

Background: The mechanisms underlying resistance of ovarian cancer cells to apoptosis by positively charged gold nanoparticles (+AuNPs) are unknown.
Results: MICU1 silencing stimulated mitochondrial membrane depolarization in the presence of +AuNPs, leading to apoptosis.
Conclusion: MICU1 confers cellular resistance to apoptosis.
Significance: This study suggests that MICU1 may be an important regulator of apoptotic resistance machineries in cancer cells.
Keywords: Apoptosis, Bcl-2, Cancer, Cancer Biology, Caspase, Mitochondrial Apoptosis
Abstract
Nanoparticles provide a potent tool for targeting and understanding disease mechanisms. In this regard, cancer cells are surprisingly resistant to the expected toxic effects of positively charged gold nanoparticles (+AuNPs). Our investigations led to the identification of MICU1, regulator of mitochondrial calcium uniporter, as a key molecule conferring cancer cells with resistance to +AuNPs. The increase in cytosolic [Ca2+]cyto in malignant cells induced by +AuNPs is counteracted by MICU1, preventing cell death. Pharmacological or siRNA-mediated inhibition of mitochondrial Ca+2 entry leads to endoplasmic reticulum stress and sensitizes cancer cells to +AuNP-induced cytotoxicity. Silencing MICU1 decreases Bcl-2 expression and increases caspase-3 activity and cytosolic cytochrome c levels, thus initiating the mitochondrial pathway for apoptosis: effects further enhanced by +AuNPs. This study highlights the potential of nanomaterials as a tool to broaden our understanding of cellular processes, establishes MICU1 as a novel regulator of the machinery in cancer cells that prevents apoptosis, and emphasizes the need to synergize nanoparticle design with understanding of mitochondrial machinery for enhancing targeted cellular toxicity.
Introduction
Nanoparticle (NP)3 biotechnology provides promising new tools for clinical applications such as diagnosis, imaging, and therapy. To realize this potential, it is necessary to design and engineer NPs that can be targeted to tissues of interest, as well as to produce specific, desired effects (with minimal toxicity) (1–3). Among inorganic cores, the importance of gold NPs (AuNPs) derives from the ability of material scientists to functionalize their surface to control uptake and physiological interactions with cells (4, 5). On the one hand, their biocompatibility and lack of core-based toxicity can be used to design AuNPs that are relatively nontoxic for targeting specific intracellular mechanisms. Conversely, AuNPs can be designed to be highly toxic, e.g. to destroy malignant cells (6, 7). Accordingly, understanding AuNP-cell interactions is critical to furthering their use in medicine. An important potential application that has not been explored so far is the utilization of NPs as tools to understand cellular processes such as calcium regulation, cell proliferation/migration, and cell death that are relevant not only to NP effects, but also to their potential use in therapy.
In a previous study, we demonstrated that positively charged AuNPs (+AuNPs) increase cytosolic Ca2+ ([Ca2+]cyto) in a variety of cells, whereas negatively charged particles are largely without effect (8). An interesting observation was that +AuNPs cause substantial cytotoxicity in normal cells (an obviously undesirable effect), yet malignant cells (e.g. ovarian cancer) are highly resistant to +AuNPs despite substantially increased [Ca2+]cyto levels induced by +AuNPs. This raises the intriguing question as to why malignant cells are resistant to +AuNP-induced cytotoxicity, and conversely provides the opportunity to utilize +AuNPs as a tool to uncover the underlying mechanisms of such resistance. In the present study, we hypothesized that mitochondria, via buffering of [Ca2+]cyto, render resistance to +AuNP-induced cytotoxicity in malignant cells (Scheme 1).
SCHEME 1.

Role of the mitochondrial uniporter regulator MICU1 in calcium buffering and ER stress. +AuNPs can be used to examine the role of MICU1. +AuNPs cause plasma membrane depolarization, allowing Ca2+ influx, which is normally buffered by mitochondria regulated by MICU1, which also has an antiapoptotic role. Separately, MICU1 may normally prevent ER stress such as that induced by +AuNPs. Thus, we propose that enhanced MICU1 expression or activity is a key mechanism that prevents the desired cytotoxic effects of nanoparticles and other agents in conditions such as cancer.
Beyond their well known role as the energy source within cells, mitochondria are involved in [Ca2+]cyto buffering and regulation of programmed apoptotic cell death (9, 10). Although mitochondria express a number of ion transporters capable of Ca2+ uptake into mitochondria, the predominant mechanism is the Ca2+ uniporter, whose identity (MICU1) has been only recently reported (11, 12). Furthermore, the role of the uniporter regulator MICU1 beyond Ca2+ uptake per se has not yet been established. Release of mitochondrial Ca2+ ([Ca2+]mito) back into the cytosol occurs through Na+/H+-dependent Ca2+ exchange or mitochondrial permeability transition pore (13, 14). Thus, alterations in the expression or function of mitochondrial Ca2+ regulatory mechanisms can have profound cellular effects. Here, the link between high levels of [Ca2+]mito and apoptosis are well established, and there is increasing recognition that mitochondria can regulate the behavior of cancer cells (15). However, the molecular machineries regulating such processes are poorly understood. It has been theorized that the apoptotic resistance of cancer cells is related to distinct properties of their mitochondria as compared with nonmalignant cells (16). One of these properties is stabilization of the mitochondrial outer membrane against permeabilization, a key event in the intrinsic apoptotic pathway (14). However, induction of mitochondrial outer membrane permeabilization is dependent on Ca2+ (17) and the proapoptotic protein Bax (18). Thus, the importance of mitochondrial Ca2+ buffering in modulating cell death cannot be underestimated, making it critical to understand how and why mitochondrial function differs in cancer cells and whether this can explain the resistance of malignant cells to +AuNP-induced cytotoxicity.
EXPERIMENTAL PROCEDURES
Materials
Sodium borohydride was from Sigma-Aldrich. Tetrachloroauric acid trihydrate was from Strem Chemicals, Newburyport, MA. [3H]Thymidine was from PerkinElmer Life Sciences. Media and PBS were from Mediatech, Manassas, VA. Primary antibodies were from Cell Signaling, Danvers, MA (phospho-ERK1/2, catalogue number 9101; total ERK1/2, catalogue number 9107; PERK (protein kinase RNA-like endoplasmic reticulum kinase), catalogue number D11A8; BiP (binding immunoglobulin protein), catalogue number C50B12; IRE1α (inositol-requiring protein 1α), catalogue number 14C10; and protein disulfide isomerase, catalogue number C81H6). BCL-2, Bax, cytochrome c, and phospho-PERK were from Santa Cruz Biotechnology (Santa Cruz, CA: sc-783, sc-6236, sc-13156, and sc-32577, respectively), as were secondary antibodies sc-2301 (anti-rabbit) and sc-2302 (anti-mouse). Scintillation mixture was through Fisher Scientific (catalogue number BP458-4). Cell-permeant dyes JC1 (T3168), Fluo4 (F-14201), Mag-Fluo4 (M14206), Rhod2 (R1245MP), MitoTracker Green (M7514), and DisBAC2(3) (B438) were from Invitrogen. Caspase 3 colorimetric assay kit (K106-25) and annexin V-FITC (K101) were from BioVision (Milpitas, CA). TUNEL assay was from Roche Applied Science (catalog number 1684795).
Particle Synthesis
Gold nanoparticles were synthesized according to methods reported by You et al. (19) In brief, 1-pentanethiol-coated gold nanoparticles (d = ∼2 nm) were prepared according to the protocol developed by Schiffrin and co-workers (20). A Murray place-exchange reaction (21) was performed by dissolving the thiolated ligand (bearing a quaternary ammonium end group), synthesized according to the reported procedure (22) in dry dichloromethane with the pentanethiol-coated gold cores and stirring for 3 days at room temperature. Then, dichloromethane was evaporated under reduced pressure, and the oily residue was dissolved in a small amount of distilled water. Dialysis was performed during 5 days (membrane molecular weight cut-off = 10,000) to remove excess ligand and salts remaining in the nanoparticle solution. After dialysis, the particles were lyophilized, redissolved in deionized water, and diluted in buffer to the necessary concentrations.
Cell Culture
Human ovarian cancer cell lines OVCAR5, OV167, and OV202 were grown in DMEM, A2780 was grown in RPMI, and SKOV3-ip was grown in McCoy's medium. All media were supplemented with 10% fetal bovine serum and 1% antibiotics, and cell lines were maintained at 37 °C in a humidified atmosphere consisting of 5% CO2 and 95% air. Ovarian surface epithelium (OSE) cells were grown in MCDB105 (Sigma, M6395)/Medium 199 (Sigma, M5017) supplemented with 15% FBS and 1% antibiotics.
siRNA Knockdown
Ovarian cancer cells were plated in 60-mm dishes with 3 ml of their respective cell culture medium. Cells were transfected with 20 μl of 20 μm siRNA (see results for siRNA targets) purchased from Abcam (Cambridge, MA) along with 40 μl of HiPerfect (Qiagen, Valencia, CA) and 500 μl of Opti-MEM (Invitrogen). Scrambled siRNA (Abcam) was used as a control. After 48 h, cells were collected to detect siRNA knockdown efficiency using RT-PCR.
Real-time PCR
Total RNA was isolated from cells using an RNA mini kit (Qiagen: 74104). Quantitative PCR primer assay for MICU1 (Qiagen: PPR50304A) was carried out using SYBR Green master mix (Applied Biosystems: 4309155). The comparative Ct method was used to calculate the relative abundance of RNA in various cell lines and compared with that with β-actin expression (-fold difference relative to OSE cells). MICU1 and β-actin primers were from Qiagen (PPH09688A and PPH0073G, respectively). The primer sequence for MT-ND1 (gene for mitochondrial Complex I) is as follows: forward, GGCTATATACAACTACGCAAAGGC, and reverse, GGTAGATGTGGCGGGTTTTAGG. MT-ND1 was used to evaluate mitochondrial copy number.
[3H]Thymidine Incorporation Assay for Cellular Proliferation
Cells transfected with siRNA were seeded (2 × 104) in 24-well plates and cultured overnight under standard conditions. Following treatment, 1 μCi of [3H]thymidine was added; 4 h later, the cells were washed with chilled PBS, fixed with 100% cold methanol, and collected for measurement of TCA-precipitable radioactivity (23). Experiments were repeated at least three separate times, with each repeat performed in triplicate.
Western Blots
Cell lysates were prepared as described previously (1), and protein concentration was determined using Bio-Rad protein assay kit (catalogue number 500-0006). Cell lysates (20 μg) were electrophoresed through 4–15% denaturing polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (Millipore). The blots were probed with the appropriate antibodies, and bound antibody was detected using enhanced chemiluminescence according to the manufacturer's protocol (Pierce). Primary antibody dilution was 1:500 for ERK1/2 (phospho and total) and 1:250 for Bcl2, Bax, and cytochrome c. The dilution factor for α-tubulin was 1:5,000. Phospho-PERK and total PERK, BiP, IRE1, and protein disulfide isomerase were diluted 1:1,000. Secondary antibodies were used at a 1:10,000 dilution.
Confocal Microscopy
A2780 cells were plated at 5,000 cells/well of a four-chamber slide (Labtek) in RPMI medium supplemented with 10% FBS and 1% antibiotics and allowed to rest overnight. Subsequently, two wells were treated with Ru360, and 1 h after treatment, +AuNPs were added to one of the Ru360 wells (Ru360/+AuNP) and to an untreated well (+AuNP) overnight. After treatment, samples were acid-stripped on ice for 1 min with stripping buffer (14.6 g of NaCl, 2.5 ml of acetic acid in 500 ml of H2O) to remove surface-bound nanoparticles. The cells were stained for 10 min with either JC1 (mitochondrial membrane potential; ΔΨm) or MitoTracker Green (mitochondrial tracker) and then fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. Separately, TUNEL stains were performed in fixed cells. Nuclei were labeled using DAPI. Samples were then mounted with VECTASHIELD and imaged using an LSM780 confocal microscope, and images were processed using the National Institutes of Health ImageJ software (24). For TUNEL analysis, the number of healthy cells and the number of TUNEL-positive nuclei were counted and totaled.
Cellular Apoptosis Assay
Cells were seeded in 60-mm dishes at 3 × 105. At ∼80% confluence, Scr siRNA and MICU siRNA cells were treated with +AuNPs overnight (medium alone for control cells). After treatment, cells were thoroughly rinsed with 1× PBS. Annexin V/propidium iodide staining flow cytometry was performed according to manufacturer-provided protocol.
Caspase 3 Colorimetric Assay
A2780 cells were seeded 1 × 106 in 10-cm dishes and allowed to grow overnight under standard conditions. Plates were treated overnight (16 h) with Ru360 only, +AuNPs only, or with both Ru360 and +AuNPs. The assay was carried out following the manufacturer's protocol, and was read at 405 nm. Treated cells were compared with nontreated cells (noninduced). The assay was repeated in triplicate on different days.
Real-time Imaging
Real-time fluorescence imaging of different parameters was performed using a Molecular Devices FlexStation 3 microplate reader with micropipetting capabilities. 1 × 104 A2780 cells were seeded onto a 96-well clear bottom plate (CoStar catalogue number 3603) and allowed to grow overnight in standard conditions. Plates were rinsed three times with Hanks' balanced salt solution and subsequently loaded with one of the following: 2 μm DisBAC2(3) (cell-permeant plasma membrane potential-sensitive dye; excitation 488/emission 530), 2 μm JC-1 (cell-permeant mitochondrial membrane potential (ΔΨm) dye; excitation 488/emission 590), 5 μm mag-Fluo-4 (ER Ca2+ [Ca2+]ER-sensitive dye; excitation 495/emission 525), 5 μm fura-2 ([Ca2+]cyto-sensitive dye; excitation 340 or 380/emission 510); or 5 μm rhod-2 ([Ca2+]mito-sensitive dye; excitation 568/emission 590). For plasma membrane potential and ΔΨm, calibration was performed in situ by exposing a subset of cells to a range of KCl concentrations (0, 2.5, 5, 10, 20, 40, 80 mm KCl in Hanks' balanced salt solution). [Ca2+]ER and [Ca2+]mito calibrations were performed as described previously (25).
Statistical Analysis
All values are expressed as means ± S.D. Statistical significance was determined using two-sided Student's t test.
RESULTS
MICU1 Enhances the Buffering Activity of Mitochondria When Cells Are Exposed to +AuNPs
To determine whether the mitochondrial Ca2+ uniporter contributes to cellular responses to AuNPs, we initially examined MICU1 mRNA expression in various ovarian cancer cells, comparing them with normal OSE, and found both A2780 and OV202 to have more than ∼3-fold higher MICU1 expression (Fig. 1A). Such high expression of MICU1 could be due to increased number of total mitochondria, a prominent feature of malignancy (26, 27). Interestingly, cells undergoing malignant transformations often increase and include ion channels, especially those that transport Ca2+ (28); however, it is unknown whether mitochondrial ion channels are similarly up-regulated. We determined the mitochondrial copy number in OSE and in different ovarian cancer cells using RT-PCR for the Complex I gene MT-ND1 (Fig. 1B). It was evident that mitochondrial copy numbers in ovarian cancer cells relative to OSE were generally higher. This is further demonstrated in Fig. 1C showing more than a 2-fold increase in green fluorescence (via MitoTracker) in A2780 cells as compared with OSE, indicative of an increased number of mitochondria in malignant cells as compared with nonmalignant cells. However, most relevant to this study, normalization of MICU1 expression to mitochondrial copy number demonstrated comparable expression of MICU1 per mitochondrion in malignant ovarian cells relative to OSE (nonmalignant cells; Fig. 1D). The functional significance of the increased number of mitochondria in malignant cells may then be that the net greater total amount of MICU1 allows for greater mitochondrial Ca2+ buffering. We tested this in A2780 cells loaded with the fluorescent [Ca2+]cyto and [Ca2+]mito indicators fura-2/AM and rhod-2/AM, respectively. Exposure of A2780 cells to 1 μm +AuNPs resulted in rapid increase in [Ca2+]cyto (as we observed previously (8), Fig. 2 A) as well as a slow sustained increase in [Ca2+]mito (Fig. 2B), the latter suggesting the involvement of the mitochondrial Ca2+ uniporter, rather than the more rapid Na+/Ca2+ exchanger. As compared with A2780, +AuNPs caused smaller [Ca2+]cyto and [Ca2+]mito responses in nonmalignant OSE cells (Fig. 2, C and D). Pretreatment with the mitochondrial Ca2+ uptake inhibitor Ru360 entirely abrogated the [Ca2+]mito response to +AuNPs, especially in A2780 cells (Fig. 2B). siRNA-mediated knockdown (MICU siRNA) had effects comparable with Ru360, whereas scrambled siRNA (Scr siRNA) was without effect (i.e. +AuNPs increased [Ca2+]mito; Fig. 2B). However, the converse effect of inhibited mitochondrial Ca2+ uptake was a substantial increase in [Ca2+]cyto in the presence of Ru360 or MICU siRNA (Fig. 2A). These differences of responses were further validated in other ovarian cancer cell lines. When normalized to OSE (nonmalignant cell line), SKOV3-ip and OV202 had a comparable response to +AuNPs and MICU inhibition analogous to A2780 (Fig. 2, E and F). This difference in amplitude (relative to OSE) demonstrates how mitochondrial buffering of cytosolic Ca2+ correlates to MICU1 expression (Fig. 1). It is also evident from Figs. 1B and 2, E and F, that when normalized with respect to mitochondrial copy number, the ability of MICU1 to regulate buffering of cytosolic calcium is higher in malignant cells as compared with normal OSE cells. Specifically, these data highlight the functional importance of MICU1 in the mitochondrial response of malignant cells, showing that increase in MICU1 expression translated to enhanced ability to buffer increases in [Ca2+]cyto induced by +AuNPs.
FIGURE 1.

MICU1 in ovarian cancer cells. A, MICU1 is expressed to a greater extent in different types of ovarian cancer cells as compared with noncancerous controls (OSE) as determined by mRNA measurements. B and C, enhanced MICU1 expression may represent more mitochondria or increased MICU1 per mitochondrion. Real-time PCR of the mitochondrial gene MT-ND1 (Complex I; B) as well as confocal imaging of cells stained with MitoTracker Green (C) show greater number of mitochondria in A2780 malignant cells (blue staining represents DAPI). A. U., absorbance units. D, however, the ratio of MICU1 to MT-ND1 is comparable between malignant and nonmalignant cells. Values are means ± S.D., n = 3.
FIGURE 2.

MICU1-mediated calcium buffering in ovarian cancer cells evaluated using +AuNPs. A and B, in A2780 cells loaded with the fluorescent dyes fura-2 ([Ca2+]cyto) and rhod-2 ([Ca2+]mito), 1 μm +AuNPs caused rapid increase in [Ca2+]cyto (A) but slower sustained increases in [Ca2+]mito (B). Inhibition of uniporter using Ru360 or MICU1 siRNA abrogated [Ca2+]mito responses to +AuNPs, and conversely enhanced [Ca2+]cyto responses. C and D, in contrast to A2780, [Ca2+]cyto and especially [Ca2+]mito responses, or the effects of uniporter inhibition, were substantially smaller in nonmalignant OSE cells. NT, nontreated controls. E and F, the greater role of uniporter in [Ca2+]cyto (E) buffering and [Ca2+]mito (F) responses to +AuNPs is valid in different types of ovarian cancer cells (A2780, OV202, and SKOV3-ip), highlighting the functional importance of MICU1 in malignant cells. Values are means ± S.D., n = 3.
+AuNPs Modulate Membrane Potential in MICU1 Knockdown Cells
Given its only recent recognition, beyond mitochondrial Ca2+ uptake, the functions of MICU1 are currently unknown. In A2780 cells loaded with the plasma membrane potential indicator DisBAC2(3), we found that +AuNPs produce substantial membrane depolarization (Fig. 3A). Interestingly, inhibition of the mitochondrial Ca2+ uniporter with Ru360, or MICU siRNA resulted in substantial enhancement of +AuNP-induced membrane depolarization. However, in OSE cells, +AuNPs induced hyperpolarization, which was further enhanced in the presence of Ru360 (Fig. 3B). Furthermore, in A2780 cells loaded with the mitochondrial membrane potential (ΔΨm) indicator JC-1, +AuNPs also produced mitochondrial depolarization to some extent, but Ru360 as well as MICU siRNA substantially enhanced such depolarization. ΔΨm is the driving force behind the influx of ions (H+, Ca2+) into mitochondria. As shown in Fig. 3C, upon depolarization, the punctate orange-red fluorescent JC1 staining was replaced by diffuse green monomer fluorescence, whereas no change was seen in OSE cells under the same conditions (Fig. 3D). Although a slight decrease in ΔΨm occurred in cells treated with Ru360 only or +AuNPs only, in the presence of both Ru360 and +AuNPs, there was substantial change in ΔΨm in A2780 cells (Fig. 3, C and E), but only a slight change in OSE cells (Fig. 3, D and F). These data suggest that inhibiting MICU1 allows +AuNPs to induce a robust mitochondrial membrane depolarization in malignant ovarian cells, substantially larger than in normal OSE. These differences may have downstream effects of sensitizing malignant cells to the antiproliferative and/or proapoptotic effects of +AuNPs.
FIGURE 3.
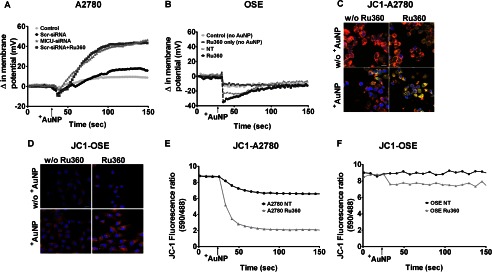
MICU1 modulates ΔΨm responses in malignant cells. A and B, in A2780 cells loaded with the fluorescent ΔΨm indicator DiBASC2(3), +AuNPs caused rapid and substantial membrane depolarization (A), whereas nonmalignant OSE cells showed slight hyperpolarization (B). +AuNP effects on ΔΨm in A2780 cells were enhanced by MICU1 blockade (siRNA). NT, nontreated controls. C–F, these +AuNP effects on ΔΨm were also evident in confocal images of the fluorescent dye JC-1 in malignant cells (C) versus normal cells (D) and corresponding measurements of JC-1 (E and F). Inhibition of uniporter by Ru360 enhanced +AuNP effects on ΔΨm. A similar effect was observed with MICU1 siRNA (not shown). Scale bar = 20 μm; n = 3. w/o +AuNP, without +AuNP.
MICU1 Influences +AuNP-induced Cellular Apoptosis
In our previous work, we had observed that unlike nonmalignant human bronchial epithelial cells and airway smooth muscle cells, cancer cells are able to tolerate the substantial increases in [Ca2+]cyto induced by +AuNPs without changes in cell proliferation or enhancement of cell death (19). Our data now suggest that the presence of higher mitochondrial number in malignant cells leading to increased expression of MICU1 and function may be contributory (Fig. 1B). One response to changes in ΔΨm is apoptosis. Therefore, we used annexin V/propidium iodide (Fig. 4A) and TUNEL (Fig. 4, B–D) assays to test whether blocking uniporter or MICU1 restores an apoptotic response of malignant cells to +AuNPs. Indeed, in the presence of Ru360, +AuNP-induced increased apoptosis in A2780 cells (Fig. 4, A, B, and D). However, only a marginal increase in apoptosis was observed in normal OSE cells (Fig. 4, A, C, and D). In separate analyses, we used [3H]thymidine incorporation to determine the role of MICU1 in cell proliferation. Complementary to the apoptosis data, A2780 cells that did not show any changes in proliferation with +AuNPs showed substantial reduction in proliferation when MICU1 was inhibited with MICU siRNA (Fig. 4E). In comparison, proliferation of OSE cells was minimally affected by MICU inhibition (Fig. 4F).
FIGURE 4.
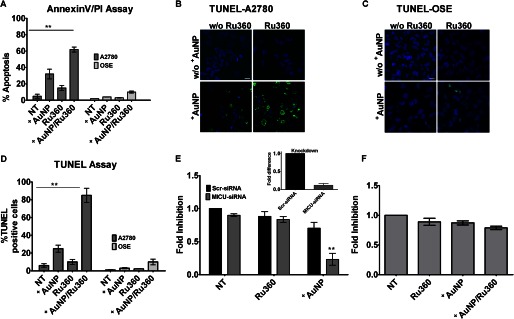
Uniporter and MICU1 regulate proliferation and apoptosis in malignant cells. A–C, exposure of A2780 cells to +AuNPs induces cellular apoptosis to a certain extent, as shown by annexin V/propidium iodide (PI) assay (A) and TUNEL stain (B and C). NT, nontreated controls. D, the ratio of TUNEL-positive nuclei (green fluorescence) to DAPI (blue fluorescence) was compared and quantified and expressed as the percentage of the total. Ru360 inhibition substantially enhances +AuNP-induced apoptosis (A–D). E, inhibition of MICU1 does not influence base-line cellular proliferation (thymidine incorporation assay) relative to nontreated controls. However, +AuNP-induced cell proliferation is substantially reduced when MICU1 is inhibited (inset shows the efficiency of MICU siRNA knockdown in A2780 cells). F, OSE cells showing a marginal effect under similar conditions. Values are means ± S.D., n = 3. ** indicates significant AuNP effect (p < 0.001). Images shown are representative. Scale bar = 20 μm.
The contribution of MICU1 to enhanced cell apoptosis/reduced proliferation was additionally tested using Western blot analysis for proapoptotic and antiproliferative proteins. Bax levels were notably increased in MICU1 siRNA-treated cells, whereas conversely Bcl-2 levels were decreased (Fig. 5A, KD) following exposure to +AuNPs as compared with the lack of effect in scrambled siRNA (Fig. 5A, Scr)-treated cells. A marked increase in cytosolic cytochrome c was induced by +AuNPs in MICU siRNA cells (Fig. 5A, KD). A decrease in phosphorylation of ERK1/2 was also observed in MICU1 siRNA-treated cells incubated with +AuNPs (Fig. 5A, KD). However, OSE cells did not show a noticeable difference under similar conditions (Fig. 5B). A time course study revealed that AuNP effect on Bcl-2 expression occurred around 2 h (Fig. 5C) in MICU1 knockdown cells. Finally, activation of caspase-3 was also initiated in A2780 cells with Ru360 exposure, whereas a modest change was observed in OSE cells (Fig. 5D). Overall, these data support the idea that MICU1 protects against +AuNP-induced cytotoxicity in malignant cells.
FIGURE 5.

Mechanisms by which MICU1 influences +AuNP-induced apoptosis. A, immunoblots of whole cell lysates from A2780 cells for markers of apoptosis (BAX), antiapoptosis (BCL-2), and proliferation (ERK1/2 and phospho-ERK1/2 (ph-ERK1/2)) showed that +AuNPs increased expression of proapoptotic factors. In the presence of MICU1 siRNA (KD), these effects were substantially enhanced. The enhancement of +AuNP-induced apoptosis by blocking MICU1 (through siRNA; KD) was also demonstrated by increased cytosolic expression of cytochrome c (CytoC). Scr, scrambled siRNA . B, these effects of +AuNPs were absent in OSE cells. NT, nontreated controls. C, the effects in A2780 cells begin after 2 h of exposure to +AuNPs in the knockdown cells. D, separately inhibition by Ru360 leads to increased activity of caspase 3 in A2780 cells with only a modest effect in OSE cells (relative to nontreated controls). Tubulin was used as a loading control. Values are means ± S.D., n = 3. ** indicates significant +AuNP effect (p < 0.001).
+AuNPs Promote Substantial Endoplasmic Reticulum Ca2+ Release
Interestingly, although peak [Ca2+]cyto responses of A2780 cells to +AuNPs increased more with MICU1 inhibition, they also rapidly declined (Fig. 2A). We tested the idea that excess [Ca2+]cyto is reabsorbed into the endoplasmic reticulum (ER). Using the fluorescent ER Ca2+ probe Mag-Fluo-4/AM, we found that the addition of +AuNPs in the presence of Ru360 resulted in a sharp decrease in fluorescence followed by an increase (Fig. 6A), indicating ER Ca2+ reuptake. In comparison, in A2780 cells not exposed to Ru360, the addition of +AuNPs did not result in fluorescence change after the initial sharp decrease. The enhanced influx of [Ca2+]cyto into the ER could be a direct result of increased levels of Bax (33) that can initiate an ER stress response (29). In accordance, as shown in Fig. 6B, both phospho-PERK expression and total PERK, IRE1α, and BiP expression were increased in the presence of +AuNPs and were further enhanced when preincubated with Ru360. Moreover, protein disulfide isomerase expression was drastically increased in cells co-incubated with Ru360 and +AuNPs. Although a small delayed effect was observed in OSE cells upon the addition of +AuNPs (Fig. 6C), Western blot analysis of the ER stress markers did not show remarkable changes.
FIGURE 6.

Uniporter contributes to ER stress in malignant cells. A, in A2780 cells loaded with the fluorescent ER Ca2+ indicator mag-fluo-4, +AuNPs produced substantial ER Ca2+ release (decreased fluorescence). In the presence of Ru360, base-line ER Ca2+ was higher, but +AuNP-induced release of ER Ca2+ was comparable with control (n = 3). However, ER uptake of Ca2+ was more rapid in Ru360-treated cells. A. U., absorbance units. B and C, these +AuNP-induced changes in ER Ca2+ were paralleled by enhanced expression of ER stress proteins such as phospho-PERK (phPERK), total PERK, BiP, and IRE1α (but not protein disulfide isomerase) in A2780 cells (B) but not in OSE cells (C). NT, nontreated controls.
DISCUSSION
The significance of the present study is two-fold. On the one hand, our results highlight the importance of the relatively novel protein MICU1, regulator of the mitochondrial calcium uniporter, in the resistance of ovarian cancer cells to cytotoxic agents wherein increased [Ca2+]cyto in malignant cells is counteracted by MICU1-regulated mitochondrial Ca2+ uptake, thus preventing cell death. Interestingly, normal OSE cells respond to MICU1 blockade with marginal changes in both plasma and mitochondrial membrane potential leading to only small increases in [Ca2+]cyto. This effect may be due to the low amount of mitochondria and hence overall low MICU1 expression, thus making the nonmalignant cells less influenced by blockade of MICU1 as compared with malignant cells. Our studies further show that MICU1 appears not only to have a role in sequestering [Ca2+]cyto but also confers protection against ER stress, apoptosis, and cytotoxicity.
The second major significance of our studies is that the mechanisms underlying protection against cytotoxicity can be revealed by the use of +AuNPs. Thus, our study highlights the ability to use nanomaterials as a tool to broaden our understanding of cellular processes that contribute to resistance of cancer cells to death. Conversely, by demonstrating that +AuNP-induced cytotoxicity per se is dependent on MICU1, we show that if nanoparticles are to be used as therapeutic avenues, synergizing nanoparticle design with understanding of mitochondrial machinery for enhancing cellular toxicity is critical.
Calcium signaling is a key aspect of most biological processes such as cellular proliferation, metabolism, and protein synthesis, making [Ca2+]i homeostasis critical to cellular function. Cells contain a diverse and complex toolkit of Ca2+ regulatory mechanisms including plasma membrane channels and intracellular organelles such as the ER. With their ability to sequester or buffer large amounts of free Ca2+, mitochondria help control [Ca2+]i and blunt any potential lethal effects of [Ca2+]cyto overload. Mitochondrial dysregulation has been shown to be involved in tumorigenesis (27, 30). This is characterized by transformation of the Ca2+ signaling pathways such that cellular proliferation, angiogenesis, and potential metastasis and evasion from apoptosis are highly favored (31). Thus, there appears to be a direct relationship between mitochondrial dysfunction, elevated [Ca2+]cyto, and tumorigenesis. Therefore, understanding the mechanisms by which mitochondria buffer [Ca2+]cyto and whether and how they contribute to tumor cell survival is key to the development of new therapies.
Although mitochondria contain multiple Ca2+ regulatory mechanisms, the importance of MICU1 has been only recently established (11, 12). MICU1 has been shown to regulate the mitochondrial Ca2+ uniporter, which pumps cytosolic Ca2+ into the mitochondria and thus can buffer Ca2+ in combination with the bidirectional mitochondrial Na+/Ca2+ exchanger (32). Considering the fact that mitochondrial Ca2+ drives mitochondrial membrane potential as well as ATP production, the importance of MICU1 is clear. Whether abnormalities in MICU1 expression and/or function are important for mitochondrial dysfunction in different pathologies or whether MICU1 can be targeted for therapy is not known. In this study, we demonstrate that increased MICU1 expression in cancer cells enhances the ability of mitochondria to buffer cytosolic Ca2+. Although enhanced MICU1 activity should reduce base-line [Ca2+]cyto (an aspect not studied here), what appears to be more important is its ability to buffer high levels of [Ca2+]cyto (e.g. that induced by +AuNPs) that would be normally cytotoxic. Interestingly, such enhanced mitochondrial buffering should have resulted in increased probability of a mitochondrial permeability transition pore and thus more apoptosis, which was obviously not observed in the malignant cells. This suggests that MICU1 may somehow prevent Ca2+-driven apoptosis in cancer cells, a feature unmasked when MICU1 expression or function is blocked.
Although the underlying mechanisms by which MICU1 prevents apoptosis are not entirely clear, MICU1 may modulate mitochondrial depolarization and thus control ATP availability. Our data further suggest that MICU1 is important for the communication between ER and mitochondria such that via MICU1 and ER stability, as well as reduced apoptosis, are promoted. The close proximity of the mitochondria to the ER allows for highly efficient communication and for either organelle to monitor Ca2+ fluxes from the other. For example, one of the central regulatory checkpoints in cells is ER inositol 1,4,5-trisphosphate receptors that are inhibited by the antiapoptotic protein Bcl-2, which is overexpressed in tumor cells (31, 33). Bcl-2 affects apoptosis/survival by regulating mitochondrial outer membrane permeability (14, 34, 35). Bax and other proapoptotic proteins form a pore in the outer mitochondrial membrane that allows release of cytochrome c into the cytosol and subsequent activation of downstream caspases. Our findings show that in tumor cells, only when MICU1 is inhibited does exposure to +AuNPs result in reduced Bcl-2 and elevated Bax, thus promoting apoptosis. Separately, perturbations in normal ER function can trigger the ER stress response, such as the unfolded protein response or changes in the ionic conditions of the ER lumen (17, 36). One of the main ER chaperones that targets misfolded proteins and regulates Ca2+ binding during ER stress response is BiP. Under normal cellular conditions, BiP binds PERK and IREα, thus rendering them inactive. However, during ER stress, BiP binds to unfolded proteins, subsequently releasing both PERK and IREα (37, 38). Upon release, PERK suppresses mRNA production of the proapoptotic protein IREα in response to the unfolded protein response. Our data demonstrate the importance of mitochondria and MICU1 in these relationships, preventing +AuNP-induced ER stress to occur in cancer cells.
Overall, the data presented in this study demonstrate how surface modified nanoparticles could be utilized to study mechanisms such as MICU1 that modulate a number of intracellular processes including Ca2+ homeostasis, apoptosis, proliferation, and ER stress. Future studies should focus on the engineering of nanoparticles to better understand the mechanism of the multidrug-resistant properties of cancer cells. This in turn may allow malignant cells to be sensitized to chemotherapeutics and further inhibit tumor growth and metastasis. Furthermore, our results highlight the importance of synergizing nanoparticle design with targeted interference with cellular mechanisms, such as mitochondria, to enhance cytotoxicity in cancer therapy.
This work was supported, in whole or in part, by National Institutes of Health Grants CA135011 and CA136494 (to P. M.), GM077173 (to V. M. R.), and HL088029 (to Y. S. P.).
This article was selected as a Paper of the Week.
- NP
- nanoparticle
- AuNP
- gold nanoparticle
- +AuNP
- positively charged AuNP
- PERK
- protein kinase RNA-like endoplasmic reticulum kinase
- OSE
- ovarian surface epithelium
- ER
- endoplasmic reticulum
- [Ca2+]mito
- mitochondrial Ca2+
- [Ca2+]cyto
- cytosolic Ca2+
- IRE1α
- inositol-requiring protein 1α
- BiP
- binding immunoglobulin protein
- DisBAC2(3)
- bis-(1,3-diethylthiobarbituric acid)trimethine oxonol
- ΔΨm
- mitochondrial membrane potential
- KD
- knockdown
- Scr
- scrambled
- MICU1
- mitochondrial calcium uptake 1.
REFERENCES
- 1. Arvizo R. R., Rana S., Miranda O. R., Bhattacharya R., Rotello V. M., Mukherjee P. (2011) Mechanism of anti-angiogenic property of gold nanoparticles: role of nanoparticle size and surface charge. Nanomedicine 7, 580–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bhattacharyya S., Singh R. D., Pagano R., Robertson J. D., Bhattacharya R., Mukherjee P. (2012) Switching the targeting pathways of a therapeutic antibody by nanodesign. Angew. Chem. Int. Ed. Engl. 51, 1563–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhirde A. A., Patel V., Gavard J., Zhang G., Sousa A. A., Masedunskas A., Leapman R. D., Weigert R., Gutkind J. S., Rusling J. F. (2009) Targeted killing of cancer cells in vivo and in vitro with EGF-directed carbon nanotube-based drug delivery. ACS Nano 3, 307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agasti S. S., Chompoosor A., You C.-C., Ghosh P., Kim C. K., Rotello V. M. (2009) Photoregulated release of caged anticancer drugs from gold nanoparticles. J. Am. Chem. Soc. 131, 5728–5729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu Z. J., Ghosh P. S., Miranda O. R., Vachet R. W., Rotello V. M. (2008) Multiplexed screening of cellular uptake of gold nanoparticles using laser desorption/ionization mass spectrometry. J. Am. Chem. Soc. 130, 14139–14143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Massich M. D., Giljohann D. A., Schmucker A. L., Patel P. C., Mirkin C. A. (2010) Cellular response of polyvalent oligonucleotide-gold nanoparticle conjugates. ACS Nano 4, 5641–5646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim C., Agasti Sarit S., Zhu Zhengjiang, Isaacs Lyle, Rotello, Vincent M. (2010) Recognition-mediated activation of therapeutic gold nanoparticles inside living cells. Nat. Chem. 2, 962–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arvizo R. R., Miranda O. R., Thompson M. A., Pabelick C. M., Bhattacharya R., Robertson J. D., Rotello V. M., Prakash Y. S., Mukherjee P. (2010) Effect of nanoparticle surface charge at the plasma membrane and beyond. Nano Lett. 10, 2543–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ni Chonghaile T., Sarosiek K. A., Vo T.-T., Ryan J. A., Tammareddi A., Moore Vdel G., Deng J., Anderson K. C., Richardson P., Tai Y.-T., Mitsiades C. S., Matulonis U. A., Drapkin R., Stone R., Deangelo D. J., McConkey D. J., Sallan S. E., Silverman L., Hirsch M. S., Carrasco D. R., Letai A. (2011) Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 334, 1129–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tait S. W. G., Green D. R. (2012) Mitochondria and cell signalling. J. Cell Sci. 125, 807–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Stefani D., Raffaello A., Teardo E., Szabò I., Rizzuto R. (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perocchi F., Gohil V. M., Girgis H. S., Bao X. R., McCombs J. E., Palmer A. E., Mootha V. K. (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kilbride S. M., Prehn J. H. M. (August 6, 2012) Central roles of apoptotic proteins in mitochondrial function. Oncogene 10.1038/onc.2012.348 [DOI] [PubMed] [Google Scholar]
- 14. Whelan R. S., Konstantinidis K., Wei A.-C., Chen Y., Reyna D. E., Jha S., Yang Y., Calvert J. W., Lindsten T., Thompson C. B., Crow M. T., Gavathiotis E., Dorn G. W., 2nd, O'Rourke B., Kitsis R. N. (2012) Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. U.S.A. 109, 6566–6571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bianchi K., Rimessi A., Prandini A., Szabadkai G., Rizzuto R. (2004) Calcium and mitochondria: mechanisms and functions of a troubled relationship. Biochim. Biophys. Acta 1742, 119–131 [DOI] [PubMed] [Google Scholar]
- 16. Kroemer G., Pouyssegur J. (2008) Tumor cell metabolism: Cancer's Achilles' heel. Cancer Cell 13, 472–482 [DOI] [PubMed] [Google Scholar]
- 17. Deniaud A., Sharaf el dein O., Maillier E., Poncet D., Kroemer G., Lemaire C., Brenner C. (2008) Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27, 285–299 [DOI] [PubMed] [Google Scholar]
- 18. Youle R. J., Strasser A. (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 19. You C. C., Miranda O. R., Gider B., Ghosh P. S., Kim I. B., Erdogan B., Krovi S. A., Bunz U. H., Rotello V. M. (2007) Detection and identification of proteins using nanoparticle-fluorescent polymer 'chemical nose' sensors. Nat. Nanotechnol. 2, 318–323 [DOI] [PubMed] [Google Scholar]
- 20. Brust M., Walker M., Bethell D., Schiffrin D. J. (1996) Synthesis of thiol derivatized gold nanoparticles in a 2-phase liquid-liquid system. J. Chem. Soc. Chem. Commun. 7, 801–802 [Google Scholar]
- 21. Hostetler M. J., Green S. J., Stokes J. J., Murray R. W. (1996) Monolayers in three dimensions: synthesis and electrochemistry of ω-functionalized alkanethiolate-stabilized gold cluster compounds. J. Am. Chem. Soc. 118, 4212–4213 [Google Scholar]
- 22. Miranda O. R., Chen H.-T., You C.-C., Mortenson D. E., Yang X.-C., Bunz U. H. F., Rotello V. M. (2010) Enzyme-amplified array sensing of proteins in solution and in biofluids. J. Am. Chem. Soc. 132, 5285–5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mukherjee P., Bhattacharya R., Wang P., Wang L., Basu S., Nagy J. A., Atala A., Mukhopadhyay D., Soker S. (2005) Antiangiogenic properties of gold nanoparticles. Clin. Cancer Res. 11, 3530–3534 [DOI] [PubMed] [Google Scholar]
- 24. Bhattacharyya S., Bhattacharya R., Curley S., McNiven M. A., Mukherjee P. (2010) Nanoconjugation modulates the trafficking and mechanism of antibody induced receptor endocytosis. Proc. Natl. Acad. Sci. U.S.A. 107, 14541–14546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delmotte P., Yang B., Thompson M. A., Pabelick C. M., Prakash Y. S., Sieck G. C. (2012) Inflammation alters regional mitochondrial Ca2+ in human airway smooth muscle cells. Am. J. Physiol. Cell Physiol. 303, C244–C256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fulda S., Galluzzi L., Kroemer G. (2010) Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 9, 447–464 [DOI] [PubMed] [Google Scholar]
- 27. Gogvadze V., Orrenius S., Zhivotovsky B. (2008) Mitochondria in cancer cells: what is so special about them? Trends Cell Biol. 18, 165–173 [DOI] [PubMed] [Google Scholar]
- 28. Prevarskaya N., Skryma R., Shuba Y. (2010) Ion channels and the hallmarks of cancer. Trends Mol. Med. 16, 107–121 [DOI] [PubMed] [Google Scholar]
- 29. Rizzuto R., Duchen M. R., Pozzan T. (2004) Flirting in little space: The ER/mitochondria Ca2+ liaison. Sci. STKE 2004, re1- [DOI] [PubMed] [Google Scholar]
- 30. Kroemer G. (2006) Mitochondria in cancer. Oncogene 25, 4630–4632 [DOI] [PubMed] [Google Scholar]
- 31. Roderick H. L., Cook S. J. (2008) Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 8, 361–375 [DOI] [PubMed] [Google Scholar]
- 32. Rizzuto R., De Stefani D., Raffaello A., Mammucari C. (2012) Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578 [DOI] [PubMed] [Google Scholar]
- 33. Rasola A., Sciacovelli M., Chiara F., Pantic B., Brusilow W. S., Bernardi P. (2010) Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. U.S.A. 107, 726–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chami M., Prandini A., Campanella M., Pinton P., Szabadkai G., Reed J. C., Rizzuto R. (2004) Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J. Biol. Chem. 279, 54581–54589 [DOI] [PubMed] [Google Scholar]
- 35. Wei M. C., Zong W.-X., Cheng E. H.-Y., Lindsten T., Panoutsakopoulou V., Ross A. J., Roth K. A., MacGregor G. R., Thompson C. B., Korsmeyer S. J. (2001) Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Breckenridge D. G., Germain M., Mathai J. P., Nguyen M., Shore G. C. (2003) Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 22, 8608–8618 [DOI] [PubMed] [Google Scholar]
- 37. Hetz C., Martinon F., Rodriguez D., Glimcher L. H. (2011) The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol. Rev. 91, 1219–1243 [DOI] [PubMed] [Google Scholar]
- 38. Ni M., Zhou H., Wey S., Baumeister P., Lee A. S. (2009) Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS One 4, e6868. [DOI] [PMC free article] [PubMed] [Google Scholar]