

Abstract
There has been significant advancement in various aspects of scientific knowledge concerning the role of cerebellum in the etiopathogenesis of autism. In the current consensus paper, we will observe the diversity of opinions regarding the involvement of this important site in the pathology of autism. Recent emergent findings in literature related to cerebellar involvement in autism are discussed, including: cerebellar pathology, cerebellar imaging and symptom expression in autism, cerebellar genetics, cerebellar immune function, oxidative stress and mitochondrial dysfunction, GABAergic and glutamatergic systems, cholinergic, dopaminergic, serotonergic, and oxytocin related changes in autism, motor control and cognitive deficits, cerebellar coordination of movements and cognition, gene-environment interactions, therapeutics in autism and relevant animal models of autism. Points of consensus include presence of abnormal cerebellar anatomy, abnormal neurotransmitter systems, oxidative stress, cerebellar motor and cognitive deficits, and neuroinflammation in subjects with autism. Undefined areas or areas requiring further investigation include lack of treatment options for core symptoms of autism, vermal hypoplasia and other vermal abnormalities as a consistent feature of autism, mechanisms underlying cerebellar contributions to cognition, and unknown mechanisms underlying neuroinflammation.
Keywords: cerebellum, autism
Introduction
Research on the biological underpinnings of autism has gathered significant momentum in the last few years. Autism is clearly a multi-system disorder that impacts the brain, the immune system, the gastrointestinal tract, and other organ systems. Here, we focus on the involvement of cerebellum in this disorder. Many experts present literature-based key findings related to abnormal cerebellar structure and function in autism. Drs. Bauman and Kemper address histopathologic abnormalities of cerebellum in autism. Drs. Welsh, Estes, and Dager provide up to date information linking cerebellar imaging findings with symptom expression in subjects with autism. Recent published work is surveyed regarding genes associated with the pathology of autism by Drs. Aldinger and Millen. Immune involvement and its impact on autism symptom progression is discussed by Dr. Ashwood. The roles of brain oxidative stress and mitochondrial dysfunction in autism is summarized by Drs. Chauhan and Chauhan. The subsequent two sections deal with the involvement of several important molecules including GABA, glutamate, Reelin, acetylcholine, dopamine, oxytocin, and serotonin in autism, and are discussed by Drs. Fatemi and Blatt. In the next two sections, abnormalities of cerebellar motor and cognition are discussed by Drs. Mosconi, Sweeney, and Heck. Dr. Persico presents novel data on examples of gene-environment interactions in the causation of autism. Therapeutic interventions and their relevance to the cerebellum in autism are discussed by Drs. Webb, Welsh, and King. Finally, several important animal models relevant to the genesis of autism are presented and discussed by Drs. Dickson, Martin, Blaha, Mittleman, and Goldowitz. Taken together, a summary of key concepts related to involvement of cerebellar circuitry in autism is presented by a panel of experts attempting to reach a consensus with regards to this important disorder.
Cerebellar Pathology in Autism (M.L. Bauman, T.L. Kemper)
Autism is a clinically complex and heterogeneous disorder. The core features include delayed and disordered language, impaired social interaction, isolated areas of interest, and an insistence on sameness [1]. Many autistic individuals exhibit dysfunction in both fine and gross motor skills. Currently, the cause of autism remains poorly defined but both genetic and environmental factors have been implicated [2].
One of the most consistently abnormal brain regions has been the cerebellum and areas related to it. Beginning with an initial case report [3], almost all of the postmortem brains of autistic individuals studied to date, regardless of age, sex, and cognitive ability, have shown a significant decrease in the number of Purkinje cells (PC), primarily in the posterolateral neocerebellar cortex and adjacent archicerebellar cortex of the cerebellar hemispheres [4–7]. More recently, Whitney et al. [6] has suggested that the presence of reduced numbers of PCs in the brains of autistic individuals may not be present in all cases. In three of the six brains of autistic individuals, the number of PCs closely approximated that of the controls. In this study the density of the PCs did not correlate with the clinical severity of the autism. Despite several detailed analyses of the vermis, our studies have been unable to identify any abnormalities of PC size or number in this region, thus failing to provide any microscopic cellular explanation for vermal hypoplasia as reported by cranial imaging studies [8].
Evidence of late developmental loss of PCs has been provided by a study assessing the number of basket cells (BC) and stellate cells (SC), cells on which PCs rely for survival [9]. These investigators found no decrease in the number of BC and SC interneurons in the cerebellar molecular layer, suggesting that once the PCs were generated, they migrated to their proper location and then died. The timing of the loss of PCs appears to be prenatal. In the brains of autistic individuals with loss of PCs there is no neuronal loss in the synaptically related inferior olive. This tight relationship is established shortly before birth. Once this bond is made, any loss of PCs thereafter results in an obligatory retrograde cell loss of inferior olivary neurons [6, 10, 11]. In inferior olive, neurons were found to be clustered along the periphery of the nuclear convolutions, a pattern of pathology that can be dated to an earlier prenatal period [12, 13].
Additional findings in the cerebellum have included abnormalities of the deep cerebellar nuclei including the fastigeal, globose and emboliform nuclei. In comparison to age and sex matched controls, the neurons of these nuclear groups appeared to differ in size according to age. All of the autistic cases over the age of 21 years showed small pale neurons that were significantly decreased in number, whereas in all of the childhood cases, ages 5–13 years, the neurons in these same nuclear groups were unusually large and plentiful in numbers [14]. These observations, in conjunction with similar findings in the nucleus of the Diagonal Band of Broca in the septum and in the inferior olivary nuclei, combined with observations of overgrowth of brain volume early in life [15, 16] and a more recent report of reduced cortical thickness with age [17], suggests that the underlying neuropathology of autism may be associated with an ongoing postnatal process [16].
It is known that the cerebellum connects with many cortical and subcortical structures in the cerebral hemispheres and acts as a modulator for many of the cognitive, language, motor, sensory and emotional functions associated with these regions [18]. It has been shown that the cerebellum communicates with the parietal lobe through the brainstem, thus providing a potential mechanism for motor dysfunction and dyspraxia in autism [19]. The cerebellum is also known to play a role in classic conditioned reflex responses, mental imagery, anticipatory planning, aspects of attention, affective behavior, visual spatial organization and the control of sensory data acquisition. Many of these functions may be disordered in autism and it is possible that the cerebellar abnormalities noted in the brains of autistic individuals contribute to these clinical features in autism.
In summary, histoanatomic abnormalities of the cerebellum are one of the most consistent neuroanatomic findings in the brains of autistic individuals. These findings support the concept that autism is a disorder of prenatal onset with neurobiological processes that extend into the postnatal period. Given what is known about many of the functions of the cerebellum and its connections with cortical and subcortical regions of the cerebral hemispheres, it is likely that abnormalities of the cerebellum contribute significantly to many of the clinical features of the disorder.
Establishing Links between Cerebellar Imaging Findings and Symptom Expression in Autistic Disorder (J.P. Welsh, A.M. Estes, S.R. Dager)
Extensive investigation of the cerebellum in autism has sought to link cerebellar abnormalities to phenotypic characteristics of autistic disorder (AD). However, the functional relevance of these proposed relationships remains obscure. Cerebellar maintenance of posture, balance, motor dexterity and coordination of movement is impaired in some individuals with AD [20, 21]. Prospective studies of infants at high genetic risk for AD implicate motor impairment as among the earliest signs of the autism phenotype [22]. Gaze linked to cerebellar function [23] is characteristically atypical in individuals with AD and the observation of subtle oculomotor alterations suggests abnormalities in cortico-cerebellar connectivity [24, 25]. Cerebellar involvement in non-motor functions may influence cognition via its broad, reciprocal and multisynaptic connectivity with the cerebrum [26]. For instance, the posterior vermis may have an important role in facilitating language function [27] which, although heterogeneous in AD, is a core impairment [28]. Particular characteristics of speech, such as phrasing, rate, stress, pitch, loudness, and resonance, are often atypical in individuals with AD [29] and may be linked to cerebellar dysfunction. Cerebellar involvement in higher-order emotional, social and cognitive processing increasingly is being recognized [30]. Acquired cerebellar lesions involving the posterior lobe of the vermis, part of a reciprocal anatomical network with the anterior limbic system, have been associated with mild cognitive impairment, deficits of executive function, expressive language impairment, and affective blunting or disinhibition [31], all behaviors recognized in AD.
The development of magnetic resonance imaging (MRI) has allowed in vivo characterization of cerebellar morphometry in AD. As recently reviewed [32], some, but not all, tracing and voxel-based morphometry studies of young children, adolescents and adults with AD find evidence for increased cerebellar volume. Cerebellar enlargement in AD, when observed, is generally proportional to overall cerebral volumes, rather than specific to the cerebellum. Specific assessment of grey-white matter involvement suggests reduced cerebellar grey matter but inconsistent white matter volumetric alterations in AD. Applications of newer imaging techniques, such as diffusion tensor imaging, that increasingly are being applied to assess white matter integrity at a cellular level, will be helpful in further assessing possible relationships between AD and white matter pathology in the cerebellum. Increasingly, attention has focused on possible links between AD and subregions of the cerebellum, primarily involving the vermis. Post-mortem reports of reduced vermal Purkinje cells in AD [33] suggest vermal lobular hypoplasia, primarily affecting posterior lobules VI-VII, as observed by MRI in both children and adults with AD [32]. However, other MRI studies have either not confirmed posterior vermal hypoplasia or suggest diffuse involvement affecting additional vermal regions such as the anterior vermis (lobules I–V) and vermal lobules VIII-X [32]. In this regard, earlier theories of regionally-specific hypoplasia have been modified to suggest a bimodal distribution of vermal alterations with the majority of individuals with AD exhibiting vermal hypoplasia and a subset having vermal hyperplasia [34]. Vermal hypoplasia also may contribute to neurological and cognitive dysfunction in a variety of childhood disorders, in addition to AD [35, 36]. Thus, vermal hypoplasia, particularly lobules VI-VII, may be associated with AD but the specificity of this relationship is not well established and there is heterogeneity within AD.
Understanding cerebellar involvement in AD is likely to require integrating new concepts that take into consideration information regarding the molecular determinants of Purkinje cell death and biophysical mechanisms of olivocerebellar function. Although imaging studies have identified intriguing relationships, they do not address why Purkinje cells are lost in some individuals with AD or the implications for information processing at a mechanistic level which will be necessary for treatment and prevention. Insights can be gained by integrating animal models of Purkinje cell death [37, 38] and on the cellular and neuronal ensemble mechanisms of information processing in the cerebellum [39–41]. Large scale post-mortem immunohistochemical mapping of the entire cerebellum will help to establish whether Purkinje cell loss in AD is patterned (topistic) and whether spatial maps of Purkinje cell loss correspond to clinical phenotypes, genetic predisposition, or exposure to environmental risk factors. This effort would be motivated by a solid foundation of understanding molecular heterogeneity among Purkinje cells [40, 41] and why highly-defined zones of Purkinje cells are most susceptible to early death by environmental risk factors, including transient ischemia [37], prenatal infection [42], and drug toxicity [43]. As accumulating evidence suggests that an impairment in long-term depression (LTD) may predispose Purkinje cells to excitotoxicity [44], genetic and epigenetic factors that influence LTD could be examined in AD, as well as perinatal factors that induce synaptic drive leading to Purkinje cell excitotoxicity [37, 43]. Evidence of normal GABAergic interneuron development in cases with Purkinje cell loss implicate the loss of Purkinje cells after their normal migration [9], suggesting a perinatal window in which to study mechanisms. Magnetic resonance spectroscopy measurement of cerebellar levels of GABA and glutamate [45, 46] can provide important insight in this regard. Last, the evolving understanding of olivocerebellar physiology may inform new models regarding the effects of Purkinje cell loss on cognition, particularly with regard to how a loss of dynamic control of membrane potential oscillations in inferior olive neurons [47] and the coordination of synchronous firing among Purkinje cell ensembles [48] may affect cognition. Recent fMRI and lesion data indicate that the olivocerebellar system is especially involved in the timing of intervals defined by successive sensory events occurring over the range of 100–600ms. Deficits in eye blink conditioning in AD [49] and after cerebellar lesions [50] provide evidence for an altered olivocerebellar “clock-signal” for sensory timing that occurs below the level of perceptual awareness [51] as illustrated in Figure 1. Future studies should determine whether impairments in implicit timing may be a biomarker for behavioral-related cerebellar dysfunctions in AD or provide a tool for early detection. Cerebellar contributions to the temporal processing of language and social cues during development also provide fertile ground for future research and would be a logical next step toward elucidating the role of cerebellar dysfunction in AD.
Figure 1.

Timing properties of the olivocerebellar network. a Organization of the olivocerebellar system. Inferior olive (IO) neurons are electrotonically coupled pacemakers that exhibit subthreshold oscillations in membrane potential and project directly to the Purkinje cells as climbing fibers. Purkinje cells are GABAergic and project to deep cerebellar nuclei, which in turn project to motor, autonomic, and limbic cerebral structures. A separate output pathway returns to the IO at the site of gap junctions and releases GABA to regulate the degree of electrotonic coupling and oscillation. b Intracellular fill showing the complex dendritic arbor of a macaque monkey IO neuron. C. An IO neuron from the macaque monkey shows subthreshold oscillations in membrane potential that entrain spiking and may provide a timing signal, as has been suggested in rodents. (After Welsh et al [47]).
Genetic evidence for cerebellar involvement in Autistic Disorder (K.A. Aldinger, K.J. Millen)
High heritability of autistic disorder (AD) has strongly implicated genetics in their etiology, but clinical heterogeneity within the broad behavioral phenotype has been a major obstacle to gene identification [52]. Despite this, genomic technology advances have uncovered several AD loci, including rare single-gene Mendelian neurodevelopmental syndromes where there is a high co-occurrence of AD (“syndromic AD”), rare chromosomal structural abnormalities, rare single gene mutations with major effects, and common gene variants with minor effects [53]. SFARI Gene curates an evolving list of AD-implicated genes [54; http://sfari.org/sfari-gene], several of which are listed in Table 1. AD genetic architecture is complex, with variants of any individual locus present in only 1–2% of patients, and collectively all known genetic causes accounting for just 15% of cases [55]. Compellingly, however, many genes converge on common biological pathways and brain circuits. These include synaptic formation, signaling and homeostasis in brain regions that process rapid and coherent integration of information from multiple, higher-level association areas, including frontal and temporal cortices, caudate, amygdala and cerebellum [55–58]. Since many AD-implicated genes are pleiotropic, disrupting their function is expected to produce a wide-rage of effects, both in the brain and in other organ systems. Here we discuss emerging genetic data providing support for specific cerebellar involvement in several ADs.
Table 1.
Emerging autism genes and available evidence for cerebellar pathology
| GENE SYMBOL | GENE NAME | GENE FUNCTION | Cerebellar Pathologyb |
Selected Human References | Selected Mouse References |
|---|---|---|---|---|---|
| Mendelian Disorders (Syndrome) | |||||
| AHI1 (Joubert syndrome) | Abelson helper integration site 1 | Encodes Jouberin, which interacts β-catenin in cilia to facilitate nuclear translocation | + | [64, 291] | [292] |
| CACNA1C (Timothy syndrome) | voltage-gated L-type calcium channel alpha 1C subunit | Encodes the alpha-1 subunit of a voltage-dependent calcium channel, which mediates calcium ion entry into excitable cells | |||
| DHCR7 (Smith- Lemli-Opitz syndrome) | 7-dehydrocholesterol reductase | Encodes the final enzyme in the cholesterol biosynthetic pathway | + | [293] | |
| FMR1 (Fragile X syndrome) | fragile X mental retardation 1 | Encodes Fragile X mental retardation protein, an RNA- binding protein that trafficks target mRNAs from the nucleus to the cytoplasm | + | [59] | [294] |
| MECP2 (Rett syndrome) | methyl CpG binding protein 2 | Encodes MeCP2, which binds methylated DNA to recruit repressor complexes for gene repression; may also activate some genes | + | [295] | [296] |
| NF1 (Neurofibromatosis 1) | neurofibromin isoform 1 | Encodes neurofibromin, a GTP-ase activator and negative regulator of the RAS signaling pathway | + | [297] | [298] |
| PTEN (Cowden disease) | phosphatase and tensin homolog | Encodes a protein tyrosine phosphatase, which negatively regulates the PI3K–AKT–mTOR pathway | + | [299] | [300] |
| TSC1,2 (Tuberous Sclerosis types I and II) | tuberous sclerosis 1 and 2 | Encodes a complex, which negatively regulates the mTOR pathway | + | [60] | [301] |
| UBE3A (Angelman syndrome) | ubiquitin protein ligase E3A | Encodes a ubiquitination ligase, which targets protein degradation system | [302] | [303] | |
| Rare Mutations (Structural Variations) | |||||
| GABRB3 (15q11–13 duplication) | GABAA receptor beta 3 | Encodes one of 18 subunits for a multisubunit chloride channel which forms the GABAA receptor | + | [70] | [71] |
| SHANK3 (22q13 deletion) | SH3 and multiple ankyrin repeat domains 3 | Encodes a multidomain scaffold protein of the postsynaptic density that connects neurotransmitter receptors, ion channels, and other proteins to the actin cytoskeleton and G-protein coupled signaling pathways. | + | [61, 62] SHANK3 – Not determined | Shank3 – Not determined |
| SEZ6L2 (16p11.2 deletion) | seizure related 6 homolog | Encodes a multidomain transmembrain protein, homologous to SRPX2 in which mutations cause epilepsy and language disorders | |||
| NLGN3 | neuroligin 3 | Encodes a member of the neuroligin family of neuronal surfact proteins, which are ligands for beta- neurexins and may be involved in synapse formation and remodeling | |||
| NLGN4X | neuroligin 4, X-linked | Encodes a member of the neuroligin family of neuronal surfact proteins, which are ligands for beta- neurexins and may be involved in synapse formation and remodeling | |||
| NRXN1 | neurexin 1 | Encodes a member of the neurexin family of cell adhesion molecules and receptors located on the neuronal cell surface; may be involved in cell recognition and adhesion by forming intracellular junctions through neuroligin binding | + | [304] | |
| Association Studiesa | |||||
| AVPR1Aa | arginine vasopressin receptor 1A | Encodes a G-protein coupled receptor for arginine vasopression, which activate a phosphatidyl- inositol-calcium second messenger system; involved in social behaviors | |||
| CNTNAP2 | contactin associated protein- like 2 | Encodes Caspr2, a neurexin family member; homozygous mutations cause cortical dysplasia-focal epilepsy syndrome | + | [305] | |
| DISC1 | disrupted in schizophrenia 1 | Encodes a large transmembrane protein involved in neurite outgrowth and cortical development | |||
| EN2a | engrailed 2 | Encodes a Homeobox protein critical for hindbrain patterning | + | [68] | |
| GRIK2 | ionotropic glutamate receptor, kainate 2 | Encodes a postsynaptic glutamatereceptor subunit; homozygous mutationscause a mental retardation syndrome | + | [306] | |
| ITGB3a | integrin beta-3 precursor | Encodes a cell-surface protein composed of an alpha and beta chain to mediate cell adhesion and cell-surface mediated signaling of platelets | |||
| METa | met proto-oncogene | Encodes a receptor tyrosine kinase involved in cell proliferation, morphogenesis and survival; influences synapse development | + | [69] | |
| OXTR* | oxytocin receptor | Encodes a G-protein coupled receptor for oxytocin; involved in social behaviors | |||
| RELN | reelin | Encodes a large secreted extracellular matrix protein involved in cell-cell interactions required for proper cell positioning and neuronal migration in brain development | + | [142, 307] | [308] |
| SLC25A12 | calcium-binding mitochondrial carrier protein | Encodes a calcium-binding mitochondrial carrier protein, which is involved in exchanging aspartate for glutamate across the inner mitochondrial membrane | [309] | ||
| SLC6A4 | serotonin transporter | Encodes the serotonin transporter, an integral membrane protein that transports serotonin from the synaptic cleft into presynaptic neurons | |||
Five or more positive association studies (https://sfari.org/sfari-gene, accessed 27 May 2011)
Presence of cerebellar effects in humans and/or mice: (blank) if no information available
Detailed phenotypic evaluations of genetically defined AD patient cohorts has minimized the confound of heterogeneity which contributed to the controversy of AD-cerebellar involvement. Analysis of syndromic AD patients now provides clear support for a cerebellar role. For example, ~30% of patients with Fragile X Syndrome (FXS), the most common inherited cause of intellectual disability, are also diagnosed with AD. FXS is also one of the most common genetic causes of AD with >2% of AD patients co-diagnosed with FXS (http://www.fragilex.org). Although FXS affects many brain regions, imaging studies have identified selective abnormalities of cerebellar vermis lobules VI-VII in FXS patients with AD that are not seen in FXS patients without the AD diagnosis. Interestingly, these lobules are also abnormal in non-syndromic AD [59]. Tuberous Sclerosis (TS) is another rare syndromic disorder presenting with cerebellar involvement and AD. TS is characterized by hamartomas in the brain and other organ systems. Approximately 40% of TS patients are co-diagnosed with AD, and those with cerebellar lesions have more severe AD features than those with lesions restricted to other brain regions [60]. As a final example, individuals harboring heterozygous chromosome 22q13 deletions (22q13DS) are often diagnosed with AD. The 22q13DS variable deletion encompasses numerous genes, including SHANK3, a gene within which mutations have independently been associated with non-syndromic AD. Cerebellar vermis hypoplasia has been noted in seven patients with 22q13DS, suggesting that SHANK3 may be involved in cerebellar development [61, 62]. However, several genes in the 22q13DS region, including PLXNB2 and MAPK8IP2 [63; http://hbatlas.org], are also expressed in the developing cerebellum, making the role for cerebellar malformation gene(s) in this region unclear.
Patients with congenital cerebellar malformation syndromes provide additional support for cerebellar involvement in AD. AD is diagnosed in up to 40% of children with Joubert Syndrome (JS), which is defined by significant cerebellar vermis hypoplasia and a distinctive brainstem malformation. Homozygous mutations in AHI1 were the first identified JS cause and common genetic variation in AHI1, represented by single nucleotide polymorphisms (SNPs), was subsequently found to be associated with non-syndromic AD [64]. Reduced expression of AHI1 has been detected in the AD brain within a large gene co-expression network that includes several other AD-implicated genes [65]. Thus, AHI1 may be more generally involved in AD etiology. There are also reports of patients with other structural cerebellar defects, including Dandy-Walker malformation, who are co-diagnosed with AD [66]. Since congenital cerebellar malformations are diagnosed early, even in utero, later cognitive or behavioral difficulties are usually attributed to the cerebellar birth defect and rigorous cognitive and behavioral evaluations are rarely performed. The reciprocal scenario also occurs. Children diagnosed with AD are not routinely evaluated for structural brain malformations, but 10% of AD patients at one center were reported to have cerebellar abnormalities consistent with a Dandy-Walker malformation diagnosis [67]. Thus, although the number of individuals diagnosed with both ADs and cerebellar malformations is small, the consistent finding of patients with both phenotypes implicates the cerebellum in autism pathogenesis and suggests that individuals with cerebellar malformations represent an important subgroup of AD patients.
Three promising AD-implicated genes (EN2, GABRB3 and MET), identified from studies of non-syndromic AD, each with independent, replicated genetic evidence, have known and specific roles in cerebellar development. EN2 intronic SNPs have been associated with AD in 5/6 studies (SFARI Gene). In mice, En2 expression is restricted to the presumptive midbrain and cerebellum and loss of En2 function causes abnormal cerebellar foliation with deficits in motor and social behavior [68]. Five studies have demonstrated an association between three MET SNPs and AD (SFARI Gene). Met is expressed postnatally in proliferating granule cell precursors and mice with a hypomorphic Met allele have cerebellar hypoplasia with reductions both in fissure size and granule cell precursor proliferation [69]. GABRB3 is found within the relatively common chromosome 15q11–13 duplication associated with AD. 9/11 studies reported a positive association between AD and either common or rare variants in GABRB3 (SFARI Gene). Additionally, GABRB3 expression is reduced in AD brains, including reduced cerebellar expression [70], and Gabrb3-/- mice exhibit cerebellar vermis hypoplasia [71]. Although mutant mouse data for these 3 genes provides compelling evidence for cerebellar pathogenesis, no human cerebellar phenotypic data currently exists for cohorts of patients with these AD-associated DNA sequence variants.
In conclusion, AD is extremely genetically heterogeneous and many AD genes are expressed throughout the brain. Disruption of their function is expected to impact many brain regions, including the cerebellum. Subsets of AD patients however, show evidence for specific cerebellar involvement, including those with syndromic AD and cerebellar malformation patients. Additionally, several AD genes are well known for their involvement in cerebellar development from mouse mutant analysis. Thorough phenotypic assessments of genetically defined AD patients will now be essential to delineate the specific pathogenesis of each AD subset.
Autism, immune dysfunction and the cerebellum (P. Ashwood)
Immune cells and the products of immune responses are able to directly alter neuronal function, migration, proliferation and synapse formation, and thus have important roles in modulating neuronal circuits that form the basis for human cognition and behavior. Inappropriate immune function or responses during early life may lead to neurodevelopmental disorders including autism. A large variety of genetic and environmental stimuli are hypothesized to play “causative” roles in AD with many having in common the ability to alter immune function [72].
While the link between immune dysfunction, neuronal dysfunction and neurodevelopment is not completely clear, several lines of evidence highlight an important role for altered immune responses in the pathogenesis of AD. One striking feature is the presence of on-going neuroinflammation in postmortem brain tissue from individuals with AD, a finding that is consistent over a broad age range (5–44 years of age) [73]. Compared with controls, brain tissue specimens from cerebellum, midfrontal and cingulate gyrus in AD show marked activation of microglia and astrocytes with the up-regulation of the cell surface major histocompatibility complex (MHC) molecule HLA-DR and, glial fibrillary acidic protein, respectively. Prominent monocyte and macrophage accumulation in the cerebellum is also detected. Although neuroinflammation is not related to symptom onset or mental retardation, microglial activation is higher in the cerebellar white matter of AD individuals with a history of epilepsy. Moreover, increases in many cytokines and chemokines are observed in the brain and cerebrospinal fluid (CSF) of individuals with AD, in particular; interleukin (IL)-6, transforming growth factor beta 1 (TGFβ1), C-C motif ligand 2 (CCL2) and CCL17 in the cerebellum [73–76].
Gene expression profiles in brain regions, including the temporal cortex, of individuals with AD show increased transcript levels of many immune system-related genes and immune signaling pathways, including the MET pathway, NF-κB, IL-1 receptor, TOLL, and TNF receptor 2 pathways [77]. Transcriptome organization patterns show that gene co-expression networks exhibit abnormalities in cortical patterning in the brain of individuals with AD and are associated with immune activation [65], highlighting a role for immune dysregulation in ongoing neurological dysfunction in AD. What the impact of these changes is and how they alter behavior is not yet known. Is the altered immune activity neurotoxic, neuroprotective, a reparative response to abnormal neuronal function, or a combination of these? Antibodies reactive to cerebellar proteins have also been described in AD [78, 79]. The precise antigenic target of these antibodies is still a mystery but strong specific reactivity is observed against cerebellar GABAergic interneurons and golgi type II cells [80, 81]. Together these findings further suggest the presence of ongoing immune activation that targets seemingly specific neuronal cells. Whether these antibodies alter cellular activity, either enhancing or blocking functions, or whether the antibodies designate cells for destruction, or are markers for possible cellular damage, requires further investigation. However, it is significant that the presence of antibodies against cerebellar proteins is strongly associated with worsening aberrant behavior [79].
Furthermore, extensive findings of immune dysfunction are frequently observed in the periphery of a substantial number of children with AD. Elevated plasma cytokine levels (IL-1β, IL-6, IL-12p40 and TNFα) [82–84], altered immunoglobulin levels [85, 86], elevated levels of complement proteins [87], and increased chemokine levels (CCL2, CCL5 and CCL11) [88] are reported in the plasma of young children with AD compared with matched typically developing children. Disrupted cellular function, under resting conditions and in response to immunological challenges is reported in AD for a number of different cell types including natural killer (NK) cells [89], monocytes [90, 91] and T cells [92, 93]. In parallel, the production of regulatory cytokines TGFβ1 and IL-10 is decreased in AD, indicating a shift towards an inflammatory profile [83, 84, 94]. As cytokines have extensive effects and can influence neural development and synaptic transmission these changes may lead to modulation of certain behavioral traits. We and others have found that many of the immunological parameters that are different in AD are associated with increased impairments in behaviors characteristic of core features of AD; in particular, deficits in social interactions and communication, as assessed using quantitative scores derived from the Autism Diagnostic Observation Schedule (ADOS), Autism Diagnostic Interview-Revised (ADI-R), as well as aberrant behaviors assessed using the aberrant behavior checklist (ABC), are linked with immune dysfunction in AD [79,82,85,86,88,89, 91, 93–95].
Taken together, the evidence thus far reported indicates a key role of immune dysfunction in the pathogenesis of AD. The exact nature of this immune dysfunction requires further investigation but potentially represents an important avenue for the development of therapies that may alleviate neuroinflammation and help in the resolution of altered behaviors.
Brain Oxidative Stress and Mitochondrial Abnormalities in Autism (A. Chauhan, V. Chauhan)
Accumulating evidence suggests that a common feature in autism cases may be oxidative stress, the mechanism through which environmental factors exert their deleterious effects, which may be further exacerbated by the interaction of genetically susceptible alleles [96–100]. Some studies support a prenatal and perinatal onset for developmental abnormalities leading to autism [101, 102].
Oxidative stress occurs when the levels of reactive oxygen species (ROS) exceed the antioxidant capacity of a cell. These ROS are highly toxic and oxidize vital cellular components such as lipids, proteins and DNA, thus causing cellular damage and subsequent cell death via apoptosis or necrosis. Oxidative stress is known to be associated with premature aging of cells, and can lead to inflammation, damaged cell membranes, autoimmunity and cell death. Several reports have suggested immunological abnormalities and inflammation in autism [96].
There is ample evidence of the presence of oxidative stress in peripheral tissues in children with autism [96, 98]. The brain is highly vulnerable to oxidative stress because of its limited antioxidant capacity, higher energy requirement and high amounts of unsaturated lipids and iron. Recent studies with postmortem brain tissues have shown elevated levels of markers of oxidative damage, coupled with reduced antioxidant status in the brain of individuals with autism as compared to age-matched control subjects. In the cerebellum, the levels of lipid hydroperoxide [103], a product of fatty acid oxidation; of malonyldialdehyde [104], an end-product of lipid peroxidation; of 8-hydroxy-2’-deoxyguanosine (8-OH-dG) [105, 106], a marker of oxidative DNA damage; of protein carbonyl [107], a marker of protein oxidation; and of 3-nitrotyrosine [108], a marker of protein nitration, were significantly increased in autism. The expression of carboxyethyl pyrrole, a marker of lipid-derived oxidative protein modification, was also increased in postmortem brain samples from autistic subjects [109]. In another study, a greater number of lipofuscin (a matrix of oxidized lipid and cross-linked protein)-containing brain cells was reported in language-related cortical areas 22, 39 and 44 in autism [110].
Glutathione (GSH) is the major endogenous antioxidant produced by cells, which neutralizes ROS, and participates in detoxification and elimination of environmental toxins. A decrease in GSH, an increase in its oxidized disulfide form (GSSG) and a decrease in the redox ratio of GSH/GSSG were observed in the cerebellum and temporal cortex of individuals with autism, suggesting a glutathione redox imbalance in autism [111]. In the cerebellum and frontal cortex of individuals with autism, we have also reported increased activities of Na+/K+- ATPase and Ca2+ /Mg2+-ATPase, the membrane-bound enzymes, which maintain intracellular gradients of ions that are essential for signal transduction [112].
The free radicals are generated endogenously during oxidative metabolism and energy production by mitochondria, and the electron transport chain (ETC) in mitochondria is a prime source for ROS generation [113]. Mitochondria produce ATP by generating a proton gradient with the help of five ETC complexes. Emerging evidence suggests increased prevalence of mitochondrial dysfunction in autism [114]. Recently, we reported a brain region–specific deficit in the levels of ETC complexes in children with autism [103]. Reduced levels of complexes III and V in the cerebellum, of complex I in the frontal cortex, and of complexes II, III, and V in the temporal cortex were observed in children with autism as compared to age-matched control subjects [103].
Our studies of different brain regions showed that oxidative stress differentially affects selective brain regions, i.e., cerebellum, temporal and frontal cortices, in autism [103–105, 107]. Increased oxidative stress and mitochondrial abnormalities were not observed in the parietal and occipital cortices in autism [103–105, 107]. There is substantial evidence from neuroimaging studies that dysfunctions in the cerebellum and possibly the temporal lobe and association cortex result in autism symptoms [115–117]. Aberrant brain structure has been reported particularly in the cerebellum of children with autism. Loss of Purkinje and granule cells has been reported throughout the cerebellar hemispheres in autism [99, 118, 119]. Alterations in neuronal size, density and dendritic branching in the cerebellum and limbic structures (hippocampus and amygdala) have also been reported in autism. In addition, neuropathological abnormalities in autism have also been suggested in the frontal and temporal cortices, cortical white matter, amygdala and brainstem [117–121].
The potential mechanisms of the role of oxidative stress and mitochondrial dysfunction in the development and pathophysiology of autism are represented in Fig. 2. The oxidative stress and intracellular redox imbalance can be induced or triggered in autism by prenatal, perinatal or postnatal exposure to certain environmental factors, which include toxins and toxicants, maternal drugs, viral and bacterial infections. Genetic factors can also modulate the threshold for vulnerability to oxidative stress in autism.
Figure 2.

Potential mechanisms depicting the role of oxidative stress and mitochondrial dysfunction in the development and pathophysiology of autism.
In conclusion, elevated oxidative stress in the cerebellum and frontal and temporal lobes of individuals with autism suggests that oxidative stress may be a contributing factor in the pathophysiology and clinical development of autism. These reports support the concept that brain oxidative stress plays an important role in autism and warrant in-depth mechanistic studies to provide new targets for therapeutic intervention.
Involvement of GABA, Glutamate, and Reelin in pathology of autism (S.H. Fatemi)
The glutamatergic and gamma-aminobutyric acid (GABA) systems are important foci of pathology in the cerebella of patients with autism. Indeed, many investigations have highlighted dysregulation of various proteins involved in this pathway; notably, GABAA and GABAB receptors, glutamic acid decarboxylase enzymes (GAD), Reelin, mGluR5, and FMRP [70, 122, 123–128].
GABA is an inhibitory neurotransmitter found in many brain regions including cerebellum. GABAA receptors are responsible for mediation of fast inhibitory action of GABA in the brain. GABAB receptors play an important role in maintaining an excitatory/inhibitory balance in the brain. Glutamic acid decarboxylase enzymes (GAD) are responsible for converting glutamate to GABA. It has been shown that GAD 65 and 67 proteins are reduced in the cerebella of subjects with autism [122, 125]. Blatt and colleagues have shown abnormalities in GAD 65 and 67 mRNAs in various cell types of autistic cerebellum, confirming previous work by Fatemi and coworkers [122, 127]. Additionally, other studies have reported reduced GABAA [129, 130] and GABAB [124, 130] receptor density and system activity in the brains of autistic patients. In cerebellum, concordant reductions have been observed in mRNA and protein levels for GABAB R1 receptor in subjects with autism [130]. Fatemi et al. have also observed concordant reductions in both GABAA and GABAB receptors in Brodmann Areas 40 and 9 [70, 124, 130]. Autoradiographic studies have confirmed these results in other brain regions of autistic individuals as well. Reduced GABAA receptor density has been observed in the anterior cingulate cortex [131]. GABAB receptor density has also been demonstrated to be reduced in the cingulate cortex and fusiform gyrus [132].
Reelin is an important protein expressed in GABAergic and glutamatergic cells [133, 134]. As a serine protease of the extracellular matrix, Reelin regulates proper lamination of neurons during embryonic development and helps in migration of neurons and maintenance of synaptic plasticity throughout life [135–137]. Reeler heterozygous mice, which are haploinsufficient for reelin, exhibit decreased GAD 67 [138] and reduced GABAA and GABAB receptors in the whole brain and hippocampus, respectively [139]. Fatemi et al. [126] was the first group to measure reductions in Reelin in post-mortem autistic cerebellum. Several studies have demonstrated abnormal reelin expression in autism and replicated these results; notably, polymorphisms of the RELN gene [140, 141], decreased Reelin mRNA in superior frontal cortex and cerebellum [89], and reduced Reelin expression in blood analyses [143].
Reelin binds to several protein receptors, including very low density lipoprotein receptor (VLDLR), apolipoprotein E receptor 2 and α3β1 integrin [144–146]. Through these receptors, Reelin is able to activate Disabled1 (Dab-1), an intracellular adapter protein that facilitates signaling between Reelin-secreting cells and pyramidal cells [147]. VLDLR is upregulated in the superior frontal cortex and cerebellum in adult patients with autism, while Dab-1 is significantly reduced in these areas [89], suggesting impaired signaling in the Reelin pathway.
Glutamate is the main excitatory neurotransmitter in the brain and it plays a key role during brain development by regulating multiple processes including neurogenesis, neuronal outgrowth, neuron survival, and synaptogenesis [148]. Glutamate is important in the acquisition of emotional behavior [149] and N-methyl-aspartic acid (NMDA)-glutamate receptors are responsible for long term potentiation [150], underlying learning and memory; two processes impaired in subjects with autism. Moreover, imbalance between GABA/glutamate can cause seizure disorders in autism. Genetic studies have found positive associations between autism and a number of polymorphisms in glutamate receptors and transporters including the mitochondrial aspartate-glutamate carrier (SLC25A12) [151] and glutamate receptor, ionotropic kainate 2 (GRIK2) [152, 153]. However, to date there have been only a limited number of studies showing altered expression of mRNA for glutamate receptors and transporters in brain [154, 155].
Mutations involving the fragile X mental retardation protein (FMRP) lead to Fragile X syndrome (FXS). Individuals with FXS and autism often share overlapping diagnoses [156]. FMRP, translated from the Fragile X mental retardation-1 (FMR1) gene, is an RNA-binding protein involved in post-transcriptional regulation of target RNAs [157] and binds approximately 4% of the mRNA expressed in the brain [158]. The absence of FMRP in mouse knockout models has demonstrated upregulation of mGluR5 and PSD-95 translation [159, 160]. Recent postmortem work has shown reductions in FMRP in cerebella and frontal cortices of subjects with autism who do not carry a mutation for FXS [123, 128]. Additionally, reductions in FMRP occurred in association with elevations in mGluR5 and decreases in GABAA β3 receptors indicating a linkage between these divergent molecular systems and pointing to potential therapeutic targets in autism.
In conclusion, the above findings highlight the role of the GABAergic system, GAD enzymes, Reelin, glutamate receptors/transporters, mGluR5, and FMRP in autism. While significant reductions in GABAA and GABAB receptors have been observed in subjects with autism, decreased GAD 65 and 67 enzymes have also been found, suggesting impaired conversion of glutamate to GABA in these patients. Moreover, Reelin signaling is also impaired in autism. In addition, FMRP has been demonstrated to be significantly reduced in the cerebellum and frontal cortex of autistic subjects, while mGluR5 is elevated in the brains of children with autism. However, more work is needed to better evaluate the role of glutamate receptors in autism (Figure 3).
Figure 3.
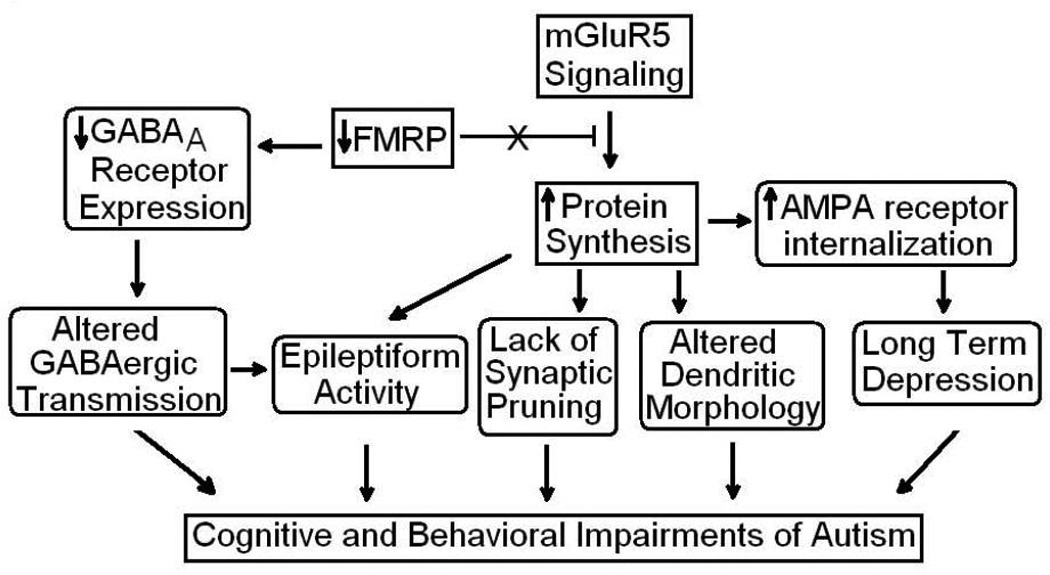
Reduced FMRP leads to a reduction of many GABAA receptor subunits, potentially contributing to altered GABAergic transmission and balance of GABA/glutamate in the brain. This effect may possibly explain the likelihood of seizure and cognitive deficits in subjects with autism and others with neuropsychiatric disorders. When activated by mGluR5, FMRP acts to inhibit protein synthesis. Without FMRP, protein synthesis is increased, resulting in internalization of AMPA receptors, leading to long term depression. In addition, increased protein synthesis may also be responsible for altered morphology of dendrites, epileptiform activity, and impaired synaptic pruning in autism. Reprinted from [289] with permission from Elsevier.
Acetylcholine, Oxytocin, Dopamine, and Serotonin in the Cerebellum and other Brain Regions with Implications to Autism (G. Blatt)
Oxytocin
Low concentrations of oxytocin have been measured in the cerebellum in rats [161] but high densities of oxytocin receptors (OTR) are localized in the amygdala and are also found in parts of the hippocampus, hypothalamus, thalamus, mesencephalon and brainstem. Research in animal models suggest a central role of oxytocin in mediating complex social behaviors with emphasis on action on OTRs in the amygdala to reduce fear and modulate aggression [162]. OTR mouse knockouts show social recognition deficits that can be remediated by oxytocin injections in the medial amygdala [163]. Experiments in prairie voles have revealed central roles for oxytocin in modulating a variety of emotional and social behaviors [164–166]. Deficiencies in social behaviors and OTRs in autism may in part be due to dysregulation of GABA via reduced reelin in autism [167]. Oxytocin may also interact with dopamine to modulate socio-affiliative behaviors and central oxytocin pathways may serve as therapeutic targets to improve mood and social behaviors [168]. Improvements in repetitive behaviors in adults with autism following oxytocin infusion have been demonstrated [169] as well as positive effects on social behaviors and cognition [170]. Intranasal administration of oxytocin may reduce repetitive behaviors, and increase performance on tasks of social recognition [171].
Acetylcholine
Key findings of 40–50% reduced nicotinic cholinergic receptor type α3, α4, and β2 measured by the high affinity agonist epibatidine in post-mortem autism cases relative to matched controls was reported in the granule cell, Purkinje cell and molecular layers in the cerebellum [172]. In contrast, a three-fold increase in nicotinic cholinergic receptor type α7 measured by receptor binding using α-bungarotoxin was found in the granule cell layer. A follow up study [173] found increased α4 mRNA expression in cerebellum but subunit protein levels and binding decreased. These authors suggested a possible relationship to decreased numbers of Purkinje cells in the autism cases. Nicotinic agonists are thought to enhance attentional processes, cognition and memory and thus may be useful as a therapeutic tool. In this regard, nicotinic cholinergic receptor type α7 is located on the surface of GABA inhibitory neurons and selective stimulation of this receptor subtype would cause GABA release and play a role in restoring inhibitory tone [174]. On the other hand, nicotinic cholinergic antagonists including some anti-depressants have helped ameliorate some autistic symptoms and are being explored as novel therapeutic agents [175]. Interestingly, no changes in muscarinic M1 or M2 type receptors were found in the cerebellum [172] but a pilot study from our laboratory [176] demonstrated a marked decrease of cholinergic M2 type receptor density in the medial accessory olive (MAO) of the inferior olivary complex (IO) but not in other olivary subfields in adult autism patients relative to age-matched controls (see Figure 4). From animal studies, the MAO projects to the anterior lobe, caudal vermis and the flocculus in cerebellar cortex and to the globose, emboliform and fastigial deep cerebellar nuclei, most of which show pathological abnormalities in autism [3]. In contrast, increased M2 cholinergic receptor expression in the MAO in normal developing infants compared to fetal binding has been reported [177], suggesting a possible role of muscarinic receptors as an early growth factor in infant development.
Figure 4.
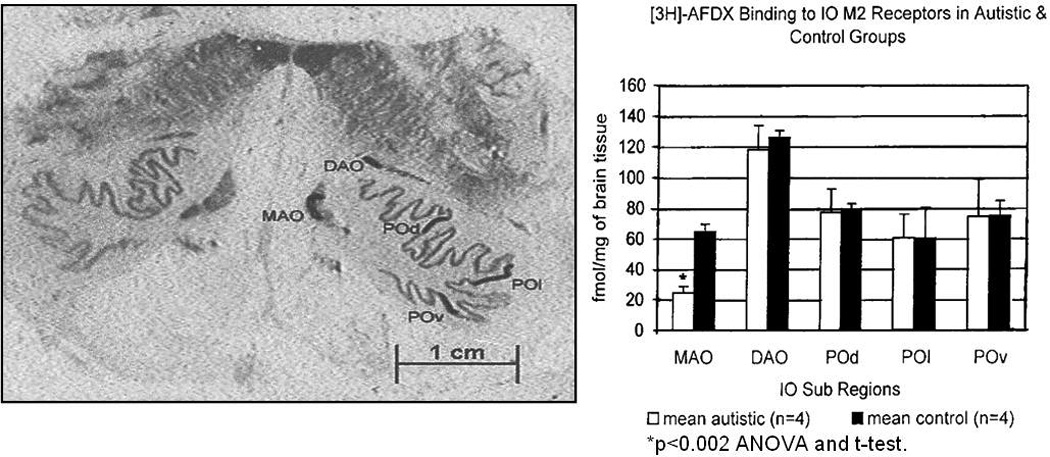
Left: Photomicrograph taken from 3[H]- sensitive film through the human inferior olivary complex (IO) from a normal control adult case. Right: In this pilot study, a statistically significant decreased density of 3[H]AFDX labeled cholinergic muscarinic Type 2 (M2) Receptors is demonstrated in the Medial Accessory Olive (MAO). Binding parameters: 5nM 3[H]-AFDX rinsed in 10mM Tris-HCl cold buffer; 10µM atropine used as a displacer ; Exposure time: 22 weeks. Abbrev.: DAO, dorsal accessory olive; fmol/mg, femtomoles per milligram protein; IO, inferior olive; MAO, medial accessory olive; POd, dorsal principal olive; POl, lateral principal olive; POv, ventral principal olive.
Dopamine
The cerebellum receives a dopaminergic pathway from the ventral tegmental area/substantia nigra pars compacta [178]. Stimulation of the dentate nuclei evokes dopamine release in the medial prefrontal cortex (mPFC). It has been recently reported that cerebellar pathology in autism and other cognitive disorders could result from the functional loss of cerebellar-mPFC circuitry due to abnormal dopaminergic activity in the mPFC [179]. The dentato-thalamo-cortical neurons are thought to be glutamatergic and thus implicate glutamate as a modulator of mPFC dopaminergic activity [179]. This was also previously shown in a lurcher mouse model by the same team of investigators [180]. In development, the physiological balance of dopamine D1 and D2 receptor activation is critical for GABA neuron migration from the basal forebrain to the cerebral cortex and may contribute to developmental disorders such as autism [181]. In a PET study, elevated dopamine transporter levels were reported in the orbitalfrontal cortex in high functioning autism subjects [182]. It is noteworthy that some selective serotonin reuptake inhibitors (SSRIs) that target 5-HTT and 5-HTR may also target some dopamine receptor types. For example, SSRIs and/or atypical antipsychotics that block both 5-HT2R and dopamine D1 and D2 receptors are especially attractive for possible clinical benefits [183].
Serotonin
Peripheral 5-HT effects in autism are well documented but central 5-HT differences are still emerging. There is evidence that activation of the serotonergic system in the inferior olive and cerebellum can contribute to the enhancement of harmaline-induced tremor in animals, once thought to only be attributable to olivocerebellar release of glutamate acting on NMDAR and AMPAR [178]. In normal post-mortem human cerebellum, there is a relatively low concentration of 5-HT transporters (SERT) compared to other brain regions except in the white matter and can be used as a reference region in binding studies looking at other brain regions with higher binding densities [184]. In contrast, a high level of 5-HT5A mRNA expression was found in the cerebellum especially localized to Purkinje cells and in the granule cells and dentate nucleus in both hemispheres and the vermis and may contribute to emotional, cognitive and motor functions associated with autism [185]. A significant reduction in SERT but not the dopamine transporter (DAT) was found in medial frontal cortex in autistic children determined by SPECT imaging [186]. In a PET imaging study, serotonin transporter binding was reduced in the anterior and posterior cingulate cortex in high functioning autistic subjects associated with impaired social cognition and, in the thalamus associated with repetitive and /or obsessive behavior and interests [182]. An increased number of 5-HT axons and terminals were reported in a variety of brain regions via immunocytochemical labeling of 5-HT transporter in post-mortem autism subjects aged 2–29 years old bringing into question the wide use of SSRIs as therapeutic agents [187]. In fact, Williams et al. [188] tested four SSRIs and concluded that there is no evidence of effect in autistic children and emerging evidence of harm, and limited evidence of effectiveness in autistic adults.
Cerebellar Dysfunctions Underlying Core Motor and Cognitive Deficits in Autistic Disorder (MW Mosconi, JA Sweeney)
Post-mortem studies have documented reduced Purkinje cell size and number in autistic disorder (AD) [5]. MRI studies have reported hypoplasia restricted to lobules I–V and vermal lobules VI-VII [189]. The majority of single-gene disorders associated with AD also are characterized by cerebellar pathology, suggesting that linking cerebellar structural abnormalities to core neurobehavioral features in AD may provide important insights into the etiology of these disorder(s) [190].
Cerebellar involvement in coordinated movements has been well described [310], and recent work indicates that the cerebellum plays an important role in non-motor functions as well [191] (Table 2). Sensorimotor pathways have been identified throughout the cerebellum, and preferential involvement of anterior lobules I–V and lobule VIII has been documented (Figure 5). In contrast, cognitive operations primarily involve posterior lobules VI-VII/Crus I–II [192]. Ascending cerebellar fibers innervate motor cortices via caudal fastigial nuclei, interpositus nuclei, and dorsal segments of dentate nuclei [193]. Posterior “cognitive” lobules synapse within ventrolateral units of the dentate nuclei [194]. There is thus a considerable anatomical basis for distinguishing motor and cognitive cerebellar systems, but the extent to which each of these systems is functionally impaired in AD remains poorly understood.
Table 2.
Motor and cognitive systems implicated by cerebellar structural abnormalities in autistic disorder
| Functions affected in AD | Cerebellar lobule |
Lateralization in cerebellum | Cerebellar dysfunction in AD? |
||
|---|---|---|---|---|---|
| Motor | Skeletomotor | Vestibular, gross and fine motor control |
I-V,VIII | Ipsi dominant | Y |
| Oculomotor | Saccade amplitude; pursuit gain | Vermis VI-VII | Contra dominant (saccades) Ipsi dominant (smooth pursuit) |
Y | |
| Cognitive | EF/Attention | Response inhibition, planning, set shifting |
VI, VIIB; Crus I |
Left dominant for spatial attention | N |
| Memory | Working memory, procedural memory |
VI, VIIB, VIIIA; Crus I |
None | N | |
| Language | Phonological processing, articulation, fluency, auditory comprehension |
VI-VII; Crus I/II |
Right dominant (for righthanders) | Y |
EF executive function, attn attention; Ipsi ipsilateral, Contra contralateral, Y yes, N no
Figure 5.
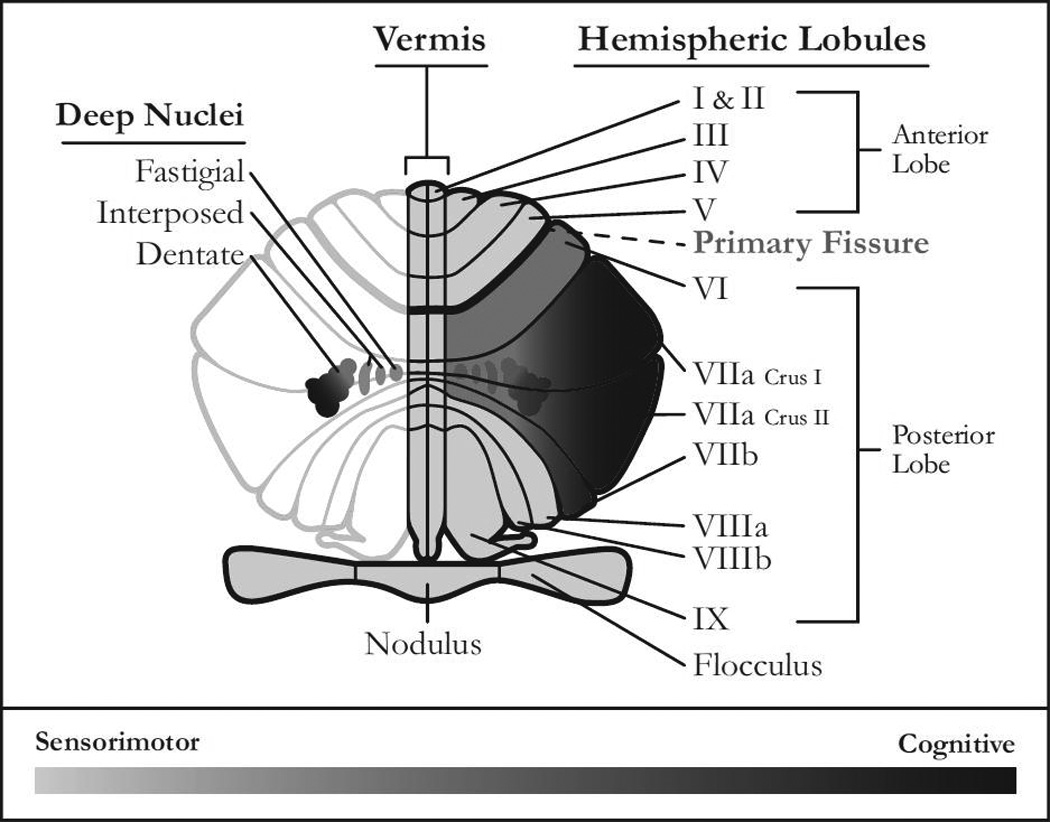
A representation of a posterior view of flattened human cerebellar cortex, vermis and deep nuclei shown in the coronal plane. Areas for which there is consistent evidence of involvement in higher cognitive functions, including executive control, memory processes, and language, are highlighted in dark gray and black. This includes posterior lobules VI-VII/Crus I–II which are separated from anterior lobules I–V by the primary fissure. Lighter shaded lobules (I-V and VIII) are dedicated to skeletomotor and oculomotor control, although it should be noted that motor control pathways have been documented throughout the cerebellum including posterior lobules, lobule IX and the flocculus-nodulus. Posterior vermian lobules VI-VII and their connections with caudal fastigial nuclei modulate conjugate eye movements. More inferior vermian lobules serve as termination sites of spinocerebellar pathways involved in proprioception and, in conjunction with the flocculus-nodulus, they organize vestibular control. The vermis also has been implicated in affect regulation [192]. Motor pathways synapse in caudal fastigial nuclei, interpositus nuclei, and dorsal segments of dentate nuclei. Cognitive systems involve ventrolateral cells within dentate nuclei. VIIAf: folium of vermis; VIIAt: tuber of vermis.
Motor impairments in AD
AD is defined by three core symptom categories: social impairment, communication abnormalities, and restricted and repetitive behaviors. Accumulating evidence indicates that the majority of affected individuals demonstrate motor impairments as well. In their original descriptions of AD, Leo Kanner [195] and Hans Asperger [196] noted awkward motility and clumsiness. Recent studies have documented impaired vestibular control [197], and gross and fine motor abnormalities similar to those demonstrated by patients with established cerebellar disease [198]. Freitag et al. [198] found that gross and fine motor impairments are associated with the severity of autistic symptoms suggesting possible common pathophysiological mechanisms.
When performing gross motor movements during fMRI studies, individuals with AD show reduced ipsilateral anterior cerebellar activation [199] and more diffuse activation across lobules VI-VII [200]. Muller et al. [201] and Mostofsky et al. [199] found concomitant increases in activation within association cortices and posterior cerebellum suggesting compensatory involvement of neocerebellar and neocortical pathways.
Conjugate eye movements are modulated by vermal lobules VI-VII and caudal fastigial nuclei. Findings of reduced smooth pursuit velocity [202], hypometric saccades, and increased trial-to-trial variability of saccade amplitude [24] implicate vermal dysfunction in AD. FMRI studies of saccades and smooth pursuit document reduced activation within the posterior vermis, and increased involvement of association cortices and dentate nuclei in AD [203]. Consistent with findings from manual motor testing, these results suggest that alterations of cerebellar motor systems may contribute to compensatory recruitment of non-motor cortical and cerebellar systems. Our recent findings that oculomotor deficits observed in individuals with AD also are present in unaffected family members suggest that dysfunctions within the oculomotor vermis may be familial and could serve as intermediate phenotypes for gene discovery [204].
Cognitive deficits and associated cerebellar findings in AD
Individuals with AD show impairments in executive function, memory and language abilities [205]. Schmahmann and Sherman [31] documented similar cognitive impairments in patients with chronic cerebellar disease. But, the primacy of the cerebellum to cognitive dysfunctions in AD remains unclear.
Executive control and attention
Executive dysfunctions and attention deficits are well characterized in AD, but few studies have linked these impairments to cerebellar abnormalities. Allen and Courchesne [200] documented reduced activation in the posterior vermis during an attention shifting task. Townsend et al. [206] found slower shift of attention was associated with reduced volumes of vermal lobules VI-VII.
Memory
Some memory functions are compromised in AD, although there is evidence that the extent to which subjects are impaired is strongly associated with the complexity of the task [205]. Verbal and spatial working memory deficits have been consistently documented in AD, but no known studies have reported an association with functional or structural abnormalities within the cerebellum.
Language
Individuals with AD typically demonstrate language impairments. Reversed structural and functional asymmetry within fronto-temporal language cortices may contribute to these impairments [207]. Also, Hodge et al. [208] reported that the typical right greater than left volume of lateral posterior cerebellar lobules VII-VIII/Crus I–II was reversed in language impaired individuals with AD. Reversed asymmetry of lobule VIIIA also was related to deficits on clinical measures of language function in AD.
In conclusion, cerebellar abnormalities are established in AD, and they may contribute to neurobehavioral deficits. Studies of motor control consistently implicate anterior lobules I–VI and posterior vermal lobules VI-VII in AD, and imaging studies have identified compensatory involvement of cortico-striatal and posterior cerebellar systems. Recent data suggests that dysfunction within posterior lobules VI-VIII may contribute to impaired higher cognitive functions as well. Future research needs to both clarify the prevalence of altered cerebellar function in AD and its contribution to broader neurobehavioral manifestations that define the clinical syndrome of AD.
Precise spatiotemporal activity patterns as key to cerebellar coordination of movements and cognitive processes (D. Heck)
In addition to a crucial cerebellar role in motor coordination, strong correlations between cerebellar neuropathology and cognitive abnormalities in disorders like autism, schizophrenia and other cerebellar cognitive syndromes have been well established [18, 209, 210]. Reciprocal connections between neocortical motor and non-motor association areas and the cerebellum have also been carefully mapped out [211, 212]. Post-mortem anatomical studies of brains from autistic individuals thus far revealed loss of Purkinje cells in the posterior vermis and intermediate cerebellar cortex as the most consistent neuropathology, suggesting a key role of the cerebellum in autism [213]. Beyond these anatomical correlations, however, the neuronal mechanisms underlying cerebellar cognitive and motor functions remain to be determined. The cerebellum’s crystalline network architecture is highly homogeneous throughout the entire structure, suggesting that one principle neuronal operation is performed in all parts of the cerebellum. This adds the interesting constraint that cerebellar contributions to motor coordination and cognitive functions are based on the same or highly similar neuronal computations. Taken one step further, this raises the question what motor and cognitive processes have in common so that both would benefit from cerebellar functional contributions. A possible answer to this question arises from considerations of cognitive functions being derived from the neuronal principles of sensory-motor control [214]. Movements, even simple ones, require precise spatiotemporal coordination of dozens of muscles and it is believed that the cerebellum is involved in coordinating the precise timing of muscle activities [215–217].
Rhythmic stereotypic movements controlled by central pattern generator circuits are among the evolutionarily oldest and most important motor patterns. Pattern generators still play major life supporting roles (breathing, swallowing, chewing, walking and running etc.) in all vertebrate nervous systems today. Yuste and colleagues recently suggested that the wiring diagram of the neocortical network is analogous to pattern generator circuits [218], an idea that is consistent with the suggestion that neocortical information processing mechanisms evolved from early neuronal mechanisms of motor control. This concept receives additional support from electrophysiological findings showing that the neocortex generates precisely timed spatiotemporal spike activity patterns during various motor and non-motor cognitive tasks [219–221]. Those patterns are comparable to muscle activity patterns in duration, temporal precision and complexity.
Here it is proposed that the precise spatiotemporal spike activity patterns generated in motor and non-motor areas of the conscious neocortex represent the cognitive equivalents of muscle activity patterns responsible for generating movements. It is furthermore suggested that the cerebellar contribution to cognition and motor control lies in the detection and coordination of spatiotemporal cortical activity patterns representing cognitive processes or motor commands. The same cerebellar input-output transformation involved in the temporal coordination of motor related cortical activity would apply to cognitive cortical activity. The nature of cerebellar input-output transformation has been suggested to involve the detection of precisely timed activity patterns [222].
The specific arrangement of parallel fibers and Purkinje cells in the cerebellar cortex forms a network that specifically detects precisely timed sequences of input activity and generates precisely timed output activity in response [223–225]. Thus precisely timed spatiotemporal cortical activity patterns, transmitted through-and potentially transformed in-the pontine nuclei would be detected by the cerebellum and trigger specific and precisely timed responses in cerebellar output activity which would–via the thalamus-contribute to the coordination of neocortical network activity involved in cognitive or motor processes (Figure 6). Molinari and colleagues also suggested that the cerebellum is crucially involved in analyzing sequential events, albeit in the behavioral domain, i.e. at a considerably slower time scale than the neuronal sequences discussed here [226]. It is interesting, that other researchers, based on clinical observations alone, suggested the terms “cognitive dysmetria” [227] or “dysmetria of thought” [31] to describe the effect of loss of cerebellar function on cognition, implying strong similarities between motor and cognitive cerebellar function. The hypothesis presented here offers a possible answer to the question of how the cerebellum could contribute to motor and cognitive processes under the constraint that the same neuronal mechanism should apply to both functions. How the specific cerebellar neuropathology observed in the brains of autistic individuals correlates with the characteristic set of cognitive deficits has yet to be determined. Important new clues, however, come from recent studies reporting cerebellar control of dopamine release in the prefrontal cortex [179, 180], a mechanism that might be triggered by spatiotemporal cortical activity patterns, as proposed here.
Figure 6.
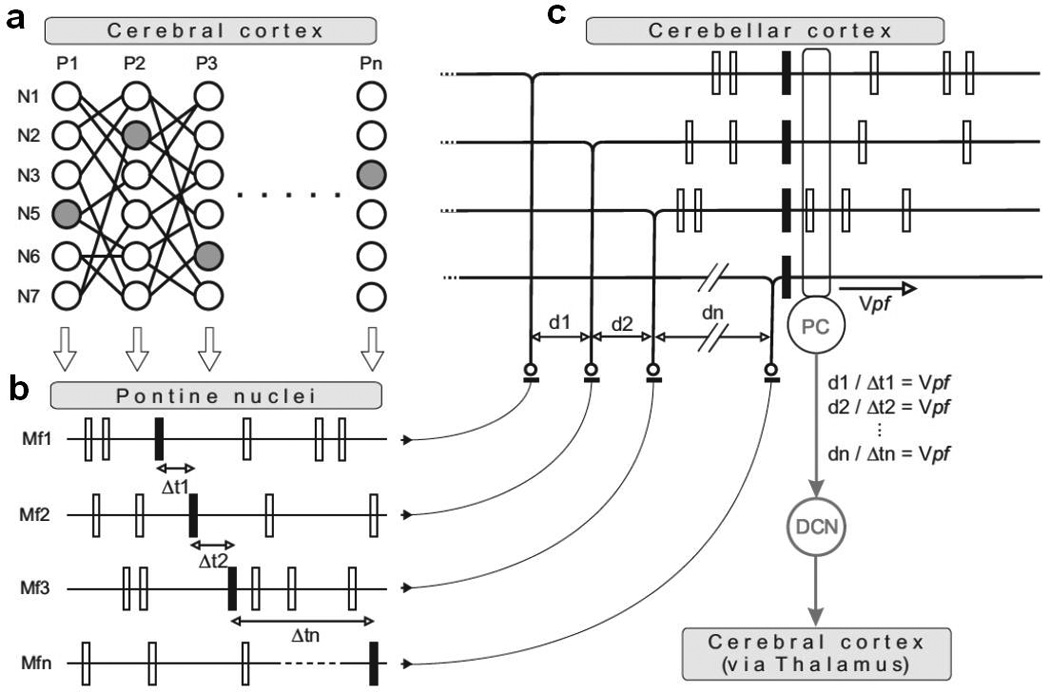
Schematic illustration of how spatiotemporal spike activity patterns in the neocortex are selectively detected by the cerebellum resulting in precisely timed cerebellar output responses. a Schematic drawing of several pools (P1 – PN) of neurons (drawn as circles, N1 –N7) in the neocortex connected through excitatory projections and propagating synchronous activity from one pool to the next. Gray filled circles represent neurons projecting to the pontine nuclei, the relay nuclei from where mossy fiber projections to the cerebellum originate. b Firing patterns of excitatory neurons in the pontine nuclei (Mf1 – Mfn) with axons projecting as mossy fibers to the cerebellum. The spatiotemporal activity patterns in the neocortex drive either unaltered or transformed spatiotemporal activity patterns in the pontine nuclei neurons. c Cerebellar granule cells receiving mossy fiber input generate action potentials that travel along the slow conducting parallelfibers (conductance velocity Vpf ≈ 0.5 m/s). If the combination of time delay and spatial separation in the mossy fiber inputs match the conductance velocity of the parallel fibers (e.g. Vpf = d1/Δt1) sequential mossy fiber activity will be synchronized by the delays introduced by parallel fibers and result in synchronized inputs to Purkinje cells (PC), triggering precisely timed Purkinje cell output to deep cerebellar nuclei (DCN) and eventually to the neocortex via the cerebellar-thalamo-cortical pathways.
Gene-environment interactions and cerebellar development in Autistic Disorder (A.M. Persico)
The cerebellum was the first brain region shown in the late 80’s to host neuroanatomical abnormalities in many autistic individuals [33, 228, 229]. The anomalies most frequently reported include a smaller vermis volume, Purkinje cell numbers reduced by up to 50%, ectopic neurons in the white matter, and patchy cytoarchitectonic ectopias in the cerebellar cortex [32, 33, 118, 228, 229]. Other brain regions also display abnormalities generally resulting from reduced programmed cell death and/or increased cell proliferation, altered cell migration, and abnormal cell differentiation with reduced neuronal size and abnormal wiring [118, 230]. These neurodevelopmental processes physiologically occurs during the first and second trimester of pregnancy [231]. Therefore, although the onset of deficits in social interaction and communication, stereotypies and insistence on sameness generally occurs after a postnatal time window of apparently normal behavior, autistic disorder is viewed by most experts as a neurodevelopmental disorder with prenatal origin or at least with essential prenatal components.
Several lines of evidence in autism research have spurred increasing interest in gene-environment interactions. The incidence of autism has dramatically risen during the last two decades from 2–5/10,000 to approximately 1–2/1,000 children [232]; in addition to broader diagnostic criteria and greater awareness, a real increase in incidence is also likely [230, 233]. Secondly, genetics strongly contributes to the pathogenesis of AD, but many patients reveal no obviously pathogenic mutations or copy number variants, and initial heritability estimates as high as >90% [234] have been challenged by more recent twin studies [235]. Finally, the validity of gene-environment interaction models is also supported by known prenatal teratological agents, such as rubella or cytomegalovirus infection, and drugs like thalidomide, misoprostol, and valproic acid, which cause autism only in a subset of presumably vulnerable individuals [230, 233].
Environmental factors potentially contributing to AD have been recently reviewed [233]. Here we shall briefly focus on three specific gene-environment interaction models especially relevant to cerebellar development:
1) RELN x PON1 x prenatal exposure to organophosphate pesticides or excessive oxidative stress
The RELN gene encodes for reelin, a stop signal protein critical to neuronal migration in the cerebral and cerebellar cortices. Among several mechanisms, this function also relies on a proteolytic activity exerted by reelin on extracellular matrix proteins, which is inhibited by organophosphate (OP) pesticides [134]. The PON1 gene encodes for a calcium-dependent enzyme frequently defined “paraoxonase”, which exerts several enzymatic activities including the inactivation of OPs, the degradation of lipid peroxides preventing atherosclerosis and vascular disease, the breakdown of bacterial endotoxins, and the prevention of protein lactonation [236]. Both Reelin protein levels and PON1 enzymatic activity (measured as “arylesterase” in these studies) are significantly reduced in AD individuals compared to controls [89, 236–238]. At the same time, autism is associated with RELN gene variants yielding reduced Reelin gene expression both in vitro and in vivo, and with PON1 gene variants responsible for lower gene expression and reduced detoxification of some OP compounds [140, 239, 240]. Hence individuals carrying genetic variants expressing reduced amounts of Reelin and PON1 enzyme, if exposed prenatally to OPs during critical periods in neurodevelopment, could be more likely to suffer from altered neuronal migration resulting in AD [230]. Very recent and exciting data indeed show: (a) an association between prenatal exposure to OPs, PON1 genotypes, and poorer neurobehavioral development in humans [241], as well as (b) abnormal CNS lamination including discontinuities in the cerebellar Purkinje cell layer and behavioral abnormalities in heterozygous reeler mice (expressing 50% lower amounts of reelin compared to wild-type mice) prenatally exposed to the OP chlorpyrifos [242]. However, the significant decrease in serum arylesterase, but not in diazoxonase PON1 activity recorded in our AD samples points toward excessive oxidative stress, previously documented in AD patients and boosted by OPs, as perhaps playing a more prominent role than acetylcholinesterase inhibition [236].
2) ATP2B2 x SLC25A12 x polychlorinated biphenyls (PCBs)
Several lines of evidence, including rare mutations affecting calcium (Ca2+) conductance in humans, indicate that excessive Ca2+ signalling plays a pivotal role in the pathophysiology of autism [243]. Ca2+ is rapidly removed from the cytoplasm into the extracellular space by plasma membrane calcium ATPases (PMCAs); the ATP2B2 gene encodes for PMCA2, whose faster kinetic and broader Ca2+ affinity range makes this PMCA critical to synaptic function and plasticity especially in the dendritic spines of Purkinje cells and in parvalbumin-positive gabaergic interneurons [244–246]. ATP2B2 gene variants possibly yielding reduced gene expression in post-mortem brains have recently been found associated with autism in males [247]. PCBs are important endocrine disruptors which, among several other effects, perturb intracellular Ca2+ signalling by promoting Ca2+ entry and by enhancing Ca2+ release from the endoplasmic reticulum through ryanodine receptors [248]. Hence, individuals carrying ATP2B2 gene variants possibly associated with reduced gene expression, if prenatally exposed to PCBs, can be predicted to develop intracellular Ca2+ spikes of greater duration and/or amplitude, likely interfering with neuronal migration, dendritic spine formation and synaptogenesis, especially in the parallel fiber-to-Purkinje cell synapse. Excessive intracellular Ca2+ spikes can in turn modulate, among several intracellular molecules and pathways, the aspartate/glutamate mitochondrial carrier AGC1 encoded by the SLC25A12 gene, leading to abnormal energy metabolism and enhanced oxidative stress [249]. Also SLC25A12 gene variants have been found associated with either liability or protection from autism [249].
3) MET and polycyclic aromatic hydrocarbons (PAHs)
the MET receptor tyrosine kinase, encoded by the MET protooncogene, exerts an important role in the nervous, gastrointestinal and immune systems, by modulating cell proliferation and migration, as well as neurite outgrowth and synaptogenesis. Its developmental role in the cerebellum is especially well established both in mammals and in zebrafish, albeit with evolutionarily determined species specificities [250]. MET mRNA and protein levels are decreased by as much as 50% in AD brains compared to matched controls [251]. Furthermore, the C allele at rs1858830, located in the MET gene promoter, is significantly associated with autism and decreases MET transcription both in vitro and in postmortem brains [251, 252]. Mice prenatally exposed to the PAH benzo(a)pyrene display blunted MET gene expression and behavioral deficits in a novelty test [253]. Therefore, individuals carrying the MET C allele at rs1858830 and prenatally exposed to PAHs may be particularly at risk of undergoing abnormal neurodevelopment leading to cognitive or autistic symptoms. Furthermore, interactions with PAHs may involve loci encoding other proteins of the MET pathway, as these show secondary changes in gene expression [252], SERPINE1 and PLAUR gene variants are also associated with autism [254], and significant gene-gene interactions have been shown between MET and PLAUR [254].
The three scenarios briefly outlined above exemplify the heuristic potential of investigating gene-environment interactions in neurodevelopmental disorders. This field is especially promising for cerebellar neuropathology, in light of the prolonged developmental time window exposing cerebellar circuits to damage by environmental agents in genetically susceptible individuals not only prenatally, but also postnatally [231].
Therapeutics in autism with relevance to the cerebellum (S.J. Webb, J. Welsh, B.H. King)
Although the cerebellum has been identified as a potential region of interest for autism and also for a variety of other neurobehavioral syndromes and symptoms that overlap with autism, it has received very little direct attention from the standpoint of being a therapeutic target. Indeed, to the extent that the therapeutic armamentarium is aimed in a specific way at autism, the specificity derives from neurotransmitter systems broadly, or in some cases receptor subtypes, such as metabotropic glutamate receptors like mGluR5 [255], that are known or believed likely to influence the development or expression of core or related autism symptoms. Strategies to target specific brain structures or neuroanatomical pathways have not yet been utilized in the service of autism therapeutics.
Nevertheless, the cerebellum is central in a wide range of functions sometimes found to be impaired in autism, including timing and coordination of movement, motor learning, evaluation of the match between intention and action, predictive learning, environmental exploration, behavioral inhibition, attention, and visual orienting. In particular, the cerebellar vermis is associated with modulation of limbic functions including emotion, sensory reactivity, and salience detection. The cerebellar hemispheres have been linked to several higher-order cerebral functions including language, working memory, planning and behavioral sequencing [e.g., 192, 256]. Moreover, the cerebellum is one of the busiest intersections in the brain--receiving signals from and sending signals back to nearly every major neural system.
In as much as motor planning and performance deficits have been demonstrated in association with cerebellar abnormality in some children with autism [e.g., 199], and restricted and repetitive behaviors in others [e.g., 257], some current behavioral targets may actually be proxies for the cerebellum. Casting a broader net, cerebellar involvement cannot be excluded from a number of behaviors often associated with autism that are very common pharmacological treatment targets -- including hyperactivity, aggression, irritability, and mood symptoms [258].
Here again, symptomatic improvement may be mediated in important ways by drug effects on the cerebellum. For example, moderate and high doses of methylphenidate modified T2 relaxation time (a measure of relative blood volume or flow) in the vermis in boys with ADHD: for boys with ADHD who were the most hyperactive T2 relaxation time increased; for boys with ADHD who were nonhyperactive, T2 relaxation time was lower [259]. Clarifying the relationship between vermal size, vermal blood flow, stimulant response, and the developmental pathophysiology of attention will be important within autism as well.
Serotonergic drugs are among the most widely prescribed medications for individuals with autism, and while not specifically studied in autism for their cerebellar effects, it is interesting that the SSRI, fluoxetine, rescues some aspects of the consequences of neuronal cell death in the cerebellum in the Weaver mouse via its effects on GIRK2 channels [260].
GABAergic dysfunction in the cerebellum in autism has been demonstrated repeatedly [70, 124, 261]. While benzodiazepine use in autism is relatively uncommon, owing to concerns of behavioral disinhibition, some anticonvulsants that likely exert GABAergic effects are used for mood stabilization [262], and there is ongoing interest in the potential role of GABAergic treatments for catatonia in autism [263]. Additionally, the GABA B receptor agonist, arbaclofen, has been reported to be helpful in preliminary trials for irritability and social withdrawal in autism and is moving forward in clinical trials [264].
Drugs that are known to have more direct cerebellar effects may deserve particular consideration in autism and related conditions based on the historical weight of evidence implicating this structure. Thus, a pharmacological approach that has not been studied in autism, but which may deserve consideration based upon cerebellar findings of Ji and colleagues [112] may be calcium channel modulating agents. Ji and colleagues found brain-region specific increases in the activities of Na(+)/K(+)-ATPase and Ca(2+)/Mg(2+)-ATPase in autism and suggested that increased activity of these enzymes in the cerebellum may be due to compensatory responses to increased intracellular calcium concentration. Calcium channelopathies have been identified as a drug development target in autism [265], and may deserve to be prioritized.
Another approach for autism therapeutics may be to address the cerebellum transynaptically. Such an approach may be uniquely applied to the cerebellum due to the fact that all output of the cerebellum is strongly regulated by the inferior olive (IO), a punctate brain structure in the medulla. IO neurons project monosynaptically to Purkinje cells, the only output neuron of the cerebellar cortex, and regulate the throughput of the mossy fiber/parallel fiber system. IO neurons are a compelling drug target, not simply because of their ability to control cerebellar output, but also due to their unique complement of ionic channels and neurotransmitter receptors. Serotonin and NMDA receptors within the IO can potently modulate cerebellar output, either by changing HCN and Cav3.1 channel activity after 5-HT2A receptor activation to modulate pacemaking [266, 267], or by changing spatial patterns of electrotonic coupling after NMDA receptor activation to regulate synchronous spiking in the cerebellum [268]. Moreover, T-type calcium channels are a powerful mechanism by which the IO regulates cerebellar output [269]. These calcium channels are an increasingly recognized target for investigational therapeutics, and are abundantly expressed in the primate IO [47]. Thus, therapeutics targeted at precerebellar afferents may present future opportunities to address cerebellar pathophysiology in children with autism.
The opportunities going forward will be to better understand the role of the cerebellum in the expression of autism symptoms, and its role in treatment response. Although Webb et al. [233] did not find any relation in 3 to 4 year old children between general indices of autism symptom severity, IQ or adaptive functioning and vermal area or cerebellum volume, this would be an early age to see children receiving pharmacotherapy. In children aged 6–14 years, cerebellar white matter abnormalities were associated with repetitive behaviors [270]; and in adults, lower fractional anisotropy in the superior cerebellar peduncles was associated with worse parent report of social behaviors in autism [271]. Kates et al. [272] also found that cerebellar white matter, specifically the discrepancy in volume between twins, was associated with discrepancies in the twins’ autism symptom scores. Taken together, cerebellar abnormalities may be a useful marker for subdividing the population with autism with common behavioral symptom targets, and a better understanding of the role of the cerebellum and its afferents in autism could importantly inform treatment selection and future drug development.
Animal models of cerebellar neuropathology relevant to autism research (P.E. Dickson*, L.A. Martin*, C.D. Blaha, G. Mittleman, D. Goldowitz; *authors contributed equally to this paper)
Autistic disorder is a behaviorally-defined, multi-causal, developmental disorder that is associated with various neuropathologies [273]. The most consistently reported neuropathology is a reduction in cerebellar Purkinje cells, the sole outflow of cerebellar cortex [6, 120]. Mounting evidence from human neuroimaging and acquired lesion studies suggests that the cerebellum may modulate executive functions [274], likely by means of reciprocal connections to cortical regions [26]. This raises the intriguing possibility that the cerebellar pathology commonly observed in AD may cause the executive dysfunction that has been proposed to contribute to, or even underlie, the core symptoms of AD by disrupting cerebellar-cortical interactions during critical periods of development [275, 276]
There are several animal models that lend support to a role for cerebellar pathology in AD. For example, at least 4 genes that have been linked to AD over the past decade have also been shown to play an important role in cerebellar development: IB2 (Islet Brain 2), Pax6 (Paired box 6), En2 (Engrailed homeobox 2) and Met (Met proto-oncogene). Of these genes, both IB2 and Pax6 are explicitly linked with syndromes that are co-morbid for AD: Phelan-McDermid syndrome and WAGR (Wilm’s tumor, Aniridia, Genitourinary malformations, and mental Retardation syndrome). Specific deletion of IB2 in mice results in altered Purkinje cell morphology and disruptions in cerebellar glutamatergic transmission [277]. While Pax6 is known to contribute widely to neurodevelopment including the retina, it has recently been shown to play a cell-intrinsic role in granule cell development [278]. Behavioral analyses of both IB2 and Pax6 animal models have also indicated a possible link between these genes and autistic-like behavior [277, 279]. Multiple studies have linked the En2 and Met genes to AD [53], and animal models involving disruptions to these genes have demonstrated their importance to normal cerebellar development [69, 250, 280]. In addition to genetic influences, AD may also be caused by environmental factors. For example, there is well-documented support for prenatal valproic acid exposure as a cause of AD [281]. Interestingly, rats prenatally exposed to valproic acid exhibit cerebellar neuropathology similar to that found in AD [282].
The above animal models provide support for a role of cerebellar pathology in AD but do not particularly address this relationship. In order to specifically explore the contribution of cerebellar pathology to the AD phenotype, we created a mouse model with variable Purkinje cell loss utilizing aggregation chimeras made from wildtype and Lurcher mouse embryos. Heterozygous Lurcher mice lose 100% of their cerebellar Purkinje cells due to a gain-of-function mutation in the δ2 glutamate receptor gene [283], while individual wildtype↔Lurcher chimeras have between 0 and 100% of normal Purkinje cell numbers depending on the percentage of mutant cells that colonize the cerebellum (Figure 7).
Figure 7.
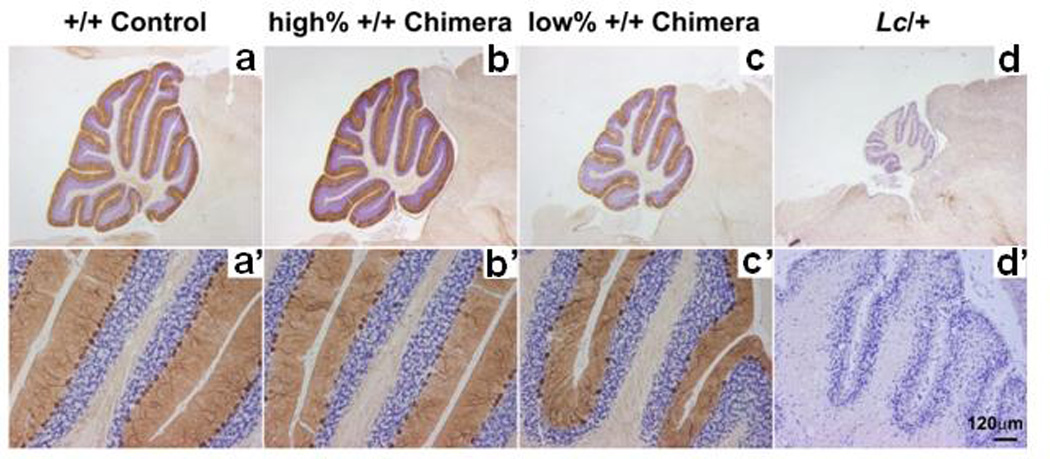
Photomicrographs of cerebellar sections from Lurcher (Lc/+), wildtype (+/+), and Lc/+↔+/+ chimeras taken at both low (top row) and high (bottom row) magnification demonstrating the loss of Purkinje cells in Lc/+ and chimeric mice compared to a control sample. Purkinje cells were stained immunocytochemically with the anti-Calbindin antibody (Chemicon) and appear dark brown in a single monolayer above the blue-stained cerebellar granule cells (cresyl violet counterstain). a Section from a +/+ control cerebellum with normal Purkinje cell number and cerebellar size. b Section from the cerebellum of a high-percentage +/+ chimera with a relatively minor loss of Purkinje cells. c Section from the cerebellum of a low-percentage +/+ chimera showing a relatively major loss of Purkinje cells. d Section from a Lc/+ cerebellum demonstrating the complete loss of Purkinje cells and extreme decrease in cerebellar size. Scale bar = 500 µm a–d, 120 µm a'–d'. Copyright © 2006 by the American Psychological Association. Reproduced with permission from [290]. The use of APA information does not imply endorsement by APA.
Through a series of experiments utilizing this model, we investigated the causal relationship between developmental loss of cerebellar Purkinje cells and AD-like phenotypes with a particular emphasis on executive dysfunction. Two principal findings resulted from these studies. First, the developmental loss of cerebellar Purkinje cells affected performance on some, but not all executive functions. As shown in Figure 8, mice with reduced Purkinje cell numbers displayed impaired performance on the later stages of a non-spatial serial reversal learning task, a measure of behavioral flexibility [284], but, surprisingly, facilitated working memory performance on a delayed matching-to-position task [285]. No relationship was observed between Purkinje cell number and performance on a sustained attention task [286]. Second, we found a negative relationship between Purkinje cell number and (1) repetitive lever pressing in a progressive ratio task, (2) activity in an open field, and (3) investigatory nose poking in a hole-board exploration task [287]. Importantly, individual chimeric mice did not show signs of motor impairment unless they lost greater than 90% of their Purkinje cells in which case they were excluded from the analyses [285]. Therefore, the observed relationships between Purkinje cells and dependent measures were not due to motor deficits.
Figure 8.
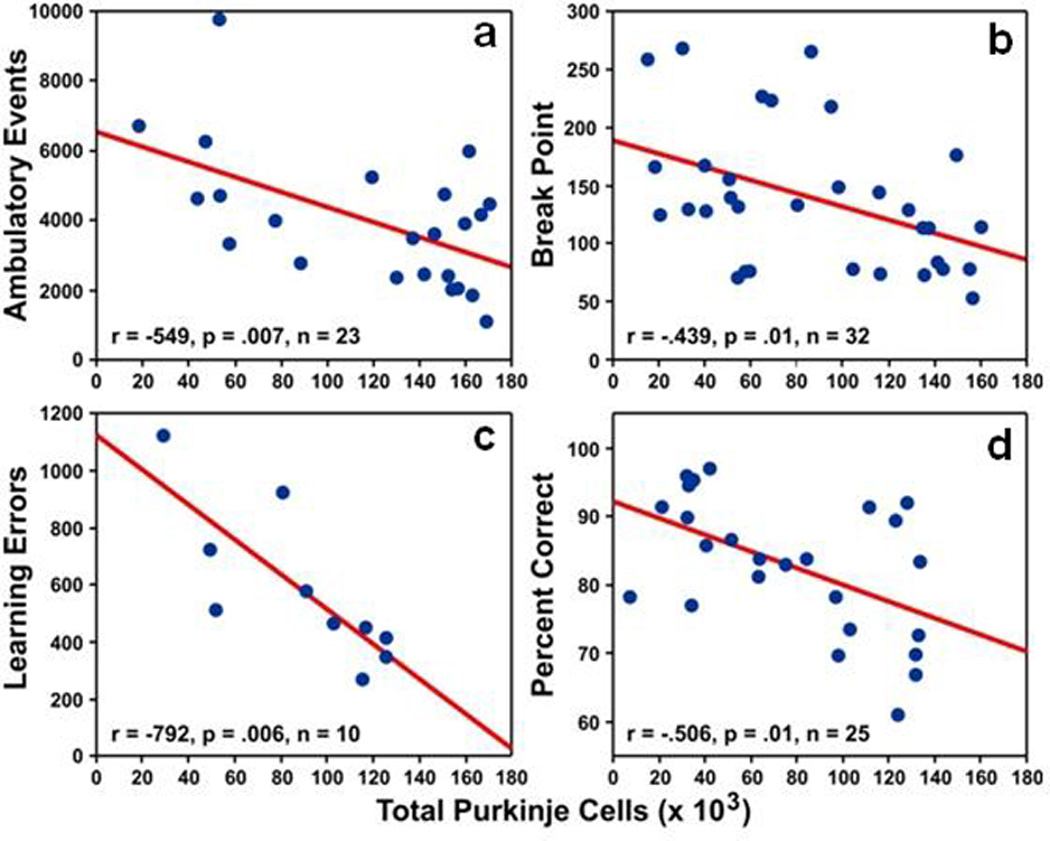
In a series of experiments using chimeric mice with varying numbers of Purkinje cells we tested the relationship between Purkinje cell number and behaviors relevant to the autism phenotype. Scatterplots depicting significant relationships between Purkinje cell number and (a) ambulatory events in an open field, (b) breakpoint in a progressive ratio task, (c) learning errors during the third reversal of a conditional visual discrimination, and (d) percent correct on a delayed matching-to-position task (24 sec delay). The best fit regression line is shown in each graph. Mice used in these analyses did not have motor deficits. Collectively these results indicate that low percentage chimeric mice are hyperactive (a), show higher levels of repetitive behavior (b), and have larger deficits in executive function (c) than high percentage chimeric mice. Reduced numbers of Purkinje cells were also surprisingly related to enhanced short term memory (d), which may be analogous to the savant skills shown by some patients with autism. a and b are reprinted from [287] with permission from John Wiley & Sons, Inc. c is reprinted from [284] with permission from Elsevier. d is reprinted from [285] with permission from John Wiley and Sons, Inc.
A second set of experiments in heterozygous Lurcher mice and wildtype controls used in vivo electrochemistry to examine possible mechanisms by which efferent projections from the cerebellum could modulate prefrontal cortex (PFC) function (Figure 9, top) [180]. Lurcher mice with a total absence of Purkinje cells showed marked attenuations in cerebellar stimulation-evoked dopamine transmission in the medial PFC (Figure 9, bottom) which could account for the altered performance on behavioral measures of executive function, repetitive behaviors and restricted interests.
Figure 9.
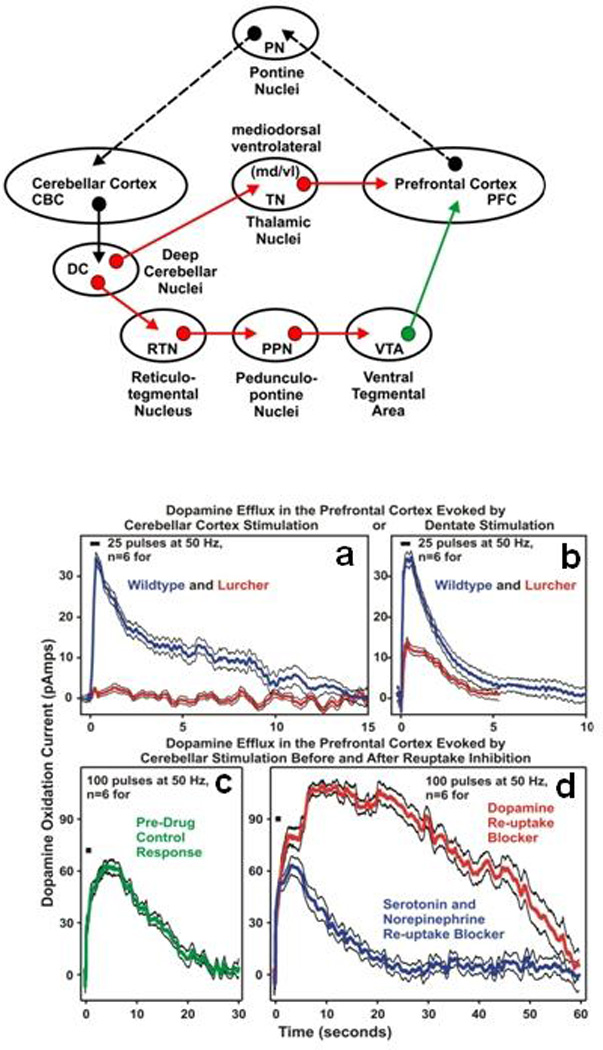
(Top) Neuronal circuitry underlying cerebellar modulation of PFC dopamine neurotransmission. Cerebellar modulation of dopamine release in the PFC may occur via polysynaptic inputs from cerebellar nuclei to dopamine-containing cells in the ventral tegmental area or via a monosynaptic input to thalamic projections making close appositions with dopamine terminals synapsing onto PFC pyramidal cells. Glutamatergic pathways are shown as red lines and the dopaminergic pathway as a green line. Dashed lines represent PFC feedback to cerebellum via the pontine nuclei. Nuclei abbreviations are shown in the ovals. To develop an animal model of autism, we used electrochemical methods to determine how and by what neural circuits cerebellar activity modulates PFC dopamine release and electrophysiological techniques to determine the impact of this modulation on PFC cellular activity in Lurcher mice with a complete loss of Purkinje cells and wildtype controls. (Bottom) Electrical stimulation of the cerebellar cortex evokes a long lasting increase in dopamine release in the PFC of wildtype mice; an effect that is absent in Lurcher mice with 100% loss in Purkinje cells. Dopamine release in the PFC evoked by 25 pulses (black bar) of stimulation (200 µA) at 50 Hz of the (a) cerebellar cortex and (b) contralateral ventromedial portion of the dentate nucleus in urethane anesthetized wildtype mice (blue lines) and Lurcher mice (red lines) bearing a 100% deficit in Purkinje cell numbers. Serotonin and norepinephrine reuptake blockade fails to alter cerebellar cortex evoked dopamine release in the PFC. Compared to the pre-drug control response (c, green line), selective blockade of dopamine reuptake in wildtype mice with nomifensine (20 mg/kg i.p.) significantly enhanced PFC dopamine release evoked by 100 pulses (black bar) of stimulation (200 µA) at 50 Hz (d, red line), whereas blockade of serotonin and norepinephrine reuptake with a combined fluoxetine and desipramine injection (20 mg/kg i.p. each), respectively, failed to alter the evoked response (d, blue line). In (a to d) the blue, red, and green center lines and outer black lines are the mean+SEM, respectively (n=6 for wildtype and Lurcher mice). Reprinted from [180] with permission from John Wiley & Sons, Inc
Collectively, the studies outlined here support a role for the cerebellum in AD and provide clues to the mechanisms by which cerebellar pathology may contribute to the overall AD phenotype. Many questions remain, however, including how the specific timing, location, and degree of a cerebellar insult affect higher order cognitive functions and lead to AD symptoms. The use of inducible and/or conditional genetically modified mice will allow future studies to explore these questions.
Conclusions
The main points of consensus derived from various areas of discussion related to the role of the cerebellum in autism include the following: 1) The anatomy of the cerebellum in autism is abnormal in at least a subset of individuals; the onset of pathology is prenatal and this process continues post-natally, affecting many cerebellar functions; 2) AD is genetically heterogeneous; the analysis of syndromic disorders with co-occurring AD, including FXS, TS, JS, and Dandy-Walker malformation, points to the involvement of cerebellar genes. Additionally, three autism-associated genes, EN2, GABRB3, and MET, have roles in cerebellar development; 3) There is ongoing neuroinflammation in the brains of subjects with autism, including cerebellum, from early age to later in life; 4) Oxidative stress is present in the cerebellum of subjects with autism; 5) Abnormalities involving several neurotransmitters, amino acids and brain proteins, including GABA, glutamate, Reelin, acetylcholine, dopamine, serotonin, and oxytocin exist in brains of subjects with autism; 6) The presence of motor and cognitive deficits in autism are reflective of specific cerebellar abnormalities; 7) gene-environmental interactions can have adverse effects on developing cerebellar circuitry as seen in autism; 8) Multiple drug treatment options, such as calcium channel modulating agents, GABAB receptor agonists, and serotonin and NMDA receptor modulating drugs may be of potential use in treatment of autism; 9) Multiple relevant animal models point to the impact of the cerebellum on dopamine fluctuations in the frontal cortex, underlying executive functions; 10) Prenatal exposure to teratologic agents, such as VPA, have been shown to contribute to autism in subsets of vulnerable individuals as well as contribute to cerebellar neuropathology similar to autism in animal models.
Despite many novel findings discussed in the preceding sections, multiple issues remain undefined and require further research in order to obtain a more complete picture of cerebellar dysfunction in autism. These items deal with: 1) Unknown mechanisms underlying neuroinflammation in the brains of subjects with autism; 2) Unknown neuronal mechanisms underlying cerebellar functions in cognition; 3) Lack of availability of specific drugs suitable for treatment of core symptoms of autism; 4) Imaging studies do not allow conclusions with respect to either total size of cerebellum or its subregions. This is especially true since there is a lack of stereological analysis of postmortem cerebellum in autism which can be correlated with the imaging studies (such a report now exists with respect to neuron number and size in prefrontal cortex in children with autism; see [288); 5) Reductions in size and number of Purkinje cells in cerebellar vermis in autism remain unresolved due to lack of stereological reports [311]; 6) Vermal hypoplasia affecting lobules VI and VII or other lobules requires further assessment; 7) It is unclear whether oxidative stress is a primary cause or a secondary consequence in autism; an; 8) More definitive behavioral tests are required to confirm the validity of animal models for autism.
Acknowledgements
1) Dr. S. Hossein Fatemi appreciates the excellent technical assistance by Rachel Elizabeth Kneeland in edition of this consensus paper, and the critical review of the manuscript by Mr. Timothy D. Folsom. Research support for Dr. Fatemi’s work is from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (5R01HD052074-05 and 3R01HD05207403-S1 supplemental grant), as well as the Alfred and Ingrid Lenz Harrison Autism Initiative; 2) Research support for work by Drs. Kimberly A. Aldinger and Kathleen J. Millen is from the Autism Speaks Foundation; 3) Research support for work by Dr. Paul Ashwood is from Autism Speaks Foundation, the Jane Botsford Johnson Foundation, National Alliance for Research on Schizophrenia and Depression, National Institute of Neurological Disorders and Stroke R21HD065269 and, the Peter Emch Foundation is gratefully acknowledged; 4) Research support for work by Drs. Margaret L. Bauman and Thomas L. Kemper is from the Nancy Lurie Marks Family Foundation, by NINDS (NS38975-05) and by NAAR/Autism Speaks. We would also like to acknowledge and thank the many families whose generous donation of postmortem brain tissue has made this research possible; 5) Research support for work by Drs. Charles D. Blaha, Price E. Dickson, Dan Goldowitz, Loren A. Martin, and Guy Mittleman is from Cure Autism Now, Autism Speaks and R01 NS063009 from NIH/NINDS; 6) Research support for work by Dr. Gene Blatt is from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (5R01HD039459-06) and The Hussman Foundation; 7) Research support for work by Drs. Abha Chauhan and Ved Chauhan is from the Department of Defense Autism Spectrum Disorders Research Program AS073224P2, the Autism Research Institute, the Autism Collaboration (autism.org), and the NYS Office for People with Developmental Disabilities; 8) Drs. Stephen R. Dager, Annette M. Estes, and John P. Welsh would like to thank Elizabeth Kelly for assistance in preparing their manuscript. Research support for their work is from Autism Centers of Excellence (NICHD P50-HD055782), Collaborative Programs of Excellence in Autism (NICHD #HD35465 NICHD, RO1-HD05571), ACE Network (NICHD RO1 supplement HD05571), American Recovery and Reinvestment Act (NICHD R01- HD065283) and National Institute for Neurological Disorders and Stroke (NINDS R01-NS31224-18). Support from Autism Speaks and the Simons Foundation is also gratefully acknowledged; 9) Research support for Dr. Detlef H. Heck is from NIH grants RO1NS060887 & R01NS063009. The content of this publication is solely the responsibility of the author and does not necessarily represent the official views of the NIH; 10) Research support for Drs. Bryan H. King, Sara J. Webb, and John P. Welsh is from the Eunice Kennedy Shriver National Institute of Child Health & Human Development (P50 HD055782; King/Webb) and the National Institute for Neurological Disorders and Stroke (R01 NS31224-18; Welsh). We want to thank the families of and individuals with autism who have participated in research; 11) Research support for work by Drs. Matthew W. Mosconi and John A. Sweeney is from the Autism Center of Excellence Award Number P50HD055751 from the Eunice Kennedy Shriver NICHD, NIMH Grant 1K23MH092696, and Autism Speaks Grant 4853. Dr. John Sweeney consults with Pfizer and Takeda, and has received a grant from the Janssen Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding institutes; 12) Research support for work by Dr. Antonio M. Persico is from the Italian Ministry of University, Research and Technology (PRIN 2006058195 and 2008BACT54), the Italian Ministry of Health (RFPS-2007-5-640174), Autism Speaks (Princeton, NJ), the Autism Research Institute (San Diego, CA), and the Fondazione Gaetano e Mafalda Luce (Milan, Italy).
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
References:
- 1.American Psychiatric Association Diagnostic and Statistical Manuel of Mental Disorders. (DSM-4) 4th ed. Washington, DC: APA; 1994. [Google Scholar]
- 2.Wassink TH, Brzustowicz LM, Bartlett CW, Szatmari P. The search for autism disease genes. Ment Retard Dev Disabil Res Rev. 2004;10:272–283. doi: 10.1002/mrdd.20041. [DOI] [PubMed] [Google Scholar]
- 3.Bauman ML, Kemper TL. Histoanatomic observations of the brain in early infantile autism. Neurology. 1985;35:866–874. doi: 10.1212/wnl.35.6.866. [DOI] [PubMed] [Google Scholar]
- 4.Arin DM, Bauman ML, Kemper TL. The distribution of Purkinje cell loss in the cerebellum in autism. Neurology. 1991;41(Suppl):307. [Google Scholar]
- 5.Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, et al. A clinicopathological study of autism. Brain. 1998;121:889–905. doi: 10.1093/brain/121.5.889. [DOI] [PubMed] [Google Scholar]
- 6.Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum. 2008;7(3):406–416. doi: 10.1007/s12311-008-0043-y. [DOI] [PubMed] [Google Scholar]
- 7.Bauman ML, Kemper TL, editors. The Neurobiology of Autism. Baltimore: Johns Hopkins University Press; 2005. [Google Scholar]
- 8.Courchesne E, Saitoh O, Yeung-Courchesne R, Press GA, Lincoln AJ, et al. Abnormality of cerebellar vermian lobules VI and VII in patients with infantile autism: identification of hypoplastic and hyperplastic subgroups by MR imaging. AJR. 1994;162:123–130. doi: 10.2214/ajr.162.1.8273650. [DOI] [PubMed] [Google Scholar]
- 9.Whitney ER, Kemper TL, Rosene DL, Bauman ML, Blatt GJ. Density of cerebellar basket and stellate cells in autism: Evidence for a late developmental loss of Purkinje cells. J Neurosci Res. 2009;87:2245–2254. doi: 10.1002/jnr.22056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes G, Stewart TG. On the connection of the inferior olives with the cerebellum in man. Brain. 1908;31:125–137. [Google Scholar]
- 11.Greenfield JG. The Spino-cerebellar Degenerations. Springfield, IL: CC Thomas; 1954. [Google Scholar]
- 12.DeBassio WA, Kemper TL, Knoefel JE. Coffin-Siris syndrome: Neuropathological findings. Arch of Neurol. 1985;42:350–353. doi: 10.1001/archneur.1985.04060040060012. [DOI] [PubMed] [Google Scholar]
- 13.Kemper TL. The developmental neuropathology of autism. In: Blatt G, editor. The Neurochemical Basis of Autism. New York: Springer; 2010. pp. 69–82. [Google Scholar]
- 14.Kemper TL, Bauman ML. Neuropathology of Infantile Autism. J Neuropath Exp Neurol. 1998;57:645–652. doi: 10.1097/00005072-199807000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Hazlett HC, Poe M, Gerig G, Smith RG, Provenzale J, Ross A, et al. Magnetic Resonance Imaging and head circumference study of brain size in autism: birth through age 2 years. Arch Gen Psychiatry. 2005;62:11366–11376. doi: 10.1001/archpsyc.62.12.1366. [DOI] [PubMed] [Google Scholar]
- 16.Lainhart JE, Bigler ED, Bocian M, Coon H, Dinh E, Dawson G, et al. Head circumference and height in autism: a study by the Collaborative Program of Excellence in Autism. Am J Med Genet A. 2006;140:2257–2274. doi: 10.1002/ajmg.a.31465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardan AY, Libove RA, Keshavan MS, Melhem NM, Minshew NJ. A preliminary longitudinal magnetic resonance imaging study of brain volume and cortical thickness in autism. Biol Psychiatry. 2009;66:313–315. doi: 10.1016/j.biopsych.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmahmann J. International Review of Neurobiology. Vol 41. SanDiego: Academic Press; 1997. The cerebellum and Cognition. [Google Scholar]
- 19.Schmahmann JD, Rosene DL, Pandya DN. Motor projections to the basis pontis in rhesus monkey. J Comp Neurol. 2004;478:248–268. doi: 10.1002/cne.20286. [DOI] [PubMed] [Google Scholar]
- 20.Fournier KA, Hass CJ, Naik SK, Lodha N, Cauraugh JH. Motor Coordination in Autism Spectrum Disorders: A Synthesis and Meta-Analysis. J Autism Dev Disord. 2010;40(10):1227–1240. doi: 10.1007/s10803-010-0981-3. [DOI] [PubMed] [Google Scholar]
- 21.Minshew NJ, Sung K, Jones B, Furman JM. Underdevelopment of the postural control system in autism. Neurology. 2004;63(11):2056–2061. doi: 10.1212/01.wnl.0000145771.98657.62. [DOI] [PubMed] [Google Scholar]
- 22.Ozonoff S, Young GS, Goldring S, Greiss-Hess L, Herrera AM, Steele J, et al. Gross motor development, movement abnormalities, and early identification of autism. J Autism Dev Disord. 2008;38:644–656. doi: 10.1007/s10803-007-0430-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brettler SC, Fuchs AF, Ling L. Discharge patterns of cerebellar output neurons in the caudal fastigial nucleus during head-free gaze shifts in primates. Ann NY Acad Sci. 2003;1004:61–68. [PubMed] [Google Scholar]
- 24.Takarae Y, Minshew NJ, Luna B, Sweeney JA. Oculomotor abnormalities parallel cerebellar histopathology in autism. J Neurol Neurosurg Psychiatry. 2004;75(9):1359–1361. doi: 10.1136/jnnp.2003.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nowinski CV, Minshew NJ, Luna B, Takarae Y, Sweeney JA. Oculomotor studies of cerebellar function in autism. Psychiatry Res. 2005;137(1–2):11–19. doi: 10.1016/j.psychres.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Strick P, Dum R, Fiez J. Cerebellum and Nonmotor Function. Ann Rev Neurosci. 2009;32(1):413–434. doi: 10.1146/annurev.neuro.31.060407.125606. [DOI] [PubMed] [Google Scholar]
- 27.Ackermann H, Wildgruber D, Daum I, Grodd W. Does the cerebellum contribute to cognitive aspects of speech production? A functional magnetic resonance imaging (fMRI) study in humans. Neurosci Lett. 1998;247(2–3):187–190. doi: 10.1016/s0304-3940(98)00328-0. [DOI] [PubMed] [Google Scholar]
- 28.Tager-Flusberg H, Caronna E. Language Disorders: Autism and Other Pervasive Developmental Disorders. Pediatr Clin North Am. 2007;54(3):469–481. doi: 10.1016/j.pcl.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Shriberg L, Paul R, Black L, van Santen J. The Hypothesis of Apraxia of Speech in Children with Autism Spectrum Disorder. J Autism Dev Disord. 2011;41(4):405–426. doi: 10.1007/s10803-010-1117-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinlin M. The cerebellum in cognitive processes: Supporting studies in children. Cerebellum. 2007;6:237–241. doi: 10.1080/14734220701344507. [DOI] [PubMed] [Google Scholar]
- 31.Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain. 1998;121(4):561–579. doi: 10.1093/brain/121.4.561. [DOI] [PubMed] [Google Scholar]
- 32.Scott JA, Schumann CM, Goodlin-Jones BL, Amaral DG. A comprehensive volumetric analysis of the cerebellum in children and adolescents with autism spectrum disorder. Autism Res. 2009;2(5):246–257. doi: 10.1002/aur.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ritvo ER, Freeman BJ, Scheibel AB, Duong T, Robinson H, Guthrie D, et al. Lower Purkinje cell counts in the cerebella of four autistic subjects: initial findings of the UCLA–NSAC autopsy research project. Am J Psychiatry. 1986;143(7):862–866. doi: 10.1176/ajp.143.7.862. [DOI] [PubMed] [Google Scholar]
- 34.Courchesne E, Saitoh O, Townsend J, Yeung-Courchesne R, Press G, Lincoln A, et al. Cerebellar hypoplasia and hyperplasia in infantile autism. Lancet. 1994;343:63–64. doi: 10.1016/s0140-6736(94)90923-7. [DOI] [PubMed] [Google Scholar]
- 35.Ciesielski KT, Harris RJ, Hart BL, Pabst HF. Cerebellar hypoplasia and frontal lobe cognitive deficits in disorders of early childhood. Neuropsychologia. 1997;35(5):643–655. doi: 10.1016/s0028-3932(96)00119-4. [DOI] [PubMed] [Google Scholar]
- 36.Webb SJ, Sparks BF, Friedman SD, Shaw DW, Giedd J, Dawson G, et al. Cerebellar vermal volumes and behavioral correlates in children with autism spectrum disorder. Psychiatry Res. 2009;172(1):61–67. doi: 10.1016/j.pscychresns.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welsh JP, Yuen G, Placantonakis DG, Vu TQ, Haiss F, O’Hearn E, et al. Why do Purkinje cells die so easily after global brain ischemia? Aldolase C, EAAT4, and the cerebellar contribution to post-hypoxic myoclonus. Adv Neurol. 2002;89:331–359. [PubMed] [Google Scholar]
- 38.Sarna JR, Hawkes R. Patterned Purkinje cell death in the cerebellum. Prog Neurobiol. 2003;70:473–507. doi: 10.1016/s0301-0082(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 39.Welsh JP, Llinas R. Some organizing principles for the control of movement based on olivocerebellar physiology. Prog Brain Res. 1997;114:449–461. doi: 10.1016/s0079-6123(08)63380-4. [DOI] [PubMed] [Google Scholar]
- 40.Hawkes R, Colonnier M, Leclerc N. Monoclonal antibodies reveal sagittal banding in the rodent cerebellar cortex. Brain Res. 1985;333(2):359–365. doi: 10.1016/0006-8993(85)91593-8. [DOI] [PubMed] [Google Scholar]
- 41.Hawkes R, Gravel C. The modular cerebellum. Prog Neurobiol. 1991;36(4):309–327. doi: 10.1016/0301-0082(91)90004-k. [DOI] [PubMed] [Google Scholar]
- 42.Williams BL, Yaddanapudi K, Hornig M, Lipkin WI. Spatiotemporal analysis of Purkinje cell degeneration relative to parasagittal expression domains in a model of neonatal viral infection. J Virol. 2007;81:2675–2687. doi: 10.1128/JVI.02245-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Hearn E, Molliver ME. The olivocerebellar projection mediates ibogaine-induced degeneration of Purkinje cells: a model of indirect, trans-synaptic excitotoxicity. J Neurosci. 1997;17:8828–8841. doi: 10.1523/JNEUROSCI.17-22-08828.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Llinas R, Lang EJ, Welsh JP. The cerebellum, LTD, and memory: alternative views. Learn Mem. 1997;3:445–455. doi: 10.1101/lm.3.6.445. [DOI] [PubMed] [Google Scholar]
- 45.Dager SR, Corrigan NM, Richards TL, Posse S. Research Applications of Magnetic Resonance Spectroscopy (MRS) to Investigate Psychiatric Disorders. Top Magn Reson Imaging. 2008;19(2):81–96. doi: 10.1097/RMR.0b013e318181e0be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dager SR, Corrigan NM, Richards TL, Shaw DWW. Brain Chemistry: Magnetic Resonance Spectroscopy. In: Amaral D, Dawson G, Geshwind D, editors. Autism Spectrum Disorders. England: Oxford University Press; 2011. [Google Scholar]
- 47.Welsh JP, Han VZ, Rossi D, Mohr C, Odagari M, Daunais J, et al. Bidirectional plasticity in the primate inferior olive induced by chronic ethanol intoxication and sustained abstinence. Proc Natl Acad Sci USA. 2011;108:10314–10319. doi: 10.1073/pnas.1017079108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Welsh JP, Lang EJ, Sugihara I, Llinas R. Dynamic organization of motor control within the olivocerebellar system. Nature. 1995;374:453–457. doi: 10.1038/374453a0. [DOI] [PubMed] [Google Scholar]
- 49.Oristaglio J, Ghaffari M, Hyman West S, Welsh JP, Malone R. A sensory timing abnormality in autism revealed by classical eyeblink conditioning. Soc Neurosci Abstr, Forthcoming. 2011 [Google Scholar]
- 50.Gerwig M, Esser AC, Guberina H, Frings M, Kolb FP, Forsting M, et al. Trace eyeblink conditioning in patients with cerebellar degeneration: comparison of short and long trace intervals. Exp Brain Res. 2008;187:85–96. doi: 10.1007/s00221-008-1283-2. [DOI] [PubMed] [Google Scholar]
- 51.Welsh JP, Ahn ES, Placantonakis DG. Is autism due to brain desynchronization? Int J Dev Neurosci. 2005;23:253–263. doi: 10.1016/j.ijdevneu.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 52.Miles JH. Autism spectrum disorders--a genetics review. Genet Med. 2011;13(4):278–294. doi: 10.1097/GIM.0b013e3181ff67ba. [DOI] [PubMed] [Google Scholar]
- 53.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banerjee-Basu S, Packer A. SFARI Gene: an evolving database for the autism research community. Dis Model Mech. 2010;3(3–4):133–135. doi: 10.1242/dmm.005439. [DOI] [PubMed] [Google Scholar]
- 55.Schaaf CP, Zoghbi HY. Solving the autism puzzle a few pieces at a time. Neuron. 2011;70(5):806–808. doi: 10.1016/j.neuron.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 56.Bill BR, Geschwind DH. Genetic advances in autism: heterogeneity and convergence on shared pathways. Curr Opin Genet Dev. 2009;19(3):271–278. doi: 10.1016/j.gde.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Geschwind DH. Autism: many genes, common pathways? Cell. 2008;135(3):391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17(1):103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 59.Kaufmann WE, Cooper KL, Mostofsky SH, Capone GT, Kates WR, Newschaffer CJ, et al. Specificity of cerebellar vermian abnormalities in autism: a quantitative magnetic resonance imaging study. J Child Neurol. 2003;18(7):463–470. doi: 10.1177/08830738030180070501. [DOI] [PubMed] [Google Scholar]
- 60.Eluvathingal TJ, Behen ME, Chugani HT, Janisse J, Bernardi B, Chakraborty P, et al. Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J Child Neurol. 2006;21(10):846–851. doi: 10.1177/08830738060210100301. [DOI] [PubMed] [Google Scholar]
- 61.Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81(6):1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Philippe A, Boddaert N, Vaivre-Douret L, Robel L, Danon-Boileau L, Malan V, et al. Neurobehavioral profile and brain imaging study of the 22q13.3 deletion syndrome in childhood. Pediatrics. 2008;122(2):e376–e382. doi: 10.1542/peds.2007-2584. [DOI] [PubMed] [Google Scholar]
- 63.Johnson MB, Kawasawa YI, Mason CE, Krsnik Z, Coppola G, Bogdanovic D, et al. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62(4):494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alvarez Retuerto AI, Cantor RM, Gleeson JG, Ustaszewska A, Schackwitz WS, Pennacchio LA, et al. Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum Mol Genet. 2008;17(24):3887–3896. doi: 10.1093/hmg/ddn291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Descipio C, Schneider L, Young TL, Wasserman N, Yaeger D, Lu F, et al. Subtelomeric deletions of chromosome 6p: molecular and cytogenetic characterization of three new cases with phenotypic overlap with Ritscher-Schinzel (3C) syndrome. Am J Med Genet A. 2005;134A(1):3–11. doi: 10.1002/ajmg.a.30573. [DOI] [PubMed] [Google Scholar]
- 67.Miles JH, Hillman RE. Value of a clinical morphology examination in autism. Am J Med Genet. 2000;91(4):245–253. [PubMed] [Google Scholar]
- 68.Cheh MA, Millonig JH, Roselli LM, Ming X, Jacobsen E, Kamdar S, et al. En2 knockout mice display neurobehavioral and neurochemical alterations relevant to autism spectrum disorder. Brain Res. 2006;1116(1):166–176. doi: 10.1016/j.brainres.2006.07.086. [DOI] [PubMed] [Google Scholar]
- 69.Ieraci A, Forni PE, Ponzetto C. Viable hypomorphic signaling mutant of the Met receptor reveals a role for hepatocyte growth factor in postnatal cerebellar development. Proc Natl Acad Sci U S A. 2002;99(23):15200–15205. doi: 10.1073/pnas.222362099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fatemi SH, Reutiman TJ, Folsom TD, Thuras PD. GABA(A) receptor downregulation in brains of subjects with autism. J Autism Dev Disord. 2009;39(2):223–230. doi: 10.1007/s10803-008-0646-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeLorey TM, Sahbaie P, Hashemi E, Homanics GE, Clark JD. Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: a potential model of autism spectrum disorder. Behav Brain Res. 2008;187(2):207–220. doi: 10.1016/j.bbr.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Careaga M, Van de Water J, Ashwood P. Immune dysfunction in autism: a pathway to treatment. Neurotherapeutics. 2010;7(3):283–292. doi: 10.1016/j.nurt.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57(1):67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 74.Chez MG, Dowling T, Patel PB, Khanna P, Kominsky M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr Neurol. 2007;36(6):361–365. doi: 10.1016/j.pediatrneurol.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 75.Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, et al. Elevated immune response in the brain of autistic patients. J Neuroimmunol. 2009;207:111–116. doi: 10.1016/j.jneuroim.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wei H, Zou H, Sheikh AM, Malik M, Dobkin C, Brown WT, et al. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflammation. 2011;8:52. doi: 10.1186/1742-2094-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K, et al. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis. 2008;30:303–311. doi: 10.1016/j.nbd.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cabanlit M, Wills S, Goines P, Ashwood P, Van de Water J. Brain-specific autoantibodies in the plasma of subjects with autistic spectrum disorder. Ann NY Acad Sci. 2007;1107:92–103. doi: 10.1196/annals.1381.010. [DOI] [PubMed] [Google Scholar]
- 79.Goines P, Haapanen L, Boyce R, Duncanson P, Braunschweig D, Delwiche L, et al. Autoantibodies to cerebellum in children with autism associate with behavior. Brain Behav Immun. 2011;25:514–523. doi: 10.1016/j.bbi.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wills S, Cabanlit M, Bennett J, Ashwood P, Amaral DG, Van de Water J. Detection of autoantibodies to neural cells of the cerebellum in the plasma of subjects with autism spectrum disorders. Brain Behav Immun. 2009;23(1):64–74. doi: 10.1016/j.bbi.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wills S, Rossi CC, Bennett J, Cerdeño VM, Ashwood P, Amaral DG, et al. Further characterization of autoantibodies to GABAergic neurons in the central nervous system produced by a subset of children with autism. Mol Autism. 2011;2:5. doi: 10.1186/2040-2392-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. 2011;25:40–45. doi: 10.1016/j.bbi.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ashwood P, Anthony A, Torrente F, Wakefield AJ. Spontaneous mucosal lymphocyte cytokine profiles in children with autism and gastrointestinal symptoms: mucosal immune activation and reduced counter regulatory interleukin-10. J Clin Immunol. 2004;24:664–673. doi: 10.1007/s10875-004-6241-6. [DOI] [PubMed] [Google Scholar]
- 84.Ashwood P, Wakefield AJ. Immune activation of peripheral blood and mucosal CD3+ lymphocyte cytokine profiles in children with autism and gastrointestinal symptoms. J Neuroimmunol. 2006;173:126–134. doi: 10.1016/j.jneuroim.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 85.Heuer L, Ashwood P, Goines P, Krakowiak P, Hertz-Picciotto I, Hansen R, et al. Reduced Levels of Immunoglobulin in Children with Autism Correlates with Behavioral Symptoms. Autism Research. 2008;1:275–283. doi: 10.1002/aur.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Enstrom A, Krakowiak P, Onore C, Pessah IN, Hertz-Picciotto I, Hansen RL, et al. Increased IgG4 levels in children with autism disorder. Brain Behav Immun. 2009;23:389–395. doi: 10.1016/j.bbi.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Corbett BA, Kantor AB, Schulman H, Walker WL, Lit L, Ashwood P, et al. A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol Psychiatry. 2007;12:292–306. doi: 10.1038/sj.mp.4001943. [DOI] [PubMed] [Google Scholar]
- 88.Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Associations of impaired behaviors with elevated plasma chemokines in autism spectrum disorders. J Neuroimmunol. 2011;232(1–2):196–201. doi: 10.1016/j.jneuroim.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Enstrom AM, Lit L, Onore CE, Gregg JP, Hansen R, Pessah IN, et al. Altered gene expression and function of peripheral blood natural killer cells in children with autism. Brain Behav Immun. 2009;23:124–133. doi: 10.1016/j.bbi.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ashwood P, Schauer J, Pessah IN, Van de Water J. Preliminary evidence of the in vitro effects of BDE-47 on innate immune responses in children with autism spectrum disorders. J Neuroimmunol. 2009;208:149–153. doi: 10.1016/j.jneuroim.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Enstrom AM, Onore CE, Van de Water JA, Ashwood P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav Immun. 2010;24:64–71. doi: 10.1016/j.bbi.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ashwood P, Corbett BA, Kantor A, Schulman H, Van de Water J, Amaral DG. search of cellular immunophenotypes in the blood of children with autism. PLoS One. 2011;6:e19299. doi: 10.1371/journal.pone.0019299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, Van de Water J. Altered T cell responses in children with autism. Brain Behav Immun. 2011;25(5):840–849. doi: 10.1016/j.bbi.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ashwood P, Enstrom A, Krakowiak P, Hertz-Picciotto I, Hansen RL, Croen LA, et al. Decreased transforming growth factor beta1 in autism: a potential link between immune dysregulation and impairment in clinical behavioral outcomes. J Neuroimmunol. 2008;204:149–153. doi: 10.1016/j.jneuroim.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Onore C, Enstrom A, Krakowiak P, Hertz-Picciotto I, Hansen R, Van de Water J, et al. Decreased cellular IL-23 but not IL-17 production in children with autism spectrum disorders. J Neuroimmunol. 2009;216:126–134. doi: 10.1016/j.jneuroim.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chauhan A, Chauhan V, Brown WT, editors. Autism: Oxidative stress, inflammation and immune abnormalities. Florida: CRC Press, Taylor and Francis Group; 2009. [Google Scholar]
- 97.Deth R, Muratore C, Benzecry J, Power-Charnitsky VA, Waly M. How environmental and genetic factors combine to cause autism: A redox/methylation hypothesis. Neurotoxicology. 2008;29:190–201. doi: 10.1016/j.neuro.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 98.Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 99.Kern JK, Jones AM. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J Toxicol Environ Health B Crit Rev. 2006;9:485–499. doi: 10.1080/10937400600882079. [DOI] [PubMed] [Google Scholar]
- 100.Wells PG, McCallum GP, Chen CS, Henderson JT, Lee CJ, Perstin J, et al. Oxidative stress in developmental origins of disease: Teratogenesis, neurodevelopmental deficits, and cancer. Toxicol Sci. 2009;108:4–18. doi: 10.1093/toxsci/kfn263. [DOI] [PubMed] [Google Scholar]
- 101.Kinney DK, Munir KM, Crowley DJ, Miller AM. Prenatal stress and risk for autism. Neurosci Biobehav Rev. 2008;32:1519–1532. doi: 10.1016/j.neubiorev.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kolevzon A, Gross R, Reichenberg A. Prenatal and perinatal risk factors for autism: a review and integration of findings. Arch Pediatr Adolesc Med. 2007;161:326–333. doi: 10.1001/archpedi.161.4.326. [DOI] [PubMed] [Google Scholar]
- 103.Chauhan A, Gu F, Essa MM, Wegiel J, Kaur K, Brown WT, Chauhan V. Brain region–specific deficit in mitochondrial electron transport chain complexes in children with autism. J Neurochem. 2011;117:209–220. doi: 10.1111/j.1471-4159.2011.07189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Muthaiyah B, Essa MM, Chauhan V, Brown WT, Wegiel J, Chauhan A. Increased lipid peroxidation in cerebellum and temporal cortex of brain in autism. J Neurochem. 2009;108(Suppl. 1):73. [Google Scholar]
- 105.Chauhan A, Audhya T, Chauhan V. Increased DNA oxidation in the cerebellum, frontal and temporal cortex of brain in autism. Transactions of the American Society for Neurochemistry. 2011:81. [Google Scholar]
- 106.Sajdel-Sulkowska EM, Xu M, Koibuchi N. Increase in cerebellar neurotrophin-3 and oxidative stress markers in autism. Cerebellum. 2009;8:366–372. doi: 10.1007/s12311-009-0105-9. [DOI] [PubMed] [Google Scholar]
- 107.Chauhan A, Essa MM, Muthaiyah B, Brown WT, Wegiel J, Chauhan V. Increased protein oxidation in cerebellum, frontal and temporal cortex in autism. International Meeting for Autism Research (Abstract) 2010 May [Google Scholar]
- 108.Sajdel-Sulkowska EM, Lipinski B, Windom H, Audhya T, McGinnis W. Oxidative stress in autism: Elevated cerebellar 3-nitrotyrosine levels. Am J Biochem Biotech. 2008;4:73–84. [Google Scholar]
- 109.Evans TA, Siedlak SL, Lu L, Fu X, Wang Z, McGinnis WR, et al. The autistic phenotype exhibits a remarkably localized modification of brain protein by products of free radical-induced lipid oxidation. Am J Biochem Biotech. 2008;4:61–72. [Google Scholar]
- 110.López-Hurtado E, Prieto JJ. A microscopic study of language-related cortex in autism. Am J Biochem Biotech. 2008;4:130–145. [Google Scholar]
- 111.Chauhan A, Audhya T, Chauhan V. Glutathione redox imbalance and increased DNA oxidation in specific brain regions in autism. International Meeting for Autism Research (Abstract) 2011 May [Google Scholar]
- 112.Ji L, Chauhan A, Brown WT, Chauhan V. Increased activities of Na+/K+-ATPase and Ca2+/Mg2+-ATPase in the frontal cortex and cerebellum of autistic individuals. Life Sci. 2009;85:788–793. doi: 10.1016/j.lfs.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2001;52:159–164. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- 114.Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012;17(4):389–401. doi: 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Courchesne E. New evidence of cerebellar and brainstem hypoplasia in autistic infants, children and adolescents: the MR imaging study by Hashimoto and colleagues. J Autism Dev Disord. 1995;25:19–22. doi: 10.1007/BF02178164. [DOI] [PubMed] [Google Scholar]
- 116.Zilbovicius M, Boddaert N, Belin P, Poline JB, Remy P, Mangin JF, et al. Temporal lobe dysfunction in childhood autism: a PET study. Positron emission tomography. Am J Psychiatry. 2000;157:1988–1993. doi: 10.1176/appi.ajp.157.12.1988. [DOI] [PubMed] [Google Scholar]
- 117.Schmitz C, Rezaie P. The neuropathology of autism: where do we stand? Neuropathol Appl Neurobiol. 2008;34:4–11. doi: 10.1111/j.1365-2990.2007.00872.x. [DOI] [PubMed] [Google Scholar]
- 118.Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci. 2005;23:183–187. doi: 10.1016/j.ijdevneu.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 119.Casanova MF. The neuropathology of autism. Brain Pathol. 2007;17:422–433. doi: 10.1111/j.1750-3639.2007.00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Palmen SJ, van Engeland H, Hof PR, Schmitz C. Neuropathological findings in autism. Brain. 2004;127:2572–2583. doi: 10.1093/brain/awh287. [DOI] [PubMed] [Google Scholar]
- 121.Wegiel J, Wisniewski T, Chauhan A, Chauhan V, Kuchna I, Nowicki K, et al. Type, topology, and sequelae of neuropathological changes shaping clinical phenotype of autism. In: Chauhan A, Chauhan V, Brown WT, editors. Autism: Oxidative stress, inflammation and immune abnormalities. CRC Press, Taylor and Francis groups; 2009. pp. 1–34. [Google Scholar]
- 122.Fatemi SH, Halt AR, Stary JM, Kanodia R, Schulz SC, Realmuto GR. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry. 2002;52(8):805–810. doi: 10.1016/s0006-3223(02)01430-0. [DOI] [PubMed] [Google Scholar]
- 123.Fatemi SH, Folsom TD. Dysregulation of fragile x mental retardation protein and metabotropic glutamate receptor 5 in superior frontal cortex of individuals with autism: a postmortem brain study. Mol Autism. 2011;2:6. doi: 10.1186/2040-2392-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fatemi SH, Folsom TD, Reutiman TJ, Thuras PD. Expression of GABA(B) receptors is altered in brains of subjects with autism. Cerebellum. 2009;8:64–69. doi: 10.1007/s12311-008-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yip J, Soghomonian JJ, Blatt GJ. Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol. 2007;113(5):559–568. doi: 10.1007/s00401-006-0176-3. [DOI] [PubMed] [Google Scholar]
- 126.Fatemi SH, Stary JM, Halt AR, Realmuto GR. Dysregulation of Reelin and Bcl-2 proteins in autistic cerebellum. J Autism Dev Disord. 2001;31:529–535. doi: 10.1023/a:1013234708757. [DOI] [PubMed] [Google Scholar]
- 127.Yip J, Soghomonian JJ, Blatt GJ. Decreased GAD65 mRNA levels in select subpopulations of neurons in the cerebellar dentate nuclei in autism: an in situ hybridization study. Autism Res. 2009;2(1):50–59. doi: 10.1002/aur.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fatemi SH, Folsom TD, Kneeland RE, Liesch SB. Metabotropic glutamate receptor 5 upregulation in children with autism is associated with underexpression of both Fragile X mental retardation protein and GABA A receptor beta 3 in adults with Autism. Anat Rec. 2011;294(10):1635–1645. doi: 10.1002/ar.21299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord. 2001;31(6):537–543. doi: 10.1023/a:1013238809666. [DOI] [PubMed] [Google Scholar]
- 130.Fatemi SH, Reutiman TJ, Folsom TD, Rooney PJ, Patel DH, Thuras PD. mRNA and protein levels for GABAAalpha4, alpha5, beta1, and GABABR1 receptors are altered in brains from subjects with autism. J Autism Dev Disord. 2010;40:743–750. doi: 10.1007/s10803-009-0924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Oblak AL, Gibbs TT, Blatt GJ. Decreased GABAA receptors and benzodiazepine binding sites in the anterior cingulate cortex in autism. Autism Res. 2009;2(4):205–219. doi: 10.1002/aur.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Oblak AL, Gibbs TT, Blatt GJ. Decreased GABA(B) receptors in the cingulate cortex and fusiform gyrus in autism. J Neurochem. 2010;114(5):1414–1423. doi: 10.1111/j.1471-4159.2010.06858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pesold C, Pisu MG, Impagnatiello F, Uzunov DP, Caruncho HJ. Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc Natl Acad Sci. 1998;95:3221–3226. doi: 10.1073/pnas.95.6.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sinagra M, Gonzalez Campo C, Verrier D, Moustié O, Manzoni OJ, Chavis P. Glutamatergic cerebellar granule neurons synthesize and secrete reelin in vitro. Neuron Glia Biol. 2008;4:189–196. doi: 10.1017/S1740925X09990214. [DOI] [PubMed] [Google Scholar]
- 135.Quattrocchi CC, Wannenes F, Persico AM, Ciafré SA, D’Arcangelo G, Farace MG, et al. Reelin is a serine protease of the extracellular matrix. J Biol Chem. 2002;277:303–309. doi: 10.1074/jbc.M106996200. [DOI] [PubMed] [Google Scholar]
- 136.Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, Sweatt JD, et al. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem. 2002;277:39944–39952. doi: 10.1074/jbc.M205147200. [DOI] [PubMed] [Google Scholar]
- 137.Forster E, Tielsch A, Saum B, Weiss KH, Johanssen C, Graus-Porta D, et al. Reelin, Disabled 1, and beta 1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proc Natl Acad Sci. 2002;99:13178–13183. doi: 10.1073/pnas.202035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nullmeier S, Panther P, Dobrowolny H, Frotscher M, Zhao S, Schwegler H, et al. Region-specific alteration of GABAergic markers in the brain of heterozygous reeler mice. Eur J Neurosci. 2011;33:689–698. doi: 10.1111/j.1460-9568.2010.07563.x. [DOI] [PubMed] [Google Scholar]
- 139.Cremer CM, Lubke JH, Palomero-Gallagher N, Zilles K. Laminar distribution of neurotransmitter receptors in different reeler mouse brain regions. Brain Struct Funct. 2011;216:201–218. doi: 10.1007/s00429-011-0303-3. [DOI] [PubMed] [Google Scholar]
- 140.Kelemenova S, Schmidtova E, Ficek A, Celec P, Kubranska A, Ostatnikova D. Polymorphisms of candidat e genes in slovak autistic patients. Psychiatr Genet. 2010;20:137–139. doi: 10.1097/YPG.0b013e32833a1eb3. [DOI] [PubMed] [Google Scholar]
- 141.Persico AM, D’Agruma L, Maiorano N, Totaro A, Militerni R, Bravaccio C, et al. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol Psychiatry. 2001;6:150–159. doi: 10.1038/sj.mp.4000850. [DOI] [PubMed] [Google Scholar]
- 142.Fatemi SH, Snow AV, Stary JM, Araghi-Niknam M, Reutiman TJ, Lee S, et al. Reelin signaling is impaired in autism. Biol Psychiatry. 2005;57(7):777–787. doi: 10.1016/j.biopsych.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 143.Fatemi SH, Stary JM, Egan EA. Reduced blood levels of reelin as a vulnerability factor in pathophysiology of autistic disorder. Cell Mol Neurobiol. 2002;22:139–152. doi: 10.1023/a:1019857620251. [DOI] [PubMed] [Google Scholar]
- 144.D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 145.Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, et al. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 146.Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, et al. Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron. 2000;27:33–44. doi: 10.1016/s0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- 147.Strasser V, Fasching D, Hauser C, Mayer H, Bock HH, Hiesberger T, et al. Receptor clustering is involved in Reelin signaling. Mol Cell Biol. 2004;24:1378–1386. doi: 10.1128/MCB.24.3.1378-1386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hoehn-Saric R, McLeod DR, Glowa JR. The effects NMDA receptor blockade on the acquisition of a conditioned emotional response. Biol Psychiatry. 1991;30:170–176. doi: 10.1016/0006-3223(91)90171-h. [DOI] [PubMed] [Google Scholar]
- 150.Lisman J. Long-term potentiation: outstanding questions and attempted synthesis. Philos Trans R Soc Lond B Biol Sci. 2003;358:829–842. doi: 10.1098/rstb.2002.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Silverman JM, Buxbaum JD, Ramoz N, Schmeidler J, Reichenberg A, Hollander E, et al. Autism-related routines and rituals associated with a mitochondrial aspartate/glutamate carrier SLC25A12 polymorphism. Am J Med Genet B Neuropsychiatr Genet. 2008;147:408–410. doi: 10.1002/ajmg.b.30614. [DOI] [PubMed] [Google Scholar]
- 152.Strutz-Seebohm N, Korniychuk G, Schwarz R, Baltaev R, Ureche ON, Mack AF, et al. Functional significance of the kainate receptor GluR6(M836I) mutation that is linked to autism. Cell Physiol Biochem. 2006;18:287–294. doi: 10.1159/000097675. [DOI] [PubMed] [Google Scholar]
- 153.Kim SA, Kim JH, Park M, Cho IH, Yoo HJ. Family-based association study between GRIK2 polymorphisms and autism spectrum disorders in Korean trios. Neurosci Res. 2007;58:332–335. doi: 10.1016/j.neures.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 154.Purcell AE, Jeon OH, Zimmerman AW, Blue ME, Pevsner J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology. 2001;57:1618–1628. doi: 10.1212/wnl.57.9.1618. [DOI] [PubMed] [Google Scholar]
- 155.Lepagnol-Bestel AM, Maussion G, Boda B, Cardona A, Iwayama Y, Delezoide AL, et al. SLC25A12 expression is associated with neurite outgrowth and is upregulated in the prefrontal cortex of autistic subjects. Mol Psychiatry. 2008;13:385–397. doi: 10.1038/sj.mp.4002120. [DOI] [PubMed] [Google Scholar]
- 156.Demark JL, Feldman MA, Holden JJ. Behavioral relationship between autism and fragile x syndrome. Am J Ment Retard. 2003;108:314–326. doi: 10.1352/0895-8017(2003)108<314:BRBAAF>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 157.De Rubeis S, Bagni C. Fragile X mental retardation protein control of neuronal mRNA metabolism: Insights into mRNA stability. Mol Cell Neurosci 2010. 2010;43:43–50. doi: 10.1016/j.mcn.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 158.Bassell GJ, Warren ST. Fragile X Syndrome: Loss of local mRNA Regulation Alters Synaptic Development and Function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile x syndrome. J Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 161.Hashimoto H, Fukui K, Noto T, Nakajima T, Kato N. Distribution of vasopressin and oxytocin in rat brain. Endocrinol Jpn. 1985;32(1):89–97. doi: 10.1507/endocrj1954.32.89. [DOI] [PubMed] [Google Scholar]
- 162.Kirsch P, Meyer-Lindenberg A. “Oxytocin and Autism”. In: Blatt Gene J., editor. The Neurochemical Basis of Autism: From Molecules to Minicolumns. New York City, NY: Springer; 2010. pp. 163–173. [Google Scholar]
- 163.Ferguson JN, Aldag JM, Insel TR, Young LJ. Oxytocin in the medial amygdala is essential for social recognition in the mouse. J Neurosci. 2001;21:8278–8285. doi: 10.1523/JNEUROSCI.21-20-08278.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.McCarthy MM, McDonald CH, Brooks PJ, Goldman D. An anxiolytic action of oxytocin is enhanced by estrogen in the mouse. Physiol Behav. 1996;60:1209–1215. doi: 10.1016/s0031-9384(96)00212-0. [DOI] [PubMed] [Google Scholar]
- 165.Insel TR, Young LJ. The neurobiology of attachment. Nat Rev Neurosci. 2001;2:129–136. doi: 10.1038/35053579. [DOI] [PubMed] [Google Scholar]
- 166.Winslow JT, Insel TR. Neuroendocrine basis of social recognition. Curr Opin Neurobiol. 2004;14:248–253. doi: 10.1016/j.conb.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 167.Liu W, Pappas GD, Carter CS. Oxytocin receptors in brain cortical regions are reduced in the haploinsufficient (+/−) reeler mice. Neurol Res. 2005;27(4):339–345. doi: 10.1179/016164105X35602. [DOI] [PubMed] [Google Scholar]
- 168.Baskerville TA, Douglas AJ. Dopamine and oxytocin interactions underlying behaviors: potential contributions to behavioral disorders. CNS Neurosci Ther. 2010;16(3):e92–e123. doi: 10.1111/j.1755-5949.2010.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hollander E, Novotny S, Hanratty M, Yaffe R, DeCaria CM, Aronowitz BR, et al. Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharm. 2003;28:193–198. doi: 10.1038/sj.npp.1300021. [DOI] [PubMed] [Google Scholar]
- 170.Hollander E, Bartz J, Chaplin W, Phillips A, Sumner J, Soorya L, et al. Oxytocin increases retention of social cognition in autism. Biol Psychiatry. 2007;61:498–503. doi: 10.1016/j.biopsych.2006.05.030. [DOI] [PubMed] [Google Scholar]
- 171.Bartz JA, Hollander E. Oxytocin and experimental therapeutics in autism spectrum disorders. Prog Brain Res. 2008;170:451–462. doi: 10.1016/S0079-6123(08)00435-4. [DOI] [PubMed] [Google Scholar]
- 172.Lee M, Martin-Ruiz C, Graham A, Court J, Jaros E, Perry R, et al. Nicotinic receptor abnormalities in the cerebellar cortex in autism. Brain. 2002;125(Pt 7):1483–1495. doi: 10.1093/brain/awf160. [DOI] [PubMed] [Google Scholar]
- 173.Martin-Ruiz CM, Lee M, Perry RH, Baumann M, Court JA, Perry EK. Molecular analysis of nicotinic receptor expression in autism. Brain Res Mol Brain Res. 2004;123(1–2):81–90. doi: 10.1016/j.molbrainres.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 174.Deutsch SI, Urbano MR, Neumann SA, Burket JA, Katz E. Cholinergic abnormalities in autism: is there a rationale for selective nicotinic agonist interventions? Clin Neuropharmacol. 2010;33(3):114–120. doi: 10.1097/WNF.0b013e3181d6f7ad. [DOI] [PubMed] [Google Scholar]
- 175.Lippiello PM. Nicotinic cholinergic antagonists: a novel approach for the treatment of autism. Med Hypotheses. 2006;66(5):985–990. doi: 10.1016/j.mehy.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 176.Blatt GJ, VanSluytman G, Marcon RG. Decreased density of 3[H]AFDX-labeled cholinergic M2 receptors in the medial accessory olive in autism. Soc Neurosci. 2004;34:116. 12. [Google Scholar]
- 177.Armstrong DD, Assman S, Kinney HC. Early developmental changes in the chemoarchitecture of the human inferior olive: A review. J Neuropathol Exp Neurol. 1999;58:1–11. doi: 10.1097/00005072-199901000-00001. [DOI] [PubMed] [Google Scholar]
- 178.Kolasiewicz W, Kuter K, Nowak P, Pastuszka A, Ossowska K. Lesion of the cerebellar noradrenergic innervation enhances the harmaline-induced tremor in rats. Cerebellum. 2011;10(2):267–280. doi: 10.1007/s12311-011-0250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rogers TD, Dickson PE, Heck DH, Goldowitz D, Mittleman G, Blaha CD. Connecting the dots of the cerebro-cerebellar role in cognitive function: Neuronal pathways for cerebellar modulation of dopamine release in the prefrontal cortex. Synapse. 2011 doi: 10.1002/syn.20960. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mittleman G, Goldowitz D, Heck DH, Blaha CD. Cerebellar modulation of frontal cortex dopamine efflux in mice: relevance to autism and schizophrenia. Synapse. 2008;62(7):544–550. doi: 10.1002/syn.20525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Crandall JE, McCarthy DM, Araki KY, Sims JR, Ren J-Q, Bhide PG. Dopamine receptor activation modulates GABA neuron migration from the basal forebrain to the cerebral cortex. J Neurosci. 2007;27(14):3813–3822. doi: 10.1523/JNEUROSCI.5124-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Nakamura K, Sekine Y, Ouchi Y, Tsujii M, Yoshikawa E, Futatsubashi M, et al. Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch Gen Psychiatry. 2010;67(1):58–68. doi: 10.1001/archgenpsychiatry.2009.137. [DOI] [PubMed] [Google Scholar]
- 183.Buitelaar JK, Willemsen-Swinkels SH. Medication treatment in subjects with autistic spectrum disorders. Eur Child Adolesc Psychiatry. 2000;9(1):185–197. doi: 10.1007/s007870070022. [DOI] [PubMed] [Google Scholar]
- 184.Kish SJ, Furukawa Y, Chang L-J, Tong J, Ginovart N, Wilson A, et al. Regional distribution of serotonin transporter protein in post-mortem human brain: Is the cerebellum a SERT-free brain region? Nuclear Medicine and Biology. 2005;32(2):123–128. doi: 10.1016/j.nucmedbio.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 185.Marazziti D. A further support to the hypothesis of a link between serotonin, autism, and the cerebellum. Biol Psychiatry. 2002;52(2):143. doi: 10.1016/s0006-3223(02)01406-3. [DOI] [PubMed] [Google Scholar]
- 186.Makkonen I, Riikonen R, Kokki H, Airaksinen MM, Kuikka JT. Serotonin and dopamine transporter binding in children with autism determined by SPECT. Dev Med Child Neurol. 2008;50(8):593–597. doi: 10.1111/j.1469-8749.2008.03027.x. [DOI] [PubMed] [Google Scholar]
- 187.Azmitia EC, Singh JS, Whitaker-Azmitia PM. Increased serotonin axons (immunoreactive to 5-HT transporter) in postmortem brains from young autism donors. Neuropharm. 2011;60:1347–1354. doi: 10.1016/j.neuropharm.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 188.Williams K, Wheeler DM, Silove N, Hazell P. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD) Cochrane Database Syst Rev. 2010;4(8):CD004677. doi: 10.1002/14651858.CD004677.pub2. [DOI] [PubMed] [Google Scholar]
- 189.Stanfield AC, McIntosh AM, Spencer MD, Philip R, Gaur S, Lawrie SM. Towards a neuroanatomy of autism: a systematic review and meta-analysis of structural magnetic resonance imaging studies. Eur Psychiatry. 2008;23(4):289–299. doi: 10.1016/j.eurpsy.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 190.Abrahams BS, Geschwind DH. Connecting genes to brain in the autism spectrum disorders. Arch Neurol. 2010;67(4):395–399. doi: 10.1001/archneurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Leiner HC, Leiner AL, Dow RS. Does the cerebellum contribute to mental skills? Behav Neurosci. 1986;100(4):443–454. doi: 10.1037//0735-7044.100.4.443. [DOI] [PubMed] [Google Scholar]
- 192.Stoodley CJ, Schmahmann JD. Functional topography in the human cerebellum: A meta-analysis of neuroimaging studies. NeuroImage. 2009;44(2):489–501. doi: 10.1016/j.neuroimage.2008.08.039. [DOI] [PubMed] [Google Scholar]
- 193.Middleton FA, Strick PL. Cerebellar output channels. Int Rev Neurobiol. 1997;41:61–82. doi: 10.1016/s0074-7742(08)60347-5. [DOI] [PubMed] [Google Scholar]
- 194.Bloedel JR, Bantli H. A spinal action of the dentate nucleus mediated by descending systems originating in the brain stem. Brain Res. 1978;153(3):602–607. doi: 10.1016/0006-8993(78)90345-1. [DOI] [PubMed] [Google Scholar]
- 195.Kanner L. Autistic disturbances of affective contact. The Nervous Child. 1943;2:217–250. [Google Scholar]
- 196.Asperger H. 'Autistic psychopathy' in childhood. In: Frith U, editor. Autism and Asperger syndrome. New York, NY: Cambridge University Press; 1991. pp. 37–92. [Google Scholar]
- 197.Molloy CA, Dietrich KN, Bhattacharya A. Postural stability in children with autism spectrum disorder. J Autism Dev Disord. 2003;33(6):643–652. doi: 10.1023/b:jadd.0000006001.00667.4c. [DOI] [PubMed] [Google Scholar]
- 198.Freitag CM, Kleser C, Schneider M, von Gontard A. Quantitative assessment of neuromotor function in adolescents with high functioning autism and Asperger Syndrome. J Autism Dev Disord. 2007;37(5):948–959. doi: 10.1007/s10803-006-0235-6. [DOI] [PubMed] [Google Scholar]
- 199.Mostofsky SH, Powell SK, Simmonds DJ, Goldberg MC, Caffo B, Pekar JJ. Decreased connectivity and cerebellar activity in autism during motor task performance. Brain. 2009 doi: 10.1093/brain/awp088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Allen G, Courchesne E. Differential effects of developmental cerebellar abnormality on cognitive and motor functions in the cerebellum: an fMRI study of autism. Am J Psychiatry. 2003;160(2):262–273. doi: 10.1176/appi.ajp.160.2.262. [DOI] [PubMed] [Google Scholar]
- 201.Muller RA, Pierce K, Ambrose JB, Allen G, Courchesne E. Atypical patterns of cerebral motor activation in autism: a functional magnetic resonance study. Biol Psychiatry. 2001;49(8):665–676. doi: 10.1016/s0006-3223(00)01004-0. [DOI] [PubMed] [Google Scholar]
- 202.Takarae Y, Minshew NJ, Luna B, Krisky CM, Sweeney JA. Pursuit eye movement deficits in autism. Brain. 2004;127(Pt 12):2584–2594. doi: 10.1093/brain/awh307. [DOI] [PubMed] [Google Scholar]
- 203.Takarae Y, Minshew NJ, Luna B, Sweeney JA. Atypical involvement of frontostriatal systems during sensorimotor control in autism. Psychiatry Res. 2007;156(2):117–127. doi: 10.1016/j.pscychresns.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mosconi MW, Kay M, D'Cruz AM, Guter S, Kapur K, Macmillan C, et al. Neurobehavioral abnormalities in first-degree relatives of individuals with autism. Arch Gen Psychiatry. 2010;67(8):830–840. doi: 10.1001/archgenpsychiatry.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Williams DL, Goldstein G, Minshew NJ. The profile of memory function in children with autism. Neuropsychology. 2006;20(1):21–29. doi: 10.1037/0894-4105.20.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Townsend J, Courchesne E, Covington J, Westerfield M, Harris NS, Lyden P, et al. Spatial attention deficits in patients with acquired or developmental cerebellar abnormality. J Neurosci. 1999;19(13):5632–5643. doi: 10.1523/JNEUROSCI.19-13-05632.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Herbert MR, Harris GJ, Adrien KT, Ziegler DA, Makris N, Kennedy DN, et al. Abnormal asymmetry in language association cortex in autism. Ann Neurol. 2002;52(5):588–596. doi: 10.1002/ana.10349. [DOI] [PubMed] [Google Scholar]
- 208.Hodge SM, Makris N, Kennedy DN, Caviness VS, Jr, Howard J, McGrath L, et al. Cerebellum, language, and cognition in autism and specific language impairment. J Autism Dev Disord. 2010;40(3):300–316. doi: 10.1007/s10803-009-0872-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Stoodley CJ. The Cerebellum and Cognition: Evidence from Functional Imaging Studies. Cerebellum. 2011 doi: 10.1007/s12311-011-0260-7. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 210.Gordon N. The cerebellum and cognition. Eur J Paediatr Neurol. 2007;11:232–234. doi: 10.1016/j.ejpn.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 211.Kelly RM, Strick PL. Cerebellar loops with motor cortex and prefrontal cortex of a nonhuman primate. Journal of Neuroscience. 2003;23:8432–8444. doi: 10.1523/JNEUROSCI.23-23-08432.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Middleton FA, Strick PL. Cerebellar projections to the prefrontal cortex of the primate. Journal of Neuroscience. 2001;21:700–712. doi: 10.1523/JNEUROSCI.21-02-00700.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Courchesne E. Brainstem, cerebellar and limbic neuroanatomical abnormalities in autism. Current Opinion in Neurobiology. 1997;7:269–278. doi: 10.1016/s0959-4388(97)80016-5. [DOI] [PubMed] [Google Scholar]
- 214.Llinas R. From Neurons to Self. Cambridge, MA: MIT Press; 2002. I of the vortex. [Google Scholar]
- 215.Sasaki K, Gemba H. Cerebro-cerebellar interactions: For fast and stable timing of voluntary movement. In: Mano N, Hamada I, DeLong MR, editors. Role of the Cerebellum and Basal Ganglia in Voluntary Movement. Amsterdam: Elsevier; 1993. pp. 41–50. [Google Scholar]
- 216.Ivry RB, Keele SW, Diener HC. Dissociation of the lateral and medial cerebellum in movement timing and movement execution. Exp Brain Res. 1988;73:167–180. doi: 10.1007/BF00279670. [DOI] [PubMed] [Google Scholar]
- 217.Braitenberg V. Progress in Brain Research Vol.25 The Cerebellum Fox CA. Amsterdam: Elsevier; 1967. Is the Cerebellar Cortex a Biological Clock in the Millisecond Range? pp. 334–346. [DOI] [PubMed] [Google Scholar]
- 218.Yuste R, MacLean JN, Smith J, Lansner A. The cortex as a central pattern generator. Nat. Rev. Neurosci. 2005;6:477–483. doi: 10.1038/nrn1686. [DOI] [PubMed] [Google Scholar]
- 219.Ayzenshtat I, Meirovithz E, Edelman H, Werner-Reiss U, Bienenstock E, Abeles M, et al. Precise spatiotemporal patterns among visual cortical areas and their relation to visual stimulus processing. J Neurosci. 2010;30:11232–11245. doi: 10.1523/JNEUROSCI.5177-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ben-Shaul Y, Drori R, Asher I, Stark E, Nadasdy Z, Abeles M. Neuronal activity in motor cortical areas reflects the sequential context of movement. Journal of Neurophysiology. 2004;91:1748–1762. doi: 10.1152/jn.00957.2003. 7. [DOI] [PubMed] [Google Scholar]
- 221.Prut Y, Vaadia E, Bergman H, Haalman I, Slovin H, Abeles M. Spatiotemporal structure of cortical activity: properties and behavioral relevance. Journal of Neurophysiology. 1998;79:2857–2874. doi: 10.1152/jn.1998.79.6.2857. [DOI] [PubMed] [Google Scholar]
- 222.Braitenberg V, Atwood RP. Morphological observations on the cerebellar cortex. Journal of Comparative Neurology. 1958;109:1–33. doi: 10.1002/cne.901090102. [DOI] [PubMed] [Google Scholar]
- 223.Heck DH. Rat cerebellar cortex in vitro responds specifically to moving stimuli. Neuroscience Letters. 1993;157:95–98. doi: 10.1016/0304-3940(93)90651-z. [DOI] [PubMed] [Google Scholar]
- 224.Heck DH. Sequential input to guinea pig cerebellar cortex in vitro strongly affects Purkinje cells via parallel fibers. Naturwissenschaften. 1995;82:201–203. doi: 10.1007/BF01143198. [DOI] [PubMed] [Google Scholar]
- 225.Braitenberg V, Heck DH, Sultan F. The detection and generation of sequences as a key to cerebellar function. Experiments and theory. Behav.Brain Sci. 1997;20:229–245. [PubMed] [Google Scholar]
- 226.Molinari M, Chiricozzi FR, Clausi S, Tedesco AM, De Lisa M, Leggio MG. Cerebellum and detection of sequences, from perception to cognition. Cerebellum. 2008;7:611–615. doi: 10.1007/s12311-008-0060-x. [DOI] [PubMed] [Google Scholar]
- 227.Andreasen NC, Paradiso S, O'Leary DS. "Cognitive dysmetria" as an integrative theory of schizophrenia: a dysfunction in cortical-subcortical-cerebellar circuitry? Schizophr.Bull. 1998;24:203–218. doi: 10.1093/oxfordjournals.schbul.a033321. [DOI] [PubMed] [Google Scholar]
- 228.Courchesne E, Yeung-Courchesne R, Press GA, Hesselink JR, Jernigan TL. Hypoplasia of cerebellar vermal lobules VI and VII in autism. N Engl J Med. 1988;318:1349–1354. doi: 10.1056/NEJM198805263182102. [DOI] [PubMed] [Google Scholar]
- 229.Bauman ML. Microscopic neuroanatomic abnormalities in autism. Pediatrics. 1991;87:791–796. [PubMed] [Google Scholar]
- 230.Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 2006;29:349–358. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 231.Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–533. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Fombonne E. Epidemiology of autistic disorder and other pervasive developmental disorders. J Clin Psychiatry. 2005;66(Suppl 10):3–8. [PubMed] [Google Scholar]
- 233.Landrigan PJ. What causes autism? Exploring the environmental contribution. Curr Opin Pediatr. 2010;22:219–225. doi: 10.1097/MOP.0b013e328336eb9a. [DOI] [PubMed] [Google Scholar]
- 234.Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 235.Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic Heritability and Shared Environmental Factors Among Twin Pairs With Autism. Arch Gen Psychiatry. 2011 doi: 10.1001/archgenpsychiatry.2011.76. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Gaita L, Manzi B, Sacco R, Lintas C, Altieri L, Lombardi F, et al. Decreased serum arylesterase activity in Autism Spectrum Disorders. Psychiatry Res. 2010;180:105–113. doi: 10.1016/j.psychres.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 237.Paşca SP, Nemeş B, Vlase L, Gagyi CE, Dronca E, Miu AC, et al. High levels of homocysteine and low serum paraoxonase 1 arylesterase activity in children with autism. Life Sci. 2006;78:2244–2248. doi: 10.1016/j.lfs.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 238.Lugli G, Krueger JM, Davis JM, Persico AM, Keller F, Smalheiser NR. Methodological factors influencing measurement and processing of plasma reelin in humans. BMC Biochem. 2003;4:9. doi: 10.1186/1471-2091-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.D'Amelio M, Ricci I, Sacco R, Liu X, D'Agruma L, Muscarella LA, et al. Paraoxonase gene variants are associated with autism in North America, but not in Italy: possible regional specificity in gene-environment interactions. Mol Psychiatry. 2005;10:1006–1016. doi: 10.1038/sj.mp.4001714. [DOI] [PubMed] [Google Scholar]
- 240.Persico AM, Levitt P, Pimenta AF. Polymorphic GGC repeat differentially regulates human reelin gene expression levels. J Neural Transm. 2006;113:1373–1382. doi: 10.1007/s00702-006-0441-6. [DOI] [PubMed] [Google Scholar]
- 241.Eskenazi B, Huen K, Marks A, Harley KG, Bradman A, Barr DB, et al. PON1 and neurodevelopment in children from the CHAMACOS study exposed to organophosphate pesticides in utero. Environ Health Perspect. 2010;118:1775–1781. doi: 10.1289/ehp.1002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Mullen B, Khialeeva E, Carpenter EM. Society for Neuroscience. San Diego CA: Dab1-lacZ reporter reveals CNS lamination defects in a mouse model for autism. Program No. 147.12, 2010 Neuroscience Meeting Planner. Online. [Google Scholar]
- 243.Krey J, Dolmetsch R. Molecular mechanisms of autism: a possible role for Ca2+ signaling. Curr Opin Neurobiol. 2007;17:112–119. doi: 10.1016/j.conb.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 244.Empson RM, Garside ML, Knöpfel T. Plasma membrane Ca2+ ATPase 2 contributes to short-term synapse plasticity at the parallel fiber to Purkinje neuron synapse. J Neurosci. 2007;27:3753–3758. doi: 10.1523/JNEUROSCI.0069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Burette AC, Strehler EE, Weinberg RJ. "Fast" plasma membrane calcium pump PMCA2a concentrates in GABAergic terminals in the adult rat brain. J Comp Neurol. 2009;512:500–513. doi: 10.1002/cne.21909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Garside ML, Turner PR, Austen B, Strehler EE, Beesley PW, Empson RM. Molecular interactions of the plasma membrane calcium ATPase 2 at pre- and post-synaptic sites in rat cerebellum. Neuroscience. 2009;162:383–395. doi: 10.1016/j.neuroscience.2009.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Carayol J, Sacco R, Tores F, Rousseau F, Lewin P, Hager J, et al. Converging evidence for an association of ATP2B2 allelic variants with autism in males. Biol Psychiatry. 2011 Jul 12; doi: 10.1016/j.biopsych.2011.05.020. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 248.Pessah IN, Cherednichenko G, Lein PJ. Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity. Pharmacol Ther. 2010;125:260–285. doi: 10.1016/j.pharmthera.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Palmieri L, Persico AM. Mitochondrial dysfunction in Autism Spectrum Disorders: cause or effect? Biochim Biophys Acta. 2010;1797:1130–1137. doi: 10.1016/j.bbabio.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 250.Elsen GE, Choi LY, Prince VE, Ho RK. The autism susceptibility gene met regulates zebrafish cerebellar development and facial motor neuron migration. Dev Biol. 2009;335:78–92. doi: 10.1016/j.ydbio.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Campbell DB, D'Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt P, et al. Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann Neurol. 2007;62:243–250. doi: 10.1002/ana.21180. [DOI] [PubMed] [Google Scholar]
- 252.Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, et al. Proc Natl Acad Sci. Vol. 103. USA: 2006. A genetic variant that disrupts MET transcription is associated with autism; pp. 16834–16839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sheng L, Ding X, Ferguson M, McCallister M, Rhoades R, Maguire M, et al. Prenatal polycyclic aromatic hydrocarbon exposure leads to behavioral deficits and downregulation of receptor tyrosine kinase, MET. Toxicol Sci. 2010;118:625–634. doi: 10.1093/toxsci/kfq304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Campbell DB, Li C, Sutcliffe JS, Persico AM, Levitt P. Genetic evidence implicating multiple genes in the MET receptor tyrosine kinase pathway in autism spectrum disorder. Autism Res. 2008;1:159–168. doi: 10.1002/aur.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Carlson GC. Glutamate receptor dysfunction and drug targets across models of autism spectrum disorders. Pharmacol Biochem Behav. 2011 doi: 10.1016/j.pbb.2011.02.003. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Steinlin M. Cerebellar disorders in childhood: cognitive problems. Cerebellum. 2008;7:607–610. doi: 10.1007/s12311-008-0083-3. [DOI] [PubMed] [Google Scholar]
- 257.Rojas DC, Peterson E, Winterrowd E, Reite ML, Rogers SJ, Tregellas JR. Regional gray matter volumetric changes in autism associated with social and repetitive behavior symptoms. BMC Psychiatry. 2006;6:56. doi: 10.1186/1471-244X-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Schanmann JD, Weilburg JB, Sherman JC. The neuropsychiatry of the cerebellum - insights from the clinic. Cerebellum. 2007;6:254–267. doi: 10.1080/14734220701490995. [DOI] [PubMed] [Google Scholar]
- 259.Anderson CM, Polcari A, Lowen SB, Renshaw PF, Teicher MH. Effects of methylphenidate on functional magnetic resonance relaxometry of the cerebellar vermis in boys with ADHD. Am J Psychiatry. 2002;159:1322–1328. doi: 10.1176/appi.ajp.159.8.1322. [DOI] [PubMed] [Google Scholar]
- 260.Takahashi T, Kobayashi T, Ozaki M, Takamatsu Y, Ogai Y, Ohta M, et al. G protein-activated inwardly rectifying K+ channel inhibition and rescue of weaver mouse motor functions by antidepressants. Neurosci Res. 2006;54:104–111. doi: 10.1016/j.neures.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 261.Blatt GJ. GABAergic cerebellar system in autism: a neuropathological and developmental perspective. Int Rev Neurobiol. 2005;71:167–178. doi: 10.1016/s0074-7742(05)71007-2. [DOI] [PubMed] [Google Scholar]
- 262.Johannessen Landmark C. Antiepileptic drugs in non-epilepsy disorders: relations between mechanisms of action and clinical efficacy. CNS Drugs. 2008;22:27–47. doi: 10.2165/00023210-200822010-00003. [DOI] [PubMed] [Google Scholar]
- 263.Fink M, Taylor MA, Ghaziuddin N. Catatonia in autistic spectrum disorders: a medical treatment algorithm. Int Rev Neurobiol. 2006;72:233–244. doi: 10.1016/S0074-7742(05)72014-6. [DOI] [PubMed] [Google Scholar]
- 264.Wang P, Erickson CA, Ginsberg G, Rathmell B, Cerubini M, Zarevics P, et al. International Meeting for Autism Research. San Diego, CA: 2011. May, [Google Scholar]
- 265.Lory P, Mezghrani A. Calcium channelopathies in inherited neurological disorders: relevance to drug screening for acquired channel disorders. IDrugs. 2010;13:467–471. [PubMed] [Google Scholar]
- 266.Placantonakis DG, Schwarz C, Welsh JP. Serotonin suppresses subthreshold and suprathreshold oscillatory activity of rat inferior olive neurons in vitro. Journal of Physiology (London) 2000;524:833–851. doi: 10.1111/j.1469-7793.2000.00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Placantonakis DG, Welsh JP. Two distinct oscillatory states determined by the NMDA receptor in rat inferior olive. Journal of Physiology (London) 2001;534:123–140. doi: 10.1111/j.1469-7793.2001.t01-1-00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Welsh JP, Han VZ. The NMDA receptor potentiates electrotonic coupling between inferior olive neurons. Society for Neuroscience Abstracts. 2010:525–525. [Google Scholar]
- 269.Park YG, Park HY, Lee CJ, Choi S, Jo S, Choi H, et al. Ca(V)3.1 is a tremor rhythm pacemaker in the inferior olive. Proceedings of the National Academy of Sciences (USA) 2010;107:10731–10736. doi: 10.1073/pnas.1002995107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Cheung C, Chua S, Cheung V, Khong P, Tai K, Wong T, et al. White matter fractional anisotrophy differences and correlates of diagnostic symptoms in autism. Journal of Child Psychology and Psychiatry. 2009;50:1102–1112. doi: 10.1111/j.1469-7610.2009.02086.x. [DOI] [PubMed] [Google Scholar]
- 271.Catani M, Jones DK, Daly E, Embiricos N, Deeley Q, Pugliese L, et al. Altered cerebellar feedback projections in Asperger syndrome. Neuroimage. 2008;41:1184–1191. doi: 10.1016/j.neuroimage.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 272.Kates WR, Burnette CP, Eliez S, Strunge LA, Kaplan D, Landa R, et al. Neuroanatomic variation in monozygotic twin pairs discordant for the narrow phenotype for autism. Am J Psychiatry. 2004;161:539–546. doi: 10.1176/appi.ajp.161.3.539. [DOI] [PubMed] [Google Scholar]
- 273.Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci. 2008;31:137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 274.Bellebaum C, Daum I. Cerebellar involvement in executive control. Cerebellum. 2007;6:184–192. doi: 10.1080/14734220601169707. [DOI] [PubMed] [Google Scholar]
- 275.Pennington BF, Ozonoff S. Executive functions and developmental psychopathology. J Child Psychol Psychiatry. 1996;37:51–87. doi: 10.1111/j.1469-7610.1996.tb01380.x. [DOI] [PubMed] [Google Scholar]
- 276.Lopez BR, Lincoln AJ, Ozonoff S, Lai Z. Examining the relationship between executive functions and restricted, repetitive symptoms of Autistic Disorder. J Autism Dev Disord. 2005;35:445–460. doi: 10.1007/s10803-005-5035-x. [DOI] [PubMed] [Google Scholar]
- 277.Giza J, Urbanski MJ, Prestori F, Bandyopadhyay B, Yam A, Friedrich V, et al. Behavioral and cerebellar transmission deficits in mice lacking the autism-linked gene islet brain-2. J Neurosci. 2010;30:14805–14816. doi: 10.1523/JNEUROSCI.1161-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Swanson DJ, Goldowitz D. Experimental Sey mouse chimeras reveal the developmental deficiencies of Pax6-null granule cells in the postnatal cerebellum. Dev Biol. 2011;351(1):1–12. doi: 10.1016/j.ydbio.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 279.Umeda T, Takashima N, Nakagawa R, Maekawa M, Ikegami S, Yoshikawa T, et al. Evaluation of Pax6 mutant rat as a model for a autism. PLoS One. 2010;5(12):e15500. doi: 10.1371/journal.pone.0015500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Kuemerle B, Gulden F, Cherosky N, Williams E, Herrup K. The mouse Engrailed genes: a window into autism. Behav Brain Res. 2007;176(1):121–132. doi: 10.1016/j.bbr.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Rasalam AD, Hailey H, Williams JH, Moore SJ, Turnpenny PD, Lloyd DJ, et al. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev Med Child Neurol. 2005;47(8):551–555. doi: 10.1017/s0012162205001076. [DOI] [PubMed] [Google Scholar]
- 282.Ingram JL, Stodgell CJ, Hyman SL, Figlewicz DA, Weitkamp LR, Rodier PM. Discovery of allelic variants of HOXA1 and HOXB1: genetic susceptibility to autism spectrum disorders. Teratology. 2000;62(6):393–405. doi: 10.1002/1096-9926(200012)62:6<393::AID-TERA6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 283.Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature. 1997;388:769. doi: 10.1038/42009. 73. [DOI] [PubMed] [Google Scholar]
- 284.Dickson PE, Rogers TD, Del Mar N, Martin LA, Heck D, Blaha CD, et al. Behavioral flexibility in a mouse model of developmental cerebellar Purkinje cell loss. Neurobiol Learn Mem. 2010;94:220–228. doi: 10.1016/j.nlm.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Martin LA, Escher T, Goldowitz D, Mittleman G. A relationship between cerebellar Purkinje cells and spatial working memory demonstrated in a lurcher/chimera mouse model system. Genes Brain Behav. 2004;3:158–166. doi: 10.1111/j.1601-183x.2004.00067.x. [DOI] [PubMed] [Google Scholar]
- 286.Martin LA, Goldowitz D, Mittleman G. Sustained attention in the mouse: a study of the relationship with the cerebellum. Behav Neurosci. 2006;120(2):477–481. doi: 10.1037/0735-7044.120.2.477. [DOI] [PubMed] [Google Scholar]
- 287.Martin LA, Goldowitz D, Mittleman G. Repetitive behavior and increased activity in mice with Purkinje cell loss: a model for understanding the role of cerebellar pathology in autism. Eur J Neurosci. 2010;31:544–555. doi: 10.1111/j.1460-9568.2009.07073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, et al. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306(18):2001–2032. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- 289.Fatemi SH, Halt AR, Realmuto G, Earle J, Kist DA, Thuras P, et al. Purkinje cell size is reduced in the cerebellum of patients with autism. Cell Mol Neurobiol. 2002;22(2):171–175. doi: 10.1023/a:1019861721160. [DOI] [PubMed] [Google Scholar]
- 290.Martin LA, Goldowitz D, Mittleman G. Sustained attention in the mouse: a study of the relationship with the cerebellum. Behav Neurosci. 2006;120(2):477–481. doi: 10.1037/0735-7044.120.2.477. [DOI] [PubMed] [Google Scholar]
- 291.Ozonoff S, Williams BJ, Gale S, Miller JN. Autism and autistic behavior in Joubert syndrome. J Child Neurol. 1999;14(10):636–641. doi: 10.1177/088307389901401003. [DOI] [PubMed] [Google Scholar]
- 292.Lancaster MA, Gopal DJ, Kim J, Saleem SN, Silhavy JL, Louie CM, et al. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat Med. 2011;17(6):726–731. doi: 10.1038/nm.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Garcia CA, McGarry PA, Voirol M, Duncan C. Neurological involvement in the Smith-Lemli-Opitz syndrome: clinical and neuropathological findings. Dev Med Child Neurol. 1973;15(1):48–55. doi: 10.1111/j.1469-8749.1973.tb04865.x. [DOI] [PubMed] [Google Scholar]
- 294.Ellegood J, Pacey LK, Hampson DR, Lerch JP, Henkelman RM. Anatomical phenotyping in a mouse model of fragile X syndrome with magnetic resonance imaging. Neuroimage. 2010;53(3):1023–1029. doi: 10.1016/j.neuroimage.2010.03.038. [DOI] [PubMed] [Google Scholar]
- 295.Bauman ML, Kemper TL, Arin DM. Microscopic observations of the brain in Rett syndrome. Neuropediatrics. 1995 Apr;26(2):105–108. doi: 10.1055/s-2007-979737. [DOI] [PubMed] [Google Scholar]
- 296.Belichenko NP, Belichenko PV, Li HH, Mobley WC, Francke U. Comparative study of brain morphology in Mecp2 mutant mouse models of Rett syndrome. J Comp Neurol. 2008;508(1):184–195. doi: 10.1002/cne.21673. [DOI] [PubMed] [Google Scholar]
- 297.Alkan A, Sigirci A, Kutlu R, Ozcan H, Erdem G, Aslan M, et al. Neurofibromatosis type 1: diffusion weighted imaging findings of brain. Eur J Radiol. 2005;56(2):229–234. doi: 10.1016/j.ejrad.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 298.van der Vaart T, van Woerden GM, Elgersma Y, de Zeeuw CI, Schonewille M. Motor deficits in neurofibromatosis type 1 mice: the role of the cerebellum. Genes Brain Behav. 2011;10(4):404–409. doi: 10.1111/j.1601-183X.2011.00685.x. [DOI] [PubMed] [Google Scholar]
- 299.Padberg GW, Schot JD, Vielvoye GJ, Bots GT, de Beer FC. Lhermitte-Duclos disease and Cowden disease: a single phakomatosis. Ann Neurol. 1991;29(5):517–523. doi: 10.1002/ana.410290511. [DOI] [PubMed] [Google Scholar]
- 300.Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, et al. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29(4):404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- 301.Reith RM, Way S, McKenna J, 3rd, Haines K, Gambello MJ. Loss of the tuberous sclerosis complex protein tuberin causes Purkinje cell degeneration. Neurobiol Dis. 2011;43(1):113–122. doi: 10.1016/j.nbd.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Asahina N, Shiga T, Egawa K, Shiraishi H, Kohsaka S, Saitoh S. [(11)C]flumazenil positron emission tomography analyses of brain gamma-aminobutyric acid type A receptors in Angelman syndrome. J Pediatr. 2008;152(4):546–549. doi: 10.1016/j.jpeds.2007.08.038. 9 e1–3. [DOI] [PubMed] [Google Scholar]
- 303.Dindot SV, Antalffy BA, Bhattacharjee MB, Beaud AL, et al. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17(1):111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- 304.Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, et al. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141(6):1068–1079. doi: 10.1016/j.cell.2010.04.035. [DOI] [PubMed] [Google Scholar]
- 305.Tan GC, Doke TF, Ashburner J, Wood NW, Frackowiak RS. Normal variation in fronto-occipital circuitry and cerebellar structure with an autism-associated polymorphism of CNTNAP2. Neuroimage. 2010;53(3):1030–1042. doi: 10.1016/j.neuroimage.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Crepel F. Developmental changes in retrograde messengers involved in depolarization-induced suppression of excitation at parallel fiber-Purkinje cell synapses in rodents. J Neurophysiol. 2007;97(1):824–836. doi: 10.1152/jn.00735.2006. [DOI] [PubMed] [Google Scholar]
- 307.Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26(1):93–96. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- 308.D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374(6524):719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 309.Sakurai T, Ramoz N, Barreto M, Gazdoiu M, Takahashi N, Gertner M, et al. Slc25a12 disruption alters myelination and neurofilaments: a model for a hypomyelination syndrome and childhood neurodevelopmental disorders. Biol Psychiatry. 2010;67(9):887–894. doi: 10.1016/j.biopsych.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Manni E, Petrosini L. A century of cerebellar somatotropy: a debated representation. Nat Rev Neurosci. 2004;5(3):241–249. doi: 10.1038/nrn1347. [DOI] [PubMed] [Google Scholar]
- 311.Fatemi SH, Halt AR, Realmuto G, Earle J, Kist DA, Thuras P, et al. Purkinje cell size is reduced in the cerebellum of patients with autism. Cell Mol Neurobiol. 2002;22(2):171–175. doi: 10.1023/a:1019861721160. [DOI] [PubMed] [Google Scholar]