

Abstract
Insulin-like growth factor (IGF) signaling greatly impacts the development and growth of the central nervous system (CNS). IGF-I and IGF-II, two ligands of the IGF system, exert a wide variety of actions both during development and in adulthood, promoting the survival and proliferation of neural cells. The IGFs also influence the growth and maturation of neural cells, augmenting dendritic growth and spine formation, axon outgrowth, synaptogenesis, and myelination. Specific IGF actions, however, likely depend on cell type, developmental stage, and local microenvironmental milieu within the brain. Emerging research also indicates that alterations in IGF signaling likely contribute to the pathogenesis of some neurological disorders. This review summarizes experimental studies and shed light on the critical roles of IGF signaling, as well as its mechanisms, during CNS development.
Keywords: IGF-I, IGF-II, IGF1R, CNS, Development, Neurons, Glial cells
1. Introduction
Experimental evidence accumulated during the past two decades has convincingly established an essential role for insulin-like growth factor (IGF) signaling in the normal growth and development of the central nervous system (CNS). IGF-I and IGF-II, two members of the IGF system, share homology with each other and with proinsulin. In developing brain, IGF signaling exerts pleiotropic actions on all major neural cell types [including neural stem cells (NSCs), lineage restricted neural precursor cells (NPCs), post-mitotic neurons, oligodendrocytes and astrocytes]. IGFs act to promote the proliferation, maturation, survival, and/or growth of neural cells, predominantly, if not exclusively, by interacting with the type 1 IGF receptor (IGF1R). The biological nature of the actions, however, appears to depend on the specific cell types in place, the local microenvironment, and the particular stage of development. IGF signaling also appears to influence specific biological processes in concert with additional neural signaling, which may provide primary instructive signals to steer NSC toward a specific cell lineage during early development.
Our current knowledge of IGF signaling in the developing brain comes predominantly from a wide variety of in vitro and in vivo experimental studies, the latter primarily derived from studies of mutant mouse models. Nevertheless, individual patients with mutation(s) in the igf-I gene (Camacho-Hubner et al., 1999; Woods et al., 1997) or in the igf1r gene (Abuzzahab et al., 2003; Kruis et al., 2010; Okubo et al., 2003; Wallborn et al., 2010; Woods et al., 1997) are found to be associated with severe body growth failure, micro-cephaly, and mental retardation, strongly arguing for a similar role for IGF signaling during CNS development in humans. Recent literature also has established that many of growth-related phenomena in neural cells, such as neurogenesis (Gage, 2002; Kelsch et al., 2010; Ming and Song, 2011), axon remodeling and de novo synaptogenesis (Bruel-Jungerman et al., 2007; Butefisch, 2006; Carmichael, 2003; Cayre et al., 2009; Gogolla et al., 2007), persist throughout adult life. In parallel, IGFs and their receptors are steadily expressed in the adult brain in a spatial-specific pattern, albeit at relatively lower levels, and are thought to have a significant role in the pathogenesis of several growth-related neurological disorders. In this article, we will review the actions of IGF signaling on brain neural cells with a focus on IGF actions during prenatal and early postnatal life.
2. Overview of the IGF system
The IGF system is traditionally comprised of IGF-I, IGF-II, the IGF1R, the type 2 IGF receptor (IGF2R), and IGF binding proteins (IGFBPs). The growth-promoting actions of IGF-I and IGF-II are pre-dominantly, if not exclusively, mediated by the IGF1R. The receptor binding and biological activities of IGFs are modulated by IGFBPs. At least 10 IGFBPs, including 6 high-affinity IGFBPs and 4 low-affinity IGFBPs, have been identified.
In mice, the actions of IGF–IGF1R signaling appear to be significantly influenced by genomic background. For example, 95% of mice carrying a null mutation (knockout, KO) in the igf-I gene (igf-I KO mice) that are on C75B/6 background die perinatally (Powell-Braxton et al., 1993), while more than 50% of igf-I KO mice on a mixed genomic background of 129/MF1 survive postnatally (Liu et al., 1993). Similar phenomena are also observed in mice with ablated IGF1R expression specifically in nestin-positive (+) neural precursors (Kappeler et al., 2008; Liu et al., 2009). Gene modification of IGF signaling may also play an important role in the development and growth of the CNS. The details, however, remain to be elucidated. Both IGF-I and IGF-II at high concentrations also can bind to the insulin receptor (InR), and InR is capable of mediating IGF-II actions (Louvi et al., 1997; Morrione et al., 1997).
2.1. IGF-I, IGF-II and derivative forms
IGF-I and IGF-II are anabolic peptides (70 and 67 amino acids, respectively), sharing homology with each other and with proinsulin (Daughaday and Rotwein, 1989; Rotwein, 1991). Each of these growth factors is produced by a single large gene (95 kb and 35 kb, respectively), with expression beginning early in embryonic development. IGF-I is produced by all types of major neural cells in the brain. In the brain, IGF-II is more abundantly expressed than IGF-I during prenatal development. During postnatal development somatic IGF-I is regulated by pituitary growth hormone (GH), mediating most of GH’s growth promoting actions. Brain IGF-I expression is also likely regulated by GH to certain extent during development (Hojvat et al., 1982; Hynes et al., 1987; Ye et al., 1997). The mechanisms regulating the expression of the igf-I and igf-II genes are still largely unclear.
Variant forms of IGF-I exist in the brain (Ballard et al., 1987). IGF-I is believed to undergo post-translational N-terminal cleavage by a specific protease into des-N-(1–3) IGF-I, which appears to be the dominant form in the brain (Ballard et al., 1987; Sara et al., 1986; Yamamoto and Murphy, 1995). In organ culture of new born rat olfactory bulb, des-N-(1–3) IGF-I is shown to be potent supporter of viability, cell survival and differentiated cell growth (Russo and Werther, 1994). Des-N-(1–3) IGF-I has been found to have an increased potency for neuroprotective (i.e. anti-apoptotic) effects on neurons. This is likely due to its lower affinity for IGFBPs caused by the absence of glutamate at position 3 (Guan et al., 1996), and thus, relatively higher local concentration of its free form. The N-terminal gly-pro-glu tripeptide fragment (GPE tripeptide), generated along with des-N-(1–3) IGF-I during proteolytic cleavage of IGF-I, also is capable of mediating neuroprotective effects both in vivo and in vitro (Guan et al., 2000; Saura et al., 1999; Shapira et al., 2009), although it remains to be determined whether GPE actions depend on IGF1R. GPE tripeptide has more localized sites of action in the adult brain, including hippocampal CA1-2, pyriform cortex, amygdala, cerebral cortex, choroid plexus and blood vessels (Ikeda et al., 1995; Saura et al., 1999).
2.2. IGF1R and IGF2R
The IGF1R is a heterotetrameric glycoprotein composed of paired, disulfide-linked α- and β-subunits. The α-subunits are extracellular and bind IGFs. The β-subunits contain a long intracytoplasmic domain which contains intrinsic tyrosine kinase activity and critical sites of tyrosine and serine phosphorylation. The IGF1R shares a 46% homology with the InR. The IGF1R and the InR can form hybrid receptors by using the α- and β-subunits of each to form heterodimers. IGF1R/InR hybrids retain capability to transduct both IGF and/or insulin signaling, although the exact functional significance of these hybrid receptors is unknown.
The IGF1R binds IGF-I with high affinity and both IGF-II and insulin with lower affinities (approximately 10-fold and 100-fold, respectively). The IGF1R is expressed in NSC and in all neural cells evaluated (Baron-Van Evercooren et al., 1991). Binding of IGFs to the α-subunit of the IGF1R induces a conformational change in the receptor that results in autophosphorylation of the β-subunit, setting signaling cascades into motion that involve phosphorylation of a series of intracellular substrate proteins (LeRoith et al., 1995), such as insulin receptor substrate (IRS)-1 and IRS-2 as described in detail in Section 6 below.
The IGF2R, a single chain transmembrane protein, is identical to the cation-independent mannose-6-phosphate receptor, and acts to translocate proteins containing mannose-6-phosphate moieties and IGF-II to lysosomes for degradation. Global ablation of IGF2R expression leads to overgrowth, resulting from an increased accumulation of IGF-II (Efstratiadis, 1998; Eggenschwiler et al., 1997). No intrinsic enzymatic activity has been observed for the intracellular domain of the IGF2R. There is also little evidence that IGF-I interact with the IGF2R, and that the IGF2R mediates IGF growth promoting activity in the brain.
2.3. IGFBP
IGF-I and IGF-II are found in circulation and in the extracellular space of most tissues almost completely bound to members of the family of IGFBPs. There are at least 10 IGFBPs, i.e., 6 high-affinity IGFBPs and 4 low-affinity IGFBPs. Six high affinity IGFBPs, designated IGFBP-1 through IGFBP-6 (Jones and Clemmons, 1995), share structural homology with each other, and bind specifically to IGF-I and IGF-II with negligible affinity for insulin. IGFBP-2, -3, -4 and -5 are the most abundant IGFBPs in brain. IGFBP-6 is also expressed, IGFBP-1, however, is not detected in brain during normal development (Ocrant et al., 1990). Each IGFBP is expressed in the CNS in a specific temporal-spatial pattern. The exact functions of IGFBPs in the CNS remain to be fully elucidated. The IGFBPs have been proposed to act: (1) as transport proteins in plasma, (2) to prolong the half lives of the IGFs in circulation, (3) to determine the tissue-and cell-specific localization of IGF-I and IGF-II, and (4) to control the biological actions of IGF-I and IGF-II by modulating their interactions with their receptors (Jones and Clemmons, 1995). In addition, some IGFBPs can also exert IGF independent actions. For example, IGFBP-1 activates integrin-mediated intracellular signaling in trophoblast (Gleeson et al., 2001), and enhances oligodendrocytes migration (Chesik et al., 2010).
Approximately 75% of IGF-I and IGF-II in circulation is carried by a complex of IGF-I or IGF-II, IGFBP-3 and a non-IGF-binding component termed acid labile subunit (ALS). Binding of IGF-I or IGF-II to IGFBP-3 in the presence of ALS produces an IGF–IGFBP-3–ALS ternary complex, which is stabilized by IGF binding (Baxter and Martin, 1989). The IGFs do not readily leave the vascular compartment, when associated with this complex, and their half lives are significantly prolonged by 70- to 90-fold (Guler et al., 1989; Hodgkinson et al., 1989). This circulating pool of bound IGFs is thought to serve as a local reservoir in times of stress. IGFBPs with lower molecular weights (e.g. IGFBP-1, IGFBP-2 and IGFBP-4) have also been implicated as IGF carrier proteins, although their mechanisms of action are less well understood. Estimates of the half lives for these IGFBPs suggest they are cleared more rapidly than the IGF–IGFBP-3–ALS ternary complex (Rechler, 1993).
3. Ontogeny of IGF-I, IGF-II, IGF1R and IGF2R in brain
In the developing CNS, IGFs and IGF receptors are widely expressed in a tempo-spatial specific manner. Table 1 summarizes the expression of IGF-I, as well as that of IGF-II, IGF1R and IGF2R, in the major brain areas during perinatal development and in adult (Ayer-le et al., 1991; Bartlett et al., 1992; Bondy and Chin, 1991; Bondy and Lee, 1993; Bondy, 1991; Cavallaro et al., 1993; Dugas et al., 2008; Folli et al., 1994; Hawkes and Kar, 2003; Kar et al., 1993; Lee et al., 1993; Logan et al., 1994; Quesada et al., 2007; Stylianopoulou et al., 1988; Walter et al., 1999; Zhang et al., 2007). While the mechanisms that regulate the expression of each of these genes still are not completely understood, IGFs and IGF1R are found to be often expressed within close proximity, strongly indicating that IGF acts locally in developing brain in an autocrine and/or paracrine fashion. Nonetheless, IGF-I can be transported across the blood brain barrier (BBB) Pulford and Ishii, 2001; Reinhardt and Bondy, 1994, and thus, it is likely that circulating IGF can influence neurogenesis and their development (Aberg et al., 2007; Anderson et al., 2002).
Table 1.
mRNA expression of IGF system proteins in neural cells in developing and adult brain.
| Brain regions | IGF-I | IGF-II | IGF1R | IGF2R |
|---|---|---|---|---|
| Cerebrum | ||||
| Cerebral cortex | + | + | + | |
| VZ/SVZ | + | + | ||
| Corpus callosum | + | + | ||
| Hippocampus | ||||
| CA | + | + | + | |
| DG | + | + | + | |
| Olfactory blub | ||||
| Mitral cell layer | + | + | + | |
| Tufted cell layer | + | + | ||
| Hypothalamus | + | + | + | |
| Periventricular nucleus | + | + | ||
| Supraoptic nucleus | + | + | ||
| Thalamus | + | + | + | |
| Basal Ganglia | + | + | ||
| Midbrain | + | + | + | |
| Brainstem | + | + | + | |
| Cerebellum | + | |||
| External granule cells | + | + | ||
| Internal granule cells | + | + | + | |
| Purkinje cells | + | + | + | |
| Deep cerebellar nuclei | + | + | ||
| Others | ||||
| Meninges | + | + | + | + |
| Choroid plexus | + | + | + | + |
| Blood vessels | + | + | ||
1. Data comprised in Table 1 are primarily derived from mRNA in situ hybridization studies of rodents at ages of E18 to adult, and adapted from references (Ayer-le et al., 1991; Bartlett et al., 1992; Bondy and Chin, 1991; Bondy and Lee, 1993; Bondy, 1991; Cavallaro et al., 1993; Lee et al., 1993; Walter et al., 1999; Zhang et al., 2007). Immunohistochemical staining and 125I-IGF-II receptor binding studies also are used for IGF1R and IGF2R (Folli et al., 1994; Hawkes and Kar, 2003; Kar et al., 1993; Quesada et al., 2007).
2. IGF-II is expressed in vascular endothelial cells (Ayer-le et al., 1991; Dugas et al., 2008; Logan et al., 1994; Stylianopoulou et al., 1988).
IGF-I and the IGF1R also are expressed in non-neural tissues in early embryonic development before neural tissues have established (Alarcon et al., 1998; Ayaso et al., 2002; Bondy et al., 1990; de et al., 1993; Morales et al., 1997; Perez-Villamil et al., 1994; Scavo et al., 1991a; Scavo et al., 1991b). In chicken embryo, IGF-I mRNA can be detected at embryonic day (E) 3, while insulin is detected earlier at E0 (Perez-Villamil et al., 1994). The InR and IGF1R are present in the blastoderm at E0 through late organogenesis at E9 (Scavo et al., 1991b). IGF-I is preferentially expressed in cephalic regions during late neurulation and throughout organogenesis, and has been shown to be compartmentalized to the epithelial cells of developing eyes (de et al., 1993). The InR and the IGF1R are localized at their highest levels in Hensen’s node, neural folds, neural tube and developing eyes (Girbau et al., 1989). In rodents, IGF-I mRNA is particularly abundant in undifferentiated mesenchymal tissue and does not become significantly evident until E14. IGF-II mRNA was also conspicuous in areas of vascular interface with the brain, such as the choroid plexus and the organum vasculosum of the lamina terminalis. IGF1R mRNA is widely distributed in embryonic tissues, but the highest levels are seen in the ventral floorplate of the hindbrain, where specialized neuroepithelial cells act as guides for axonal targeting (Bondy et al., 1990).
3.1. IGF-I
IGF-I is expressed in all regions of the CNS. In rodents, significant IGF-I expression is detected as early as E11 in most regions of rodent brain (Ayer-le et al., 1991). Brain IGF-I expression in rodents peaks in the second week of postnatal life, gradually decreases, but continues throughout life (Bach et al., 1991; Bartlett et al., 1991; Ye et al., 1997). The peak expression of local IGF-I is often spatially correlated with the active proliferation, development and growth of neural cells. During normal development, IGF-I production is predominately located in neurons, and to a lesser extent in glial cells (Bondy and Lee, 1993; Bondy, 1991; Bondy and Lee, 1993; Bondy et al., 1990; Shinar and McMorris, 1995). IGF-I mRNA also is detected in the postnatal subventricular zone (SVZ) Bartlett et al., 1992; Perez-Martin et al., 2003 and in the Ki67+ proliferating precursors in the dentate gyrus (DG) Zhang et al., 2007, strongly suggesting that IGF-I is likely to be produced by proliferating neural precursors.
3.2. IGF-II
IGF-II is highly expressed in mesenchymal tissues. In brain, unlike IGF-I which peaks during postnatal development, the highest levels of brain IGF-II expression are observed during embryonic development (Ayer-le et al., 1991; Bondy et al., 1990). With increasing age, parenchymal IGF-II mRNA expression is gradually reduced, and its expression becomes restricted to meninges and choroid plexus in adult (Hynes et al., 1988; Logan et al., 1994; Zhang et al., 2007). Consistent with studies using RNA in situ hybridization, analysis using laser capture microdissection followed by polymerase chain reaction (PCR) demonstrated that there is little IGF-II mRNA expression, if any, in the postnatal hippocampal neurons (Zhang et al., 2007). Meninges and choroid plexus are major sources of cerebro-spinal fluid (CSF) IGF-II, which appears to be a critically important stimulator for NSC proliferation in the ventricular zone (VZ) during embryonic development (Lehtinen et al., 2011). Nonetheless, IGF-II protein immunoreactivity is also observed in other brain regions in adult (Logan et al., 1994), suggesting that IGF-II is likely produced at meninges and choroid plexus and transferred to these locations.
3.3. IGF1R
The IGF1R also is ubiquitously expressed in all neural cell types, including NSC and NPC (Popken et al., 2005), and its abundance is positively correlated with cell proliferation and growth. In a detailed study of mouse hippocampus, we show that DG proliferating progenitors have the most abundant amount of IGF1R mRNA, although all hippocampal neurons express it (Zhang et al., 2007). An appropriate expression pattern of the IGF1R is likely to pertinently contribute to its proper functionality. During prenatal development when neuronal processes actively outgrow, IGF1R is significantly enriched in the growth cone (Quiroga et al., 1995). In the cortical VZ of murine embryonic brains, IGF1R expression is restricted to the apical domain of neural precursors, likely by protein associated with Lin 7 (Pals1) and phosphatease and tensin homolog (Pten) Lehtinen et al., 2011. Disruption of Pals1 or Pten expression in embryos alters the expression pattern of IGF1R and the rate of cell proliferation and brain growth (Lehtinen et al., 2011). Focal adhesion kinase (FAK) also stabilizes IGF-1 receptor in cultured mouse embryonic fibroblasts (Andersson et al., 2009), whether FAK acts same in neural cells remains to be determined.
4. IGF-I actions in the CNS
The development of the mammalian brain occurs along specific stages, including neurulation, neurogenesis, differentiation into neurons and glia, neuronal migration, dendritic and axon out-growth, naturally occurring cell death, synaptogenesis, and myelination. The time course for these stages differs among species. In general, there is a caudal-to-rostral gradient in the time course of these developmental stages for individual regions in a given brain. The growth promoting actions of IGF-I–IGF1R signaling in the developing brain have been documented at virtually every stage of CNS development.
4.1. Early embryonic development
IGF-I has been shown to increase the survival of mammalian embryos by increasing the proportion of preimplantation embryos that eventually become blastocysts. This occurs during normal development (Lima et al., 2006; Sirisathien et al., 2003) and during abnormal development in response to various stressors such as heat shock, oxidative stress, tumor necrosis factor-α and toxicity (Byrne et al., 2002; Fabian et al., 2004; Jousan and Hansen, 2007; Jousan et al., 2008; Moss et al., 2009). These protective actions of IGF-I appear to be developmentally regulated. Bovine embryos before implantation (≥16 cells) treated with IGF-I at gestational day 5 exhibit altered gene expression, including upregulation of 5 anti-apoptotic genes (il6st, dyrk3, nfatc3, anp32, and eif3a), and downregulation of 5 pro-apoptotic genes (dpysl4, mst1, tnfsf11a, and arhgef10l), and upregulation of 2 genes involved in protection from reactive oxygen species (gstm2 and coo9) Bonilla et al., 2011. Interestingly, these same embryos also exhibited downregulation of genes involved in neural development and differentiation. In animal models of maternal diabetes, hyperglycemic developmental conditions in utero have been shown to result in downregulation of IGF1R expression, a 40% drop in the number of preimplantation embryos surviving to become blastocysts, delayed onset and progression of gastrulation, and increased numbers of apoptotic cells in the embryonic disk (Ramin et al., 2010).
The default fate of embryonic neuroectoderm is to become neural tissue, and this process is inhibited in early stages of development (De Robertis et al., 2000) by bone morphogenic protein (BMP-4). In Xenopus embryos injection of IGF-I, IGF-II or IGFBP-5 mRNA promotes neural induction and head induction, likely by increasing the expression of the anterior neural transcription factors Six-3, Rx2a, Pax-6, Otx-2, while injection of a secreted dominant-negative IGF1R mRNA has the opposite effect (Pera et al., 2001). Injection of IGF mRNA also causes induction of ectopic eyes and ectopic head-like structures containing brain tissue. Conversely, blockage of IGF1R signaling in zebrafish by a dominant- negative IGF1R or specific IGF1R inhibitors not only delays the emergence of GnRH2 and GnRH3 neurons, but also results in an abnormal appearance of GnRH3 neurons (Onuma et al., 2011). This IGF action is developmental stage-dependent because IGF signaling blockade in advanced embryos has no such effect (Onuma et al., 2011). These data support a critical role for IGF signaling in anterior neural induction in non-mammalian animals. Whether IGF signaling is also capable of inducing neural tissues in mammalian remains to be determined. Current data, however, indicate otherwise. In mutant mice with ablated IGF-I–IGF1R signaling, brain cytoarchitecture in general appears to be normal, although its growth is severely retarded (Beck et al., 1995; Liu et al., 1993). However, it is possible that signaling through InR or other receptor(s) can compensate for the loss of IGF1R signaling during the early developmental stages.
During neural induction in Xenopus embryos, cortical rotation triggered by fertilization leads to an increase in β-catenin on the dorsal side, inhibiting BMP-4 transcription (Baker et al., 1999a), and predisposing cells in the ectoderm toward neural fates. The inhibition of Wnt signaling at the gastrula stage mediates the process of head induction (Glinka et al., 1998; Glinka et al., 1997). It has been shown that overexpression of IGF-I produces an anterior expansion of head neural tissues, while blunting IGF-I expression reduces head structures, by antagonizing the activity of the Wnt signaling pathway in the embryo at the level of β-catenin (Richard-Parpaillon et al., 2002). Furthermore, the IGF may act in concert with other potent inducers of neural induction, including the fibroblast growth factors and Chordin, through the inhibitory phosphorylation of SMAD1 (Pera et al., 2003), culminating in a downstream inhibition of BMP-4.
4.2. Brain weight, regional brain volume and CNS growth
Scientific interest in IGF-I as a neuronal growth factor stemmed from in vitro studies demonstrating IGF-I’s ability to promote proliferation in neural cells and to inhibit apoptosis and cell death in various culture systems (see Section 4.3 below). The true magnitude of IGF-I’s role in promoting growth and development of the brain became apparent following in vivo studies using mutant mouse models (Fig. 1). Multiple lines of mice with genomic alterations in IGF system proteins have been generated and studied. Table 2 lists those mouse lines and changes in brain growth and cellular phenotypes. Briefly, these include transgenic (Tg) mice that overexpress IGF-I or IGF-II, mutant mice with a null mutation for IGF-I, IGF-II or IGF1R, and Tg mice with altered IGFBP expression.
Fig. 1.
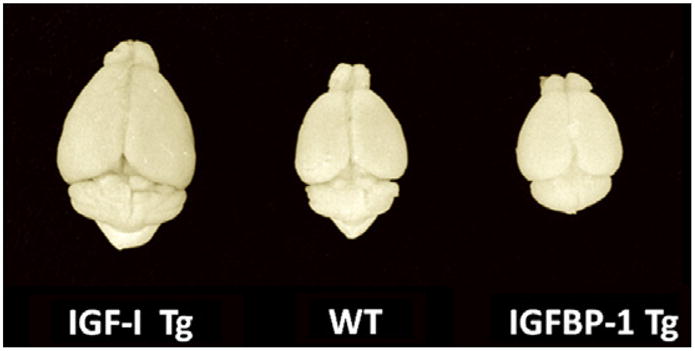
Brains from an IGF-IMT-I overexpressing Tg mouse (IGF-I Tg), a wild type control mouse (WT), and an IGFBP-1 Tg mouse that ectopically express brain IGFBP-1, which reduces IGF availability.
Table 2.
Phenotype changes in IGF system protein mutant mice.
| Protein | Abbreviation | Neural outcomes | References |
|---|---|---|---|
| IGF-I | |||
| IGF-IMT-I Tg | ↑ Brain growth, ↑ myelination | (Behringer et al., 1990; Carson et al., 1993; Mathews et al., 1988) | |
| IGF-IMT-I Tga | ↑ Brain growth, ↑ myelination, ↑ oligodendrocyte and oligodendrocyte precursor number, ↑ oligodendrocyte and oligodendrocytes precursor proliferation, ↑ neuron number, ↓ neuronal apoptosis, ↑ synaptogenesis, ↑ uptake of [3H]2-deoxyglucose | Gutierrez-Ospina et al., 1996; Gutierrez-Ospina et al., 1997; Mason et al., 2000; Ye et al., 1995a; Ye et al., 1995b | |
| IGF-INestin Tg | ↑ Brain growth, ↓ mitotic cell cycle duration, ↑ neuron number | Popken et al., 2004 | |
| IGF-IIGF-II Tg | ↑ Brain growth, ↑ neuronal proliferation, ↑ neuron number, ↓ neuronal apoptosis, ↑ synaptogenesis, ↓ levels of 14-3-3 | Chrysis et al., 2001; Dentremont et al., 1999; O’Kusky et al., 2000; O’Kusky et al., 2003; Ye et al., 1996; Zhang et al., 2003 | |
| IGF-IGFAP Tg | ↑ Brain growth, ↑ neuron number, ↑ neuronal proliferation, ↑ astrocyte number, ↑ GFAP expression, ↑ myelination, ↑ histone H3 and H4 acetylation | Lehtinen et al., 2011; Sun and D’Ercole, 2006; Ye et al., 2004 | |
| igf-I KO | ↓ Brain growth, ↓ myelination, ↓ oligodendrocyte number, ↓ neuron number | Ye et al., 2002b | |
| igf-I KO | ↓ Brain growth, ↓ myelination, ↓ oligodendrocyte number, ↓ neuron number ↓ dendritic length and branching | Beck et al., 1995; Cheng et al., 2003 | |
| igf-Im/m KOb | Brain growth not reported | Lembo et al., 1996 | |
| igf-IAlb-KOc | No growth changes in body, liver, spleen, kidney and heart. Brain growth not reported | Sjogren et al., 1999; Yakar et al., 1999 | |
| IGF-II | |||
| igf-II KO | ↓ Brain growth, ↓ neuron number | Lehtinen et al., 2011 | |
| IGF-IIH-2Kb Tg | No obvious brain phenotypic changes | Reijnders et al., 2004; Smink et al., 1999; Van Buul-Offers et al., 1995 | |
| IGF1R | |||
| igf1r KO | |||
| Heterozygous | No phenotypic changes | Liu et al., 1993 | |
| Homozygous | ↓ Brain growth | (Liu et al., 1993) | |
| igf1rmini-KO b | ↓ Brain growth | (Holzenberger et al., 2000) | |
| igf1rMeu KO d | ↓ Brain growth | Holzenberger et al., 2001 | |
| igf1rNestin-KO | ↓ Brain growth, ↓ neuron number, ↓ neuronal proliferation, ↑ neuronal apoptosis | (Liu et al., 2011; Liu et al., 2009) | |
| igf1rNestin-KO | ↓ Brain growth, ↓ neuron number | (Kappeler et al., 2008) | |
| igf1rOlig1-KO | ↓ Brain growth, ↓ myelination, ↓ oligodendrocytes and oligodendrocytes precursor number | (Zeger et al., 2007) | |
| igf1rPLP-KO | ↓ Brain growth, ↓ oligodendrocytes number, ↓ myelination | (Zeger et al., 2007) | |
| igf1rGnRH-KO | ↓ dendritic branching, ↓ spine formation | (DiVall et al., 2010) | |
| IGFBPs | |||
| IGFBP-1MT Tg | ↓ Brain growth, ↓ myelination, ↓ oligodendrocyte and oligodendrocytes precursor number, ↓ neuron number | (Gutierrez-Ospina et al., 1996; Ye et al., 1995a; Ye et al., 1995b) | |
| IGFBP-1PGK Tg | ↓ Brain growth, ↓ myelination, ↓ neuron number | (Ni et al., 1997; Rajkumar et al., 1995) | |
| IGFBP-2CMV Tg | ↓ Brain growth | (Hoeflich et al., 2001; Hoeflich et al., 1999) | |
| IGFBP-3GKG Tg | ↓ Brain growth | (Modric et al., 2001) | |
| IGFBP-3GKG mini Tge | ↓ Brain growth, ↓ neural proliferation in the periventricular zone | (Silha et al., 2005) | |
| ALSCMV Tg | ↔ Brain growth | (Modric et al., 2001) | |
| IGFBP-5Actin Tg | ↓ Body growth, ↑ relative brain growth | (Salih et al., 2004) | |
| IGFBP-5MT Tg | ↔ Brain growth | f | |
| IGFBP-6GFAP Tg | ↓ Brain growth, ↓ astrocyte number | Bienvenu et al., 2004 | |
Transgenic mice were generated using the same transgene construct that was employed for the original line of mice described in Behringer et al. (1990), Carson et al. (1993), Mathews et al. (1988).
Unlike IGF-I KO and IGF1R KO mice in which IGF-I and IGF1R expression is completely ablated, igf-Im/m-KO and igf1rmini-KO mutant mice exhibit marked reduction in IGF-I or IGF1R expression, respectively, due to an insertion mutation.
igf-IAlb-KO mice carry a igf-I null mutation specifically in liver, and exhibit a severe reduction (by 70%) in serum IGF-I.
igf1rMeu KO mice carry a null mutation on one igf1r allele and a partial inducible null mutation on the other igf1r allele during late embryonic development.
A human Gly(56)/Gly(80)/Gly(81)-mutant IGFBP-3 cDNA was inserted downstream of a GKG promoter. The mutant has a markedly reduced affinity for the IGFs, but retains the IGF-independent effects. No phenotypic changes, except for brain, are observed.
Our unpublished data.
Specific IGF actions likely depend on cell type, developmental stage, and local microenvironmental milieu within the brain, and the magnitude of altered brain regional growth may also in part reflect regional differences in IGF signaling in each specific mutant mouse line. In several lines of Tg mice that overexpress a metallthionein- I (MT-I) promoter-driven IGF-I transgene (IGF-IMT-I Tg mice), which exhibit increased expression of IGF-I during early postnatal development, brain weight is increased from 22% to 91% with no significant change in body weight in adult (Gutierrez-Ospina et al., 1996). Consistently, morphometric studies of the primary somatosensory cortex in one IGF-IMT-I Tg mouse line report an 81% increase in the volume of the cerebral cortex (Gutierrez-Ospina et al., 1996).
In Tg mice expressing an IGF-I transgene driven by IGF-II 5′ regulatory sequences (IGF-IIGF-II Tg mice), transgene expression and increased levels of IGF-I can only be detected in the brain, beginning at E18 and gradually increasing to plateau levels by postnatal days (P) 20 Ye et al., 1996. Although transgene expression occurs throughout the brain in these Tg mice, the highest level of transgene expression is found in the cerebellum (Ye et al., 1996). At P30, total brain weight was increased by 28%, with regional increases observed in the cerebellum (43%), hippocampus (34%), diencephalon (28%), brainstem (28%) and cerebral cortex (9%) Ye et al., 1996. Cerebellum weight was further increased by 90% in adult IGF-IIGF-II Tg mice (Ye et al., 1996). The total volume of the medulla was found to be significantly increased in Tg mice by 27% at P35 (Dentremont et al., 1999), while individual medullary nuclei exhibited differential increases in volume, ranging from 29% in the facial nucleus to 84% in the dorsal motor nucleus of the vagus. In the hippocampal DG, the volumes of both the granule cell layer and the molecular layer were significantly greater in Tg mice after P7, exceeding control volumes by 55–66% at P130 (O’Kusky et al., 2000).
In IGF-INestin Tg mice, IGF-I transgene expression is only observed in the brain, beginning in embryonic development as early as E13 and continuing throughout postnatal life (Popken et al., 2004). Brain weights of IGF-INestin Tg mice were significantly greater than their non-transgenic littermate controls at E18 (6.5%) and P45 (23%), with no significant differences in body weight. Morphometric analysis of IGF-INestin Tg embryos at E16 revealed a 25% increase in the volume of the dorsolateral telencephalic wall, corresponding to the primordial cerebral cortex. However, differential increases in tissue volume were observed in the cortical plate (52%), the intermediate zone (12%) and the combined volumes of the VZ and SVZ (26%) in IGF-INestin embryos. Interestingly, the volume of the caudate-putamen anlage did not differ significantly between IGF-INestin Tg and control embryos. At P12 regional tissue volumes in IGF-INestin Tg mice were found to be significantly greater than controls in the forebrain (26%), cerebral cortex (29%), subcortical white matter (52%), caudate-putamen (37%), hippocampus (49%), DG (71%), and habenular complex (48%) Popken et al., 2004. The relatively greater increases in volume, observed in the hippocampus, DG and habenular complex, were consistent with a significantly increased expression of the transgene in these regions as detected by in situ hybridization. The neocortical overgrowth in IGF-INestin Tg mice at P12 was not uniform, differing as a function of cytoarchitectonic area. For example, significantly greater increases in cortical volume were found for the motor cortex (42%) compared to the somatosensory cortex (35%) Hodge et al., 2005.
The brain overgrowth observed in IGF-I overexpressing Tg mice also likely requires a continuous presence of IGF-I transgene expression. In a line of conditional IGF-I Tg mice, in which the expression of an IGF-I transgene in glial fibrillary acidic protein (GFAP)+ astrocytes (IGF-IGFAP Tg mice) can be regulated by doxycycline (Ye et al., 2004), brains continuously overgrow from P5 to P110 when IGF-I transgene is allowed to express throughout the experimental period. Brain overgrowth, however, ceases after transgene expression is suppressed with doxycycline treatment (Ye et al., 2004).
In contrast, total IGF-I deficiency resulting from targeted disruption of the igf-I gene produces severe brain growth retardation. At 2 months of age mice homozygous for the null mutation (igf-I KO mice) exhibit a 38% decrease in brain weight and a 74% decrease in body weight relative to wild type controls (Beck et al., 1995). Decreased tissue volumes were reported in the pyramidal cell layer of the hippocampus (38%), the granule cell layer of the DG (59%), and the striatum (28%). Disproportionately greater decreases were detected in white matter regions, with a 69% decrease in the area of the anterior commissure and a 70% decrease in the thickness of the corpus callosum, due to decreased numbers of axons and oligodendrocytes (Beck et al., 1995). This preferential involvement of white matter in reduced brain volumes has been further confirmed in subsequent studies (Cheng et al., 1998).
Mice with a null mutation of the gene encoding the IGF1R (igf1r KO mice) invariably die at birth presumably due to respiratory failure and exhibit a 55% decrease in body weight compared to wild type controls (Liu et al., 1993). At E14 to E18, igf1r KO mutants exhibit corresponding growth retardation of the brain, as well as reduced volume and increased cell density in the mantle zone of the brainstem and spinal cord (Liu et al., 1993). These results suggest that brain growth retardation in these mice results predominantly from a reduction in the volume of neuropil in the CNS regions examined.
In mutant mice in which IGF1R expression is conditionally ablated in nestin-expressing neural precursors (igf1rNestin-KO mice), the whole-brain abundance of IGF1R protein is only ~52% and <2% of control values in heterozygous and homozygous igf1rNestin-KO mice, respectively (Liu et al., 2009). Homozygotes usually die within 48 h after birth. Surviving heterozygotes exhibit significant reductions in brain weight at P0 (56%), P5 (56%), P20 (59%) and P90 (60%), without significant changes in body weight (Liu et al., 2009). Regional tissue volumes for the hippocampus (CA 1–3), DG and granule cell layer of the DG were significantly reduced in heterozygotes by 44–54% at P0 and by 65–69% at P90.
Our unpublished morphometric and stereological analyses demonstrate that the volume of the telencephalic wall, corresponding to the primordial cerebral cortex, measured from the ventricular surface to the pial surface, is significantly reduced in both homozygotes (64%) and heterozygotes (31%) compared to controls by E16. In homozygous igf1rNestin-KO embryos, gross malformations of the telencephalon were observed at E16, but not at E12 or E14 (Fig. 2). In the worst cases, the malformation appears to result from an almost complete elimination of the developing telencephalic wall (Fig. 3). This cortical loss in homozygotes was preceded by a transient spike of apoptosis at E14. In heterozygotes at E14, a transient wave of apoptosis was also observed, although the density of apoptotic cells was reduced by 67%, when compared to homozygotes. Compared to controls, the volume of hippocampus in homozygous igf1rNestin-KO mice also was reduced by ~69%.
Fig. 2.

Representative sections through the head and brain of an E16 homozygous igf1rNestin-KO embryo (right panels) and its heterozygous littermate (left panels), stained with hematoxylin and eosin. In heterozygous embryos the brain exhibits relatively normal morphology at all levels, although smaller in size compared to littermate controls (not illustrated). In homozygous embryos the brain exhibits gross malformations in some mice. There is a failure to close the longitudinal suture of the skull in homozygous embryos, accompanied by the extrusion of gray matter through the skull and sloping forward (Panels B and C, arrows). In heterozygous embryos the developing hippocampus and DG are located along the medial edge of the telencephalic wall as in controls (E, arrow). In homozygous embryos the hippocampus and DG rotated to the lateral edge of the telencephalic wall (E, arrows) following extensive apoptosis in the dorsolateral wall at E14 and protrusion of the underlying telencephalic structures.
Fig. 3.
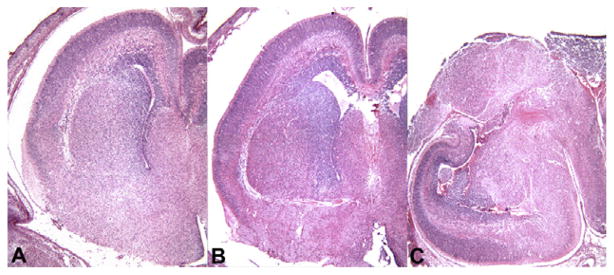
Representative sections of the right cerebral hemisphere in a control embryo (A), a heterozygous igf1rNestin-KO embryo (B), and a homozygous igf1rNestin-KO embryo at E16.
IGFBP-1 inhibits the interactions of IGF-I and IGF-II with their cell surface receptors when present in molar excess. In Tg mice with ectopic brain IGFBP-1 expression driven by a MT-I promoter (IGFBP-1MT-I Tg mice), brain weight was significantly decreased by 8–16% at P14 (D’Ercole et al., 1994). This brain growth retardation is most obvious in the cerebral cortex (18%), hippocampus (20%) and diencephalons (12%), respectively Ye et al., 1995a. Decreased density of myelinated axons is prominent in the cerebral cortex, anterior commissure, corpus callosum and diencephalon, although the cerebellum and brainstem are relatively spared (Ye et al., 1995a). Morphometric analysis of the primary somatosensory cortex in these Tg mice revealed a 24% decrease in the volume of somatosensory barrels in cortical layer IV. In another line of IGFBP-1 Tg mice (IGFBP-1PGK Tg mice), in which the IGFBP-1 transgene expression is driven by a mouse phosphoglycerate kinase (PGK) promoter, the authors reported more significant decreases in body weight (12%) and brain weight (40%), likely due to a more abundant and earlier expression of the transgene. Brain DNA content (16%) and total protein (23%) also are reduced in adult mice (Ni et al., 1997), consistent with a reduction in cell number. On representative histological sections, cross-sectional areas of the hippocampus and DG were significantly reduced in IGFBP-1PGK Tg mice by 55% and 72%, respectively, with a decrease of 33% for the brain as a whole. White matter and fiber tracts were less well developed, and the thickness of the corpus callosum was decreased by 62% (Ni et al., 1997). Interestingly, in a line of Tg mice with hepatic IGFBP-1 overexpression and increased circulating levels of IGFBP-1, there was a significant reduction in brain weight by 2 months of age (Doublier et al., 2000), in line with the view that circulating IGF can pass across the BBB and regulate the brain growth. In IGFBP-2 Tg mice, as well as Tg mice with an increased expression of IGFBP-3 or IGFBP-5, modest reductions of brain weight were also reported (see Table 2).
4.3. Neurogenesis and apoptosis
The capacity of IGF-I to promote neuron progenitor proliferation and differentiation has been well-documented in vitro (Arsenijevic and Weiss, 1998; Arsenijevic et al., 2001; Cicco-Bloom and Black, 1988; Drago et al., 1991; Torres-Aleman et al., 1990b; Werther et al., 1993; Zackenfels et al., 1995). Additional in vitro studies have shown that IGF-I promotes cell survival through its anti-apoptotic actions (Russell et al., 1998; Takadera et al., 1999; Torres-Aleman et al., 1990a; Torres-Aleman et al., 1990b; Yamada et al., 2001). Morphometric and stereological analyses of the developing brain in IGF-I overexpressing Tg mice have reported substantial increases in the total number of neurons in the cerebral cortex (Gutierrez-Ospina et al., 1996; Hodge et al., 2005), cerebellar cortex (Ye et al., 1996), DG of the hippocampus (O’Kusky et al., 2000; Ye et al., 2004), and selected brainstem nuclei (Dentremont et al., 1999). By contrast, in igf-I KO mutants and IGFBP-1 overexpressing Tg mice, significant decreases in neuron number have been reported in the cerebral cortex, hippocampus, DG striatum, and cochlear nucleus (Beck et al., 1995; Camarero et al., 2001; Gutierrez-Ospina et al., 1996; Ni et al., 1997). Both in vitro and in vivo studies show that des-N-(1–3) IGF-I can block neuronal apoptosis in response to hypoxia/ischemia or excitotoxicity (Guan et al., 1996; Saura et al., 1999).
Morphometric analysis of IGF-IMT-I mice revealed a 24% increase in the total number of neurons in somatosensory barrels in cortical layer IV by P90 (Gutierrez-Ospina et al., 1996). There is a 39% decrease in the numerical density of neurons (NV, number per unit volume), indicating an increase in the volume of neuropil separating individual neuronal cell bodies, and a 33% increase in mean neuronal profile area. Neurons in layer IV of the cerebral cortex in mouse are generated during prenatal development (Hicks and D’Amato, 1968), while apoptotic neuron death occurs predominantly from birth to P10 (Spreafico et al., 1995; Verney et al., 2000). Given that the transgene in IGF-IMT-I mice is significantly expressed after birth (Ye et al., 1995a), it would appear that increased neuron number in these adult Tg mice results from decreased apoptosis during the regressive phase of neurogenesis. A similar morphometric analysis of the somatosensory cortex in IGFBP-1MT-I Tg mice revealed a 15% decrease in the total number of neurons in somatosensory barrels and a 39% increase in the NV of neurons (Gutierrez-Ospina et al., 1996). Decreased cortical volume in these Tg mice results from a decrease in both the number of neurons and the volume of neuropil separating individual cell bodies. Since the transgene expression in these mice also is driven by the same promoter and is observed first after birth (Ye et al., 1995a), decreased neuron number likely results from increased apoptosis during the regressive phase of neurogenesis.
In the cerebellar cortex of IGF-IIGF-II Tg mice at P50, the total number of Purkinje cells and granule cells is increased by 20% and 82%, respectively (Ye et al., 1996). When bromo-2-deoxyuridine (BrdU) was employed to label proliferation of granule cell progenitors in the external granule cell layer, the total number of labeled cells was increased by 38% in IGF-IIGF-II Tg mice at P15. Purkinje cells of the cerebellar cortex originate from mitotic neuroepithelial cells from E11 to E13, while granule cells originate from E17 to P15 (Mares and Lodin, 1970; Miale and Sidman, 1961). Given this protracted period of proliferation for granule cells and the increase in BrdU-labeled progenitors, it appears most likely that elevated levels of IGF-I during early postnatal development act to increase the rate of mitosis in the external granular layer. In a subsequent study, the anti-apoptotic effects of elevated IGF-I were investigated in the cerebellum of IGF-IIGF-II Tg mice (Chrysis et al., 2001). Morphometric analysis of apoptotic cells in the cerebellum, detected by terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL), revealed a 47% decrease in Tg mice when compared to controls. The abundance of procaspase-3 and caspase-3 is also decreased in IGF-IIGF-II Tg mice, accompanied by increased expression of the anti-apoptotic Bcl genes, bcl-xL and bcl-2. Consistent with these studies, BCL-2 was found to be increased in immunohistochemical studies of cerebellum in these Tg mice (Baker et al., 1999b). These results provide direct evidence that elevated IGF-I acts to inhibit apoptosis during early postnatal development in a developmentally specific manner.
Increased growth of the hippocampal DG has been studied in IGF-IIGF-II Tg mice throughout postnatal development (O’Kusky et al., 2000). In control mice the total number of neurons in the granule cell layer increased by 113% from P7 to P35, with no additional increase in neuron number by P130. In IGF-IIGF-II Tg mice there was a 172% increase in neuron number from P7 to P35, with an additional and significant increase of 17% between P35 and P130, suggesting a protracted period of accelerated neurogenesis. The total number of neurons in the granule cell layer was significantly greater in IGF-IIGF-II Tg mice by 56% at P35 and by 61% at P130. In IGFBP-1GKP Tg mice with inhibited IGF-I actions, BrdU labeling of proliferating cells in the granule cell layer of the DG revealed a 41% decrease in the number of labeled cells in Tg mice at P3, as compared to controls (Ni et al., 1997). In the VZ and SVZ of the lateral ventricle, BrdU-labeled cells were decreased by 19%. The number of TUNEL-labeled apoptotic cells throughout the hippocampus was found to be increased by 55% in IGFBP-1GKP Tg mice (Ni et al., 1997). These studies indicate that IGF-I acts to increase neuron proliferation while inhibiting apoptosis in a region of the brain where progressive and regressive phases of neurogenesis are known to occur throughout the life of the organism.
In the brainstem of IGF-IIGF-II Tg mice at P35, the total number of neurons is significantly increased in the nucleus of the solitary tract (50%) and in the dorsal motor nucleus of the vagus (53%), but not in the hypoglossal nucleus or the facial nucleus (Dentremont et al., 1999). Neuron proliferation in mice occurs from E9 to E12 for the nucleus of the solitary tract and from E9 to E10 for the dorsal motor nucleus of the vagus, the hypoglossal nucleus and the facial nucleus (Pierce, 1973). Given that transgene expression in IGF-IIGF-II Tg mice is very low prior to birth, increased rates of neuronal proliferation are unlikely to account for increased neuron numbers. An inhibition of naturally occurring neuron death (apoptosis) appears to be more likely. Apoptotic death of motor neurons in rodent hypoglossal nucleus occurs exclusively from E16 to E21 (Friedland et al., 1995), as it does for motor neurons in the spinal cord (Oppenheim, 1986). Motor neurons in the facial nucleus are likely to undergo similarly early programmed cell death. Thus, the lack of effect of elevated IGF-I on neuron number in the hypoglossal and facial nuclei in these mice appears to stem from the age at which the transgene is expressed. Interestingly, although the total number of neurons did not differ significantly between Tg and control mice in the hypoglossal and facial nuclei, the NV of neurons was significantly decreased in both regions while the mean neuronal profile area is significantly increased. Changes in these variables indicate an increased volume of neuropil and possibly more complex arborizations of the dendritic trees on individual neurons within these regions.
IGF also significantly increases neural precursor proliferation in vivo. In IGF-INestin Tg embryos a 54% increase in the total number of cells in the dorsolateral wall of the telencephalon was observed within the cortical plate at E16 (Popken et al., 2004). Cumulative S phase labeling with BrdU revealed a significant decrease in total cell cycle length (TC) in Tg embryos at E14 (Hodge et al., 2004). This decrease in TC was found to result entirely from a reduction in the length of the G1 phase of the cell cycle from 10.66 h to 8.81 h, with no significant changes in the lengths of the S, G2 and M phases. Additionally, the proportion of daughter cells reentering the cell cycle was significantly increased by 15% in IGF-INestin Tg embryos, compared with littermate controls (Hodge et al., 2004). These results demonstrate that IGF-I accelerates progenitor cell division in the VZ by reducing G1 phase length and decreasing TC but increases cell cycle re-entry.
In IGF-INestin Tg mice at P12, significant increases in the total number of neurons were determined in the cerebral cortex (27%), caudate-putamen (27%), DG of the hippocampus (69%), the medial habenular nucleus (61%), and the lateral habenular nucleus (36%), when compared to littermate controls (Hodge et al., 2005; Popken et al., 2004). In the cerebral cortex of IGF-INestin Tg and control mice, apoptotic cells were observed in all cortical layers from P0 to P7 by virtue of their immunoreactivity to antibodies against cleaved caspase-3 (Hodge et al., 2007; Popken et al., 2004). The vast majority of these cells (>80%) appeared to be neurons because of their atrophic dendritic arborizations. In vivo studies using these IGF-INestin Tg mice clearly demonstrate that elevated levels of IGF-I in the developing brain, beginning as early as E13, can simultaneously accelerate mitosis in neural progenitors and inhibit apoptosis in post-mitotic neurons.
In igf-I KO mutant mice, immunohistochemical studies (Beck et al., 1995) have reported significant decreases in the number of parvalbumin-immunoreactive neurons in the striatum (52%), hippocampus (32%) and DG (59%). Interestingly, the numbers of cholinergic neurons in both the striatum and basal forebrain and dopaminergic neurons in the mesencephalon did not change, suggesting a differential susceptibility of neurotransmitter-specific neuron populations to the effects of IGF-I (Beck et al., 1995).
Preliminary results from ongoing immunohistochemical studies of adult heterozygous igf1rNestin-KO mice in our laboratories indicate a disproportionate loss of GABAergic neurons in the ventral prefrontal cortex, when compared to controls (Fig. 4). In the cerebral cortex of igf1rNestin-KO mice, the NV of all neurons did not differ significantly from controls, although the total number of neurons was reduced by 26% due to decreases in cortical volume. In the ventral prefrontal cortex, the NV of GABAergic neurons in heterozygous igf1rNestin-KO mice (15,042 ± 536, mean ± standard error) was significantly reduced (25%, P < 0.001), when compared to controls (20,093 ± 857). In the dorsal prefrontal cortex, the NV of GABAergic neurons did not differ significantly between heterozygous igf1rNestin- KO mice (16,294 ± 452) and controls (15,772 ± 697). Interestingly, this disproportionate loss of GABAergic neurons was due entirely to a loss of calbindin-immunoreactive GABAergic neurons (NV in heterozygous igf1rNestin-KO mice: 13,484 ± 745; compared to NV in controls: 17,508 ± 432, P < 0.001), with no significant change in the proportion of calretinin-immunoreactive GABAergic neurons (NV in heterozygous igf1rNestin-KO mice and controls was 7309 ± 389 and 7765 ± 520, respectively), as illustrated in Fig. 4.
Fig. 4.
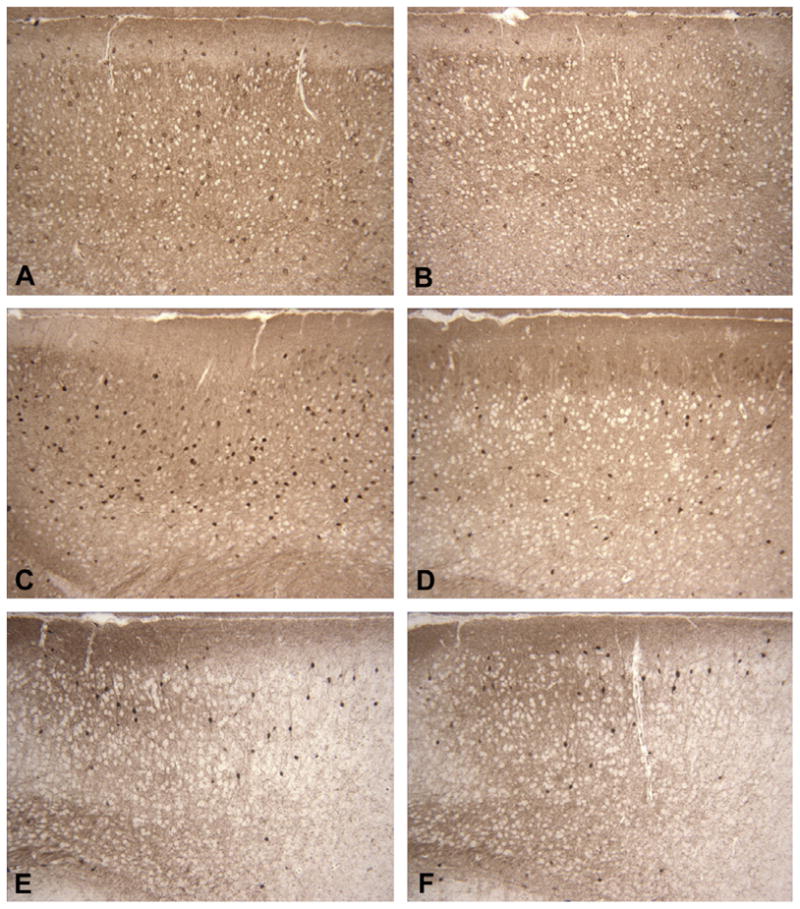
Immunostaining for GAD67, calbindin, and calretinin in the ventral prefrontal cortex of an adult heterozygous igf1rNestin-KO mouse (B, D, and F) and its littermate control (A, C, and E). Coronal sections through the ventral prefrontal cortex have been stained with antibodies against GAD67 (A and B), calbindin (C and D) and calretinin (E and F). Note the decreased density of GAD67-immunoreactive GABAergic neurons in (B) and the decreased density of calbindin-immunoreactive GABAergic neurons in (D) with no change in the density of calretinin-immunoreactive GABAergic neurons in (F), when compared to controls.
Delayed maturation of the inner ear and neuron loss have been reported in homozygous igf-I KO mice during early postnatal development (Camarero et al., 2001). The volumes of the cochlea and cochlear ganglion were reduced by 34% at P20, accompanied by a 19% loss of cochlear neurons and a 31% decrease in the volume of the surviving neuron body. The number of apoptotic neurons, demonstrated by TUNEL labeling and caspase-3, also was increased in igf-I KO mutant mice (Camarero et al., 2001).
Clearly, IGF-I is a potent factor in the control of neurogenesis and cell survival through its pro-mitotic and anti-apoptotic actions. IGF-I continues to be a modulator of neurogenesis in the adult hippocampus (Aberg et al., 2000; Trejo et al., 2008). IGF-I signaling is also a potent mediator of axonal and dendritic circuit remodeling during postnatal development.
4.4. Neurite outgrowth and synaptogenesis
In vitro studies of fetal hypothalamic cells in culture (Torres-Aleman et al., 1990b) have reported that in the presence of IGF-I neurons have a more differentiated morphology and express significantly higher levels of protein kinase C, an enzyme that increases during neurite outgrowth and synaptogenesis. These data suggest that IGF-I can regulate neurite and synopsis genesis.
In vivo studies of mice support the findings derived from culture studies. Injection of an IGF-I antisense nucleotide in the inferior olive of adult rats results in a significant decrease of IGF-I levels in the contralateral cerebellum and a marked reduction in the size of dendritic spines and in the numerical density of dendritic spines on Purkinje cells (Nieto-Bona et al., 1997). Morphometric analyses of the developing fronto-parietal cortex of igf-I KO mice (Cheng et al., 2003) and the hypothalamus of mice with a null mutation specifically in gonadotropin-releasing hormone (GnRH) neurons (igf1r GnRH-KO mice) (DiVall et al., 2010) demonstrate that blunting IGF-I–IGF1R signaling decreases dendritic length, branching complexity, and dendritic spine formation. Interestingly, both male and female igf1rGnRH-KO mice exhibit delayed onset of puberty, although reduced IGF1R signaling during development produces no change in the distribution and total number of GnRH neurons (DiVall et al., 2010). Mice with a mutation in the InR gene specifically in GnRH neurons, however, do not exhibit delayed puberty and GnRH neuronal pathology (DiVall et al., 2010). Therefore, these data suggest that IGF1R signaling, but not InR signaling, in GnRH neurons is necessary for normal pubertal onset, likely involving dendritic tree growth and the prepubertal maturation of afferent input to these neurons.
In igf-I KO mice, there is also a 16% reduction in the density of dendritic spines, correlating with a 30% reduction in synaptotagmin levels. Consistently, P20 igf-I KO mice exhibit an abnormal distribution of synaptophysin in the organ of Corti (Camarero et al., 2001), as the pattern of its immunoreactivity in the cell bodies of cochlear ganglion neurons and sensory hair cells more closely resemble control mice at P5. These data indicate the persistence of an immature pattern of synapse distribution in the absence of IGF-I, and suggest that IGF-I deficiency is associated with a reduction of synaptogenesis in the developing brain.
More direct evidence that IGF-I promotes synaptogensis comes from studies of IGF-I Tg mice and IGFBP-1MT-I Tg mice (Gutierrez-Ospina et al., 2004; O’Kusky et al., 2003). The density and total number of synapses in individual barrels of the somatosensory cortex were determined using histochemical methods for the oxidative enzyme succinic dehydrogenase. The density of synapses did not differ significantly among Tg and control mice. However, the total number of synapses was found to be 38% greater in IGF-IMT-I Tg mice and 43% less in IGFBP-1MT-I Tg mice (Gutierrez-Ospina et al., 2004). These changes resulted from the increased or decreased volumes of individual barrels in IGF-IMT-I Tg and IGFBP-1MT-I Tg mice, respectively, rather than from changes in the packing density of individual synaptic contacts (Gutierrez-Ospina et al., 2004).
The effects of elevated IGF-I on both the progressive and regressive phases of synaptogenesis in the hypoglossal nucleus were also investigated in IGF-IIGF-II mice (O’Kusky et al., 2000; O’Kusky et al., 2003). In control mice the total number of synapses in the hypoglossal nucleus was increased by 354% from P7 to peak values at P21, followed by a significant decrease of 33% by P130 (O’Kusky et al., 2003). IGF-IIGF-II Tg mice exhibited a similar trend of changes. When compared to control mice, however, IGF-IIGF-II Tg mice had 42% and 52% more synapses at P21 and P130, respectively (O’Kusky et al., 2003). Given that the total number of hypoglossal neurons does not differ significantly between Tg and control mice at the ages examined, the synapse-to-neuron ratio is significantly greater in IGF Tg mice after P14. Thus, the increased in vivo expression of IGF-I during postnatal development augments the progressive phase of synaptogenesis, although it does not prevent synapse elimination during the regressive phase.
Similarly, in the hippocampal DG of IGF-IIGF-II Tg mice, significant increases in the total number of synapses in the molecular layer were observed at P14 (by 61%), P21 (42%), P28 (105%), P35 (96%), and P130 (54%) O’Kusky et al., 2000. Interestingly, the density of synapses is significantly greater only at P28 (35%) and P35 (21%) in IGF-IIGF-II Tg mice, and accordingly, a greater synapse-to-neuron ratio, which returns to normal values by P130. The increase in synapse number in the DG tends to reflect the increased number of neurons in the granule cell layer in IGF-IIGF- II Tg mice. It has been shown that reduced neurogenesis and synaptogenesis in the hippocampus of animal models of gestational–neonatal iron deficiency is secondary to reduced levels of IGF-I during development (Tran et al., 2012), suggesting that IGFI can mediate the effects of iron on synaptogenesis.
Taken together, these data strongly indicate that IGF signaling plays an important role in neurite outgrowth and synaptogenesis throughout postnatal development and in adult. In addition, IGF signaling may act by regulating the expression of synaptic proteins, such as synaptophysin, because synaptophysin, by binding synaptobrevin, is believed to play an important role in regulating SNARE assembly, vesicle fusion (Edelmann et al., 1995; Valtorta et al., 2004), and synapse formation (Tarsa and Goda, 2002).
4.5. Oligodendrocytes and myelination
IGF actions on oligodendrocyte lineage cells have been well-documented. In cultures both IGF-I and IGF-II are capable of promoting the proliferation, survival and development of rodent and human oligodendrocyte lineage cells (Armstrong et al., 1992; Barres et al., 1992; Cui et al., 2012; Masters et al., 1991a; McMorris et al., 1986; Mozell and McMorris, 1991; Ye and D’Ercole, 1999). Many of these results have been confirmed in studies of mutant mice.
Overexpression of IGF-I in the brain of Tg mice results in dramatic increases in brain growth, myelin content, the number of oligodendrocytes and their precursors (Carson et al., 1993; Ye et al., 1995a; Ye et al., 1995b; Ye et al., 2007; Ye et al., 2004). Compared to control mice, adult IGF-IMT-I Tg mice have 130% more myelin content (Carson et al., 1993) and 2–3-fold more abundant mRNAs for myelin-basic protein (MBP) and proteolipid protein (PLP) Ye et al., 1995a, the two most abundant myelin-specific proteins. The increased myelin is the result of increases in the thickness of myelin sheath and in the number of myelinated axons, which does not proportionally depend on axon size (Ye et al., 1995a; Ye et al., 1995b). Taken together with the fact that the number of PLP+ oligodendrocytes is only modestly increased (by 18% in corpus callosum and 36% in cerebral cortex) (Ye et al., 1995a), these data suggest that IGF-I promotes the production of myelin by each oligodendrocyte. In contrast, blunting IGF-I expression in igf-I KO mice results in a dramatic reduction in oligodendrocytes and their precursors, and much less abundant MBP and PLP during early postnatal development (Beck et al., 1995; Ye et al., 2002b). The abundance of the myelin-specific proteins, however, gradually increases with increasing age, and becomes normal in adult (Ye et al., 2002b). Because the abundance of IGF-II is significantly increased during the same developmental period (Ye et al., 2002b), it is likely that the recovery of reduced myelin-specific protein expression induced by IGF-I deficiency is, at least in part, due to the compensatory effects of IGF-II. This speculation is supported by the findings that myelination and the expression of MBP and PLP remain suppressed in the adult mice with an IGF1R null mutation specifically in the cells of oligodendrocyte lineage (see below), or in the adult mice that ectopically express IGFBP-1 (Ni et al., 1997; Ye et al., 1995a), an IGF binding protein that inhibits IGF-I and IGF-II actions.
Direct interactions of IGF with the IGF1R on the cell surface of oligodendrocyte lineage cells are essential to the normal oligodendrocyte development and myelination, although indirect IGF effects via neurons and other neural cell types also are likely to be involved. When IGF1R expression is specifically blunted in the Olig1+ oligodendrocyte precursors and their progeny, the volume of corpus callosum and anterior commissure, two regions that are rich in oligodendrocyte lineage cells, and the number of oligodendrocytes and their precursors are markedly decreased (Zeger et al., 2007). The reduction in these cells apparently is a result of decreased proliferation and increased apoptosis (Zeger et al., 2007).
While IGF signaling has been convincingly shown to promote the development of oligodendrocyte precursors, the details about its role in each of the developmental stages remain to be delineated. In cultured oligodendrocyte precursors from adult corpus callosum, IGF-I, in the presence of insulin at a concentration that also stimulates IGF1R, promotes the development of O4+ oligodendrocyte precursors to O1+ early oligodendrocytes, but has no effects on the progression of A2B5+ early oligodendrocyte precursors to O4+ precursors (Mason and Goldman, 2002). These data suggest that IGF signaling preferentially promotes the differentiation of adult oligodendrocyte precursors at one or more stages of their development. The actions of IGF signaling on oligodendrocyte specification also have been reported, but conclusion remains discordant. In cultured adult rat hippocampal NSC IGF-I appears to preferentially direct NSC towards oligodendrocyte lineage (Hsieh et al., 2004). In contrast, a more recent study of cultured NSC from neonatal mouse forebrain shows that overexpression of an IGF-I transgene in the cells markedly increases neuronal number without an appreciable change in the number of oligodendrocytes (Kouroupi et al., 2010). The exact reason(s) for the discrepancy between these two studies is not known. Possible explanations likely include different NSC sources and culture conditions employed. It is worth noting that in both studies GFAP was used as a marker for astrocytes. Because a subpopulation of NSC express GFAP, thus, the reduction in GFAP reported in the both studies may also reflect a reduction in the NSC population due to their development to mature neural cells.
Currently, there is no available data from mutant mice that support an essential role for IGF in oligodendrocyte specification in vivo. In igf1rOlig1-KO mice that carry an igf1r null mutation in the oligodendrocyte precursors positive for Olig1, a transcription factor that is specifically expressed in early oligodendrocyte precursors and motor neurons (Lu et al., 2002; Woodruff et al., 2001), both mature oligodendrocytes and their precursors continue to exist, although markedly reduced in number (Zeger et al., 2007). In addition, the total number of astrocytes and neurons is not inversely increased, rather modestly decreased. In contrast, in the developing brains of each of the 4 IGF-I overexpressing Tg mouse lines that we have studied (see Table 2), including IGF-INestin Tg mice which overexpress an IGF-I transgene from E13, the number of neurons, astrocytes and oligodendrocytes are all increased, although the magnitude of the increase in each neural cell type varies depending on specific lines (Dentremont et al., 1999; Gutierrez-Ospina et al., 1996; Popken et al., 2004; Ye et al., 1995a; Ye et al., 2000; Ye et al., 2004; Ye et al., 1996). In addition, mice that conditionally overexpress IGF-I in GFAP+ cells exhibit a proportional increase in the number of astrocytes, oligodendrocytes and neurons in hippocampus (Ye et al., 2004).
4.6. Astrocytes and microglia
There are several lines of evidence indicating that IGF also promotes the development and growth of astrocytes. In astrocyte cultures, IGF-I significantly increases their proliferation (Aberg et al., 2003b; Ballotti et al., 1987; Chernausek, 1993; Han et al., 1987; Han et al., 1992). Consistently, injection of IGF-1 into developing sheep brains following ischemic injury (Cao et al., 2003) or overexpressing IGF-I in the brain of IGF-IGFAP Tg mice (Ye et al., 2004) increases the number of astrocytes. Intriguingly, unlike IGF-IMT-I Tg mice that show a predominate expression of IGF-I transgene in neurons, adult IGF-IGFAP Tg mice have 50–270% more GFAP protein (depending on the brain regions) (Ye et al., 2004), suggesting that astrocyte-derived IGF-I can differentially influence astrocyte functions. In line with this speculation, IGF-I treatment of cultured astrocytes also increases the expression of the gap junction protein connexin43 (Aberg et al., 2003b), alpha 1 isoform of Na-ATPase (Matsuda et al., 1993), glucose transporter (Masters et al., 1991b), and glial glutamate transporter GLAST (Suzuki et al., 2001), as well as their activity (Masters et al., 1991b; Matsuda et al., 1993). These data also suggest that IGF-I is capable of promoting the cross-talk between astrocytes and other neural cells. Taken together, these data suggest that IGF signaling is likely to promote the development of both oligodendrocyte, neuron and astrocyte lineages in the developing brain, and that the site of IGF-I expression influences the nature and magnitude of its actions.
Following various brain injuries in adult, IGF expression is significantly increased in astrocytes (Garcia-Estrada et al., 1992; Gudi et al., 2011; Komoly et al., 1992; Li et al., 1998; Liu et al., 1994), parallel to an increased astroglial activation and proliferation. Decrease in IGF-I availability by ectopic expression of IGFBP-1 in the brain of IGFBP-1PGK Tg mice significantly reduced astrocyte responses to injury (Ni et al., 1997). These data indicate that IGF signaling is critically important to astrocyte proliferation and functions following injury.
Microglia are derived from hemangioblastic mesoderm (Streit, 2001), and populate the developing neuroectoderm as early as E8 in rodents. As with the case in astrocytes, there are few reports about IGF actions in microglia generation and development during prenatal and postnatal development. Like astrocytes, IGF production is markedly increased in activated microglial cells following various brain injuries (Beilharz et al., 1998; Breese et al., 1996; Choi et al., 2008; Gudi et al., 2011; O’Donnell et al., 2002). In cultures, IGF-I increases microglia proliferation (O’Donnell et al., 2002), and microglia-derived IGF-II protects oligodendrocytes from tumor necrosis factor-α induced cell death (Nicholas et al., 2002). These data suggest that microglial IGF-I acts on neural cells in a paracrine and/or autocrine pattern.
5. IGF-II actions
In cultures, IGF-II exerts actions on the development and growth of neural cells in a manner that is similar to that of IGF-I. Genetic studies also clearly demonstrate that IGF-II has an important role in growth during early development. Global ablation of IGF-II gene expression in igf-II KO mice results in a marked retardation in body growth (Baker et al., 1993; DeChiara et al., 1990; Liu et al., 1993) and in brain growth, leading to a 24% reduction in brain weight at postnatal day 8 (Lehtinen et al., 2011). The decreased brain growth, at least in part, is a result of reduced cell proliferation of NSC/NPC during early development (Lehtinen et al., 2011). In contrast, adding exogenous IGF-II to culture medium significantly increases the number of neurospheres derived from NSC/NPC and the number of proliferating cells in the VZ of embryonic explants (Lehtinen et al., 2011). Similarly, during late embryonic development in mice, IGF-II, derived from choroid plexus and signaling through the IGF1R, appears to be a dominate growth factor that stimulates the proliferation of VZ NSC (Lehtinen et al., 2011). Nonetheless, postnatal overexpression of IGF-II in mutant mice does not alter brain growth (Van Buul-Offers et al., 1995). It is possible that during postnatal development IGF1R signaling derived from abundantly-expressed IGF-I, which has 10–100 fold higher affinity for IGF1R compared to IGF-II, has already operated at its maximal level. Thus, increasing IGF-II postnatally may not be able to further increase signaling through the IGF1R and to elicit additional growth actions.
However, during late brain development or in the adult brain, IGF-II is likely to have an important role in modulation of non-growth neuronal functions. For example, in adult rodents injection of IGF-II into hippocampus improves memory (Chen et al., 2011). The IGF-II action appears to be specifically mediated by IGF2R, because co-injection of an IGF2R inhibitor, but not IGF1R inhibitors, abolishes the memory improvement induced by IGF-II (Chen et al., 2011). In addition, Leu27-IGF-II, an IGF-II analog that preferentially binds to the IGF2R, attenuates depolarization-evoked GABA release in cultured adult hippocampal and cortex (Amritraj et al., 2010), further supporting that IGF-II, via the IGF2R, can affect neuronal functions in adult.
6. IGF signaling
6.1. Signaling through IGF1R
Most IGF growth actions, if not all, are mediated by IGF1R. Direct interaction of IGF with the IGF1R in CNS neural cells is essential for the normal neural development and the proper brain cytoarchitecture, as summarized in the sections above. While our understanding of IGF intracellular signaling has been significantly advanced in recent years, the intracellular signaling pathways that mediate each of IGF actions in the CNS remain to be precisely elucidated. Fig. 5 depicts largely simplified IGF signaling pathways in the CNS.
Fig. 5.

IGF signaling in the CNS. In this largely simplified diagram the IGF signaling pathways in the CNS are schematically depicted. Other signaling molecules and pathways described in non-neural cells systems are not included in the Figure, although it is possible that they are also involved. ⊥ = inhibitory modification, and ↓ = stimulatory modification.
It is known that binding IGF-I or IGF-II to the IGF1R leads to activation of the tyrosine kinase in the β-subunits, which in turn autophosphorylates its tyrosine residues and recruits docking proteins (LeRoith et al., 1995). Docking proteins that are involved in IGF signaling include five isoforms of IRS [IRS-1 to IRS-4 and Grb2-associated binder-1(Gab-1)], Ras, and Src homology domain- containing proteins. Phosphorylated docking proteins then subsequently recruit downstream signaling molecules and transduce IGF signaling. The Ras–Raf-MAP and PI3–Akt kinase pathways are two extensively studied signaling pathways that play key roles in IGF neural actions. There is also evidence that G protein mediates some of IGF neural actions (Keummerle and Murthy, 2001). More recently, it has been reported that in cultured non-neural cells IGF1R can be modified by small ubiquitin-like modifier protein- 1 (SUMO), and transported into the nucleus, where the SUMO-modified IGF1R acts as a transcription factor to directly regulate its target gene expression (Sehat et al., 2010). It is highly possible that the IGF1R can also act as a transcription factor in neural cells, because neural cells are capable of internalizing the IGF1R (Romanelli et al., 2007). It is worth noting that most, if not all, IGF signaling molecules and pathways described below also are involved in many other signaling pathways. For example, IGF and insulin share many signaling molecules and pathways, although how they exerts distinct functions remains to be understood.
6.1.1. IRS
IRSs, molecules that are also involved in insulin signaling, are widely expressed in the CNS. IRS-1, -2, -4 and Gab-1 are expressed in a tempo-spatial specific pattern (Fantin et al., 1999; Folli et al., 1994; Holgado-Madruga et al., 1996; Numan and Russell, 1999; Sciacchitano and Taylor, 1997; Ye et al., 2002a), and little IRS-3 is detected in adult brain (Sciacchitano and Taylor, 1997). It appears that while it mediates some IGF-I neural actions, IRS-1 is not essential in IGF neural signaling. Brain growth (Pete et al., 1999; Schubert et al., 2003; Ye et al., 2002a) and myelination (Engleka et al., 1996) are modestly reduced in irs-1 KO mice. Unlike skeletal muscle in which IGF-I dependent growth is impaired by IRS-1 ablation (Pete et al., 1999), blunting IRS-1 expression does not block IGF-I-stimulated brain growth and myelin-specific protein expression (Ye et al., 2002a). The abundance of IRS-2 and IRS-4 is significantly increased in the brain of irs-1 KO mice, suggesting that increased IRS-2 and/or IGF-4 may compensate for the loss of IRS-1.
On the other hand, blunting IRS-2 expression globally (Schubert et al., 2003) or specifically in nestin+ neural cells (Taguchi et al., 2007) results in a much more significant reduction in brain weight (by 30–38% in adult mice), compared to that in irs-1 KO mice. These data suggest that IRS-2 appears to be more important in mediating IGF/insulin signaling in the CNS. It is worthwhile to note that in irs-2 KO mice blunting one igf1r allele further reduces brain weight by 15%, while brain growth retardation is similar in igf1r KO and irs-2/igf1r double KO embryos (Schubert et al., 2003). These data strongly indicate that IGF–IGF1R signaling can act through molecule(s) other than IRS-2. At E14-E16, irs-2 KO mice have 37% less proliferating neuronal cells without apparent changes in cell apoptosis (Schubert et al., 2003). This suggests that at this developmental stage IRS-2 primarily transduces proliferation signaling, but is not essential to pro-survival signaling. IRS-2, however, is capable of mediating pro-survival signaling, at least during postnatal development. For example, the number of apoptotic photoreceptor cells is increased by 50% in the retina of 2 week old irs-2 KO mice (Yi et al., 2005).
IRS-2 also appears to play an important role in the growth of oligodendrocyte lineage cells. Blunting IRS-2 expression transiently decreases the abundance of multiple myelin/oligodendrocyte-specific proteins during the first 2 weeks of postnatal life (Freude et al., 2008). The expression of these proteins then gradually recovers with increasing age, likely due to the compensatory actions of increased IRS-1 and/or Akt (Freude et al., 2008). The details of IRS-2 actions in the generation and differentiation of oligodendrocyte lineage cells, as well as other neural cells remain to be further defined.
Taken together, IRS-2 appears to mediate a significant portion of growth signaling from IGF, as well as insulin, in a cell- and development- dependent manner, and one or more IRS molecules are capable of compensating for the loss of others.
6.1.2. Phosphoinositide-3 (PI3) kinase–Akt kinase pathway
IGF treatment rapidly increases Akt phosphorylation and activation in neural cultures. Unlike the transient activation of Raf–Erk (see below), however, IGF-I-stimulated PI3–Akt activation is sustained for at least 24 h, at least in cultured oligodendrocytes (Ness and Wood, 2002; Romanelli et al., 2007; Ye et al., 2010). In line with these in vitro studies, IGF-I overexpression markedly increases the abundance of phosphorylated Akt in developing brains, while reducing IGF availability decreases its abundance during the same developmental period (Sun and D’Ercole, 2006).
It has been well-documented that the PI3–Akt kinase pathway plays a key role in the survival of neural cells, including NSC/NPC, neurons (Johnson-Farley et al., 2007; Johnson-Farley et al., 2006) and oligodendrocytes (Dudek et al., 1997; Ebner et al., 2000; Sinor and Lillien, 2004; Vemuri and McMorris, 1996; Yao and Cooper, 1995). Multiple downstream signaling mechanisms are involved in these survival-promoting actions, including: (1) inhibition of the activity of the pro-apoptotic protein Bad (Kulik and Weber, 1998) and the forkhead transcription factor FKHRL1 (Brunet et al., 1999), which regulates the transcription of proapoptotic protein genes; (2) suppression of Bax translocation (Ness et al., 2004); (3) enhancement of the expression of the pro-survival proteins Bcl2 (Baker et al., 1999b; Chrysis et al., 2001; Minshall et al., 1999) and inhibitor of apoptosis proteins (IAPs) Liu et al., 2011; (4) reduction of the expression of the apoptotic enzymes caspase-3 and caspase-9 (Cui et al., 2005; Ness et al., 2004); and (5) increase in the activity of mammalian target of rapamycin (mTOR) Sinor and Lillien, 2004.
Accumulating data also show that the PI3–Akt pathway is required for neural proliferation and IGF-stimulated proliferation. In culture cells inhibition of PI3 kinase significantly reduces the incorporation of 3H-thymidine into oligodendrocyte precursors (Ebner et al., 2000), and markedly suppresses the IGF-I-stimulated proliferation in cortical NSC/NPC (Mairet-Coello et al., 2009) and cerebellar granule cell precursors (Cui et al., 1998). Conversely, overexpression of Akt in cortical precursor cells increases PCNA and BrdU labeling (Sinor and Lillien, 2004), markers of cell proliferation.
Glycogen synthase kinase 3 β (GSK3β) and β-catenin appear to be two critical downstream effectors in the PI3–Akt pathway that mediates IGF proliferation actions. Phosphorylation and inactivation of GSK3β by PI3–Akt kinases leads to stabilization of cyclin D1 protein (Ryves and Harwood, 2003; Takahashi-Yanaga and Sasaguri, 2008), a molecule key to the cell cycle progression through the G1/S phases. IGF can also signal through β-catenin, a key member of Wnt signaling pathway. In studies of cultured oligodendrocytes we (Ye et al., 2010) have show that IGF-I promotes Akt kinase and GSK3β phosphorylation. As a result, the abundance of β-catenin and the mRNA expression of its target gene cyclin D1 are increased. Inhibiting PI3–Akt kinase activity significantly suppresses the IGF-I-stimulated expression of β-catenin mRNA and its protein. Importantly, knocking down β-catenin expression markedly inhibits the IGF-I actions on cyclin D1 mRNA expression, cell proliferation and survival. These data suggest that β-catenin is a common effector mediating a portion of IGF-I and Wnt signaling to promote neural cell proliferation and survival.
Mammalian target of rapamycin (mTOR) is yet another important downstream effector of the IGF–PI3–Akt pathway. In recent years, details about the role of mTOR in IGF pleiotropic neural actions begin to emerge. It appears that mTOR is mainly involved in signals promoting cell maturation and function, because blocking mTOR signaling suppresses the development of cultured immature oligodendrocytes (Guardiola-Diaz et al., 2012) and myelination in mice (Narayanan et al., 2009). mTOR activation, as well as that of PI3 and Akt kinases, is also required for the IGF-I stimulated protein synthesis in cultured oligodendrocyte precursors (Bibollet-Bahena and Almazan, 2009). Similarly, insulin at a concentration that also activates the IGF1R promotes the differentiation of cultured neuronal precursors via mTOR (Han et al., 2008). PI3K-Akt-mTOR signaling appears also to be involved in IGF-I enhanced GLUT3 expression in P12 neuronal cells (Yu et al., 2012), long-term potentiation in the medial prefrontal cortex (Sui et al., 2008), and both cell growth and dendritic elaboration in hippocampus neurons (Jaworski et al., 2005). These data suggest that IGF-mTOR signaling likely promotes cellular maturation and functions by increasing protein synthesis. Intriguingly, it is reported that in cultured oligodendrocyte precursors mTOR is directly downstream of PI3 kinase rather than Akt kinase (Bibollet-Bahena and Almazan, 2009). Whether this signaling pathway also occurs in vivo or in other neural cell types, however, in not known.
6.1.3. Ras–Raf MAP pathway
IGF signaling through mitogen-activated protein (MAP) kinases is another pathway that has been abundantly studied. MAP kinases, a family of serine/threonine protein kinases, are conserved and widely distributed throughout the body. MAP kinase signaling cascades are organized in three-tiers. MAP signaling is initiated by the activation of MAP kinase kinase kinases, which in turn activates MAP kinase kinases and subsequently MAP kinases. MAP kinases include Erk1/2, p38, SAPK/JNK1/2/3, and Erk5/BMK1. While there are few reports on the role(s) of SAPK/JNK1/2/3 or Erk5/BMK1 in IGF neural actions, evidence has indicated that IGF can act in part by signaling through the Erk1/2 and p38 MAP pathways.
In cultured oligodendroglial cells and neurons, IGF-I transiently induces a rapid phosphorylation and activation of Erk1/2 and p38 MAP kinases within minutes (Cui and Almazan, 2007; Feldman et al., 1997; Hallak et al., 2001; Ye et al., 2010). Intriguingly, IGF-I treatment also leads to a delayed repression of Erk1/2 activity in cerebellar granule cells (Subramaniam et al., 2005). The biological significance of this late Erk suppression induced by IGF-I is not fully understood, although Erk suppression or ablation in adult neuronal cells has been associated with an enhanced cell survival (Subramaniam et al., 2005; Subramaniam et al., 2004) and synaptic plasticity, learning and memory abilities in mice (Mazzucchelli et al., 2002).
Studies of cultured neural cells indicate that Erk and p38 MAP signaling play an important role in IGF actions on cell maturation and differentiation. In oligodendroglial cell cultures, suppressing Erk activity blocks IGF-I stimulated protein synthesis (Bibollet-Bahena and Almazan, 2009), and halts the progression of early oligodendrocyte precursors to late oligodendrocyte precursors (Guardiola-Diaz et al., 2012). Similarly, suppression of p38 MAP pathway arrests the development of oligodendrocytes and myelination (Fragoso et al., 2007; Haines et al., 2008). The role of the Raf–Erk pathway in IGF promotive actions on oligodendrocyte maturation has been confirmed in mutant mice. Blunting the expression of brain B-Raf (the dominant activating isoform of the 3 Rafs in brain) decreases the abundance of phosphorylated Erk1 and Erk2 by more than 60% (Galabova-Kovacs et al., 2008; Zhong et al., 2007), and markedly reduces the number of mature oligodendrocytes and myelination in mutant mice (Galabova-Kovacs et al., 2008).
Based on culture studies using pharmacological inhibitors, activation of the Raf–Erk pathway has been believed to be key to IGF-stimulated cell proliferation (D’Ercole et al., 1996). In line with this concept, in cultured NSC inhibiting Erk MAP activity also ablates IGF-I-stimulated proliferation (Aberg et al., 2003a). Furthermore, in cultured oligodendrocytes IGF-I can synergize FGF stimulatory effects on Erk MAP kinase activation and on the expression of cyclin D1 (Frederick et al., 2007; Frederick and Wood, 2004), a factor that is involved in cell proliferation and the cell cycle progression. These data suggest that in neural cells the Raf–Erk pathway plays an important role in mediating IGF proliferative signaling. In these culture studies, however, Erk inhibitors and IGFs remain in medium for the entire culture period, rather than transiently during the initial IGF-induced Erk activation. Thus, it remains unclear about the nature of IGF-I-induced initial Erk activation. In addition, because the inhibitors employed have multiple actions, inhibition of IGF-stimulated activation of other signaling pathways cannot be excluded in these studies.
The view that the Raf–Erk pathway plays a dominant role in the IGF-dependent proliferation has been further challenged by recent genetic studies, at least in oligodendrocyte precursors. IGF-I overexpressing Tg mice, which exhibit an increased neural proliferation, have a brain abundance of phosphorylated Erk similar to that in control mice (Sun and D’Ercole, 2006). Similarly the abundance of phosphorylated Erk does not change in the brain of E17 igf1rNestin-KO embryos (our unpublished data). In contrast, embryos with a global Erk2 KO exhibit normal progenitor proliferation (Yao et al., 2003). Because further ablation of Erk1 (i.e. the other known Erk), in cultured Erk2-null stem cells does not affect this outcome, the normal proliferation in Erk2 null mutants is unlikely due to Erk1 compensation. Studies of mice with an Erk deletion specifically in neural precursors have proven inconclusive (Heffron et al., 2009; Samuels et al., 2008; Satoh et al., 2007; Satoh et al., 2011). These data suggest that Erk is not essential in cell proliferation mediated by IGF signaling and/or other growth signaling.
In addition, data from mice with a Raf deletion specifically in neural precursors argue strongly against the idea that the Raf–Erk pathway mediates the proliferative actions of IGF and/or other signaling. As indicated above, blunting the expression of brain B-Raf significantly decreases phosphorylated Erk1 and Erk2 (Galabova-Kovacs et al., 2008; Zhong et al., 2007). Despite B-Raf ablation and haplo-insufficiency in phosphorylated Erk1/2, however, the brain of these mice exhibits minimal undergrowth (by 5–10%) and an increased number of oligodendrocyte precursors. Because IGF-I increases oligodendrocyte precursor proliferation, these findings, albeit indirectly, do not support an essential role for the Raf–Erk pathway in IGF proliferation signaling. Further evidence supporting this conclusion comes from a recent report showing that inhibition of Erk does not suppress IGF-I-stimulated proliferation of NSC/NPC in cultured cortex (Mairet-Coello et al., 2009). However, these data do not preclude the possibility that IGF signals through Erk to stimulate neural proliferation under pathological conditions.
Taking together, the MAP kinase pathway in neural IGF signaling, although not fully comprehended, appears to be primarily involved in cellular maturation and survival. In addition, the actions of the IGF-MAP kinase pathway in brain are likely cell-dependent. More detailed studies are needed.
6.2. Signaling through IGF2R
There is little evidence that the IGF2R mediates the growth actions of both IGF-I and IGF-II. Several studies, however, have strongly suggested that the IGF2R is capable of mediating some of IGF biological actions. For example, specific stimulation of the IGF2R potentiates acetylcholine release from cultured neurons (Hawkes et al., 2006), while suppressing IGF2R signaling by an IGF2R specific antibody blocks IGF-II-enhanced choline acetyltransferase activity in cultured neurons (Konishi et al., 1994) and memory in adult rodents (Chen et al., 2011).
As indicated in Section 2.2 above, there is no enzymatic activity in the intracellular domain of the IGF2R, and at present the IGF2R intracellular signaling pathway(s) remains largely unclear. Multiple studies have identified G protein as a key signaling molecule to IGF-II–IGF2R actions in neuronal cells (Hawkes et al., 2006). Involvement of G protein activity in IGF2R signaling also has been observed in non-neuronal cells (McKinnon et al., 2001). Protein kinase C (Hawkes et al., 2006), MAP kinase (McKinnon et al., 2001), and GSK3 (Chen et al., 2011) are also likely to participate IGF2R signaling.
7. IGF actions in the human CNS
IGF-I, IGF-II, IGF1R and IGF2R are widely expressed in human brain and CSF (Carlsson-Skwirut et al., 1986; Chesik et al., 2006; Connor et al., 1997; De Keyser et al., 1994; Mashayekhi et al., 2010; Wilczak et al., 2000; Wilczak and De, 1997). There is, however, little information available about the actions of IGF signaling in human neural development. Several individuals with a mutation in the igf-I gene or the igf1r gene have been reported. The available findings from these patients are consistent with the CNS abnormalities seen in mutant rodents, as summarized above in Section 4, and clearly point to an important role for IGF signaling in the human CNS.
Three homozygous igf-I gene mutations have been identified in three individuals, respectively. These mutations include a deletion of exons 4 and 5 of the igf-I gene (Camacho-Hubner et al., 1999; Woods et al., 1997), and a single nucleotide substitution (G → A) mutation each at a distinct location in the igf-I gene, resulting in a change of a valine residue at position 44 of the mature IGF-I to a methionine (Denley et al., 2005; Walenkamp et al., 2005) or an arginine residue at position 36 to an glutamine (Netchine et al., 2009), respectively. The mutations lead to a severe deficit in IGFI expression (Camacho-Hubner et al., 1999; Woods et al., 1997), or to a marked reduction in its affinity for the IGF1R (Denley et al., 2005; Netchine et al., 2009; Walenkamp et al., 2005). Another patient with a homozygous T → A nucleotide substitution within exon 6 of the igf-I gene also has been reported (Bonapace et al., 2003), but a later study indicate that the change also is observed as polymorphism in normal persons and is not associated with growth retardation observed (Coutinho et al., 2007).
Each of these three patients exhibited intrauterine growth retardation, with a birth weight from 1400–2350 g [−4 to −2.5 standard derivation scores (SDSs)] and birth length 37.8–44 cm (−4.9 to −3.7 SDS). They had microcephaly with a head circumference 27–32 cm (−4.9 to −2.5 SDS). During postnatal development, the children persistently exhibited growth failure, and had severe deafness and mental retardation (Camacho-Hubner et al., 1999; Denley et al., 2005; Walenkamp et al., 2005; Woods et al., 1997). Despite a smaller head and likely smaller brain size, magnetic resonance imaging revealed that myelination and hippocampal architecture seemed to be normal (Camacho-Hubner et al., 1999; Netchine et al., 2009; Woods et al., 1997). While serum IGF-II was increased in the patients with igf-I gene mutations, it is not clear whether brain IGF-II expression also was increased, nor whether IGF-II could compensate for the IGF-I deficiency in the children, as in the apparent case in mice (Ye et al., 2002b).
More recently, a heterozygous duplication mutation in the igf-I gene has been reported in two patients from the same family (van Duyvenvoorde et al., 2010). The duplication mutation of four nucleotides at the position 35 of the mature IGF-I results in a frame shift and a premature termination of protein synthesis. The two patients exhibited postnatal growth retardation, but their nature, however, remains to be investigated.
There have been at least 8 individuals with a heterozygous mutation in the igf1r gene reported. However, there are no reports of humans born with mutations leading to absence of IGF1R expression. Heterozygous igf1r mutation (Abuzzahab et al., 2003; Inagaki et al., 2007; Kawashima et al., 2005; Kruis et al., 2010; Okubo et al., 2003; Raile et al., 2006; Walenkamp et al., 2006; Wallborn et al., 2010) or monoallelic IGF1R expression due to a deletion of the terminal 15q segment (Hammer et al., 2004) results in reduced IGF1R abundance and/or aberrant IGF1R functions (Abuzzahab et al., 2003; Inagaki et al., 2007; Kawashima et al., 2005; Kruis et al., 2010; Okubo et al., 2003; Raile et al., 2006; Wallborn et al., 2010), associated with an increased serum IGF-I (Inagaki et al., 2007; Raile et al., 2006; Wallborn et al., 2010). As with the individuals carrying an igf-I gene mutation, patients with an igf1r gene mutation exhibited prenatal and postnatal growth retardation, microcephaly and mild mental retardation, learning disorders, and/or altered behavioral characteristics (Abuzzahab et al., 2003; Inagaki et al., 2007; Kawashima et al., 2005; Kruis et al., 2010; Okubo et al., 2003; Raile et al., 2006; Walenkamp et al., 2006; Wallborn et al., 2010).
Taken together, these date strongly indicate that IGF–IGF1R signaling plays a key role in the developing CNS in human. Further support for this conclusion comes from limited studies of cultured human neural cells, showing that IGF exerts pro-growth actions similar to those observed in rodents (Armstrong et al., 1992; Cui et al., 2012; Satoh and Kim, 1994; Wilson et al., 2003). Nonetheless, the details of IGF actions in the development and growth of each of the human neural cell lineages require further study. More precise brain imagining and functional studies performed during the course of development are needed to better understand the actions of IGF signaling in human brain during development.
8. Injuries and neurological disorders
Emerging data have shown that the expression of IGF system proteins is significantly altered in a variety of brain injuries and neurological disorders (Popken et al., 2005), which are characterized by a marked loss of neural cells due to either increased cell death and/or reduced proliferation. Given that IGF signaling has a critical role in proliferation, survival and differentiation of neural cells, IGFs (primarily IGF-I) have been implicated in the pathogenesis of many neurological disorders during development.
8.1. Hypoxia/ischemia (H/I) brain injury
Associations of IGF and H/I brain injury are relatively well-documented. H/I in the prenatal brain is a major cause of neural cell loss that leads to cerebral palsy, epilepsy, cognitive deficits and growth retardation in affected children (Vannucci, 1990). Following H/I brain injury, the expression of IGF-I and its downstream signaling molecules (Akt and β-catenin) sharply declines in injured and surrounding areas within 24 h following H/I (Beilharz et al., 1993; Brywe et al., 2005; Costain et al., 2008; Lee et al., 1996; Lee et al., 1992; Mastroiacovo et al., 2009), concurrent with significant neuronal apoptosis during this time (Clawson et al., 1999). IGF-I expression, followed by IGF-II expression, then progressively increases in astrocytes (Lee et al., 1996) and/or microglia (O’Donnell et al., 2002), in association with tissue recovery.
Direct evidence that IGF signaling plays an important role in protecting neural cells from H/I injury comes from studies of rodents administered with IGF-I or IGF-II. In rodents IGF-I administered directly to the H/I brain prevents the loss of neurons and oligodendrocytes (Cao et al., 2003; Guan et al., 2001; Lin et al., 2005), increases the number of newly born cells in the DG (Dempsey et al., 2003), and improves neurological signs (Zhong et al., 2009). Increasing the abundance of β-catenin, a molecular in both IGF and Wnt signaling pathways, by lithium treatment or by blunting the expression of the Wnt antagonist DICKKOPF-1 markedly ameliorates ischemic cortical damage (Mastroiacovo et al., 2009).
More recently, ithas been reported that peripherally administered IGF-I through subcutaneous injection (Zhong et al., 2009) or nasal insufflation (Lin et al., 2009) mitigates H/I brain injury significantly, in part by promoting the survival of neuronal cells and proliferation of neural precursor cells. Notably, IGF-I remains effectivewhen given by subcutaneous injection 24 and 48 h after H/I injury (Zhong et al., 2009), a finding that supports the clinical utility of IGF-I in treating ischemic neural injury. Consistent with these findings, an early clinical trial shows that IGF-I, given intravenously, appears to improve clinical outcome in patients with moderate-to-severe head injury (Hatton et al., 1997), whose BBB was likely compromised, facilitating translocation of blood IGF-I into the brain.
The capacity of IGF-I to promote neuronal cell survival during H/I injury is, at least in part, mediated by IGF1R. Blunting the IGF1R expression in neuronal cells exacerbates H/I-induced neuronal apoptosis (Liu et al., 2011). Mice with ablated neural IGF1R expression also exhibit a reduction in the expression of both cellular IAP- 1 (cIAP-1) and X-linked IAP (XIAP) Liu et al., 2011, two members of IAP family proteins that are capable of suppressing the activity of caspases by either enhancing caspase degradation (cIAP) or directly inhibiting their activity (XIAP) Eckelman and Salvesen, 2006; Eckelman et al., 2006. These data suggest that IGF-stimulated IAP expression partially contributes to IGF–IGF1R protection against neuronal apoptosis following H/I injury.
8.2. Schizophrenia
Substantial evidence suggests that schizophrenia is a developmental disorder of the brain, reflecting a genetically conferred vulnerability combined with early insults from the environment (Boksa, 2004; Fatemi and Folsom, 2009; Tsuang, 2000). Epidemiological studies have provided evidence that exposure to certain obstetric complications is associated with a significantly increased risk for the later development of schizophrenia (Boksa, 2004; Fatemi and Folsom, 2009). Obstetric complications with the greatest effect size for schizophrenia include maternal diabetes, infections during pregnancy, preeclampsia, intrauterine growth retardation, and fetal/neonatal hypoxia.
Plasma levels of IGF-I are significantly decreased in schizophrenic patients (Kyle and Pichard, 2006; Venkatasubramanian et al., 2007; Wu et al., 2008).Microarray studies of the cerebral cortex and hippocampus fromcases of schizophrenia and of the DG in amouse model of human 22q11 deletion have revealed decreased gene expression andproteins for mitochondrial oxidative energymetabolismand specific cytoskeletal proteins implicated in synaptic functions (Altar et al., 2005; Jurata et al., 2006; Mirnics et al., 2000; Prabakaran et al., 2004). IGF-I and insulin have been shown to modulate these genes in a dose-dependent fashion, increasing the expression of those genes that are decreased in schizophrenia (Altar et al., 2008).
Brain imaging and postmortem histological studies of schizophrenia patients show gross pathological abnormalities, including marked reductions in the volumes of cortical gray matter, the hippocampal formation, and subcortical myelin (Andreone et al., 2007; Bartzokis et al., 2003; Di et al., 2009; Fornito et al., 2009; Friedman et al., 2008; Goldstein et al., 1999; Harrison, 1999; van Haren et al., 2008; Voineskos et al., 2010). Perhaps the most robust lesion found in human schizophrenia is a preferential loss of a particular subset of GABAergic interneurons in the dorsal prefrontal cortex (DPFC). By immunohistochemical analysis, this neuron loss involves those GABAergic interneurons which are GABA+/parvalbumin+ and GABA+/calbindin+ interneurons, but not those that are GABA+/calretinin+ (Beasley and Reynolds, 1997; Beasley et al., 2002; Benes et al., 1986; Benes et al., 1991; Reynolds and Beasley, 2001).
In heterozygous igf1rNestin-KO mice, whose IGF1R expression is conditionally ablated in nestin+ neural cells, our ongoing morphometric and stereological studies have revealed a loss of GABAergic neurons that are positive for calbindin, but not for calretinin, in the ventral prefrontal cortex (VPFC) (see Section 4.3. and Fig. 4). The VPFC in rodents corresponds to human DPFC (Heidbreder and Groenewegen, 2003), which is known to be associated with schizophrenia pathogenesis. Our unpublished behavioral studies further show that adult heterozygous igf1rNestin-KO mice exhibit behavioral abnormalities. These abnormalities include impaired learning and memory and abnormal acoustic startle/pre-pulse inhibition (AS/PPI), which are consistent with hallmark signs of schizophrenia in mouse models (Arguello and Gogos, 2006). These data suggest that reduced IGF-I signaling during prenatal and early postnatal development may play a role in the pathogenesis of schizophrenia during development.
9. Conclusion
Over the past 20 years or more, literature illustrates that IGF signaling plays critical roles in the growth and development of the CNS at virtually every stage of development. IGF-I increases the survival of pre-implantation embryos reaching the blastocyst stage through its anti-apoptotic actions and through its ability to protect against reactive oxygen species. Both IGF-I and IGF-II promote NSC/NPC proliferation during development, and IGF-I accelerates neuronogenesis by reducing G1 phase length of the mitotic cell cycle while increasing cell cycle re-entry. IGF-I promotes neural cell survival during normal development and in response to pathological insults. In postmitotic neurons, IGF-I acts to promote elongation and branching of dendrites and the relative density of dendritic spines. IGF-I also is capable of augmenting the progressive phase of synaptogenesis and the myelination of maturing axons. Clearly, the IGFs and their related IGF family proteins as therapeutic targets have tremendous potential in the treatment of injuries and developmental disorders of the brain and in various neurological diseases in the adult brain.
Acknowledgments
Data from our laboratories reported in this work were partially supported by NIH Grants RO1 NS038891, RO1 NS048868, and Multiple Sclerosis Society Grant PP1531.
References
- Aberg MA, Aberg ND, Hedbacker H, Oscarsson J, Eriksson PS. Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J Neurosci. 2000;20:2896–2903. doi: 10.1523/JNEUROSCI.20-08-02896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberg MA, Aberg ND, Palmer TD, Alborn AM, Carlsson-Skwirut C, Bang P, Rosengren LE, Olsson T, Gage FH, Eriksson PS. IGF-I has a direct proliferative effect in adult hippocampal progenitor cells. Mol Cell Neurosci. 2003a;24:23–40. doi: 10.1016/s1044-7431(03)00082-4. [DOI] [PubMed] [Google Scholar]
- Aberg ND, Blomstrand F, Aberg MA, Bjorklund U, Carlsson B, Carlsson-Skwirut C, Bang P, Ronnback L, Eriksson PS. Insulin-like growth factor-I increases astrocyte intercellular gap junctional communication and connexin43 expression in vitro. J Neurosci Res. 2003b;74:12–22. doi: 10.1002/jnr.10734. [DOI] [PubMed] [Google Scholar]
- Aberg ND, Johansson UE, Aberg MA, Hellstrom NA, Lind J, Bull C, Isgaard J, Anderson MF, Oscarsson J, Eriksson PS. Peripheral infusion of insulin-like growth factor-I increases the number of newborn oligodendrocytes in the cerebral cortex of adult hypophysectomized rats. Endocrinology. 2007;148:3765–3772. doi: 10.1210/en.2006-1556. [DOI] [PubMed] [Google Scholar]
- Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, Kiess W, Klammt J, Kratzsch J, Osgood D, Pfaffle R, Raile K, Seidel B, Smith RJ, Chernausek SD. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. New Engl J Med. 2003;349:2211–2222. doi: 10.1056/NEJMoa010107. [DOI] [PubMed] [Google Scholar]
- Alarcon C, Morales AV, Pimentel B, Serna J, de PF. (Pro)insulin and insulin-like growth factor I complementary expression and roles in early development. Comp Biochem Physiol B: Biochem Mol Biol. 1998;121:13–17. doi: 10.1016/s0305-0491(98)10105-0. [DOI] [PubMed] [Google Scholar]
- Altar CA, Hunt RA, Jurata LW, Webster MJ, Derby E, Gallagher P, Lemire A, Brockman J, Laeng P. Insulin, IGF-1, and muscarinic agonists modulate schizophrenia-associated genes in human neuroblastoma cells. Biol Psychiatry. 2008;64:1077–1087. doi: 10.1016/j.biopsych.2008.08.031. [DOI] [PubMed] [Google Scholar]
- Altar CA, Jurata LW, Charles V, Lemire A, Liu P, Bukhman Y, Young TA, Bullard J, Yokoe H, Webster MJ, Knable MB, Brockman JA. Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol Psychiatry. 2005;58:85–96. doi: 10.1016/j.biopsych.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Amritraj A, Rauw G, Baker GB, Kar S. Leu27 insulin-like growth factor-II, an insulin-like growth factor-II analog, attenuates depolarization-evoked GABA release from adult rat hippocampal and cortical slices. Neuroscience. 2010;170:722–730. doi: 10.1016/j.neuroscience.2010.07.026. [DOI] [PubMed] [Google Scholar]
- Anderson MF, Aberg MAI, Nilsson M, Eriksson PS. Insulin-like growth factor-I and neurogenesis in the adult mammalian brain. Dev Brain Res. 2002;134:115–122. doi: 10.1016/s0165-3806(02)00277-8. [DOI] [PubMed] [Google Scholar]
- Andersson S, D’Arcy P, Larsson O, Sehat B. Focal adhesion kinase (FAK) activates and stabilizes IGF-1 receptor. Biochem Biophys Res Commun. 2009;387:36–41. doi: 10.1016/j.bbrc.2009.06.088. [DOI] [PubMed] [Google Scholar]
- Andreone N, Tansella M, Cerini R, Rambaldelli G, Versace A, Marrella G, Perlini C, Dusi N, Pelizza L, Balestrieri M, Barbui C, Nose M, Gasparini A, Brambilla P. Cerebral atrophy and white matter disruption in chronic schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2007;257:3–11. doi: 10.1007/s00406-006-0675-1. [DOI] [PubMed] [Google Scholar]
- Arguello PA, Gogos JA. Modeling madness in mice: one piece at a time. Neuron. 2006;52:179–196. doi: 10.1016/j.neuron.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Dorn HH, Kufta CV, Friedman E, Dubois-Dalcq ME. Pre-oligodendrocytes from adult human CNS. J Neurosci. 1992;12:1538–1547. doi: 10.1523/JNEUROSCI.12-04-01538.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic Y, Weiss S. Insulin-like growth factor-I is a differentiation factor for postmitotic CNS stem cell-derived neuronal precursors: distinct actions from those of brain-derived neurotrophic factor. J Neurosci. 1998;18:2118–2128. doi: 10.1523/JNEUROSCI.18-06-02118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic Y, Weiss S, Schneider B, Aebischer P. Insulin-like growth factor-1 is necessary for neural stem cell proliferation and demonstrates distinct actions of epidermal growth factor and fibroblast growth factor-2. J Neurosci. 2001;21:7194–7202. doi: 10.1523/JNEUROSCI.21-18-07194.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayaso E, Nolan CM, Byrnes L. Zebrafish insulin-like growth factor-I receptor: molecular cloning and developmental expression. Mol Cell Endocrinol. 2002;191:137–148. doi: 10.1016/s0303-7207(02)00083-7. [DOI] [PubMed] [Google Scholar]
- Ayer-le LC, Stahlbom PA, Sara VR. Expression of IGF-I and -II mRNA in the brain and craniofacial region of the rat fetus. Development. 1991;111:105–115. doi: 10.1242/dev.111.1.105. [DOI] [PubMed] [Google Scholar]
- Bach MA, Shen-Orr Z, Lowe WL, Jr, Roberts CT, Jr, LeRoith D. Insulin-like growth factor I mRNA levels are developmentally regulated in specific regions of the rat brain. Brain Res Mol Brain Res. 1991;10:43–48. doi: 10.1016/0169-328x(91)90054-2. [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- Baker JC, Beddington RS, Harland RM. Wnt signaling in Xenopus embryos inhibits bmp4 expression and activates neural development. Genes Dev. 1999a;13:3149–3159. doi: 10.1101/gad.13.23.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NL, Carlo R, Bernard VO, D’Ercole AJ, Werther GA. Interactions between bcl-2 and the IGF system control apoptosis in the developing mouse brain. Brain Res Dev Brain Res. 1999b;118:109–118. doi: 10.1016/s0165-3806(99)00136-4. [DOI] [PubMed] [Google Scholar]
- Ballard FJ, Francis GL, Ross M, Bagley CJ, May B, Wallace JC. Natural and synthetic forms of insulin-like growth factor-1 (IGF-1) and the potent derivative, destripeptide IGF-1: biological activities and receptor binding. Biochem Biophys Res Commun. 1987;149:398–404. doi: 10.1016/0006-291x(87)90380-9. [DOI] [PubMed] [Google Scholar]
- Ballotti R, Nielsen FC, Pringle N, Kowalski A, Richardson WD, Van OE, Gammeltoft S. Insulin-like growth factor I in cultured rat astrocytes: expression of the gene, and receptor tyrosine kinase. EMBO J. 1987;6:3633–3639. doi: 10.1002/j.1460-2075.1987.tb02695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron-Van Evercooren A, Olichon-Berthe C, Kowalski A, Visciano G, Van Obberghen E. Expression of IGF-I and insulin receptor genes in the rat central nervous system: a developmental, regional, and cellular analysis. J Neurosci Res. 1991;28:244–253. doi: 10.1002/jnr.490280212. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Bartlett WP, Li XS, Williams M, Benkovic S. Localization of insulin-like growth factor-1 mRNA in murine central nervous system during postnatal development. Dev Biol. 1991;147:239–250. doi: 10.1016/s0012-1606(05)80021-1. [DOI] [PubMed] [Google Scholar]
- Bartlett WP, Li XS, Williams M. Expression of IGF-I mRNA in the murine subventricular zone during postnatal development. Mol Brain Res. 1992;12:285–291. doi: 10.1016/0169-328x(92)90131-t. [DOI] [PubMed] [Google Scholar]
- Bartzokis G, Nuechterlein KH, Lu PH, Gitlin M, Rogers S, Mintz J. Dysregulated brain development in adult men with schizophrenia: a magnetic resonance imaging study. Biol Psychiatry. 2003;53:412–421. doi: 10.1016/s0006-3223(02)01835-8. [DOI] [PubMed] [Google Scholar]
- Baxter RC, Martin JL. Binding proteins for the insulin-like growth factors: structure, regulation and function. Prog Growth Factor Res. 1989;1:49–68. doi: 10.1016/0955-2235(89)90041-0. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Reynolds GP. Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophr Res. 1997;24:349–355. doi: 10.1016/s0920-9964(96)00122-3. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry. 2002;52:708–715. doi: 10.1016/s0006-3223(02)01360-4. [DOI] [PubMed] [Google Scholar]
- Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F. Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron. 1995;14:717–730. doi: 10.1016/0896-6273(95)90216-3. [DOI] [PubMed] [Google Scholar]
- Behringer RR, Lewin TM, Quaife CJ, Palmiter RD, Brinster RL, D’Ercole AJ. Expression of insulin-like growth factor I stimulates normal somatic growth in growth hormone-deficient transgenic mice. Endocrinology. 1990;127:1033–1040. doi: 10.1210/endo-127-3-1033. [DOI] [PubMed] [Google Scholar]
- Beilharz EJ, Klempt ND, Klempt M, Sirimanne E, Dragunow M, Gluckman PD. Differential expression of insulin-like growth factor binding proteins (IGFBP) 4 and 5 mRNA in the rat brain after transient hypoxic-ischemic injury. Mol Brain Res. 1993;18:209–215. doi: 10.1016/0169-328x(93)90191-q. [DOI] [PubMed] [Google Scholar]
- Beilharz EJ, Russo VC, Butler G, Baker NL, Connor B, Sirimanne ES, Dragunow M, Werther GA, Gluckman PD, Williams CE, Scheepens A. Co-ordinated and cellular specific induction of the components of the IGF/IGFBP axis in the rat brain following hypoxic–ischemic injury. Brain Res Mol Brain Res. 1998;59:119–134. doi: 10.1016/s0169-328x(98)00122-3. [DOI] [PubMed] [Google Scholar]
- Benes FM, Davidson J, Bird ED. Quantitative cytoarchitectural studies of the cerebral cortex of schizophrenics. Arch Gen Psychiatry. 1986;43:31–35. doi: 10.1001/archpsyc.1986.01800010033004. [DOI] [PubMed] [Google Scholar]
- Benes FM, McSparren J, Bird ED, SanGiovanni JP, Vincent SL. Deficits in small interneurons in prefrontal and cingulate cortices of schizophrenic and schizoaffective patients. Arch Gen Psychiatry. 1991;48:996–1001. doi: 10.1001/archpsyc.1991.01810350036005. [DOI] [PubMed] [Google Scholar]
- Bibollet-Bahena O, Almazan G. IGF-1-stimulated protein synthesis in oligodendrocyte progenitors requires PI3K/mTOR/Akt and MEK/ERK pathways. J Neurochem. 2009;109:1440–1451. doi: 10.1111/j.1471-4159.2009.06071.x. [DOI] [PubMed] [Google Scholar]
- Bienvenu G, Seurin D, Grellier P, Froment P, Baudrimont M, Monget P, Le Bouc Y, Babajko S. Insulin-like growth factor binding protein-6 transgenic mice: postnatal growth, brain development, and reproduction abnormalities. Endocrinology. 2004;145:2412–2420. doi: 10.1210/en.2003-1196. [DOI] [PubMed] [Google Scholar]
- Boksa P. Animal models of obstetric complications in relation to schizophrenia. Brain Res Brain Res Rev. 2004;45:1–17. doi: 10.1016/j.brainresrev.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Bonapace G, Concolino D, Formicola S, Strisciuglio P. A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J Med Genet. 2003;40:913–917. doi: 10.1136/jmg.40.12.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy C, Chin E. IGF-I mRNA localization in trigeminal and sympathetic nerve target zones during rat embryonic development. Adv Exp Med Biol. 1991;293:431–437. doi: 10.1007/978-1-4684-5949-4_38. [DOI] [PubMed] [Google Scholar]
- Bondy C, Lee WH. Correlation between insulin-like growth factor (IGF)-binding protein 5 and IGF-I gene expression during brain development. J Neurosci. 1993;13:5092–5104. doi: 10.1523/JNEUROSCI.13-12-05092.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy CA. Transient IGF-I gene expression during the maturation of functionally related central projection neurons. J Neurosci. 1991;11:3442–3455. doi: 10.1523/JNEUROSCI.11-11-03442.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy CA, Lee WH. Patterns of insulin-like growth factor and IGF receptor gene expression in the brain. Functional implications. Ann NY Acad Sci. 1993;692:33–43. doi: 10.1111/j.1749-6632.1993.tb26203.x. [DOI] [PubMed] [Google Scholar]
- Bondy CA, Werner H, Roberts CT, Jr, LeRoith D. Cellular pattern of insulin-like growth factor-I (IGF-I) and type I IGF receptor gene expression in early organogenesis: comparison with IGF-II gene expression. Mol Endocrinol. 1990;4:1386–1398. doi: 10.1210/mend-4-9-1386. [DOI] [PubMed] [Google Scholar]
- Bonilla AQ, Oliveira LJ, Ozawa M, Newsom EM, Lucy MC, Hansen PJ. Developmental changes in thermoprotective actions of insulin-like growth factor-1 on the preimplantation bovine embryo. Mol Cell Endocrinol. 2011;332:170–179. doi: 10.1016/j.mce.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Breese CR, D’Costa A, Rollins YD, Adams C, Booze RM, Sonntag WE, Leonard S. Expression of insulin-like growth factor-1 (IGF-1) and IGF-binding protein 2 (IGF-BP2) in the hippocampus following cytotoxic lesion of the dentate gyrus. J Comp Neurol. 1996;369:388–404. doi: 10.1002/(SICI)1096-9861(19960603)369:3<388::AID-CNE5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Bruel-Jungerman E, Davis S, Laroche S. Brain plasticity mechanisms and memory: a party of four. Neuroscientist. 2007;13:492–505. doi: 10.1177/1073858407302725. [DOI] [PubMed] [Google Scholar]
- Brunet A, Nonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Andersen MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Brywe KG, Mallard C, Gustavsson M, Hedtjarn M, Leverin AL, Wang X, Blomgren K, Isgaard J, Hagberg H. IGF-I neuroprotection in the immature brain after hypoxia–ischemia, involvement of Akt and GSK3beta? Eur J Neurosci. 2005;21:1489–1502. doi: 10.1111/j.1460-9568.2005.03982.x. [DOI] [PubMed] [Google Scholar]
- Butefisch CM. Neurobiological bases of rehabilitation. Neurol Sci. 2006;27 (Suppl 1):S18–S23. doi: 10.1007/s10072-006-0540-z. [DOI] [PubMed] [Google Scholar]
- Byrne AT, Southgate J, Brison DR, Leese HJ. Effects of insulin-like growth factors I and II on tumour-necrosis-factor-alpha-induced apoptosis in early murine embryos. Reprod Fertil Dev. 2002;14:79–83. doi: 10.1071/rd01015. [DOI] [PubMed] [Google Scholar]
- Camacho-Hubner C, Woods KA, Miraki-Moud F, Hindmarsh PC, Clark AJ, Hansson Y, Johnston A, Baxter RC, Savage MO. Effects of recombinant human insulin-like growth factor I (IGF-I) therapy on the growth hormone-IGF system of a patient with a partial IGF-I gene deletion. J Clin Endocrinol Metab. 1999;84:1611–1616. doi: 10.1210/jcem.84.5.5649. [DOI] [PubMed] [Google Scholar]
- Camarero G, Avendano C, Fernandez-Moreno C, Villar A, Contreras J, De Pablo F, Pichel JG, Varela-Nieto I. Delayed inner ear maturation and neuronal loss in postnatal Igf-1-deficient mice. J Neurosci. 2001;21:7630–7641. doi: 10.1523/JNEUROSCI.21-19-07630.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Gunn AJ, Bennet L, Wu D, George S, Gluckman PD, Shao XM, Guan J. Insulin-like growth factor (IGF)-1 suppresses oligodendrocyte caspase-3 activation and increases glial proliferation after ischemia in near-term fetal sheep. J Cereb Blood Flow Metab. 2003;23:739–747. doi: 10.1097/01.WCB.0000067720.12805.6F. [DOI] [PubMed] [Google Scholar]
- Carlsson-Skwirut C, Jörnvall H, Holmgren A, Andersson C, Bergman T, Lundquist G, Sjögren B, Sara VR. Isolation and characterization of variant IGF-1 as well as IGF-2 from adult human brain. FEBS Lett. 1986;201:46–50. doi: 10.1016/0014-5793(86)80568-3. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Plasticity of cortical projections after stroke. Neuroscientist. 2003;9:64–75. doi: 10.1177/1073858402239592. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Behringer RR, Brinster RL, McMorris FA. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron. 1993;10:729–740. doi: 10.1016/0896-6273(93)90173-o. [DOI] [PubMed] [Google Scholar]
- Cavallaro T, Martone RL, Stylianopoulou F, Herbert J. Differential expression of the insulin-like growth factor II and transthyretin genes in the developing rat choroid plexus. J Neuropathol Exp Neurol. 1993;52:153–162. doi: 10.1097/00005072-199303000-00008. [DOI] [PubMed] [Google Scholar]
- Cayre M, Canoll P, Goldman JE. Cell migration in the normal and pathological postnatal mammalian brain. Prog Neurobiol. 2009;88:41–63. doi: 10.1016/j.pneurobio.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Stern SA, Garcia-Osta A, Saunier-Rebori B, Pollonini G, Bambah-Mukku D, Blitzer RD, Alberini CM. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–497. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CM, Joncas G, Reinhardt RR, Farrer R, Quarles R, Janssen J, McDonald MP, Crawley JN, Powell-Braxton L, Bondy CA. Biochemical and morphometric analyses show that myelination in the insulin-like growth factor 1 null brain is proportionate to its neuronal composition. J Neurosci. 1998;18:5673–5681. doi: 10.1523/JNEUROSCI.18-15-05673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CM, Mervis RF, Niu SL, Salem N, Jr, Witters LA, Tseng V, Reinhardt R, Bondy CA. Insulin-like growth factor 1 is essential for normal dendritic growth. J Neurosci Res. 2003;73:1–9. doi: 10.1002/jnr.10634. [DOI] [PubMed] [Google Scholar]
- Chernausek SD. Insulin-like growth factor-I (IGF-I) production by astroglial cells: regulation and importance for epidermal growth factor-induced cell replication. J Neurosci Res. 1993;34:189–197. doi: 10.1002/jnr.490340206. [DOI] [PubMed] [Google Scholar]
- Chesik D, De KJ, Bron R, Fuhler GM. Insulin-like growth factor binding protein-1 activates integrin-mediated intracellular signaling and migration in oligodendrocytes. J Neurochem. 2010;113:1319–1330. doi: 10.1111/j.1471-4159.2010.06703.x. [DOI] [PubMed] [Google Scholar]
- Chesik D, De KJ, Glazenburg L, Wilczak N. Insulin-like growth factor binding proteins: regulation in chronic active plaques in multiple sclerosis and functional analysis of glial cells. Eur J Neurosci. 2006;24:1645–1652. doi: 10.1111/j.1460-9568.2006.05034.x. [DOI] [PubMed] [Google Scholar]
- Choi YS, Cho HY, Hoyt KR, Naegele JR, Obrietan K. IGF-1 receptor-mediated ERK/MAPK signaling couples status epilepticus to progenitor cell proliferation in the subgranular layer of the dentate gyrus. Glia. 2008;56:791–800. doi: 10.1002/glia.20653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrysis D, Calikoglu AS, Ye P, D’Ercole AJ. Insulin-like growth factor-I overexpression attenuates cerebellar apoptosis by altering the expression of Bcl family proteins in a developmentally specific manner. J Neurosci. 2001;21:1481–1489. doi: 10.1523/JNEUROSCI.21-05-01481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicco-Bloom E, Black IB. Insulin growth factors regulate the mitotic cycle in cultured rat sympathetic neuroblasts. Proc Natl Acad Sci USA. 1988;85:4066–4070. doi: 10.1073/pnas.85.11.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clawson TF, Vannucci SJ, Wang GM, Seaman LB, Yang XL, Lee WH. Hypoxia–ischemia-induced apoptotic cell death correlates with IGF-I mRNA decrease in neonatal rat brain. Biol Signals Recept. 1999;8:281–293. doi: 10.1159/000014599. [DOI] [PubMed] [Google Scholar]
- Connor B, Beilharz EJ, Williams C, Synek B, Gluckman PD, Faull RL, Dragunow M. Insulin-like growth factor-I (IGF-I) immunoreactivity in the Alzheimer’s disease temporal cortex and hippocampus. Brain Res Mol Brain Res. 1997;49:283–290. doi: 10.1016/s0169-328x(97)00192-7. [DOI] [PubMed] [Google Scholar]
- Costain WJ, Rasquinha I, Sandhu JK, Rippstein P, Zurakowski B, Slinn J, MacManus JP, Stanimirovic DB. Cerebral ischemia causes dysregulation of synaptic adhesion in mouse synaptosomes. J Cereb Blood Flow Metab. 2008;28:99–110. doi: 10.1038/sj.jcbfm.9600510. [DOI] [PubMed] [Google Scholar]
- Coutinho DC, Coletta RR, Costa EM, Pachi PR, Boguszewski MC, Damiani D, Mendonca BB, Arnhold IJ, Jorge AA. Polymorphisms identified in the upstream core polyadenylation signal of IGF1 gene exon 6 do not cause pre- and postnatal growth impairment. J Clin Endocrinol Metab. 2007;92:4889–4892. doi: 10.1210/jc.2007-1661. [DOI] [PubMed] [Google Scholar]
- Cui H, Meng Y, Bulleit RF. Inhibition of glycogen synthase kinase 3beta activity regulates proliferation of cultured cerebellar granule cells. Brain Res Dev Brain Res. 1998;111:177–188. doi: 10.1016/s0165-3806(98)00136-9. [DOI] [PubMed] [Google Scholar]
- Cui QL, Almazan G. IGF-I-induced oligodendrocyte progenitor proliferation requires PI3K/Akt, MEK/ERK, and Src-like tyrosine kinases. J Neurochem. 2007;100:1480–1493. doi: 10.1111/j.1471-4159.2006.04329.x. [DOI] [PubMed] [Google Scholar]
- Cui QL, D’Abate L, Fang J, Leong SY, Ludwin S, Kennedy T, Antel J, Almazan G. Human fetal oligodendrocyte progenitor cells from different gestational stages exhibit substantially different potential to myelinate. Stem Cells Dev. 2012 doi: 10.1089/scd.2011.0494. http://dx.doi.org/10.1089/scd.2011.0494. [DOI] [PubMed]
- Cui QL, Zheng WH, Quirion R, Almazan G. Inhibition of Src-like kinases reveals Akt-dependent and -independent pathways in insulin-like growth factor I-mediated oligodendrocyte progenitor survival. J Biol Chem. 2005;280:8918–8928. doi: 10.1074/jbc.M414267200. [DOI] [PubMed] [Google Scholar]
- D’Ercole AJ, Dai Z, Xing Y, Boney C, Wilkie MB, Lauder JM, Han VK, Clemmons DR. Brain growth retardation due to the expression of human insulin-like growth factor binding protein-1 (IGFBP-1) in transgenic mice. An invivo model for the analysis of IGF function in the brain. Brain Res Dev Brain Res. 1994;82:213–222. doi: 10.1016/0165-3806(94)90164-3. [DOI] [PubMed] [Google Scholar]
- D’Ercole AJ, Ye P, Calikoglu AS, Gutierrez-Ospina G. The role of the insulin-like growth factors in the central nervous system. Mol Neurobiol. 1996;13:227–255. doi: 10.1007/BF02740625. [DOI] [PubMed] [Google Scholar]
- Daughaday WH, Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989;10:68–91. doi: 10.1210/edrv-10-1-68. [DOI] [PubMed] [Google Scholar]
- De Keyser J, Wilczak N, De Backer JP, Herroelen L, De Backer JP, Vauquelin G. Insulin-like growth factor-I receptors in human brain and pituitary gland: an autoradiographic study. Synapse. 1994;17:196–202. doi: 10.1002/syn.890170309. [DOI] [PubMed] [Google Scholar]
- De Robertis EM, Larrain J, Oelgeschlager M, Wessely O. The establishment of Spemann’s organizer and patterning of the vertebrate embryo. Nat Rev Genet. 2000;1:171–181. doi: 10.1038/35042039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de PF, Perez-Villamil B, Serna J, Gonzalez-Guerrero PR, Lopez-Carranza A, de la Rosa EJ, Alemany J, Caldes T. IGF-I and the IGF-I receptor in development of nonmammalian vertebrates. Mol Reprod Dev. 1993;35:427–432. doi: 10.1002/mrd.1080350418. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature. 1990;345:78–80. doi: 10.1038/345078a0. [DOI] [PubMed] [Google Scholar]
- Dempsey RJ, Sailor KA, Bowen KK, Tureyen K, Vemuganti R. Stroke-induced progenitor cell proliferation in adult spontaneously hypertensive rat brain: effect of exogenous IGF-1 and GDNF. J Neurochem. 2003;87:586–597. doi: 10.1046/j.1471-4159.2003.02022.x. [DOI] [PubMed] [Google Scholar]
- Denley A, Wang CC, Mcneil KA, Walenkamp MJ, van DH, Wit JM, Wallace JC, Norton RS, Karperien M, Forbes BE. Structural and functional characteristics of the Val44Met insulin-like growth factor I missense mutation: correlation with effects on growth and development. Mol Endocrinol. 2005;19:711–721. doi: 10.1210/me.2004-0409. [DOI] [PubMed] [Google Scholar]
- Dentremont KD, Ye P, D’Ercole AJ, O’Kusky JR. Increased insulin-like growth factor-I (IGF-I) expression during early postnatal development differentially increases neuron number and growth in medullary nuclei of the mouse. Brain Res Dev Brain Res. 1999;114:135–141. doi: 10.1016/s0165-3806(99)00024-3. [DOI] [PubMed] [Google Scholar]
- Di RE, Crow TJ, Walker MA, Black G, Chance SA. Reduced neuron density, enlarged minicolumn spacing and altered ageing effects in fusiform cortex in schizophrenia. Psychiatry Res. 2009;166:102–115. doi: 10.1016/j.psychres.2008.04.007. [DOI] [PubMed] [Google Scholar]
- DiVall SA, Williams TR, Carver SE, Koch L, Bruning JC, Kahn CR, Wondisford F, Radovick S, Wolfe A. Divergent roles of growth factors in the GnRH regulation of puberty in mice. J Clin Invest. 2010;120:2900–2909. doi: 10.1172/JCI41069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublier S, Duyckaerts C, Seurin D, Binoux M. Impaired brain development and hydrocephalus in a line of transgenic mice with liver-specific expression of human insulin-like growth factor binding protein-1. Growth Horm IGF Res. 2000;10:267–274. doi: 10.1054/ghir.2000.0168. [DOI] [PubMed] [Google Scholar]
- Drago J, Murphy M, Carroll SM, Harvey RP, Bartlett PF. Fibroblast growth factor-mediated proliferation of central nervous system precursors depends on endogenous production of insulin-like growth factor I. Proc Natl Acad Sci USA. 1991;88:2199–2203. doi: 10.1073/pnas.88.6.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg MG. Regulation of neuronal survival by the serine–threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Mandemakers W, Rogers M, Ibrahim A, Daneman R, Barres BA. A novel purification method for CNS projection neurons leads to the identification of brain vascular cells as a source of trophic support for corticospinal motor neurons. J Neurosci. 2008;28:8294–8305. doi: 10.1523/JNEUROSCI.2010-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner S, Dunbar M, McKinnon RD. Distinct roles for PI3K in proliferation and survival of oligodendrocyte progenitor cells. J Neurosci Res. 2000;62:336–345. doi: 10.1002/1097-4547(20001101)62:3<336::AID-JNR3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281:3254–3260. doi: 10.1074/jbc.M510863200. [DOI] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann L, Hanson PI, Chapman ER, Jahn R. Synaptobrevin binding to synaptophysin: a potential mechanism for controlling the exocytotic fusion machine. EMBO J. 1995;14:224–231. doi: 10.1002/j.1460-2075.1995.tb06995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstratiadis A. Genetics of mouse growth. Int J Dev Biol. 1998;42:955–976. [PubMed] [Google Scholar]
- Eggenschwiler J, Ludwig T, Fisher P, Leighton PA, Tilghman SM, Efstratiadis A. Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith–Wiedemann and Simpson–Golabi–Behmel syndromes. Genes Dev. 1997;11:3128–3142. doi: 10.1101/gad.11.23.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engleka MJ, Folli F, Winnay J, Kahn CR, McMorris FA. Insulin-receptor substrate-1 is required for normal myelination. J Neurochem. 1996;66:S20. [Google Scholar]
- Fabian D, Il’kova G, Rehak P, Czikkova S, Baran V, Koppel J. Inhibitory effect of IGF-I on induced apoptosis in mouse preimplantation embryos cultured in vitro. Theriogenology. 2004;61:745–755. doi: 10.1016/s0093-691x(03)00254-1. [DOI] [PubMed] [Google Scholar]
- Fantin VR, Lavan BE, Wang Q, Jenkins NA, Gilbert DJ, Copeland NG, Keller SR, Lienhard GE. Cloning, tissue expression, and chromosomal location of the mouse insulin receptor substrate 4 gene. Endocrinology. 1999;140:1329–1337. doi: 10.1210/endo.140.3.6578. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull. 2009;35:528–548. doi: 10.1093/schbul/sbn187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman EL, Sullivan KA, Kim B, Russell JW. Insulin-like growth factors regulate neuronal differentiation and survival. Neurobiol Dis. 1997;4:201–214. doi: 10.1006/nbdi.1997.0156. [DOI] [PubMed] [Google Scholar]
- Folli F, Bonfanti L, Renard E, Kahn CR, Merighi A. Insulin receptor substrate-1 (IRS-1) distribution in the rat central nervous system. J Neurosci. 1994;14 (Pt 1):6412–6422. doi: 10.1523/JNEUROSCI.14-11-06412.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornito A, Yucel M, Dean B, Wood SJ, Pantelis C. Anatomical abnormalities of the anterior cingulate cortex in schizophrenia: bridging the gap between neuroimaging and neuropathology. Schizophr Bull. 2009;35:973–993. doi: 10.1093/schbul/sbn025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragoso G, Haines JD, Roberston J, Pedraza L, Mushynski WE, Almazan G. P38 mitogen-activated protein kinase is required for central nervous system myelination. Glia. 2007;55:1531–1541. doi: 10.1002/glia.20567. [DOI] [PubMed] [Google Scholar]
- Frederick TJ, Min J, Altieri SC, Mitchell NE, Wood TL. Synergistic induction of cyclin D1 in oligodendrocyte progenitor cells by IGF-I and FGF-2 requires differential stimulation of multiple signaling pathways. Glia. 2007;55:1011–1022. doi: 10.1002/glia.20520. [DOI] [PubMed] [Google Scholar]
- Frederick TJ, Wood TL. IGF-I and FGF-2 coordinately enhance cyclin D1 and cyclin E-cdk2 association and activity to promote G1 progression in oligodendrocyte progenitor cells. Mol Cell Neurosci. 2004;25:480–492. doi: 10.1016/j.mcn.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Freude S, Leeser U, Muller M, Hettich MM, Udelhoven M, Schilbach K, Tobe K, Kadowaki T, Kohler C, Schroder H, Krone W, Bruning JC, Schubert M. IRS-2 branch of IGF-1 receptor signaling is essential for appropriate timing of myelination. J Neurochem. 2008;107:907–917. doi: 10.1111/j.1471-4159.2008.05631.x. [DOI] [PubMed] [Google Scholar]
- Friedland DR, Eden AR, Laitman JT. Naturally occurring motoneuron cell death in rat upper respiratory tract motor nuclei: a histological, fast DiI and immunocytochemical study in the hypoglossal nucleus. J Neurobiol. 1995;27:520–534. doi: 10.1002/neu.480270407. [DOI] [PubMed] [Google Scholar]
- Friedman JI, Tang C, Carpenter D, Buchsbaum M, Schmeidler J, Flanagan L, Golembo S, Kanellopoulou I, Ng J, Hof PR, Harvey PD, Tsopelas ND, Stewart D, Davis KL. Diffusion tensor imaging findings in first-episode and chronic schizophrenia patients. Am J Psychiatry. 2008;165:1024–1032. doi: 10.1176/appi.ajp.2008.07101640. [DOI] [PubMed] [Google Scholar]
- Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22:612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galabova-Kovacs G, Catalanotti F, Matzen D, Reyes GX, Zezula J, Herbst R, Silva A, Walter I, Baccarini M. Essential role of B-Raf in oligodendrocyte maturation and myelination during postnatal central nervous system development. J Cell Biol. 2008;180:947–955. doi: 10.1083/jcb.200709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Estrada J, Garcia-Segura LM, Torres-Aleman I. Expression of insulin-like growth factor I by astrocytes in response to injury. Brain Res. 1992;592:343–347. doi: 10.1016/0006-8993(92)91695-b. [DOI] [PubMed] [Google Scholar]
- Girbau M, Bassas L, Alemany J, de PF. In situ autoradiography and ligand-dependent tyrosine kinase activity reveal insulin receptors and insulin-like growth factor I receptors in prepancreatic chicken embryos. Proc Natl Acad Sci USA. 1989;86:5868–5872. doi: 10.1073/pnas.86.15.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson LM, Chakraborty C, McKinnon T, Lala PK. Insulin-like growth factor-binding protein 1 stimulates human trophoblast migration by signaling through alpha 5 beta 1 integrin via mitogen-activated protein Kinase pathway. J Clin Endocrinol Metab. 2001;86:2484–2493. doi: 10.1210/jcem.86.6.7532. [DOI] [PubMed] [Google Scholar]
- Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature. 1998;391:357–362. doi: 10.1038/34848. [DOI] [PubMed] [Google Scholar]
- Glinka A, Wu W, Onichtchouk D, Blumenstock C, Niehrs C. Head induction by simultaneous repression of Bmp and Wnt signalling in Xenopus. Nature. 1997;389:517–519. doi: 10.1038/39092. [DOI] [PubMed] [Google Scholar]
- Gogolla N, Galimberti I, Caroni P. Structural plasticity of axon terminals in the adult. Curr Opin Neurobiol. 2007;17:516–524. doi: 10.1016/j.conb.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Goldstein JM, Goodman JM, Seidman LJ, Kennedy DN, Makris N, Lee H, Tourville J, Caviness VS, Jr, Faraone SV, Tsuang MT. Cortical abnormalities in schizophrenia identified by structural magnetic resonance imaging. Arch Gen Psychiatry. 1999;56:537–547. doi: 10.1001/archpsyc.56.6.537. [DOI] [PubMed] [Google Scholar]
- Guan J, Krishnamurthi R, Waldvogel HJ, Faull RL, Clark R, Gluckman P. N-terminal tripeptide of IGF-1 (GPE) prevents the loss of TH positive neurons after 6-OHDA induced nigral lesion in rats. Brain Res. 2000;859:286–292. doi: 10.1016/s0006-8993(00)01988-0. [DOI] [PubMed] [Google Scholar]
- Guan J, Miller OT, Waugh KM, McCarthy DC, Gluckman PD. Insulin-like growth factor-1 improves somatosensory function and reduces the extent of cortical infarction and ongoing neuronal loss after hypoxia–ischemia in rats. Neuroscience. 2001;105:299–306. doi: 10.1016/s0306-4522(01)00145-2. [DOI] [PubMed] [Google Scholar]
- Guan J, Williams CE, Skinner SJ, Mallard EC, Gluckman PD. The effects of insulin-like growth factor (IGF)-1, IGF-2, and des-IGF-1 on neuronal loss after hypoxic-ischemic brain injury in adult rats: evidence for a role for IGF binding proteins. Endocrinology. 1996;137:893–898. doi: 10.1210/endo.137.3.8603600. [DOI] [PubMed] [Google Scholar]
- Guardiola-Diaz HM, Ishii A, Bansal R. Erk1/2 MAPK and mTOR signaling sequentially regulates progression through distinct stages of oligodendrocyte differentiation. Glia. 2012;60:476–486. doi: 10.1002/glia.22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudi V, Skuljec J, Yildiz O, Frichert K, Skripuletz T, Moharregh-Khiabani D, Voss E, Wissel K, Wolter S, Stangel M. Spatial and temporal profiles of growth factor expression during CNS demyelination reveal the dynamics of repair priming. PLoS One. 2011;6:e22623. doi: 10.1371/journal.pone.0022623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guler HP, Zapf J, Schmid C, Froesch ER. Insulin-like growth factors I and II in healthy man. Estimations of half-lives and production rates. Acta Endocrinol (Copenh) 1989;121:753–758. doi: 10.1530/acta.0.1210753. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Ospina G, Calikoglu AS, Ye P, D’Ercole AJ. In vivo effects of insulin-like growth factor-I on the development of sensory pathways: analysis of the primary somatic sensory cortex (S1) of transgenic mice. Endocrinology. 1996;137:5484–5492. doi: 10.1210/endo.137.12.8940375. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Ospina G, Saum L, Calikoglu AS, az-Cintra S, Barrios FA, D’Ercole AJ. Increased neural activity in transgenic mice with brain IGF-I overexpression: a [3H]2DG study. Neuroreport. 1997;8:2907–2911. doi: 10.1097/00001756-199709080-00021. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Ospina G, Uribe-Querol E, Sanchez N, Geovannini H, Padilla P, Hernandez-Echeagaray E. Similar synapse density in layer IV columns of the primary somatosensory cortex of transgenic mice with different brain size: implications for mechanisms underlying the differential allocation of cortical space. Brain Behav Evol. 2004;64:61–69. doi: 10.1159/000079116. [DOI] [PubMed] [Google Scholar]
- Haines JD, Fragoso G, Hossain S, Mushynski WE, Almazan G. p38 Mitogen-activated protein kinase regulates myelination. J Mol Neurosci. 2008;35:23–33. doi: 10.1007/s12031-007-9011-0. [DOI] [PubMed] [Google Scholar]
- Hallak H, Seiler AE, Green JS, Henderson A, Ross BN, Rubin R. Inhibition of insulin-like growth factor-I signaling by ethanol in neuronal cells. Alcohol Clin Exp Res. 2001;25:1058–1064. [PubMed] [Google Scholar]
- Hammer E, Kutsche K, Haag F, Ullrich K, Sudbrak R, Willig RP, Braulke T, Kubler B. Mono-allelic expression of the IGF-I receptor does not affect IGF responses in human fibroblasts. Eur J Endocrinol. 2004;151:521–529. doi: 10.1530/eje.0.1510521. [DOI] [PubMed] [Google Scholar]
- Han J, Wang B, Xiao Z, Gao Y, Zhao Y, Zhang J, Chen B, Wang X, Dai J. Mammalian target of rapamycin (mTOR) is involved in the neuronal differentiation of neural progenitors induced by insulin. Mol Cell Neurosci. 2008;39:118–124. doi: 10.1016/j.mcn.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Han VK, Lauder JM, D’Ercole AJ. Characterization of somatomedin/insulin-like growth factor receptors and correlation with biologic action in cultured neonatal rat astroglial cells. J Neurosci. 1987;7:501–511. doi: 10.1523/JNEUROSCI.07-02-00501.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han VKM, Smith A, Myint W, Nygard K, Bradshaw S. Mitogenic activity of epidermal growth factor on newborn rat astroglia: interaction with insulin-like growth factors. Endocrinology. 1992;131:1134–1142. doi: 10.1210/endo.131.3.1380434. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122 (Pt 4):593–624. doi: 10.1093/brain/122.4.593. [DOI] [PubMed] [Google Scholar]
- Hatton J, Rapp RP, Kudsk KA, Brown RO, Luer MS, Bukar JG, Chen SA, McClain CJ, Gesundheit N, Dempsey RJ, Young B. Intravenous insulin-like growth factor-I (IGF-I) in moderate-to-severe head injury: a phase II safety and efficacy trial. J Neurosurg. 1997;86:779–786. doi: 10.3171/jns.1997.86.5.0779. [DOI] [PubMed] [Google Scholar]
- Hawkes C, Jhamandas JH, Harris KH, Fu W, MacDonald RG, Kar S. Single transmembrane domain insulin-like growth factor-II/mannose-6-phosphate receptor regulates central cholinergic function by activating a G-protein-sensitive, protein kinase C-dependent pathway. J Neurosci. 2006;26:585–596. doi: 10.1523/JNEUROSCI.2730-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes C, Kar S. Insulin-like growth factor-II/mannose-6-phosphate receptor: widespread distribution in neurons of the central nervous system including those expressing cholinergic phenotype. J Comp Neurol. 2003;458:113–127. doi: 10.1002/cne.10578. [DOI] [PubMed] [Google Scholar]
- Heffron DS, Landreth GE, Samuels IS, Mandell JW. Brain-specific deletion of extracellular signal-regulated kinase 2 mitogen-activated protein kinase leads to aberrant cortical collagen deposition. Am J Pathol. 2009;175:2586–2599. doi: 10.2353/ajpath.2009.090130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidbreder CA, Groenewegen HJ. The medial prefrontal cortex in the rat: evidence for a dorso-ventral distinction based upon functional and anatomical characteristics. Neurosci Biobehav Rev. 2003;27:555–579. doi: 10.1016/j.neubiorev.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Hicks SP, D’Amato CJ. Cell migrations to the isocortex in the rat. Anat Rec. 1968;160:619–634. doi: 10.1002/ar.1091600311. [DOI] [PubMed] [Google Scholar]
- Hodge RD, D’Ercole AJ, O’Kusky JR. Insulin-like growth factor-I accelerates the cell cycle by decreasing G1 phase length and increases cell cycle reentry in the embryonic cerebral cortex. J Neurosci. 2004;24:10201–10210. doi: 10.1523/JNEUROSCI.3246-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, D’Ercole AJ, O’Kusky JR. Insulin-like growth factor-I (IGF-I) inhibits neuronal apoptosis in the developing cerebral cortex in vivo. Int J Dev Neurosci. 2007;25:233–241. doi: 10.1016/j.ijdevneu.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, D’Ercole AJ, O’Kusky JR. Increased expression of insulin-like growth factor-I (IGF-I) during embryonic development produces neocortical overgrowth with differentially greater effects on specific cytoarchitectonic areas and cortical layers. Dev Brain Res. 2005;154:227–237. doi: 10.1016/j.devbrainres.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Hodgkinson SC, Davis SR, Moore LG, Henderson HV, Gluckman PD. Metabolic clearance of insulin-like growth factor-II in sheep. J Endocrinol. 1989;123:461–468. doi: 10.1677/joe.0.1230461. [DOI] [PubMed] [Google Scholar]
- Hoeflich A, Nedbal S, Blum WF, Erhard M, Lahm H, Brem G, Kolb HJ, Wanke R, Wolf E. Growth inhibition in giant growth hormone transgenic mice by overexpression of insulin-like growth factor-binding protein-2. Endocrinology. 2001;142:1889–1898. doi: 10.1210/endo.142.5.8149. [DOI] [PubMed] [Google Scholar]
- Hoeflich A, Wu MY, Mohan S, Fl J, Wanke R, Froehlich T, Arnold GJ, Lahm H, Kolb HJ, Wolf E. Overexpression of insulin-like growth factor-binding protein-2 in transgenic mice reduces postnatal body weight gain. Endocrinology. 1999;140:5488–5496. doi: 10.1210/endo.140.12.7169. [DOI] [PubMed] [Google Scholar]
- Hojvat S, Baker G, Kirsteins L, Lawrence AM. Growth hormone immunoreactivity in the rodent and primate CNS: distribution, characterization and presence post hypophysectomy. Brain Res. 1982;239:543–557. doi: 10.1016/0006-8993(82)90529-7. [DOI] [PubMed] [Google Scholar]
- Holgado-Madruga M, Emlet DR, Moscatello DK, Godwin AK, Wong AJ. A Grb2-associated docking protein in EGF- and insulin-receptor signalling. Nature. 1996;379:560–564. doi: 10.1038/379560a0. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Hamard G, Zaoui R, Leneuve P, Ducos B, Beccavin C, Perin L, Le BY. Experimental IGF-I receptor deficiency generates a sexually dimorphic pattern of organ-specific growth deficits in mice, affecting fat tissue in particular. Endocrinology. 2001;142:4469–4478. doi: 10.1210/endo.142.10.8461. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Leneuve P, Hamard G, Ducos B, Perin L, Binoux M, Le Bouc Y. A targeted partial invalidation of the insulin-like growth factor I receptor gene in mice causes a postnatal growth deficit. Endocrinology. 2000;141:2557–2566. doi: 10.1210/endo.141.7.7550. [DOI] [PubMed] [Google Scholar]
- Hsieh J, Aimone JB, Kaspar BK, Kuwabara T, Nakashima K, Gage FH. IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J Cell Biol. 2004;164:111–122. doi: 10.1083/jcb.200308101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes MA, Brooks PJ, Van Wyk JJ, Lund PK. Insulin-like growth factor II messenger ribonucleic acids are synthesized in the choroid plexus of the rat brain. Mol Endocrinol. 1988;2:47–54. doi: 10.1210/mend-2-1-47. [DOI] [PubMed] [Google Scholar]
- Hynes MA, Van Wyk JJ, Brooks PJ, D’Ercole AJ, Jansen M, Lund PK. Growth hormone dependence of somatomedin-C/insulin-like growth factor-I and insulin-like growth factor-II messenger ribonucleic acids. Mol Endocrinol. 1987;1:233–242. doi: 10.1210/mend-1-3-233. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Waldbillig RJ, Puro DG. Truncation of IGF-I yields two mitogens for retinal Muller glial cells. Brain Res. 1995;686:87–92. doi: 10.1016/0006-8993(95)00473-4. [DOI] [PubMed] [Google Scholar]
- Inagaki K, Tiulpakov A, Rubtsov P, Sverdlova P, Peterkova V, Yakar S, Terekhov S, LeRoith D. A familial insulin-like growth factor-I receptor mutant leads to short stature: clinical and biochemical characterization. J Clin Endocrinol Metab. 2007;92:1542–1548. doi: 10.1210/jc.2006-2354. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Farley NN, Patel K, Kim D, Cowen DS. Interaction of FGF-2 with IGF-1 and BDNF in stimulating Akt, ERK, and neuronal survival in hippocampal cultures. Brain Res. 2007;1154:40–49. doi: 10.1016/j.brainres.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Farley NN, Travkina T, Cowen DS. Cumulative activation of akt and consequent inhibition of glycogen synthase kinase-3 by brain-derived neurotrophic factor and insulin-like growth factor-1 in cultured hippocampal neurons. J Pharmacol Exp Ther. 2006;316:1062–1069. doi: 10.1124/jpet.105.094433. [DOI] [PubMed] [Google Scholar]
- Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- Jousan FD, Hansen PJ. Insulin-like growth factor-I promotes resistance of bovine preimplantation embryos to heat shock through actions independent of its anti-apoptotic actions requiring PI3K signaling. Mol Reprod Dev. 2007;74:189–196. doi: 10.1002/mrd.20527. [DOI] [PubMed] [Google Scholar]
- Jousan FD, Oliveira LJ, Hansen PJ. Short-term culture of in vitro produced bovine preimplantation embryos with insulin-like growth factor-i prevents heat shock-induced apoptosis through activation of the phosphatidylinositol 3-kinase/Akt pathway. Mol Reprod Dev. 2008;75:681–688. doi: 10.1002/mrd.20830. [DOI] [PubMed] [Google Scholar]
- Jurata LW, Gallagher P, Lemire AL, Charles V, Brockman JA, Illingworth EL, Altar CA. Altered expression of hippocampal dentate granule neuron genes in a mouse model of human 22q11 deletion syndrome. Schizophr Res. 2006;88:251–259. doi: 10.1016/j.schres.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Kappeler L, De Magalhaes FC, Dupont J, Leneuve P, Cervera P, Perin L, Loudes C, Blaise A, Klein R, Epelbaum J, Le BY, Holzenberger M. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008;6:e254. doi: 10.1371/journal.pbio.0060254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S, Chabot JG, Quirion R. Quantitative autoradiographic localization of [125I]insulin-like growth factor I, [125I]insulin-like growth factor II, and [125I]insulin receptor binding sites in developing and adult rat brain. J Comp Neurol. 1993;333:375–397. doi: 10.1002/cne.903330306. [DOI] [PubMed] [Google Scholar]
- Kawashima Y, Kanzaki S, Yang F, Kinoshita T, Hanaki K, Nagaishi J, Ohtsuka Y, Hisatome I, Ninomoya H, Nanba E, Fukushima T, Takahashi S. Mutation at cleavage site of insulin-like growth factor receptor in a short-stature child born with intrauterine growth retardation. J Clin Endocrinol Metab. 2005;90:4679–4687. doi: 10.1210/jc.2004-1947. [DOI] [PubMed] [Google Scholar]
- Kelsch W, Sim S, Lois C. Watching synaptogenesis in the adult brain. Annu Rev Neurosci. 2010;33:131–149. doi: 10.1146/annurev-neuro-060909-153252. [DOI] [PubMed] [Google Scholar]
- Keummerle JF, Murthy KS. Coupling of the insulin-like growth factor-I receptor tyrosine kinase to G12 in human intestinal smooth muscle – Gbgamma-Dependent mitogen-activated protein kinase activation and growth. J Biol Chem. 2001;276:7187–7194. doi: 10.1074/jbc.M011145200. [DOI] [PubMed] [Google Scholar]
- Komoly S, Hudson LD, Webster HD, Bondy CA. Insulin-like growth factor I gene expression is induced in astrocytes during experimental demyelination. Proc Natl Acad Sci USA. 1992;89:1894–1898. doi: 10.1073/pnas.89.5.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Takahashi K, Chui DH, Rosenfeld RG, Himeno M, Tabira T. Insulin-like growth factor II promotes in vitro cholinergic development of mouse septal neurons: comparison with the effects of insulin-like growth factor I. Brain Res. 1994;649:53–61. doi: 10.1016/0006-8993(94)91048-0. [DOI] [PubMed] [Google Scholar]
- Kouroupi G, Lavdas AA, Gaitanou M, Thomaidou D, Stylianopoulou F, Matsas R. Lentivirus-mediated expression of insulin-like growth factor-I promotes neural stem/precursor cell proliferation and enhances their potential to generate neurons. J Neurochem. 2010;115:460–474. doi: 10.1111/j.1471-4159.2010.06939.x. [DOI] [PubMed] [Google Scholar]
- Kruis T, Klammt J, Galli-Tsinopoulou A, Wallborn T, Schlicke M, Muller E, Kratzsch J, Korner A, Odeh R, Kiess W, Pfaffle R. Heterozygous mutation within a kinase-conserved motif of the insulin-like growth factor I receptor causes intrauterine and postnatal growth retardation. J Clin Endocrinol Metab. 2010;95:1137–1142. doi: 10.1210/jc.2009-1433. [DOI] [PubMed] [Google Scholar]
- Kulik G, Weber MJ. Akt-dependent and -independent survival signaling pathways utilized by insulin-like growth factor I. Mol Cell Biol. 1998;18:6711–6718. doi: 10.1128/mcb.18.11.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle UG, Pichard C. The Dutch Famine of 1944–1945: a pathophysiological model of long-term consequences of wasting disease. Curr Opin Clin Nutr Metab Care. 2006;9:388–394. doi: 10.1097/01.mco.0000232898.74415.42. [DOI] [PubMed] [Google Scholar]
- Lee WH, Wang GM, Seaman LB, Vannucci SJ. Coordinate IGF-I and IGFBP5 gene expression in perinatal rat brain after hypoxia–ischemia. J Cereb Blood Flow Metab. 1996;16:227–236. doi: 10.1097/00004647-199603000-00007. [DOI] [PubMed] [Google Scholar]
- Lee WH, Clemens JA, Bondy CA. Insulin-like growth factors in the response to cerebral ischemia. Mol Cell Neurosci. 1992;3:36–43. doi: 10.1016/1044-7431(92)90006-n. [DOI] [PubMed] [Google Scholar]
- Lee WH, Michels KM, Bondy CA. Localization of insulin-like growth factor binding protein-2 messenger RNA during postnatal brain development: correlation with insulin-like growth factors I and II. Neuroscience. 1993;53:251–265. doi: 10.1016/0306-4522(93)90303-w. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, D’Ercole AJ, Wong ET, LaMantia AS, Walsh CA. The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron. 2011;69:893–905. doi: 10.1016/j.neuron.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembo G, Rockman HA, Hunter JJ, Steinmetz H, Koch WJ, Ma L, Prinz MP, Ross J, Jr, Chien KR, Powell-Braxton L. Elevated blood pressure and enhanced myocardial contractility in mice with severe IGF-1 deficiency. J Clin Invest. 1996;98:2648–2655. doi: 10.1172/JCI119086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoith D, Werner H, Beitner-Johnson D, Roberts CT., Jr Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- Li XS, Williams M, Bartlett WP. Induction of IGF-1 mRNA expression following traumatic injury to the postnatal brain. Brain Res Mol Brain Res. 1998;57:92–96. doi: 10.1016/s0169-328x(98)00075-8. [DOI] [PubMed] [Google Scholar]
- Lima PF, Oliveira MA, Santos MH, Reichenbach HD, Weppert M, Paula-Lopes FF, Neto CC, Goncalves PB. Effect of retinoids and growth factor on in vitro bovine embryos produced under chemically defined conditions. Anim Reprod Sci. 2006;95:184–192. doi: 10.1016/j.anireprosci.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Lin S, Fan LW, Pang Y, Rhodes PG, Mitchell HJ, Cai Z. IGF-1 protects oligodendrocyte progenitor cells and improves neurological functions following cerebral hypoxia–ischemia in the neonatal rat. Brain Res. 2005;1063:15–26. doi: 10.1016/j.brainres.2005.09.042. [DOI] [PubMed] [Google Scholar]
- Lin S, Fan LW, Rhodes PG, Cai Z. Intranasal administration of IGF-1 attenuates hypoxic–ischemic brain injury in neonatal rats. Exp Neurol. 2009;217:361–370. doi: 10.1016/j.expneurol.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- Liu W, D’Ercole AJ, Ye P. Blunting type 1 Insulin-like growth factor receptor expression exacerbates neuronal apoptosis following hypoxic/ischemic injury. BMC Neurosci. 2011;12:64. doi: 10.1186/1471-2202-12-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Ye P, O’Kusky JR, D’Ercole AJ. Type 1 insulin-like growth factor receptor signaling is essential for the development of the hippocampal formation and dentate gyrus. J Neurosci Res. 2009;87:2821–2832. doi: 10.1002/jnr.22129. [DOI] [PubMed] [Google Scholar]
- Liu X, Yao DL, Bondy CA, Brenner M, Hudson LD, Zhou J, Webster HD. Astrocytes express insulin-like growth factor-I (IGF-I) and its binding protein, IGFBP-2, during demyelination induced by experimental autoimmune encephalomyelitis. Mol Cell Neurosci. 1994;5:418–430. doi: 10.1006/mcne.1994.1052. [DOI] [PubMed] [Google Scholar]
- Logan A, Gonzalez AM, Hill DJ, Berry M, Gregson NA, Baird A. Coordinated pattern of expression and localization of insulin-like growth factor-II (IGF-II) and IGF-binding protein-2 in the adult rat brain. Endocrinology. 1994;135:2255–2264. doi: 10.1210/endo.135.5.7525264. [DOI] [PubMed] [Google Scholar]
- Louvi A, Accili D, Efstratiadis A. Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev Biol. 1997;189:33–48. doi: 10.1006/dbio.1997.8666. [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu ZM, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- Mairet-Coello G, Tury A, DiCicco-Bloom E. Insulin-like growth factor-1 promotes G(1)/S cell cycle progression through bidirectional regulation of cyclins and cyclin-dependent kinase inhibitors via the phosphatidylinositol 3-kinase/Akt pathway in developing rat cerebral cortex. J Neurosci. 2009;29:775–788. doi: 10.1523/JNEUROSCI.1700-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mares V, Lodin Z. The cellular kinetics of the developing mouse cerebellum. II The function of the external granular layer in the process of gyrification. Brain Res. 1970;23:343–352. doi: 10.1016/0006-8993(70)90061-2. [DOI] [PubMed] [Google Scholar]
- Mashayekhi F, Mirzajani E, Naji M, Azari M. Expression of insulin-like growth factor-1 and insulin-like growth factor binding proteins in the serum and cerebrospinal fluid of patients with Parkinson’s disease. J Clin Neurosci. 2010;17:623–627. doi: 10.1016/j.jocn.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Mason JL, Goldman JE. A2B5+ and O4+ cycling progenitors in the adult forebrain white matter respond differentially to PDGF-AA, FGF-2, and IGF-1. Mol Cell Neurosci. 2002;20:30–42. doi: 10.1006/mcne.2002.1114. [DOI] [PubMed] [Google Scholar]
- Mason JL, Ye P, Suzuki K, D’Ercole AJ, Matsushima GK. Insulin-like growth factor-1 inhibits mature oligodendrocyte apoptosis during primary demyelination. J Neurosci. 2000;20:5703–5708. doi: 10.1523/JNEUROSCI.20-15-05703.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters BA, Werner H, Roberts CT, Jr, LeRoith D, Raizada MK. Insulin-like growth factor I (IGF-I) receptors and IGF-I action in oligodendrocytes from rat brains. Regul Pept. 1991a;33:117–131. doi: 10.1016/0167-0115(91)90207-w. [DOI] [PubMed] [Google Scholar]
- Masters BA, Werner H, Roberts CT, Jr, LeRoith D, Raizada MK. Developmental regulation of insulin-like growth factor-I-stimulated glucose transporter in rat brain astrocytes. Endocrinology. 1991b;128:2548–2557. doi: 10.1210/endo-128-5-2548. [DOI] [PubMed] [Google Scholar]
- Mastroiacovo F, Busceti CL, Biagioni F, Moyanova SG, Meisler MH, Battaglia G, Caricasole A, Bruno V, Nicoletti F. Induction of the Wnt antagonist, Dickkopf-1, contributes to the development of neuronal death in models of brain focal ischemia. J Cereb Blood Flow Metab. 2009;29:264–276. doi: 10.1038/jcbfm.2008.111. [DOI] [PubMed] [Google Scholar]
- Mathews LS, Hammer RE, Brinster RL, Palmiter RD. Expression of insulin-like growth factor I in transgenic mice with elevated levels of growth hormone is correlated with growth. Endocrinology. 1988;123:433–437. doi: 10.1210/endo-123-1-433. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Murata Y, Kawamura N, Hayashi M, Tamada K, Takuma K, Maeda S, Baba A. Selective induction of alpha 1 isoform of (Na++ K+)-ATPase by insulin/insulin-like growth factor-I in cultured rat astrocytes. Arch Biochem Biophys. 1993;307:175–182. doi: 10.1006/abbi.1993.1576. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouyssegur J, Brambilla R. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- McKinnon T, Chakraborty C, Gleeson LM, Chidiac P, Lala PK. Stimulation of human extravillous trophoblast migration by IGF-II is mediated by IGF type 2 receptor involving inhibitory G protein(s) and phosphorylation of MAPK. J Clin Endocrinol Metab. 2001;86:3665–3674. doi: 10.1210/jcem.86.8.7711. [DOI] [PubMed] [Google Scholar]
- McMorris FA, Smith TM, DeSalvo S, Furlanetto RW. Insulin-like growth factor I/somatomedin C: a potent inducer of oligodendrocyte development. Proc Natl Acad Sci USA. 1986;83:822–826. doi: 10.1073/pnas.83.3.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miale IL, Sidman RL. An autoradiographic analysis of histogenesis in the mouse cerebellum. Exp Neurol. 1961;4:277–296. doi: 10.1016/0014-4886(61)90055-3. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall C, Arkins S, Dantzer R, Freund GG, Kelley KW. Phosphatidylinositol 3′-kinase, but not S6-kinase, is required for insulin-like growth factor-I and IL-4 to maintain expression of Bcl-2 and promote survival of myeloid progenitors. J Immunol. 1999;162:4542–4549. [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28:53–67. doi: 10.1016/s0896-6273(00)00085-4. [DOI] [PubMed] [Google Scholar]
- Modric T, Silha JV, Shi Z, Gui Y, Suwanichkul A, Durham SK, Powell DR, Murphy LJ. Phenotypic manifestations of insulin-like growth factor-binding protein-3 overexpression in transgenic mice. Endocrinology. 2001;142:1958–1967. doi: 10.1210/endo.142.5.8165. [DOI] [PubMed] [Google Scholar]
- Morales AV, Serna J, Alarcon C, de la Rosa EJ, de PF. Role of prepancreatic (pro)insulin and the insulin receptor in prevention of embryonic apoptosis. Endocrinology. 1997;138:3967–3975. doi: 10.1210/endo.138.9.5396. [DOI] [PubMed] [Google Scholar]
- Morrione A, Valentinis B, Xu SQ, Yumet G, Louvi A, Efstratiadis A, Baserga R. Insulin-like growth factor II stimulates cell proliferation through the insulin receptor. Proc Natl Acad Sci USA. 1997;94:3777–3782. doi: 10.1073/pnas.94.8.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss JI, Pontes E, Hansen PJ. Insulin-like growth factor-1 protects preimplantation embryos from anti-developmental actions of menadione. Arch Toxicol. 2009;83:1001–1007. doi: 10.1007/s00204-009-0458-3. [DOI] [PubMed] [Google Scholar]
- Mozell RL, McMorris FA. Insulin-like growth factor I stimulates oligodendrocyte development and myelination in rat brain aggregate cultures. J Neurosci Res. 1991;30:382–390. doi: 10.1002/jnr.490300214. [DOI] [PubMed] [Google Scholar]
- Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29:6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness JK, Scaduto RC, Jr, Wood TL. IGF-I prevents glutamate-mediated bax translocation and cytochrome C release in O4+ oligodendrocyte progenitors. Glia. 2004;46:183–194. doi: 10.1002/glia.10360. [DOI] [PubMed] [Google Scholar]
- Ness JK, Wood TL. Insulin-like growth factor I, but not neurotrophin-3, sustains Akt activation and provides long-term protection of immature oligodendrocytes from glutamate-mediated apoptosis. Mol Cell Neurosci. 2002;20:476–488. doi: 10.1006/mcne.2002.1149. [DOI] [PubMed] [Google Scholar]
- Netchine I, Azzi S, Houang M, Seurin D, Perin L, Ricort JM, Daubas C, Legay C, Mester J, Herich R, Godeau F, Le BY. Partial primary deficiency of insulin-like growth factor (IGF)-I activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J Clin Endocrinol Metab. 2009;94:3913–3921. doi: 10.1210/jc.2009-0452. [DOI] [PubMed] [Google Scholar]
- Ni W, Rajkumar K, Nagy JI, Murphy LJ. Impaired brain development and reduced astrocyte response to injury in transgenic mice expressing IGF binding protein-1. Brain Res. 1997;769:97–107. doi: 10.1016/s0006-8993(97)00676-8. [DOI] [PubMed] [Google Scholar]
- Nicholas RS, Stevens S, Wing MG, Compston DA. Microglia-derived IGF-2 prevents TNFalpha induced death of mature oligodendrocytes in vitro. J Neuroimmunol. 2002;124:36–44. doi: 10.1016/s0165-5728(02)00011-5. [DOI] [PubMed] [Google Scholar]
- Nieto-Bona MP, Garcia-Segura LM, Torres-Aleman I. Transynaptic modulation by insulin-like growth factor I of dendritic spines in Purkinje cells. Int J Dev Neurosci. 1997;15:749–754. doi: 10.1016/s0736-5748(97)00021-x. [DOI] [PubMed] [Google Scholar]
- Numan S, Russell DS. Discrete expression of insulin receptor substrate-4 mRNA in adult rat brain. Brain Res Mol Brain Res. 1999;72:97–102. doi: 10.1016/s0169-328x(99)00160-6. [DOI] [PubMed] [Google Scholar]
- O’Donnell SL, Frederick TJ, Krady JK, Vannucci SJ, Wood TL. IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia. 2002;39:85–97. doi: 10.1002/glia.10081. [DOI] [PubMed] [Google Scholar]
- O’Kusky JR, Ye P, D’Ercole AJ. Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J Neurosci. 2000;20:8435–8442. doi: 10.1523/JNEUROSCI.20-22-08435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Kusky JR, Ye P, D’Ercole AJ. Increased expression of insulin-like growth factor I augments the progressive phase of synaptogenesis without preventing synapse elimination in the hypoglossal nucleus. J Comp Neurol. 2003;464:382–391. doi: 10.1002/cne.10794. [DOI] [PubMed] [Google Scholar]
- Ocrant I, Fay CT, Parmelee JT. Characterization of insulin-like growth factor binding proteins produced in the rat central nervous system. Endocrinology. 1990;127:1260–1267. doi: 10.1210/endo-127-3-1260. [DOI] [PubMed] [Google Scholar]
- Okubo Y, Siddle K, Firth H, O’Rahilly S, Wilson LC, Willatt L, Fukushima T, Takahashi S, Petry CJ, Saukkonen T, Stanhope R, Dunger DB. Cell proliferation activities on skin fibroblasts from a short child with absence of one copy of the type 1 insulin-like growth factor receptor (IGF1R) gene and a tall child with three copies of the IGF1R gene. J Clin Endocrinol Metab. 2003;88:5981–5988. doi: 10.1210/jc.2002-021080. [DOI] [PubMed] [Google Scholar]
- Onuma TA, Ding Y, Abraham E, Zohar Y, Ando H, Duan C. Regulation of temporal and spatial organization of newborn GnRH neurons by IGF signaling in zebrafish. J Neurosci. 2011;31:11814–11824. doi: 10.1523/JNEUROSCI.6804-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. The absence of significant postnatal motoneuron death in the brachial and lumbar spinal cord of the rat. J Comp Neurol. 1986;246:281–286. doi: 10.1002/cne.902460211. [DOI] [PubMed] [Google Scholar]
- Pera EM, Ikeda A, Eivers E, De Robertis EM. Integration of IGF, FGF, and anti-BMP signals via Smad1 phosphorylation in neural induction. Genes Dev. 2003;17:3023–3028. doi: 10.1101/gad.1153603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pera EM, Wessely O, Li SY, De Robertis EM. Neural and head induction by insulin-like growth factor signals. Dev Cell. 2001;1:655–665. doi: 10.1016/s1534-5807(01)00069-7. [DOI] [PubMed] [Google Scholar]
- Perez-Martin M, Cifuentes M, Grondona JM, Bermudez-Silva FJ, Arrabal PM, Perez-Figares JM, Jimenez AJ, Garcia-Segura LM, Fernandez-Llebrez P. Neurogenesis in explants from the walls of the lateral ventricle of adult bovine brain: role of endogenous IGF-1 as a survival factor. Eur J Neurosci. 2003;17:205–211. doi: 10.1046/j.1460-9568.2003.02432.x. [DOI] [PubMed] [Google Scholar]
- Perez-Villamil B, de la Rosa EJ, Morales AV, de PF. Developmentally regulated expression of the preproinsulin gene in the chicken embryo during gastrulation and neurulation. Endocrinology. 1994;135:2342–2350. doi: 10.1210/endo.135.6.7988416. [DOI] [PubMed] [Google Scholar]
- Pete G, Fuller CR, Oldham JM, Smith DR, D’Ercole AJ, Kahn CR, Lund PK. Postnatal growth responses to insulin-like growth factor I in insulin receptor substrate-1-deficient mice. Endocrinology. 1999;140:5478–5487. doi: 10.1210/endo.140.12.7219. [DOI] [PubMed] [Google Scholar]
- Pierce ET. Time of origin of neurons in the brain stem of the mouse. Prog Brain Res. 1973;40:53–65. doi: 10.1016/S0079-6123(08)60679-2. [DOI] [PubMed] [Google Scholar]
- Popken GJ, Dechert-Zeger M, Ye P, D’Ercole AJ. Brain development. Adv Exp Med Biol. 2005;567:187–220. doi: 10.1007/0-387-26274-1_8. [DOI] [PubMed] [Google Scholar]
- Popken GJ, Hodge RD, Ye P, Zhang J, Ng W, O’Kusky JR, D’Ercole AJ. In vivo effects of insulin-like growth factor-I (IGF-I) on prenatal and early postnatal development of the central nervous system. Eur J Neurosci. 2004;19:2056–2068. doi: 10.1111/j.0953-816X.2004.03320.x. [DOI] [PubMed] [Google Scholar]
- Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N, Stewart TA. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993;7:2609–2617. doi: 10.1101/gad.7.12b.2609. [DOI] [PubMed] [Google Scholar]
- Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, Wayland M, Freeman T, Dudbridge F, Lilley KS, Karp NA, Hester S, Tkachev D, Mimmack ML, Yolken RH, Webster MJ, Torrey EF, Bahn S. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 2004;9 (643):684–697. doi: 10.1038/sj.mp.4001511. [DOI] [PubMed] [Google Scholar]
- Pulford BE, Ishii DN. Uptake of circulating insulin-like growth factors (IGFs) into cerebrospinal fluid appears to be independent of the IGF receptors as well as IGF-binding proteins. Endocrinology. 2001;142:213–220. doi: 10.1210/endo.142.1.7894. [DOI] [PubMed] [Google Scholar]
- Quesada A, Romeo HE, Micevych P. Distribution and localization patterns of estrogen receptor-beta and insulin-like growth factor-1 receptors in neurons and glial cells of the female rat substantia nigra: localization of ERbeta and IGF-1R in substantia nigra. J Comp Neurol. 2007;503:198–208. doi: 10.1002/cne.21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroga S, Garofalo RS, Pfenninger KH. Insulin-like growth factor I receptors of fetal brain are enriched in nerve growth cones and contain a beta-subunit variant. Proc Natl Acad Sci USA. 1995;92:4309–4312. doi: 10.1073/pnas.92.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raile K, Klammt J, Schneider A, Keller A, Laue S, Smith R, Pfaffle R, Kratzsch J, Keller E, Kiess W. Clinical and functional characteristics of the human Arg59Ter insulin-like growth factor i receptor (IGF1R) mutation: implications for a gene dosage effect of the human IGF1R. J Clin Endocrinol Metab. 2006;91:2264–2271. doi: 10.1210/jc.2005-2146. [DOI] [PubMed] [Google Scholar]
- Rajkumar K, Barron D, Lewitt MS, Murphy LJ. Growth retardation and hyperglycemia in insulin-like growth factor binding protein-1 transgenic mice. Endocrinology. 1995;136:4029–4034. doi: 10.1210/endo.136.9.7544274. [DOI] [PubMed] [Google Scholar]
- Ramin N, Thieme R, Fischer S, Schindler M, Schmidt T, Fischer B, Navarrete SA. Maternal diabetes impairs gastrulation and insulin and IGF-I receptor expression in rabbit blastocysts. Endocrinology. 2010;151:4158–4167. doi: 10.1210/en.2010-0187. [DOI] [PubMed] [Google Scholar]
- Rechler MM. Insulin-like growth factor binding proteins. Vitam Horm. 1993;47:1–114. doi: 10.1016/s0083-6729(08)60444-6. [DOI] [PubMed] [Google Scholar]
- Reijnders CM, Koster JG, Buul-Offers SC. Overexpression of human IGF-II mRNA in the brain of transgenic mice modulates IGFBP-2 gene expression in the medulla oblongata. J Endocrinol. 2004;182:445–455. doi: 10.1677/joe.0.1820445. [DOI] [PubMed] [Google Scholar]
- Reinhardt RR, Bondy CA. Insulin-like growth factors cross the blood-brain barrier. Endocrinology. 1994;135:1753–1761. doi: 10.1210/endo.135.5.7525251. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Beasley CL. GABAergic neuronal subtypes in the human frontal cortex-development and deficits in schizophrenia. J Chem Neuroanat. 2001;22:95–100. doi: 10.1016/s0891-0618(01)00113-2. [DOI] [PubMed] [Google Scholar]
- Richard-Parpaillon L, Heligon C, Chesnel F, Boujard D, Philpott A. The IGF pathway regulates head formation by inhibiting Wnt signaling in Xenopus. Dev Biol. 2002;244:407–417. doi: 10.1006/dbio.2002.0605. [DOI] [PubMed] [Google Scholar]
- Romanelli RJ, LeBeau AP, Fulmer CG, Lazzarino DA, Hochberg A, Wood TL. Insulin-like growth factor type-I receptor internalization and recycling mediate the sustained phosphorylation of Akt. J Biol Chem. 2007;282:22513–22524. doi: 10.1074/jbc.M704309200. [DOI] [PubMed] [Google Scholar]
- Rotwein P. Structure, evolution, expression and regulation of insulin-like growth factors I and II. Growth Factors. 1991;5:3–18. doi: 10.3109/08977199109000267. [DOI] [PubMed] [Google Scholar]
- Russell JW, Windebank AJ, Schenone A, Feldman EL. Insulin-like growth factor-I prevents apoptosis in neurons after nerve growth factor withdrawal. J Neurobiol. 1998;36:455–467. doi: 10.1002/(sici)1097-4695(19980915)36:4<455::aid-neu1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Russo VC, Werther GA. Des (1–3) IGF-I potently enhances differentiated cell growth in olfactory bulb organ culture. Growth Factors. 1994;11:301–311. doi: 10.3109/08977199409011003. [DOI] [PubMed] [Google Scholar]
- Ryves WJ, Harwood AJ. The interaction of glycogen synthase kinase-3 (GSK-3) with the cell cycle. Prog Cell Cycle Res. 2003;5:489–495. [PubMed] [Google Scholar]
- Salih DA, Tripathi G, Holding C, Szestak TA, Gonzalez MI, Carter EJ, Cobb LJ, Eisemann JE, Pell JM. Insulin-like growth factor-binding protein 5 (Igfbp5) compromises survival, growth, muscle development, and fertility in mice. Proc Natl Acad Sci USA. 2004;101:4314–4319. doi: 10.1073/pnas.0400230101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara VR, Carlsson-Skwirut C, Andersson C, Hall E, Sjogren B, Holmgren A, Jornvall H. Characterization of somatomedins from human fetal brain: identification of a variant form of insulin-like growth factor I. Proc Natl Acad Sci USA. 1986;83:4904–4907. doi: 10.1073/pnas.83.13.4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J, Kim SU. Proliferation and differentiation of fetal human oligodendrocytes in culture. J Neurosci Res. 1994;39:260–272. doi: 10.1002/jnr.490390304. [DOI] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S, Takishima K. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Kobayashi Y, Takeuchi A, Pages G, Pouyssegur J, Kazama T. Deletion of ERK1 and ERK2 in the CNS causes cortical abnormalities and neonatal lethality: Erk1 deficiency enhances the impairment of neurogenesis in Erk2-deficient mice. J Neurosci. 2011;31:1149–1155. doi: 10.1523/JNEUROSCI.2243-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura J, Curatolo L, Williams CE, Gatti S, Benatti L, Peeters C, Guan J, Dragunow M, Post C, Faull RL, Gluckman PD, Skinner SJ. Neuroprotective effects of Gly-Pro-Glu, the N-terminal tripeptide of IGF-1, in the hippocampus in vitro. Neuroreport. 1999;10:161–164. doi: 10.1097/00001756-199901180-00031. [DOI] [PubMed] [Google Scholar]
- Scavo L, Shuldiner AR, Serrano J, Dashner R, Roth J, de PF. Genes encoding receptors for insulin and insulin-like growth factor I are expressed in Xenopus oocytes and embryos. Proc Natl Acad Sci USA. 1991a;88:6214–6218. doi: 10.1073/pnas.88.14.6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scavo LM, Serrano J, Roth J, de PF. Genes for the insulin receptor and the insulin-like growth factor I receptor are expressed in the chicken embryo blastoderm and throughout organogenesis. Biochem Biophys Res Commun. 1991b;176:1393–1401. doi: 10.1016/0006-291x(91)90441-9. [DOI] [PubMed] [Google Scholar]
- Schubert M, Brazil DP, Burks DJ, Kushner JA, Ye J, Flint CL, Farhang-Fallah J, Dikkes P, Warot XM, Rio C, Corfas G, White MF. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci. 2003;23:7084–7092. doi: 10.1523/JNEUROSCI.23-18-07084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacchitano S, Taylor SI. Cloning, tissue expression, and chromosomal localization of the mouse IRS-3 gene. Endocrinology. 1997;138:4931–4940. doi: 10.1210/endo.138.11.5518. [DOI] [PubMed] [Google Scholar]
- Sehat B, Tofigh A, Lin Y, Trocme E, Liljedahl U, Lagergren J, Larsson O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci Signal. 2010;3:ra10. doi: 10.1126/scisignal.2000628. [DOI] [PubMed] [Google Scholar]
- Shapira S, Mathai S, Zhang R, Guan J. Delayed peripheral administration of the N-terminal tripeptide of IGF-1 (GPE) reduces brain damage following microsphere induced embolic damage in young adult and aged rats. Neurosci Lett. 2009;454:53–57. doi: 10.1016/j.neulet.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Shinar Y, McMorris FA. Developing oligodendroglia express mRNA for insulin-like growth factor-I, a regulator of oligodendrocyte development. J Neurosci Res. 1995;42:516–527. doi: 10.1002/jnr.490420410. [DOI] [PubMed] [Google Scholar]
- Silha JV, Gui Y, Mishra S, Leckstrom A, Cohen P, Murphy LJ. Overexpression of gly56/gly80/gly81-mutant insulin-like growth factor-binding protein-3 in transgenic mice. Endocrinology. 2005;146:1523–1531. doi: 10.1210/en.2004-0905. [DOI] [PubMed] [Google Scholar]
- Sinor AD, Lillien L. Akt-1 expression level regulates CNS precursors. J Neurosci. 2004;24:8531–8541. doi: 10.1523/JNEUROSCI.1470-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirisathien S, Hernandez-Fonseca HJ, Brackett BG. Influences of epidermal growth factor and insulin-like growth factor-I on bovine blastocyst development in vitro. Anim Reprod Sci. 2003;77:21–32. doi: 10.1016/s0378-4320(02)00272-5. [DOI] [PubMed] [Google Scholar]
- Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Tornell J, Isaksson OG, Jansson JO, Ohlsson C. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci USA. 1999;96:7088–7092. doi: 10.1073/pnas.96.12.7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smink JJ, Koster JG, Hendriks-Stegeman BI, Van Buul-Offers SC. Insulin-like growth factor (IGF) II induced changes in expression of IGF binding proteins in lymphoid tissues of hIGF-II transgenic mice. Endocrinology. 1999;140:5876–5882. doi: 10.1210/endo.140.12.7174. [DOI] [PubMed] [Google Scholar]
- Spreafico R, Frassoni C, Arcelli P, Selvaggio M, De BS. In situ labeling of apoptotic cell death in the cerebral cortex and thalamus of rats during development. J Comp Neurol. 1995;363:281–295. doi: 10.1002/cne.903630209. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia and macrophages in the developing CNS. Neurotoxicology. 2001;22:619–624. doi: 10.1016/s0161-813x(01)00033-x. [DOI] [PubMed] [Google Scholar]
- Stylianopoulou F, Efstratiadis A, Herbert J, Pintar J. Pattern of the insulin-like growth factor II gene expression during rat embryogenesis. Development. 1988;103:497–506. doi: 10.1242/dev.103.3.497. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Shahani N, Strelau J, Laliberte C, Brandt R, Kaplan D, Unsicker K. Insulin-like growth factor 1 inhibits extracellular signal-regulated kinase to promote neuronal survival via the phosphatidylinositol 3-kinase/protein kinase A/c-Raf pathway. J Neurosci. 2005;25:2838–2852. doi: 10.1523/JNEUROSCI.5060-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Zirrgiebel U, von Bohlen Und HO, Strelau J, Laliberte C, Kaplan DR, Unsicker K. ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J Cell Biol. 2004;165:357–369. doi: 10.1083/jcb.200403028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui L, Wang J, Li BM. Role of the phosphoinositide 3-kinase-Akt-mammalian target of the rapamycin signaling pathway in long-term potentiation and trace fear conditioning memory in rat medial prefrontal cortex. Learn Mem. 2008;15:762–776. doi: 10.1101/lm.1067808. [DOI] [PubMed] [Google Scholar]
- Sun LY, D’Ercole AJ. Insulin-like growth factor-I stimulates histone H3 and H4 acetylation in the brain in vivo. Endocrinology. 2006;147:5480–5490. doi: 10.1210/en.2006-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Ikegaya Y, Matsuura S, Kanai Y, Endou H, Matsuki N. Transient upregulation of the glial glutamate transporter GLAST in response to fibroblast growth factor, insulin-like growth factor and epidermal growth factor in cultured astrocytes. J Cell Sci. 2001;114:3717–3725. doi: 10.1242/jcs.114.20.3717. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317:369–372. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- Takadera T, Matsuda I, Ohyashiki T. Apoptotic cell death and caspase-3 activation induced by N-methyl-D-aspartate receptor antagonists and their prevention by insulin-like growth factor I. J Neurochem. 1999;73:548–556. doi: 10.1046/j.1471-4159.1999.0730548.x. [DOI] [PubMed] [Google Scholar]
- Takahashi-Yanaga F, Sasaguri T. GSK-3beta regulates cyclin D1 expression: a new target for chemotherapy. Cell Signal. 2008;20:581–589. doi: 10.1016/j.cellsig.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Tarsa L, Goda Y. Synaptophysin regulates activity-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci USA. 2002;99:1012–1016. doi: 10.1073/pnas.022575999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Aleman I, Naftolin F, Robbins RJ. Trophic effects of basic fibroblast growth factor on fetal rat hypothalamic cells: Interactions with insulin-like growth factor I. Dev Brain Res. 1990a;52:253–257. doi: 10.1016/0165-3806(90)90242-q. [DOI] [PubMed] [Google Scholar]
- Torres-Aleman I, Naftolin F, Robbins RJ. Trophic effects of insulin-like growth factor-I on fetal rat hypothalamic cells in culture. Neuroscience. 1990b;35:601–608. doi: 10.1016/0306-4522(90)90332-x. [DOI] [PubMed] [Google Scholar]
- Tran PV, Fretham SJ, Wobken J, Miller BS, Georgieff MK. Gestationalneonatal iron deficiency suppresses and iron treatment reactivates IGF signaling in developing rat hippocampus. Am J Physiol Endocrinol Metab. 2012;302:E316–E324. doi: 10.1152/ajpendo.00369.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo JL, Llorens-Martin MV, Torres-Aleman I. The effects of exercise on spatial learning and anxiety-like behavior are mediated by an IGF-I-dependent mechanism related to hippocampal neurogenesis. Mol Cell Neurosci. 2008;37:402–411. doi: 10.1016/j.mcn.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Tsuang M. Schizophrenia: genes and environment. Biol Psychiatry. 2000;47:210–220. doi: 10.1016/s0006-3223(99)00289-9. [DOI] [PubMed] [Google Scholar]
- Valtorta F, Pennuto M, Bonanomi D, Benfenati F. Synaptophysin: leading actor or walk-on role in synaptic vesicle exocytosis? Bioessays. 2004;26:445–453. doi: 10.1002/bies.20012. [DOI] [PubMed] [Google Scholar]
- Van Buul-Offers SC, de Haan K, Reijnen-Gresnigt MG, Meinsma D, Jansen M, Oei SL, Bonte EJ, Sussenbach JS, Van den Brande JL. Overexpression of human insulin-like growth factor-II in transgenic mice causes increased growth of the thymus. J Endocrinol. 1995;144:491–502. doi: 10.1677/joe.0.1440491. [DOI] [PubMed] [Google Scholar]
- van Duyvenvoorde HA, van Setten PA, Walenkamp MJ, van DJ, Koenig J, Gauguin L, Oostdijk W, Ruivenkamp CA, Losekoot M, Wade JD, De MP, Karperien M, Noordam C, Wit JM. Short stature associated with a novel heterozygous mutation in the insulin-like growth factor 1 gene. J Clin Endocrinol Metab. 2010;95:E363–E367. doi: 10.1210/jc.2010-0511. [DOI] [PubMed] [Google Scholar]
- van Haren NE, Hulshoff Pol HE, Schnack HG, Cahn W, Brans R, Carati I, Rais M, Kahn RS. Progressive brain volume loss in schizophrenia over the course of the illness: evidence of maturational abnormalities in early adulthood. Biol Psychiatry. 2008;63:106–113. doi: 10.1016/j.biopsych.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Vannucci RC. Experimental biology of cerebral hypoxia–ischemia: relation to perinatal brain damage. Pediatr Res. 1990;27:317–326. doi: 10.1203/00006450-199004000-00001. [DOI] [PubMed] [Google Scholar]
- Vemuri GS, McMorris FA. Oligodendrocytes and their precursors require phosphatidylinositol 3-kinase signaling for survival. Development. 1996;122:2529–2537. doi: 10.1242/dev.122.8.2529. [DOI] [PubMed] [Google Scholar]
- Venkatasubramanian G, Chittiprol S, Neelakantachar N, Naveen MN, Thirthall J, Gangadhar BN, Shetty KT. Insulin and insulin-like growth factor-1 abnormalities in antipsychotic-naive schizophrenia. Am J Psychiatry. 2007;164:1557–1560. doi: 10.1176/appi.ajp.2007.07020233. [DOI] [PubMed] [Google Scholar]
- Verney C, Takahashi T, Bhide PG, Nowakowski RS, Caviness VS., Jr Independent controls for neocortical neuron production and histogenetic cell death. Dev Neurosci. 2000;22:125–138. doi: 10.1159/000017434. [DOI] [PubMed] [Google Scholar]
- Voineskos AN, Lobaugh NJ, Bouix S, Rajji TK, Miranda D, Kennedy JL, Mulsant BH, Pollock BG, Shenton ME. Diffusion tensor tractography findings in schizophrenia across the adult lifespan. Brain. 2010;133:1494–1504. doi: 10.1093/brain/awq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walenkamp MJ, Karperien M, Pereira AM, Hilhorst-Hofstee Y, van DJ, Chen JW, Mohan S, Denley A, Forbes B, van Duyvenvoorde HA, van Thiel SW, Sluimers CA, Bax JJ, de Laat JA, Breuning MB, Romijn JA, Wit JM. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab. 2005;90:2855–2864. doi: 10.1210/jc.2004-1254. [DOI] [PubMed] [Google Scholar]
- Walenkamp MJ, van der Kamp HJ, Pereira AM, Kant SG, van Duyvenvoorde HA, Kruithof MF, Breuning MH, Romijn JA, Karperien M, Wit JM. A variable degree of intrauterine and postnatal growth retardation in a family with a missense mutation in the insulin-like growth factor I receptor. J Clin Endocrinol Metab. 2006;91:3062–3070. doi: 10.1210/jc.2005-1597. [DOI] [PubMed] [Google Scholar]
- Wallborn T, Wuller S, Klammt J, Kruis T, Kratzsch J, Schmidt G, Schlicke M, Muller E, van de Leur HS, Kiess W, Pfaffle R. A heterozygous mutation of the insulin-like growth factor-I receptor causes retention of the nascent protein in the endoplasmic reticulum and results in intrauterine and postnatal growth retardation. J Clin Endocrinol Metab. 2010;95:2316–2324. doi: 10.1210/jc.2009-2404. [DOI] [PubMed] [Google Scholar]
- Walter HJ, Berry M, Hill DJ, Cwyfan-Hughes S, Holly JMP, Logan A. Distinct sites of insulin-like growth factor (IGF)-II expression and localization in lesioned rat brain: possible roles of IGF binding proteins (IGFBPs) in the mediation of IGF-II activity. Endocrinology. 1999;140:520–532. doi: 10.1210/endo.140.1.6463. [DOI] [PubMed] [Google Scholar]
- Werther GA, Cheesman H, Russo V. Olfactory bulb organ culture is supported by combined insulin-like growth factor-I and basic fibroblast growth factor. Brain Res. 1993;617:339–342. doi: 10.1016/0006-8993(93)91102-x. [DOI] [PubMed] [Google Scholar]
- Wilczak N, De BP, Luiten P, Geerts A, Teelken A, De KJ. Insulin-like growth factor II receptors in human brain and their absence in astrogliotic plaques in multiple sclerosis. Brain Res. 2000;863:282–288. doi: 10.1016/s0006-8993(00)02153-3. [DOI] [PubMed] [Google Scholar]
- Wilczak N, De KJ. Insulin-like growth factor-I receptors in normal appearing white matter and chronic plaques in multiple sclerosis. Brain Res. 1997;772:243–246. doi: 10.1016/s0006-8993(97)00940-2. [DOI] [PubMed] [Google Scholar]
- Wilson HC, Onischke C, Raine CS. Human oligodendrocyte precursor cells in vitro: phenotypic analysis and differential response to growth factors. Glia. 2003;44:153–165. doi: 10.1002/glia.10280. [DOI] [PubMed] [Google Scholar]
- Woodruff RH, Tekki-Kessaris N, Stiles CD, Rowitch DH, Richardson WD. Oligodendrocyte development in the spinal cord and telencephalon: common themes and new perspectives. Int J Dev Neurosci. 2001;19:379–385. doi: 10.1016/s0736-5748(00)00083-6. [DOI] [PubMed] [Google Scholar]
- Woods KA, Camacho-Hubner C, Barter D, Clark AJL, Savage MO. Insulin-like growth factor I gene deletion causing intrauterine growth retardation and severe short stature. Acta Paediatr. 1997;86:39–45. doi: 10.1111/j.1651-2227.1997.tb18367.x. [DOI] [PubMed] [Google Scholar]
- Wu MK, Huang CY, Liou YJ, Wang CK, Lee SD. Glucose-insulin homeostasis, lipid profiles and GH–IGF–IGFBP axis in clozapine-treated schizophrenic obesity versus non-psychiatric obesity. Int J Obes (Lond) 2008;32:436–442. doi: 10.1038/sj.ijo.0803750. [DOI] [PubMed] [Google Scholar]
- Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA. 1999;96:7324–7329. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Tanabe K, Wada K, Shimoke K, Ishikawa Y, Ikeuchi T, Koizumi S, Hatanaka H. Differences in survival-promoting effects and intracellular signaling properties of BDNF and IGF-1 in cultured cerebral cortical neurons. J Neurochem. 2001;78:940–951. doi: 10.1046/j.1471-4159.2001.00497.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Murphy LJ. Enzymatic conversion of IGF-I to des(1–3)IGF-I in rat serum and tissues: a further potential site of growth hormone regulation of IGF-I action. J Endocrinol. 1995;146:141–148. doi: 10.1677/joe.0.1460141. [DOI] [PubMed] [Google Scholar]
- Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- Yao Y, Li W, Wu J, Germann UA, Su MS, Kuida K, Boucher DM. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc Natl Acad Sci USA. 2003;100:12759–12764. doi: 10.1073/pnas.2134254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Carson J, D’Ercole AJ. In vivo actions of insulin-like growth factor-I (IGF-I) on brain myelination: studies of IGF-I and IGF binding protein-1 (IGFBP-1) transgenic mice. J Neurosci. 1995a;15:7344–7356. doi: 10.1523/JNEUROSCI.15-11-07344.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Carson J, D’Ercole AJ. Insulin-like growth factor-I influences the initiation of myelination: studies of the anterior commissure of transgenic mice. Neurosci Lett. 1995b;201:235–238. doi: 10.1016/0304-3940(95)12194-3. [DOI] [PubMed] [Google Scholar]
- Ye P, D’Ercole AJ. Insulin-like growth factor I protects oligodendrocytes from tumor necrosis factor-alpha-induced injury. Endocrinology. 1999;140:3063–3072. doi: 10.1210/endo.140.7.6754. [DOI] [PubMed] [Google Scholar]
- Ye P, Hu Q, Liu H, Yan Y, D’Ercole AJ. Beta-catenin mediates insulin-like growth factor-I actions to promote cyclin D1 mRNA expression, cell proliferation and survival in oligodendroglial cultures. Glia. 2010;58:1031–1041. doi: 10.1002/glia.20984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Kollias G, D’Ercole AJ. Insulin-like growth factor-I ameliorates demyelination induced by tumor necrosis factor-alpha in transgenic mice. J Neurosci Res. 2007;85:712–722. doi: 10.1002/jnr.21181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Lee KH, D’Ercole AJ. Insulin-like growth factor-I (IGF-I) protects myelination from undernutritional insult: studies of transgenic mice overexpressing IGF-I in brain. J Neurosci Res. 2000;62:700–708. doi: 10.1002/1097-4547(20001201)62:5<700::AID-JNR9>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Ye P, Li L, Lund PK, D’Ercole AJ. Deficient expression of insulin receptor substrate-1 (IRS-1) fails to block insulin-like growth factor-I (IGF-I) stimulation of brain growth and myelination. Brain Res Dev Brain Res. 2002a;136:111–121. doi: 10.1016/s0165-3806(02)00355-3. [DOI] [PubMed] [Google Scholar]
- Ye P, Li L, Richards RG, DiAugustine RP, D’Ercole AJ. Myelination is altered in insulin-like growth factor-I null mutant mice. J Neurosci. 2002b;22:6041–6051. doi: 10.1523/JNEUROSCI.22-14-06041.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Popken GJ, Kemper A, McCarthy K, Popko B, D’Ercole AJ. Astrocyte-specific overexpression of insulin-like growth factor-I promotes brain overgrowth and glial fibrillary acidic protein expression. J Neurosci Res. 2004;78:472–484. doi: 10.1002/jnr.20288. [DOI] [PubMed] [Google Scholar]
- Ye P, Umayahara Y, Ritter D, Bunting T, Auman H, Rotwein P, D’Ercole AJ. Regulation of insulin-like growth factor I (IGF-I) gene expression in brain of transgenic mice expressing an IGF-I-luciferase fusion gene. Endocrinology. 1997;138:5466–5475. doi: 10.1210/endo.138.12.5600. [DOI] [PubMed] [Google Scholar]
- Ye P, Xing Y, Dai Z, D’Ercole AJ. In vivo actions of insulin-like growth factor-I (IGF-I) on cerebellum development in transgenic mice: evidence that IGF-I increases proliferation of granule cell progenitors. Brain Res Dev Brain Res. 1996;95:44–54. doi: 10.1016/0165-3806(96)00492-0. [DOI] [PubMed] [Google Scholar]
- Yi X, Schubert M, Peachey NS, Suzuma K, Burks DJ, Kushner JA, Suzuma I, Cahill C, Flint CL, Dow MA, Leshan RL, King GL, White MF. Insulin receptor substrate 2 is essential for maturation and survival of photoreceptor cells. J Neurosci. 2005;25:1240–1248. doi: 10.1523/JNEUROSCI.3664-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Li J, Zhang S, Xu X, Zheng M, Jiang G, Li F. IGF-1 induces hypoxia-inducible factor 1alpha-mediated GLUT3 expression through PI3K/Akt/mTOR dependent pathways in PC12 cells. Brain Res. 2012;1430:18–24. doi: 10.1016/j.brainres.2011.10.046. [DOI] [PubMed] [Google Scholar]
- Zackenfels K, Oppenheim RW, Rohrer H. Evidence for an important role of IGF-I and IGF-II for the early development of chick sympathetic neurons. Neuron. 1995;14:731–741. doi: 10.1016/0896-6273(95)90217-1. [DOI] [PubMed] [Google Scholar]
- Zeger M, Popken G, Zhang J, Xuan S, Lu QR, Schwab MH, Nave KA, Rowitch D, D’Ercole AJ, Ye P. Insulin-like growth factor type 1 receptor signaling in the cells of oligodendrocyte lineage is required for normal in vivo oligodendrocyte development and myelination. Glia. 2007;55:400–411. doi: 10.1002/glia.20469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Moats-Staats BM, Ye P, D’Ercole AJ. Expression of insulin-like growth factor system genes during the early postnatal neurogenesis in the mouse hippocampus. J Neurosci Res. 2007;85:1618–1627. doi: 10.1002/jnr.21289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Popken GJ, Ye P, D’Ercole AJ. Down-regulation of 14-3-3 eta gene expression by IGF-I in mouse cerebellum during postnatal development. Brain Res Dev Brain Res. 2003;143:199–206. doi: 10.1016/s0165-3806(03)00132-9. [DOI] [PubMed] [Google Scholar]
- Zhong J, Li X, McNamee C, Chen AP, Baccarini M, Snider WD. Raf kinase signaling functions in sensory neuron differentiation and axon growth in vivo. Nat Neurosci. 2007;10:598–607. doi: 10.1038/nn1898. [DOI] [PubMed] [Google Scholar]
- Zhong J, Zhao L, Du Y, Wei G, Yao WG, Lee WH. Delayed IGF-1 treatment reduced long-term hypoxia–ischemia-induced brain damage and improved behavior recovery of immature rats. Neurol Res. 2009;31:483–489. doi: 10.1179/174313208X338133. [DOI] [PubMed] [Google Scholar]