | 50 | 50 | 1 | Y | |||
| Ptx3 | Pentraxin-related gene | 50 | 148 | 271 | 50 | 50 | |||||
| Tnfrsf13c | TNF receptor superfamily, member 13c; BAFF receptor | 50 | 221 | 394 | 50 | 74 | Y | Y | |||
| Dtx2 | Deltex 2 homolog (Drosophila) | 183 | 172 | 299 | 179 | 165 | 3 | Y | Y | ||
| Ccl3 | Chemokine (C-C motif) ligand 3; MIP-1α | 1,909 | 3,258 | 5,293 | 1,931 | 2,647 | 1 | Y | Y | ||
| Alas1 | Aminolevulinic acid synthase 1 | 793 | 1,819 | 2,921 | 1,650 | 1,687 | 4 | Y | Y | ||
| Expression 1.5-fold lower | |||||||||||
| Thbs1 | Thrombospondin 1 | 686 | 5,421 | 3,169 | 6,118 | 5,052 | Y | Y | |||
| Ifitm1 | Interferon-induced transmembrane protein 1 | 774 | 1,663 | 698 | 1,884 | 1,416 | 5 | Y | |||
| B3gnt5 | UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 5 | 153 | 1,662 | 478 | 1,029 | 1,069 | Y | Y | |||
| F13a1 | Coagulation factor XIII, A1 subunit | 199 | 1,421 | 628 | 1,787 | 1,298 | Y | Y | |||
| Crem | cAMP-responsive element modulator | 230 | 1,123 | 614 | 1,030 | 1,431 | 1 | Y | Y | ||
| F5 | Coagulation factor V | 50 | 832 | 208 | 1,057 | 1,104 | Y | Y | Y | ||
| Ets1 | E26 avian leukemia oncogene 1, 5′ domain | 880 | 491 | 179 | 382 | 759 | Y | 4 | Y | Y | |
| Ctla2a | Cytotoxic T lymphocyte-associated protein 2 alpha | 378 | 1,040 | 428 | 1,357 | 678 | Y | ||||
| Egln3 | EGL nine homolog 3 (Caenorhabditis elegans) | 90 | 414 | 189 | 358 | 435 | 3 | Y | Y | ||
| Fcgr4 | Fc receptor, IgG, low affinity IV | 2,673 | 1,643 | 965 | 3,016 | 1,641 | Y | Y | |||
A list of host BMDM genes that are expressed at least 1.5-fold higher or lower in response to infection with the II+ROP16I strain compared to all other type II strains (strains type II, II Δgra15, II Δgra15 +ROP16I). The mouse gene symbol, gene name, and relative gene expression values (MAS5) determined by microarray analysis following 18 h of infection are indicated (17). Gene expression values from uninfected macrophages are also listed, and those below 50 were considered background.
MIP-1α, macrophage inflammatory protein 1α; cAMP, cyclic AMP.
Indicates whether the ROP16-modulated host gene in infected BMDMs is influenced by either STAT6 or STAT3 transcription factors, as described in Table S3 in the supplemental material (Y, yes).
Listed is the number of STAT5A/B TFBSs found within −2,000 to +200 bp of the transcriptional start site for each mouse gene, according to the TRANSFAC Professional database.
Indicates whether a V$STAT or V$NFKB transcription factor family binding site is found within a 2,000-bp window of the promoter region using standard weight matrices in Genomatix (Y, yes).
Peyer's patch genes modulated by strain II+ROP16I are enriched for STAT5 TFBSs.
To deduce host signaling pathways that characterize the in vivo immune response to the II+ROP16I strain and to uncover other potential host targets of ROP16, the gene expression profile during oral infection was determined. We noticed that during early time points of infection, Toxoplasma could be detected within Peyer's patches (see Fig. S6A in the supplemental material). When monitoring infection with a nonlethal type III strain, the Peyer's patch was the only place where the parasite was observed in the intestine (see Fig. S6A in the supplemental material). Whether Toxoplasma has tropism for this lymphoid follicle is unknown but is consistent with an earlier finding that the Peyer's patch was the first place Toxoplasma could be detected following oral infection (35). Thus, the Peyer's patch was explored as a point of regulation for Toxoplasma.
The difference in the transcriptional response of whole Peyer's patches with equivalent parasite burdens between type II and II+ROP16I strain infections was determined at day 5 after oral challenge (see Fig. S6B and C in the supplemental material). Expression of genes for IFN-γ (Ifng) and STAT1 (Stat1), Th1-associated chemokines (Cxcl9, Cxcl10), and IFN-γ-induced mediators of Toxoplasma killing (the IRGs Iigbp1, Irgm2, Igtp, and Tgtp1/2 and the GBPs Gbp2, Gbp3, Gbp6, and Gbp8) in the Peyer's patch were similarly induced between strains (Fig. 4C). Peyer's patch genes that were equally and highly upregulated between strains were significantly enriched in interferon regulatory factor (IRF) TFBSs (Distant Regulatory Elements of co-regulated genes [DiRE]). Importantly, STAT5B was the most significantly enriched TFBS in the 278 genes that were upregulated (1.7-fold) in response to II+ROP16I strain infections compared to type II strain infections (DiRE) (Fig. 4D). Thus, the STAT5 signaling pathway may be exploited by Toxoplasma ROP16 to regulate host gene expression in infected BMDMs and Peyer's patches. We also noted that Hif1a and its downstream targets, Egln3 and Bnip3, were upregulated in response to the II+ROP16I strain. Likewise, pathway enrichment analysis positively correlated the hypoxia gene signature in strain II+ROP16I-induced genes (Fig. 4E). The cellular hypoxic response has been shown to negatively influence parasite growth in vitro (36). Finally, the M2 versus M1 gene signature correlated with strain II+ROP16I upregulated Peyer's patch genes (Fig. 4E), findings that are consistent with ROP16's ability to induce M2 activation (17).
Infected small intestines with similar parasite burdens were also analyzed (see Fig. S6D and E in the supplemental material). As observed in the Peyer's patch, the Th1 gene expression profile in the intestine was equally induced between strain infections on day 5 (not shown). In contrast, the gene for the retinoid X receptor, Rxrb, was highly induced following strain II+ROP16I infection, and RORA2 was the most significantly enriched TFBS in the 196 genes that were upregulated (2-fold) in response to the II+ROP16I strain (Fig. 4D). Importantly, several genes were similarly regulated in both the Peyer's patch and intestine following oral challenge; the Mast cell proteases Mcpt1 and Mcpt2, the hypoxia regulator Egln3, and the Th2-associated cysteine-rich secreted factor Retnlb were all upregulated in response to strain II+ROP16I oral infection. Whether these genes mediate resistance to Toxoplasma infection is unknown.
DISCUSSION
In this report, we provide a variety of data demonstrating that Toxoplasma GRA15, which activates NF-κB, and ROP16, which activates the transcription factors STAT3/6 and, as shown in this report, STAT5 (Fig. 4), promote host resistance to oral Toxoplasma infection. The expression of both virulence factors promoted host resistance to the type II strain by lowering intestinal parasite numbers, which in turn reduced intestinal pathology in susceptible C57BL/6 mice. For pathogens that spread by achieving chronic infection, it is probably not surprising that Toxoplasma carries within its arsenal of virulence effectors those that quell its own replication or assist host immune defenses. This has been demonstrated for the Toxoplasma serine protease inhibitor TgPI1 (37), the surface proteins SRS9 and SRS29C (38, 39), and possibly many others that regulate parasite invasion, replication, and metabolism.
Our initial hypothesis was that host susceptibility to oral infection would correlate with the degree to which our type II engineered strains could promote M1/M2 activation (17). We anticipated that type II strains expressing GRA15II would cause more pathology because these strains activate NF-κB and induce the proinflammatory M1 response; conversely, we reasoned that strains expressing the STAT6-activating version of ROP16 (type I/III) should induce less pathology and inflammation because they promote M2 activation. However, this hypothesis was not supported by the data, as the M2-inducing type II Δgra15 +ROP16I strain and the M1-inducing type II strain were equally virulent. Rather, host survival to the type II strain was dependent on the synergistic expression of both GRA15 and ROP16 parasite effectors. GRA15 and ROP16 were discovered by screening candidate genes in Toxoplasma virulence loci (Vir1-5) that determined mouse mortality to i.p. infection (22). The genes in the Vir loci that encode GRA15 and ROP16 had weak genetic association peaks; hence, any effect on virulence would require a particular combination of these or other genetic factors. For GRA15, after both i.p. (16) and oral challenge, the II Δgra15 strain produced an infection characterized by elevated parasite numbers on day 5 (see Fig. S4 in the supplemental material), but as the infection progressed, parasite burden and host susceptibility were no different from those observed following type II strain control infections. The data presented here gave us the first indication that GRA15 did indeed affect mouse mortality, as the II Δgra15 +ROP16I and II+ROP16I strains differed considerably in intestinal parasite burden, pathology, and virulence (Fig. 1). Likewise, earlier demonstrations that transgenic expression of ROP16 (type I or III) in the type II strain promoted host survival (17, 22) were possible only because GRA15 is endogenously expressed in the type II strain.
These findings highlight why sexual recombination in the gut of the feline, which produces progeny with different combinations of virulence factors, is so important for the spread of Toxoplasma gondii in nature. Chronic infection in resistant intermediate hosts with stronger innate immune responses or with previous immunity to Toxoplasma might require progeny that express increased numbers of virulence factors that antagonize host resistance mechanisms. Conversely, in hosts that have weakened innate or adaptive immune systems, virulence factors like ROP16 and GRA15 might assist host survival by limiting parasite replication. This scenario played out in laboratory mice. Compared to acute infection in C57BL/6 mice, the A/J strain was able to control type II replication in the intestine (Fig. 1) and in the liver (25). Importantly, expression of ROP16 and GRA15 can correct for this genetic susceptibility in B6 mice, leading to chronic infection and cyst generation (not shown). The intriguing possibility that inflammation-related pathologies surrounding the type II strain in HIV-AIDS patients with reactive encephalitis (10) are due to polymorphisms present in type II ROP16 remains.
In our attempts to determine host signaling pathways and molecular players that promote host control of parasite replication, we found that ROP16 targets B7-H1 and STAT6 inhibits rather than promots parasite clearance mechanisms in vivo. Furthermore, it was not obvious from these screens that the STAT3/6 signaling pathways were controlling the relevant resistance mechanisms activated by the II+ROP16I strain. Thus, we explored the transcriptional response of infected intestinal samples and BMDMs to search for novel targets and functions of ROP16. This led to the finding that ROP16 induces the phosphorylation and nuclear translocation of STAT5 and STAT5 TFBSs are significantly enriched in differentially expressed genes from Peyer's patches with similar type II and II+ROP16I parasite burdens. Many growth factors like erythropoietin and prosurvival cytokines like IL-2, IL-7, and IL-15 signal through STAT5. Regulatory T cells (Tregs), which dampen proinflammatory responses mediated by Th1/Th17 cells following Toxoplasma infection (40), require endogenous IL-2 for maintenance of function, and IL-2 therapeutics promote host survival to oral Toxoplasma infection (41). Granulocyte-macrophage colony-stimulating factor (GM-CSF) also signals via STAT5, can ameliorate dextran sodium sulfate-induced colitis in rats, and is being investigated in clinical trials for the treatment of Crohn's disease (42). Whether STAT5 plays a role in the functional outcome of the infected host due to ROP16I expression remains to be determined.
Recently, the function of SOCS3, a host gene that is highly induced by type I ROP16 (15, 17), has been studied during i.p. Toxoplasma infection. SOCS3 binds directly to the gp130 subunit of the IL-6 receptor complex and inhibits IL-6-induced STAT3 signaling (43, 44). In the absence of SOCS3, gp130 activation of STAT3, which typically lasts minutes, becomes protracted over hours, a situation that is similar to the longer STAT3 activation by the type I ROP16 kinase and the IL-10 receptor. When SOCS3 is deleted from macrophages and neutrophils, IL-6 mimics IL-10 and turns off IL-12 production, resulting in a delayed IFN-γ response. As a consequence, LysM-cre Soc3fl/fl mice were no longer able to control parasite replication following i.p. Toxoplasma infection (45). In contrast, although ROP16 induces SOCS3 expression and strain II+ROP16I-infected BMDMs produce less IL-12, there was a decrease rather than an increase in II+ROP16I parasite numbers in vivo. In fact, macrophage proinflammatory cytokine production did not correlate at all with the oral virulence of the engineered strains, nor did we find any evidence that these strains were unable to elicit a mucosal IFN-γ response in either the Peyer's patch (Fig. 4C) or the small intestine (see Fig. S4C in the supplemental material). Thus, GRA15 and ROP16 may have very little to do with influencing Th1 responses through modulation of NF-κB. Consistent with this supposition, NF-κB and the upstream signaling activator MyD88 regulate the Th1 response during Toxoplasma infection primarily through their action in T cells as opposed to other cell types (26, 46). Instead, differences in parasite burden are evident in cytokine-stimulated macrophages (Fig. 3), suggesting that ROP16 and GRA15 may work directly on host mechanisms that regulate parasite survival or replication within infected cells.
A recent report by Butcher and colleagues has identified such a mechanism. When l-arginine is limiting in the medium of infected cells, type I ROP16 quells parasite growth through STAT6-dependent induction of the host arginase 1 enzyme (47), which metabolizes l-arginine. Because Toxoplasma does not synthesize its own l-arginine, it relies on the host's cellular sources for this required amino acid. Whether arginine becomes limiting during oral infection remains an intriguing possibility. Initial attempts to rescue intestinal replication of the II+ROP16I strain through administration of l-arginine in the drinking water or by daily i.p. injection proved unsuccessful (not shown). The strain type likely matters with these experiments. Whereas both arginase 1 (43) and STAT6 have a negative impact on host survival during type II infections (Fig. 2; not shown), the STAT6 pathway limits type I replication in vitro and inhibits type I dissemination in a macrophage i.p. transfer model in vivo (47). Uniform Stat6 deletion can also have pleiotropic effects in a variety of cell types that may alter host susceptibility to Toxoplasma infection. It remains to be determined whether the timing and quality of the Th1 response to Toxoplasma are altered in Stat6−/− mice. STAT6 binds and represses the Ifng locus in mouse Th2 cells but does not affect Th1 IFN-γ secretion (48). IL-5, a Th2-associated cytokine which signals through STAT6, was produced in the intestine following oral infection (see Fig. S4 in the supplemental material). Thus, in Stat6−/− mice, uninhibited IFN-γ secretion from (IL-5-positive) Th2-like cells might lead to increased host resistance. Enhanced Th1 responses may also underlie the increased resistance of B7-H1-deficient mice to acute (Fig. 2) and chronic infection (29).
A second possible alternative is the way in which GBP recruitment to the parasitophorous vacuole is modulated by the combined expression of GRA15 and ROP16. ROP16 inhibited but GRA15 promoted GBP1 recruitment to the parasitophorous vacuole in IFN-γ-stimulated fibroblasts (49). In contrast, these effectors did not alter IRG coating in IFN-γ-stimulated mouse embryonic fibroblasts (50), nor did IFN-γ-stimulated macrophages elicit differences in parasite numbers between the engineered strains. Importantly, the effect of GRA15 and ROP16 on parasite numbers in macrophages was seen only upon stimulation with IFN-γ and TNF-α, cytokines which activate STAT1 and NF-κB, respectively. Exactly how GRA15 and ROP16 work through the GBP system to alter host toxoplasmacidal mechanisms is still not clear and warrants future study. An interesting avenue for investigation is determining whether the NF-κB or STAT3/5/6 signaling pathways intersect the GBP system to regulate parasite replication and GBP recruitment to the parasitophorous vacuole.
Finally, synergistic expression of ROP16 and GRA15 induces maximal gene expression of the chemoattractants CCL3 and CCL1. Thus, in addition to promoting parasite killing mechanisms in stimulated macrophages, the induced chemokine profile might elicit greater inflammatory monocyte recruitment to the intestine, amplifying parasite clearance of the II+ROP16I strain. These are testable hypotheses.
In conclusion, Toxoplasma has evolved ways to limit parasite replication in the infected host. Toxoplasma-induced ileitis in C57BL/6 mice could be ameliorated by reducing the intestinal parasite burden through the action of GRA15 and ROP16. The convergence on the STAT and NF-κB signaling pathways by Toxoplasma effectors and host proinflammatory cytokines points us to the interface between host and pathogen, where the fate of both is decided.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by RO1 grant AI080621 from the National Institutes of Health (NIH) and the Pew Charitable Trust (to J.P.J.S.) and by the state of Pennsylvania and NIH grants AI071302 and AI084882 (to C.A.H.); K.D.C.J. was supported by postdoctoral fellowships from the Cancer Research Institute and the Charles A. King Trust; M.A.H. was supported by a Wellcome Trust-MIT postdoctoral fellowship.
We also thank the MIT Biomicro Center for technical assistance with the microarray experiments.
Footnotes
Published ahead of print 1 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01185-12.
REFERENCES
- 1. Schreiner M, Liesenfeld O. 2009. Small intestinal inflammation following oral infection with Toxoplasma gondii does not occur exclusively in C57BL/6 mice: review of 70 reports from the literature. Mem. Inst. Oswaldo Cruz 104:221–233 [DOI] [PubMed] [Google Scholar]
- 2. Darde ML, Villena I, Pinon JM, Beguinot I. 1998. Severe toxoplasmosis caused by a Toxoplasma gondii strain with a new isoenzyme type acquired in French Guyana. J. Clin. Microbiol. 36:324 (Letter.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Melo MB, Jensen KD, Saeij JP. 2011. Toxoplasma gondii effectors are master regulators of the inflammatory response. Trends Parasitol. 27:487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adams LB, Hibbs JB, Jr, Taintor RR, Krahenbuhl JL. 1990. Microbiostatic effect of murine-activated macrophages for Toxoplasma gondii. Role for synthesis of inorganic nitrogen oxides from l-arginine. J. Immunol. 144:2725–2729 [PubMed] [Google Scholar]
- 5. Zhao Y, Ferguson DJ, Wilson DC, Howard JC, Sibley LD, Yap GS. 2009. Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. J. Immunol. 182:3775–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, Howard JC. 2005. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 1:e24 doi:10.1371/journal.ppat.0010024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamamoto M, Okuyama M, Ma JS, Kimura T, Kamiyama N, Saiga H, Ohshima J, Sasai M, Kayama H, Okamoto T, Huang DC, Soldati-Favre D, Horie K, Takeda J, Takeda K. 2012. A cluster of interferon-gamma-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity 37:302–313 [DOI] [PubMed] [Google Scholar]
- 8. Suzuki Y, Sher A, Yap G, Park D, Neyer LE, Liesenfeld O, Fort M, Kang H, Gufwoli E. 2000. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J. Immunol. 164:5375–5382 [DOI] [PubMed] [Google Scholar]
- 9. Egan CE, Cohen SB, Denkers EY. 2012. Insights into inflammatory bowel disease using Toxoplasma gondii as an infectious trigger. Immunol. Cell Biol. 90:668–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lindstrom I, Kaddu-Mulindwa DH, Kironde F, Lindh J. 2006. Prevalence of latent and reactivated Toxoplasma gondii parasites in HIV-patients from Uganda. Acta Trop. 100:218–222 [DOI] [PubMed] [Google Scholar]
- 11. Suzuki Y, Joh K. 1994. Effect of the strain of Toxoplasma gondii on the development of toxoplasmic encephalitis in mice treated with antibody to interferon-gamma. Parasitol. Res. 80:125–130 [DOI] [PubMed] [Google Scholar]
- 12. Liesenfeld O, Kosek J, Remington JS, Suzuki Y. 1996. Association of CD4+ T cell-dependent, interferon-gamma-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J. Exp. Med. 184:597–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ong YC, Reese ML, Boothroyd JC. 2010. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J. Biol. Chem. 285:28731–28740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamamoto M, Standley DM, Takashima S, Saiga H, Okuyama M, Kayama H, Kubo E, Ito H, Takaura M, Matsuda T, Soldati-Favre D, Takeda K. 2009. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J. Exp. Med. 206:2747–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445:324–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, Jensen KD, Saeij JP. 2011. Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J. Exp. Med. 208:195–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jensen KD, Wang Y, Wojno ED, Shastri AJ, Hu K, Cornel L, Boedec E, Ong YC, Chien YH, Hunter CA, Boothroyd JC, Saeij JP. 2011. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe 9:472–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martinez FO, Helming L, Gordon S. 2009. Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27:451–483 [DOI] [PubMed] [Google Scholar]
- 19. Schneider DS, Ayres JS. 2008. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 8:889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liesenfeld O, Kang H, Park D, Nguyen TA, Parkhe CV, Watanabe H, Abo T, Sher A, Remington JS, Suzuki Y. 1999. TNF-alpha, nitric oxide and IFN-gamma are all critical for development of necrosis in the small intestine and early mortality in genetically susceptible mice infected perorally with Toxoplasma gondii. Parasite Immunol. 21:365–376 [DOI] [PubMed] [Google Scholar]
- 21. Munoz M, Heimesaat MM, Danker K, Struck D, Lohmann U, Plickert R, Bereswill S, Fischer A, Dunay IR, Wolk K, Loddenkemper C, Krell HW, Libert C, Lund LR, Frey O, Holscher C, Iwakura Y, Ghilardi N, Ouyang W, Kamradt T, Sabat R, Liesenfeld O. 2009. Interleukin (IL)-23 mediates Toxoplasma gondii-induced immunopathology in the gut via matrixmetalloproteinase-2 and IL-22 but independent of IL-17. J. Exp. Med. 206:3047–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saeij JP, Boyle JP, Coller S, Taylor S, Sibley LD, Brooke-Powell ET, Ajioka JW, Boothroyd JC. 2006. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314:1780–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. 2008. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4:337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rath HC, Wilson KH, Sartor RB. 1999. Differential induction of colitis and gastritis in HLA-B27 transgenic rats selectively colonized with Bacteroides vulgatus or Escherichia coli. Infect. Immun. 67:2969–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McLeod R, Eisenhauer P, Mack D, Brown C, Filice G, Spitalny G. 1989. Immune responses associated with early survival after peroral infection with Toxoplasma gondii. J. Immunol. 142:3247–3255 [PubMed] [Google Scholar]
- 26. Mason NJ, Artis D, Hunter CA. 2004. New lessons from old pathogens: what parasitic infections have taught us about the role of nuclear factor-kappaB in the regulation of immunity. Immunol. Rev. 201:48–56 [DOI] [PubMed] [Google Scholar]
- 27. McLeod R, Skamene E, Brown CR, Eisenhauer PB, Mack DG. 1989. Genetic regulation of early survival and cyst number after peroral Toxoplasma gondii infection of A × B/B × A recombinant inbred and B10 congenic mice. J. Immunol. 143:3031–3034 [PubMed] [Google Scholar]
- 28. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. 2008. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26:677–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bhadra R, Gigley JP, Weiss LM, Khan IA. 2011. Control of Toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1–PDL-1 blockade. Proc. Natl. Acad. Sci. U. S. A. 108:9196–9201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Charles E, Joshi S, Ash JD, Fox BA, Farris AD, Bzik DJ, Lang ML, Blader IJ. 2010. CD4 T-cell suppression by cells from Toxoplasma gondii-infected retinas is mediated by surface protein PD-L1. Infect. Immun. 78:3484–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huber S, Hoffmann R, Muskens F, Voehringer D. 2010. Alternatively activated macrophages inhibit T-cell proliferation by Stat6-dependent expression of PD-L2. Blood 116:3311–3320 [DOI] [PubMed] [Google Scholar]
- 32. Schulthess J, Meresse B, Ramiro-Puig E, Montcuquet N, Darche S, Begue B, Ruemmele F, Combadiere C, Di Santo JP, Buzoni-Gatel D, Cerf-Bensussan N. 2012. Interleukin-15-dependent NKp46(+) innate lymphoid cells control intestinal inflammation by recruiting inflammatory monocytes. Immunity 37:108–121 [DOI] [PubMed] [Google Scholar]
- 33. Dunay IR, Damatta RA, Fux B, Presti R, Greco S, Colonna M, Sibley LD. 2008. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 29:306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Butcher BA, Kim L, Panopoulos AD, Watowich SS, Murray PJ, Denkers EY. 2005. IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-alpha in host macrophages. J. Immunol. 174:3148–3152 [DOI] [PubMed] [Google Scholar]
- 35. Sumyuen MH, Garin YJ, Derouin F. 1995. Early kinetics of Toxoplasma gondii infection in mice infected orally with cysts of an avirulent strain. J. Parasitol. 81:327–329 [PubMed] [Google Scholar]
- 36. Spear W, Chan D, Coppens I, Johnson RS, Giaccia A, Blader IJ. 2006. The host cell transcription factor hypoxia-inducible factor 1 is required for Toxoplasma gondii growth and survival at physiological oxygen levels. Cell. Microbiol. 8:339–352 [DOI] [PubMed] [Google Scholar]
- 37. Pszenny V, Davis PH, Zhou XW, Hunter CA, Carruthers VB, Roos DS. 2012. Targeted disruption of Toxoplasma gondii serine protease inhibitor 1 increases bradyzoite cyst formation in vitro and parasite tissue burden in mice. Infect. Immun. 80:1156–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim SK, Karasov A, Boothroyd JC. 2007. Bradyzoite-specific surface antigen SRS9 plays a role in maintaining Toxoplasma gondii persistence in the brain and in host control of parasite replication in the intestine. Infect. Immun. 75:1626–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wasmuth JD, Pszenny V, Haile S, Jansen EM, Gast AT, Sher A, Boyle JP, Boulanger MJ, Parkinson J, Grigg ME. 2012. Integrated bioinformatic and targeted deletion analyses of the SRS gene superfamily identify SRS29C as a negative regulator of Toxoplasma virulence. mBio 3(6):e00321–12 doi:10.1128/mBio.00321-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, Harris TH, Grainger J, Wojno ED, Wagage S, Roos DS, Scott P, Turka LA, Cherry S, Reiner SL, Cua D, Belkaid Y, Elloso MM, Hunter CA. 2012. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 37:511–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O'Brien S, Blank R, Lamb E, Natarajan S, Kastenmayer R, Hunter C, Grigg ME, Belkaid Y. 2009. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31:772–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Egea L, Hirata Y, Kagnoff MF. 2010. GM-CSF: a role in immune and inflammatory reactions in the intestine. Expert Rev. Gastroenterol. Hepatol. 4:723–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, Basaraba RJ, Konig T, Schleicher U, Koo MS, Kaplan G, Fitzgerald KA, Tuomanen EI, Orme IM, Kanneganti TD, Bogdan C, Wynn TA, Murray PJ. 2008. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 9:1399–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR, Yoshimura A. 2003. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immunol. 4:551–556 [DOI] [PubMed] [Google Scholar]
- 45. Whitmarsh RJ, Gray CM, Gregg B, Christian DA, May MJ, Murray PJ, Hunter CA. 2011. A critical role for SOCS3 in innate resistance to Toxoplasma gondii. Cell Host Microbe 10:224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raetz M, Hwang S-H, Wilhelm CL, Kirkland D, Benson A, Sturge CR, Mirpuri J, Vaishnava S, Hou B, DeFranco AL, Gilpin CJ, Hooper LV, Yarovinsky F. 2013. Parasite-induced TH1 cells and intestinal dysbiosis cooperate in IFN-[gamma]-dependent elimination of Paneth cells. Nat. Immunol. 14:136–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, Yarovinsky F, Herbert DR, Bzik DJ, Denkers EY. 2011. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog. 7:e1002236 doi:10.1371/journal.ppat.1002236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chang S, Aune TM. 2007. Dynamic changes in histone-methylation ‘marks’ across the locus encoding interferon-gamma during the differentiation of T helper type 2 cells. Nat. Immunol. 8:723–731 [DOI] [PubMed] [Google Scholar]
- 49. Virreira Winter S, Niedelman W, Jensen KD, Rosowski EE, Julien L, Spooner E, Caradonna K, Burleigh BA, Saeij JP, Ploegh HL, Frickel EM. 2011. Determinants of GBP recruitment to Toxoplasma gondii vacuoles and the parasitic factors that control it. PLoS One 6:e24434 doi:10.1371/journal.pone.0024434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Niedelman W, Gold DA, Rosowski EE, Sprokholt JK, Lim D, Farid Arenas A, Melo MB, Spooner E, Yaffe MB, Saeij JP. 2012. The rhoptry proteins ROP18 and ROP5 mediate Toxoplasma gondii evasion of the murine, but not the human, interferon-gamma response. PLoS Pathog. 8:e1002784 doi:10.1371/journal.ppat.1002784 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.