

This paper studies the regulation of alternative splicing of the myelin-associated glycoprotein pre-mRNA. The authors show that regulation of exon 12 skipping involves hnRNP A1 interacting with a binding site that overlaps the 5′ splice site. A conserved secondary structure around the binding site contributes to regulation.
Keywords: alternative splicing, myelin-associated glycoprotein, secondary structure, hnRNP A1, Mag, myelin
Abstract
Myelin-associated glycoprotein (MAG) is a major component of myelin in the vertebrate central nervous system. MAG is present in the periaxonal region of the myelin structure, where it interacts with neuronal proteins to inhibit axon outgrowth and protect neurons from degeneration. Two alternatively spliced isoforms of Mag mRNA have been identified. The mRNA encoding the shorter isoform, known as S-MAG, contains a termination codon in exon 12, while the mRNA encoding the longer isoform, known as L-MAG, skips exon 12 and produces a protein with a longer C-terminal region. L-MAG is required in the central nervous system. How inclusion of Mag exon 12 is regulated is not clear. In a previous study, we showed that heteronuclear ribonucleoprotein A1 (hnRNP A1) contributes to Mag exon 12 skipping. Here, we show that hnRNP A1 interacts with an element that overlaps the 5′ splice site of Mag exon 12. The element has a reduced ability to interact with the U1 snRNP compared with a mutant that improves the splice site consensus. An evolutionarily conserved secondary structure is present surrounding the element. The structure modulates interaction with both hnRNP A1 and U1. Analysis of splice isoforms produced from a series of reporter constructs demonstrates that the hnRNP A1-binding site and the secondary structure both contribute to exclusion of Mag exon 12.
INTRODUCTION
Axons in the vertebrate nervous system are surrounded by myelin, a complex structure comprised of layers of plasma membrane with specific protein components located at specific sites. Myelin insulates axons and enhances the saltatory propagation of electrical impulses. In addition, myelin is required to maintain axonal integrity. Communication between the glial cell and the neuron occurs through the action of proteins located at the periaxonal surface.
MAG, a transmembrane IgG superfamily member, is one such protein. MAG has several functions, including inhibition of axon outgrowth and regeneration (McKerracher et al. 1994; Mukhopadhyay et al. 1994), protecting neurons from excitation-induced toxicity (Lopez et al. 2011), and protecting neurons from age-related degeneration (Lassmann et al. 1997). Although it is largely normal, the nervous system of Mag-deficient mice has several subtle defects, such as decreased axon diameter and an increased number of unmyelinated axons (Li et al. 1994; Montag et al. 1994; Bartsch et al. 1997; Yin et al. 1998). The mice also exhibit age-related axonal degeneration (Lassmann et al. 1997) and enhanced excitotoxicity (Lopez et al. 2011). MAG interacts with several receptors on the surface of the axon, including ganglioside receptors (GD1a and GT1b) (Yang et al. 1996), Nogo receptors (Liu et al. 2002; Venkatesh et al. 2005), and the paired immunoglobulin-like receptor B (PIR-B) (Atwal et al. 2008). PIR-B, at least in part, mediates the effects of MAG in neurite outgrowth inhibition (Atwal et al. 2008; Fujita et al. 2011). Additionally, mice with a null mutation in the B4galnt1 gene, which is responsible for synthesis of the ganglioside receptors GD1a and GT1b, have defects similar to those seen in the Mag null mouse, highlighting the importance of these receptors for MAG function (Sheikh et al. 1999).
Mag is alternatively spliced to produce two isoforms, S-MAG and L-MAG, which are regulated developmentally and spatially (Lai et al. 1987; Tropak et al. 1988; Wu et al. 2002). Both isoforms contain the extracellular IgG domain and the transmembrane domain. They differ at the C-terminal tail, which protrudes into the cytoplasmic space. S-MAG contains an alternative exon (exon 12) that contains a stop codon, producing a truncated protein. L-MAG has a longer C-terminal tail. The functional differences between the isoforms are unclear. A mutant mouse, in which the longer isoform is prematurely truncated to mimic the shorter isoform, exhibits similar defects in the central nervous system (CNS) to the Mag null mouse (Fujita et al. 1998). Additionally, L-MAG has been reported to be the isoform responsible for promoting outgrowth of neurites in the CNS (Shimizu-Okabe et al. 2001). Therefore, it is possible that L-MAG is the functionally important isoform in the CNS, and that alternative splicing controls the amount of L-MAG available.
hnRNP A1 has been shown to repress inclusion of exons by binding to nearby elements (Mayeda and Krainer 1992; Blanchette and Chabot 1999; Del Gatto-Konczak et al. 1999). Recently, we and others showed that hnRNP A1 contributes to the alternative splicing of Mag exon 12 (Zhao et al. 2010; Zearfoss et al. 2011). Moreover, we showed that the sequence UAGGU is enriched within and adjacent to exons that show alternative splicing changes upon hnRNP A1 knockdown in oligodendrocyte precursor cells (Zearfoss et al. 2011). UAGGU, UAGGGU, and similar sequences have been shown to interact with hnRNP A1 (Burd and Dreyfuss 1994; An and Grabowski 2007; Michlewski et al. 2008). Examination of the sequences surrounding Mag exon 12 revealed the presence of this element at the 5′ splice site (Zearfoss et al. 2011). In the current study, we asked whether the UAGGU element and its surrounding sequences interact with hnRNP A1 and control alternative splicing of Mag exon 12.
RESULTS
hnRNP A1 binds an element at the Mag exon 12 5′ splice site
To determine whether hnRNP A1 interacts with the UAGGU sequence at the exon 12 5′ splice site (Fig. 1A), we used a pull-down assay where streptavidin-coated magnetic beads and biotinylated RNA fragments were used to recover specifically associated proteins from HeLa nuclear lysate. Recovered proteins were detected by Western blotting. A 29-nucleotide fragment corresponding to the Mag 5′ splice site, numbered −12 to 17, relative to the exon–intron junction (Fig. 1A), efficiently pulled down hnRNP A1 in this assay. In contrast, a mutant version of the RNA in which UAGGU is mutated to UAAGU (G4A) did not pull down hnRNP A1 (Fig. 1B). Neither RNA pulled down Quaking, an RNA-binding protein that does not recognize this sequence. (Fig. 1B). To determine whether association of hnRNP A1 with UAGGU at the 5′ splice site anticorrelates with association of the spliceosome, we probed the blot for U1A, a component of U1 snRNP that recognizes the 5′ splice site during pre-mRNA splicing. U1A is expected to associate indirectly with the splice site via base-pairing between the splice site and the U1 snRNA. In direct contrast to hnRNP A1, we observe that U1A is efficiently recovered by the G4A mutant RNA, but not the wild-type sequence (Fig. 1B).
FIGURE 1.

hnRNP A1 interacts with the sequence at the 5′ splice site of Mag exon 12. (A) Diagram of exons 11–13 from rat MAG. Exons are represented with boxes and introns are represented with lines. The sequence surrounding the 5′ splice site is indicated below the diagram. (*) Position of the exon 12 stop codon. Exon sequence is capitalized and intron sequence is lowercase. The putative hnRNP A1-binding element is shaded gray. These conventions are used throughout the remainder of the figures. (B) Streptavidin pulldown assay. Biotin-labeled synthetic RNAs of 29 nucleotides were incubated with a HeLa nuclear lysate, and bound proteins were detected by Western blotting. RNA sequences are presented beneath the image of the blots. (C) Fluorescence polarization and gel electromobility shift analysis of recombinant hnRNP A1 interaction with nine-nucleotide synthetic fluorescent RNAs of the indicated sequence. The apparent dissociation constant for the interaction (Kd, app) is indicated on the figure.
To confirm the interaction between hnRNP A1 and the Mag exon 12 5′ splice site using an independent method, we purified recombinant MBP-tagged hnRNP A1 and incubated it with synthetic fluorescent RNAs corresponding to the exon–intron junction. We measured the apparent equilibrium dissociation constant using two complementary methods, fluorescence polarization (FP) and electrophoretic mobility shift assays (EMSA), as described by Pagano et al. (2011). hnRNP A1 binds to a 10-nucleotide boundary fragment (−2 to 8) with an apparent Kd of 51 ± 5 nM by FP and 145 ± 12 nM by EMSA (Fig. 1C). When G4 is mutated to A, which improves the 5′ splice site consensus sequence, hnRNP A1 binding was reduced to >500 nM by FP and was not detected by EMSA (Fig. 1C). The apparent threefold disparity in affinity between FP and EMSA is likely due to dissociation during electrophoresis, which can lead to underestimation of binding affinity if the dissociation rate constant is fast. Together, the data reveal a specific interaction between hnRNP A1 and the Mag exon 12 5′ splice site, and suggest that binding of hnRNP A1 may interfere with U1 snRNP association. Our observations of an interaction between hnRNP A1 and Mag RNA is consistent with the observation by Zhao et al. (2010) that the Mag pre-mRNA coimmunoprecipitates with hnRNP A1.
A conserved secondary structure is present at the Mag exon 12 5′ splice site
When examining the sequences surrounding the Mag exon 12 5′ splice site, we noticed a region of possible self-complementarity. We used M-fold to predict secondary structures in mouse, rat, human, and several other vertebrate species (Zuker 2003). The results revealed a possible hairpin structure that is conserved across seven vertebrate species (Fig. 2A). To assess whether this structure is formed in vitro, a series of truncated synthetic RNAs were analyzed by native and denaturing gel electrophoresis (Fig. 2B). Fifteen- and 29-nucleotide RNA fragments (−4 to 11 and −12 to 17, respectively), which are predicted to contain a portion of the base-paired region, migrate with faster than expected mobility in native gels, but not in denaturing gels (Fig. 2C). This is consistent with the presence of a folded RNA structure. In contrast, we did not observe differential mobility when analyzing a 10-nt RNA that retains only 1 bp of the predicted stem (Fig. 2C).
FIGURE 2.

A conserved secondary structure is present at the Mag exon 12 5′ splice site. (A) Putative secondary structure of the region surrounding the splice site from seven vertebrate species. Exon sequence is capitalized, and intron sequence is in lowercase. The hnRNP A1-interacting region is shown in gray. Base pairs are indicated by a horizontal bar. (B) Sequences of the RNAs analyzed in the secondary structure assays in C and D. RNAs are based on the rat sequence. (C) Gel electrophoresis of truncation mutants. RNAs have a higher mobility than expected on a native gel, but migrate according to size on a denaturing gel. (D) Thermal melting analysis of 15 nucleotide RNAs. Mutation of bases predicted to be paired in the stem–loop structure results in a decrease in the melting temperature of the RNA. The predicted structure is presented to the right of the melting curves.
To confirm the presence of the structure and the requirement for the bases in the predicted stem, we analyzed a series of mutant RNAs using UV thermal melting analysis. The 15-nucleotide RNA exhibited a melting temperature (Tm) of 56 ± 0.2°C (Fig. 2D). Mutation of two cytidines (C7, C8) to G7G8 in the predicted base-paired region reduced the Tm to 36 ± 0.5°C, and U7U8 reduced the Tm to 30 ± 1°C (Fig. 2D). Mutation of C7 and C8 to A7A8 also modifies the thermal melting characteristics of the RNA, although no folding transition is observed, suggesting that the RNA is unfolded even at low temperature (Supplemental Fig. 1). The data show that a stem–loop structure can form around the 5′ splice site and that the structure can be perturbed by mutating nucleotides in the stem.
Secondary structure affects hnRNP A1 binding and spliceosome association in vitro
We wished to determine whether the conserved secondary structure at the Mag exon 12 5′ splice site affects binding of hnRNP A1. We tested the ability of the folded RNAs to associate with recombinant hnRNP A1 by FP and EMSA (Fig. 3A; Table 1). The 15-nucleotide fragment binds to hnRNP A1 with an estimated Kd,app of >1 µM (Fig. 3B). The 29-nucleotide RNA fragment also does not bind (Supplemental Fig. 2). This result contrasts with nonquantitative pull-down experiments performed in HeLa nuclear extracts in which hnRNP A1 is observed to interact with the 29-nucleotide RNA fragment (Fig. 1C).
FIGURE 3.

Mag secondary structure modulates association of hnRNP A1 and U1A. (A) Diagram of RNAs used in binding assays. Predicted folding patterns and names used in the text are shown. (B) Fluorescence polarization assay. (Circles) 10-nt rat wild-type RNA; (triangles) 15-nt rat wild-type RNAs. Binding curves were fit to the Hill equation. The fit is shown as a solid line. Error bars indicate the standard deviation of five reads of the microtiter plate. The data were normalized to the maximum and minimum of the fit 10-nt RNA for display purposes. (C) Fluorescence polarization assay. (Triangles) Mouse 15-nt wild-type RNA; (circles) G7G8 RNA. Data were normalized to the maximum and minimum of the fit of the G7G8 RNA for display purposes. (D) Streptavidin pull-down assay. RNAs are as indicated in A. Biotinylated RNAs of the indicated sequence were incubated with a HeLa nuclear lysate, and bound proteins were analyzed by Western blotting. After blotting for U1A, the blot was stripped and reprobed for hnRNP A1.
TABLE 1.
Affinity of hnRNP A1 for 15-nucleotide RNAs
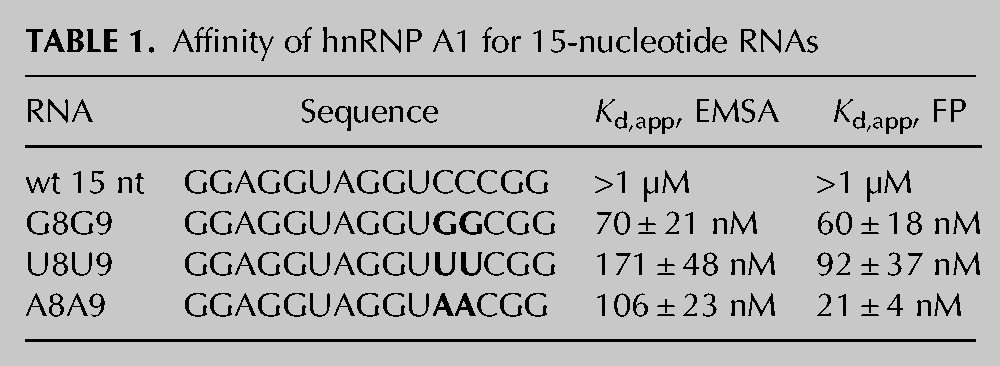
Next, we tested hnRNP A1 binding to the C7C8 mutant 15-nucleotide RNAs. Binding to the G7G8 mutant is improved relative to wild type (Kd, app, FP= 60 ± 18 nM, Kd, app, EMSA= 70 ± 21 nM) (Fig. 3C). The A7A8 and U7U8 mutations also increase association with recombinant hnRNP A1 (Table 1). We then used the streptavidin pull-down assay to test whether the stem–loop mutants affect hnRNP A1 or U1A association in HeLa nuclear extract. Mutation of C7C8 to G7G8, A7A8, or U7U8 increased the ability of the RNA to pull down U1A from a HeLa nuclear lysate (Fig. 3D; Supplemental Fig. 3). We also detect binding of A1 to the mutant RNAs in this assay (Fig. 3D; Supplemental Fig. 3). Together, the results demonstrate that the folded structure decreases the affinity of hnRNP A1 for the Mag exon–intron junction, at least in vitro using recombinant protein, and show that the secondary structure also inhibits association with the U1 snRNP in nuclear lysate.
Secondary structure and the hnRNP A1-binding site determine the Mag isoform ratio
To determine whether secondary structure and the hnRNP A1-binding element at the 5′ splice site modulate alternative splicing of Mag exon 12, we developed a dual luciferase-splicing reporter assay. The reporter is based on the expression vector psiCheck2 (Promega), which encodes both firefly and Renilla luciferase on the same vector. In the reporter, Mag intron 11, exon 12, and intron 12 are positioned upstream of the start codon of firefly luciferase, but downstream from the transcription start site (Fig. 4A). Methionine and glutamate codons were added upstream of Mag intron 11. This provides a minimal 5′ exon sequence that enables translation initiation and contains a strong splicing consensus at the exon–intron junction. After construction of the reporter, we deleted the methionine at the beginning of firefly luciferase in order to reduce the likelihood of reporter translation caused by scanning through the Mag insert. The reporter provides two ways in which to monitor alternative splicing. First, the isoform ratio can be directly measured using RT-PCR primers that detect exon 12 inclusion. Second, because Mag exon 12 contains a translation termination codon, inclusion of this exon precludes expression of firefly luciferase. Therefore, firefly luciferase activity reports on the amount of the exon excluded isoform. We used the CG-4 cell line as our assay system. CG-4 cells are a rat oligodendrocyte precursor cell line that expresses both isoforms of MAG. We previously showed that knocking down hnRNP A1 expression with siRNA in these cells causes an increase in the Mag splice isoform ratio (Zearfoss et al. 2011).
FIGURE 4.

The hnRNP A1-binding element and secondary structure control inclusion of Mag exon 12. (A) Diagram of the reporter construct. Mag exon 12 and its flanking introns were cloned upstream of firefly luciferase. The positions of the genomic insertion, stop codon, secondary structure, methionine deletion, and 5′ artificial exon are shown. (B) Diagram of the mutant sequences used in the reporter assay. The positions of the mutations are indicated. (C) Luciferase and RT-PCR analysis of upper right stem mutations. For the luciferase activity assay, firefly activity was divided by Renilla to control for transfection efficiency. Data were normalized to the expression of the wild-type construct. Data are the average and standard deviation of at least three independent transfections. For the RT-PCR assays, the ratio of the upper to the lower isoform after gel electrophoresis is shown. Error bars indicate the standard deviation of at least three replicates per construct. Data were normalized to the ratio of the wild-type construct. (D) Luciferase and RT-PCR analysis of loop mutations. Mutations are as indicated. Data were processed as in C. (E) Luciferase and RT-PCR analysis of combined upper stem and loop mutations. Data were processed as in C. (F) Luiferase and RT-PCR analysis of structural mutations. Data were processed as in C. (C–F) Data from all experimental replicates were aggregated for each construct. Expression data for constructs appearing on multiple graphs are the same from panel to panel, represented multiple times for clarity.
To assess the contribution of the stem–loop structure and the hnRNP A1 binding element to exon 12 inclusion, we constructed a series of mutations in the stem and terminal loop structures (Fig. 4B). First, we mutated the base-paired C residues of the upper stem. Specifically, we changed C7C8 to G7G8, U7U8, and A7A8. Both the G7G8 and A7A8 mutations increased the isoform ratio by 1.8- and 2.7-fold, respectively (P = 2 × 10−5 and 7 × 10−4) (Fig. 4C). The U7U8 mutation had an insignificant effect (1.3-fold, P = 0.08) (Fig. 4C). The corresponding luciferase activity assays revealed that the G7G8 mutation reduced activity by 1.4-fold (P = 0.03), while the A7A8 mutation reduced activity by twofold (P = 2 × 10−11) (Fig. 4C). The U7U8 mutation did not cause a significant change in expression. The data show that mutation of C7 and C8 to A or G, which destabilizes the stem–loop, increases Mag exon 12 inclusion. Note that when C7 and C8 are mutated to U in the context of the lower stem, GU base pairs could form with G-3 and G-4 (Fig. 4B). While the U7U8 mutations are sufficient to destabilize a minimal 15-nucleotide stem–loop fragment in vitro (Fig. 2D), they may not be sufficient to destabilize the structure in the context of the full stem in vivo.
To assess the contribution of the sequence at the loop, we mutated the G4 position to A, C, or U. Due to the overlap in the hnRNP A1 binding element and the 5′ splice site, mutations in the hnRNP A1 site also change the region of the 5′ splice site that recognizes the U1 snRNA through base-pair interactions. In the wild-type sequence, G4 is opposite a pseudouridine (Ψ), which has the potential to form non-Watson–Crick interactions with G. Ψ is opposed by an A, its Watson–Crick partner, in the A4 mutant. In the C4 and U4 mutants, neither C nor U would be predicted to maintain pairing with Ψ. The overlap of the recognition elements makes it impossible to determine whether the observed effects are due to alternations in hnRNP A1 binding, spliceosome association, or a combination of the two.
In the RT-PCR-based splicing assay, mutation of G4 to A caused an increase in the isoform ratio of 2.6-fold (P = 2 × 10−11) (Fig. 4D). This result corroborates a previously published report using an independent minigene reporter (Wu et al. 2002). Mutation of G4 to U or C did not cause a statistically significant change. In the luciferase activity assay, the U4 mutation caused a decrease in expression of 1.5-fold (P = 0.03), the A4 mutation caused a decrease of 1.8-fold (P = 1 × 10−4), and the C4 mutation did not cause a significant change. The results show that the sequence at the loop functions as a weakened splice site.
To determine whether the mutations in the loop act in combination with the base-pairing in the stem, we combined the A4 mutation with the A7A8 mutation in the base-paired region of the stem (construct A4A7A8). In the RT-PCR assay, we observed a 10-fold increase in the isoform ratio in this sample (P = 2 × 10−12) (Fig. 4E). Similarly, luciferase activity was decreased by fourfold (P = 3 × 10−13) (Fig. 4E). The data show that the loop sequence and the stem both contribute to the observed isoform ratio and are not redundant.
To determine whether base-pairing in the stem contributes to the resulting isoform ratio, we generated two additional sets of mutations. These sets included compensatory mutations that restored base-pairing and maintained the GC content of the stem while changing the primary sequence. First, we mutated each of the base-paired G residues in the stem to a C (Fig. 4B, stem disruption construct). We also generated the compensatory mutation, in which the opposing C bases were changed to G to restore base-pairing (Fig. 4B, comp construct). We observed an 11-fold increase in the isoform ratio with the stem disruption construct (P = 0.008) in the PCR based assay and a fivefold decrease in the luciferase activity assay (P = 3 × 10−13) (Fig. 4F). When base-pairing was restored, in the comp mutant we observed nearly wild-type isoform ratio and luciferase expression (Fig. 4F). The result suggests that base-pairing of the stem inhibits exon recognition. To confirm this result, we made smaller mutations to the lower stem, again designed to alter the primary sequence of this region while maintaining the GC content. These mutations interrupt the G and C tracts present in the lower stem (Fig. 4F). Mutating G12 and G14 to C12 and C14 caused an increase in the isoform ratio of 5.8-fold (P = 2 × 10−12) in the PCR based assay and a decrease of 2.4-fold in the luciferase activity assay (P = 2 × 10−11) (Fig. 4F). The corresponding base changes on the opposite strand, G-7 and G-9, caused changes in the same direction, with an increase of 2.8-fold (P = 3 × 10−9) in the PCR assay and a decrease of 1.4-fold (P = 2 × 10−5) in the luciferase assay (Fig. 4F). The compensatory mutant, containing the G-7G-9C12C14, produced an increase of 3.6-fold in the PCR assay (P = 3 × 10−12) and was equivalent to the wild-type construct in the luciferase assay (f.c. = 1.1, P = 0.1). Together, the results show that the mutations on opposing sides of the stem do not act in an additive fashion, and that restoring the base-pairing brings the level of splicing closer to the wild-type level. The results are consistent with a model in which the structure of the stem–loop restricts access of the splicing machinery to the splice site.
Numerous Mag-like 5′ splice sites are present in the human genome
We wished to determine whether the presence of hairpin structures at UAGGU 5′ splice sites is a unique characteristic of Mag or whether it is widespread. To find out whether other splice sites with similar characteristics exist, we obtained the genomic coordinates of all human 5′ splice sites from the UCSC genome browser hg19 assembly (Kent et al. 2002) and downloaded sequences of 39-nucleotide fragments surrounding the splice site, centered at the UAGGU element. The region corresponds to positions −17 to 22 relative to the splice site. We then folded the sequences using the Vienna RNA package (Hofacker 2009) and filtered the results to obtain sequences that contained a predicted hairpin with a free energy of folding of −8 kcal/mol or less and containing at least 8 base pairs, with a loop at the UAGGU element. We identified 525 sequences that passed the criteria (Supplemental Table 1). For each of these sequences, we obtained PhyloP placental mammal conservation scores from the UCSC genome browser in order to obtain a measure of the conservation of the sequence. We summed the scores across the element at the 5′ splice site as well as the entire 39-nucleotide length. Conservation varied for the predicted hairpins (Supplemental Table 1). The identification of these splice sites suggests that hairpins at the 5′ splice site may affect alternative splicing in other genes in addition to Mag.
We used two methods to determine whether 5′ splice sites with UAGGU elements are more likely to be present in structured regions of RNA. First, we constructed three artificial sets of sequences based upon intron and exon nucleotide frequencies. More specifically, we determined frequency of each nucleotide for the intron and exon regions of all 5′ splice-site sequences, omitting the conserved splice-site sequence between positions −3 and +7. Artificial sets were constructed based on the calculated intron and exon nucleotide frequencies. Splice sites containing the UAGGU element are predicted to fold into more stable structures than the random sets (Fig. 5). At −10 kcal/mol, twofold more sequences were identified in the UAGGU set than in the random sets (f.c. = 2.2, P = 0.0002) (Fig. 5). Second, we calculated the nucleotide frequency independently for each nucleotide position and generated a set of 100,000 random sequences based on these frequencies (Fig. 5). Sites containing a UAGGU at the 5′ splice site were also predicted to fold into a more stable structure than this control group.
FIGURE 5.

GUAGGU 5′ splice sites are located in predicted secondary structure-rich sequences. (Left) The 39-nucleotide sequences surrounding the 5′ splice site of exons with a GUAGGU 5′ splice site or three sets of permuted artificial exon sequences were folded using the Vienna RNA package. The fraction of sequences folding at a given energy level was plotted versus the predicted free energy of folding. Error bars on the permuted set indicate the standard deviation of the three artificial sets. See the Materials and Methods for details regarding the construction of this set. A dashed line indicates the position of −8 kcal/mol. (Right) The predicted free energy of folding of GUAGGU exons was plotted against a background set of 100,000 random 39 mers, in which the nucleotide bias of each individual position relative to the 5′ splice site was preserved.
DISCUSSION
We have shown that hnRNP A1 interacts with a sequence at the Mag exon 12 5′ splice site, that a secondary structure is present at this region, and that mutations that destabilize the secondary structure can increase the ability of the RNA to be recognized by both the U1 snRNP and hnRNP A1. hnRNP A1 is known to function as a splicing silencer, inhibiting inclusion of exons near its binding sites on a pre-mRNA (Mayeda and Krainer 1992; Del Gatto-Konczak et al. 1999). A simple model would predict that disruptions of secondary structure that increase hnRNP A1 association would lead to repression of exon inclusion. However, in addition to modulating association with hnRNP A1, the secondary structure also modulates association with the U1 snRNP. The data, taken together, suggest a model in which stem–loop functions as a bipartite control element in which the base-pairing of the stem and the primary sequence of the loop are required elements that define the ratio of splice isoforms (Fig. 6).
FIGURE 6.

A model for Mag exon 12 alternative splicing. A hairpin loop at the 5′ splice site inhibits association of hnRNP A1 and the spliceosome. Relaxation of the structure facilitates association between both hnRNPA1 and the spliceosome, allowing competition between the two to determine whether the exon is included or excluded.
Our in vitro binding assays, conducted using purified hnRNP A1 protein and synthetic RNAs, provide evidence that the hairpin structure inhibits the association of hnRNP A1 with the sequence at the terminal loop. Recently, however, several examples of the association between hnRNP A1 and hairpin RNA structures have been described. For example, hnRNP A1 has been shown to interact with the pri-miR18A small RNA, and the site of association has been narrowed to the loop and one-half of the stem (Guil and Cáceres 2007). hnRNP A1 binding sites have also been proposed to lie near the terminal loops of several other pri-miRNAs (Michlewski et al. 2008). hnRNP A1 has also been shown to bind folded RNA elements that repress alternative splicing. For example, hnRNP A1 interacts with HIV RNA hairpin loop sequences by RNase footprinting and NMR experiments (Damgaard et al. 2002; Levengood et al. 2012). In the case of HIV ESS3 and a synthetic MS2 hairpin structure, association between hnRNP A1 and the folded RNA induces unfolding of the structure (Okunola and Krainer 2009; Levengood et al. 2012). In contrast to these observations, we do not observe unfolding of the RNA upon addition of purified recombinant protein to the folded RNAs. There are at least two possible explanations for this observation. First, the MBP-tagged and purified protein, while possessing RNA-binding ability, may not retain all of the functional capabilities of endogenous hnRNP A1. Second, the Mag hairpin may be sufficiently stable to prevent this mode of action.
Notably, we are able to detect an association between hnRNP A1 and the folded RNAs that does not interact by EMSA in a HeLa nuclear lysate. We would predict that additional RNA-binding proteins present in the lysate also interact with the hairpin structure used in this assay. It is possible that association of one or more of these factors may facilitate association with hnRNP A1 in the extract. The tract of G bases in the lower stem could serve as a binding site for hnRNP H or F. However, we have tested these factors in our pull-down assay and have not detected an association.
Secondary structure has been demonstrated to affect alternative splicing in a variety of ways. Cases exist in which secondary structures facilitate the bringing together of exons, and in which structure inhibits the association of splicing factors. For example, base-pairing interactions in Drosophila Dscam control the selection of several mutually exclusive 6th exons, facilitating splicing (Graveley 2005). In contrast, base-pairing at the 5′ splice site of Hnrnpa1 exon 7B represses exon inclusion (Blanchette and Chabot 1997). Additionally, MBNL1 interacts with a stem–loop structure at the 3′ end of intron 4 of cardiac troponin T; the spliceosomal protein U2AF is able to interact with the stem–loop structure when in its unfolded form but not its folded form (Warf et al. 2009). The mechanism we describe in this article, in which secondary structure controls the Mag isoform ratio, bears resemblance to this last mechanism.
The presence of the stop codon in exon 12 is notable given that stop codons positioned in internal exons are known to target the transcript for nonsense-mediated decay (NMD). We would not predict that this would be the case for Mag, however. NMD generally requires a spacing of 50–55 nucleotides between the stop codon and the exon junction for efficient degradation (Nagy and Maquat 1998); in Mag exon 12, only 12 nucleotides are present.
Recently, Feng and coworkers reported that hnRNP A1 enhances Mag exon 12 inclusion in HEK293T cells through an interaction with an element within exon 13 (Zhao et al. 2010). The splicing reporter used in the current study does not include this exon. While we cannot exclude the possibility of an effect mediated by exon 13, a deletion analysis conducted by Wu and coworkers shows a large effect on exon 12 inclusion when regions of intron 12 are deleted and a comparatively minor effect when exon 13 is replaced with a heterologous exon (Wu et al. 2002). Additionally, the mutational analysis described herein generated large changes in the isoform ratios observed, demonstrating the importance of the sequences included in the reporter.
In conclusion, the binding and splicing reporter experiments presented herein reveal (1) that a sequence at the Mag exon 12 5′ splice site promotes hnRNP A1 association and inhibits exon inclusion, and (2) a secondary structure at the splice site inhibits both hnRNP A1 association and U1 snRNP association, with a net effect of inhibiting exon recognition. Over 500 exon–intron junctions in the human genome are predicted to fold into a hairpin structure that contains a GUAGGU 5′ splice site in the loop, suggesting that 5′ splice site sequestration may be a relatively common mechanism to regulate exon inclusion. It will be interesting to determine whether such exons are alternatively spliced, and if so, to what extent hnRNP A1 contributes to their regulation.
MATERIALS AND METHODS
Purification of recombinant hnRNP A1
The coding sequence of mouse hnRNP A1 was amplified by PCR from a cDNA clone (Open Biosystems, clone ID 6308746) and inserted 3′ of MBP into the bacterial expression vector pHMTC. Expression in CodonPlus cells was induced with 1 mM IPTG, cells were lysed, and the lysate was passed over an amylose column. Bound proteins were eluted with 10 mM maltose. hnRNP A1 was further purified over an Hi-Trap S-column in MOPS (pH 6.0) and eluted with a 20 mM to 2 M gradient of sodium chloride, followed by a Source Q column in Tris (pH 8.8), followed by elution with a 20 mM to 2 M gradient of sodium chloride. Fractions were analyzed by gel electrophoresis, and fractions containing a single band of the expected molecular weight were pooled and concentrated using Amicon spin concentrators for further use.
Streptavidin pull-down assays
Synthetic RNAs were biotinylated at the 5′ end using biotinamidocaproyl hydrazide (BACH). Briefly, RNAs were oxidized at their 3′ ends by incubating with sodium periodate. The oxidized RNA was then reacted with BACH, and unreacted BACH was removed by ethanol precipitation, followed by purification over a Sephadex G25 (GE Healthcare) spin column. Streptavidin-coated magnetic beads were obtained from Invitrogen and were used according to the manufacturer’s instructions. Briefly, beads were washed twice in an equal volume of 0.1 M NaOH and 0.05 M NaCl, then twice in 0.1 M NaCl. Beads were then incubated with a 1:1 mixture of a RNA and 2 × Bind and Wash buffer (2.5 µM RNA final), incubated for 15 min with gentle shaking, and washed three times in 1 × bind and wash buffer. Beads were then incubated for 2 h with HeLa nuclear extract (Protein One) diluted 1:1 in buffer to provide final concentrations of 0.01 mg/mL tRNA, 0.01% NP40, 0.1 mg/mL BSA, 50 mM Tris-Cl (pH 8.0), 100 mM NaCl, and 1/20 SuperaseIn (Ambion). Beads were washed four times in 100 mM NaCl, 50 mM Tris-Cl (pH 8.0), 0.01% NP40, and 0.01 mg/mL tRNA and were resuspended in sample buffer and analyzed by Western blotting using antibodies for hnRNP A1 (Sigma), U1A (Abcam), or Quaking (Bethyl Laboratories) using established procedures (Sambrook and Russell 2001).
Native gel electrophoresis
RNAs were diluted in a buffer that contains 100 mM NaCl and 50 mM Tris-Cl (pH 8.0), heated to 60°C, and cooled to ambient temperature. They were then run on a 6% slab polyacrylamide gel in 1 × TBE.
Thermal denaturation experiments
Synthetic RNAs (IDT) were prepared at 5 µM in a buffer containing 100 mM NaCl, 0.2 mM EDTA, and 20 mM sodium cacodylate (pH 7.0). The absorbance of each RNA was detected in a Beckman DU Series 600 spectrophotometer equipped with a temperature controller. Temperature was increased from 5°C to 80°C with a ramp rate of 1°/min, and absorbance values were collected at 0.5°C intervals. Melting temperature was calculated by fitting to a mathematical model of a two-state unimolecular transition as described in Proctor et al. (2004).
Construction of splicing reporter
QuikChange mutagenesis was used to remove the HindIII site upstream of Renilla luciferase in the vector psiCheck2 (Promega). After mutagenesis, the plasmid contained a single HindIII site upstream of firefly luciferase. Mag genomic sequence spanning intron 11–12, exon 12, and intron 12–13 was amplified from mouse genomic DNA by PCR using Pfu polymerase. Restriction sites were added to the ends by a second round of PCR using the primers 5′-ggggcgtctcaagcttgggatggaggtgagagggccacaccc-3′ and 5′-ccccaagcttgagacgcctatggagaggagaag-3′. The primers were designed to be recognized by BsmB1, which cuts asymmetrically to yield HindIII-compatible overhangs. The construct was digested with BsmB1 and cloned into the mutated psiCheck2 vector cut with HindIII. The forward primer contains an initiatior methionine codon, followed by a single amino acid upstream of the first Mag intron sequence, to provide an upstream exon to which either exon 12 or the downstream luciferase sequence can be spliced. The initiator methionine of firefly luciferase was deleted using QuikChange mutagenesis. All mutations indicated in the text were made using QuikChange mutagenesis, and the presence of the desired mutation was confirmed by sequencing.
Cell culture and transfections
CG-4 and B104 cells were a gift from Lynn Hudson (NINDS) and were cultured as described (Louis et al. 1992). B104 cells were used to produce conditioned medium, which contained growth factors that prevent CG-4 cells from differentiating. Cells were transfected using Lipofectamine 2000 (Invitrogen) or Dharmafect (Thermo Scientific) as described by the manufacturer. After transfection, cells were incubated for 2 d prior to harvesting.
Reporter analyses
RNA was isolated from cells using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. RNA was further cleaned through an RNeasy (Qiagen) spin column. For RT-PCR analysis, RNA was reverse transcribed and amplified using a One-Step RT-PCR kit (Invitrogen) with the primers 5′-GCTTAAAAGCTTGGGATGGA-3′ and 5′-CAGGGCAGGTGTCCACTC-3′. The reverse primer was labeled with 5′-FAM (IDT). Dual luciferase reporter assays were conducted using the Dual-luciferase reporter assay system (Promega) following the instructions from the manufacturer. Firefly luciferase was divided by the Renilla activity encoded on the same plasmid as a transfection efficiency control.
Genomic analysis
Genomic coordinates of human exons were obtained directly from the UCSC Genome Browser from the hg19 assembly (Kent et al. 2002). Coordinates of the region surrounding the 5′ splice site were obtained using custom Perl scripts, and the genomic sequence corresponding to the region was extracted from hg19 genomic sequence using Galaxy (Giardine et al. 2005; Blankenberg et al. 2010; Goecks et al. 2010). Folding, free energy analysis, and filtering were conducted using a local installation of the Vienna RNA package (Hofacker 2009) using Perl scripts. PhyloP conservation scores were obtained directly from the UCSC Genome Browser, and scores were added to obtain the measures presented in Supplemental Table 1.
For the frequency analysis, four of six bases in the GUAGGU region were required to be unpaired for inclusion in the analysis; however, no other pairing requirements were set. For the background, three sets were constructed of a similar length to the GUAGGU set. Because the nucleotide frequency in the intron and exon portions of the sequence differs, we calculated the nucleotide frequency of the intron and exon regions independently and excluded the conserved region surrounding the splice site, from positions −3 to +7, from the calculations. The artificial sets preserved the nucleotide frequency in the intron and exon regions. The three randomized sets were folded, and the mean and standard deviation were calculated for each free energy level. For the background set in which the nucleotide frequency was calculated for each position independently, the background consisted of a single set of 100,000 artificial sequences.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Brian Farley for assistance with Perl scripts. This work was supported by NIGMS grant number GM081422 to S.P.R.
REFERENCES
- An P, Grabowski PJ 2007. Exon silencing by UAGG motifs in response to neuronal excitation. PLoS Biol 5: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, Tessier-Lavigne M 2008. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322: 967–970 [DOI] [PubMed] [Google Scholar]
- Bartsch S, Montag D, Schachner M, Bartsch U 1997. Increased number of unmyelinated axons in optic nerves of adult mice deficient in the myelin-associated glycoprotein (MAG). Brain Res 762: 231–234 [DOI] [PubMed] [Google Scholar]
- Blanchette M, Chabot B 1997. A highly stable duplex structure sequesters the 5′ splice site region of hnRNP A1 alternative exon 7B. RNA 3: 405–419 [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Chabot B 1999. Modulation of exon skipping by high-affinity hnRNP A1-binding sites and by intron elements that repress splice site utilization. EMBO J 18: 1939–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Kuster Von G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J 2010. Galaxy: A web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol Chapter 19: Unit 19.10.1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CG, Dreyfuss G 1994. RNA binding specificity of hnRNP A1: Significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J 13: 1197–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard CK, Tange TO, Kjems J 2002. hnRNP A1 controls HIV-1 mRNA splicing through cooperative binding to intron and exon splicing silencers in the context of a conserved secondary structure. RNA 8: 1401–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Gatto-Konczak F, Olive M, Gesnel MC, Breathnach R 1999. hnRNP A1 recruited to an exon in vivo can function as an exon splicing silencer. Mol Cell Biol 19: 251–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Kemper A, Dupree J, Nakayasu H, Bartsch U, Schachner M, Maeda N, Suzuki K, Popko B 1998. The cytoplasmic domain of the large myelin-associated glycoprotein isoform is needed for proper CNS but not peripheral nervous system myelination. J Neurosci 18: 1970–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Endo S, Takai T, Yamashita T 2011. Myelin suppresses axon regeneration by PIR-B/SHP-mediated inhibition of Trk activity. EMBO J 30: 1389–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. 2005. Galaxy: A platform for interactive large-scale genome analysis. Genome Res 15: 1451–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J, Galaxy Team 2010. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11: R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR 2005. Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell 123: 65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guil S, Cáceres JF 2007. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat Struct Mol Biol 14: 591–596 [DOI] [PubMed] [Google Scholar]
- Hofacker IL 2009. RNA secondary structure analysis using the Vienna RNA package. Curr Protoc Bioinformatics 10.1002/0471250953.bif202s26 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, Brow MA, Nave KA, Noronha AB, Quarles RH, Bloom FE, Milner RJ, Sutcliffe JG 1987. Two forms of 1B236/myelin-associated glycoprotein, a cell adhesion molecule for postnatal neural development, are produced by alternative splicing. Proc Natl Acad Sci 84: 4337–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, Bartsch U, Montag D, Schachner M 1997. Dying-back oligodendrogliopathy: A late sequel of myelin-associated glycoprotein deficiency. Glia 19: 104–110 [PubMed] [Google Scholar]
- Levengood JD, Rollins C, Mishler CHJ, Johnson CA, Miner G, Rajan P, Znosko BM, Tolbert BS 2012. Solution structure of the HIV-1 exon splicing silencer 3. J Mol Biol 415: 680–698 [DOI] [PubMed] [Google Scholar]
- Li C, Tropak MB, Gerlai R, Clapoff S, Abramow-Newerly W, Trapp B, Peterson A, Roder J 1994. Myelination in the absence of myelin-associated glycoprotein. Nature 369: 747–750 [DOI] [PubMed] [Google Scholar]
- Liu B, Fournier A, GrandPré T 2002. Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 297: 1190–1193 [DOI] [PubMed] [Google Scholar]
- Lopez PHH, Ahmad AS, Mehta NR, Toner M, Rowland EA, Zhang J, Doré S, Schnaar RL 2011. Myelin-associated glycoprotein protects neurons from excitotoxicity. J Neurochem 116: 900–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis JC, Magal E, Muir D, Manthorpe M, Varon S 1992. CG-4, a new bipotential glial cell line from rat brain, is capable of differentiating in vitro into either mature oligodendrocytes or type-2 astrocytes. J Neurosci Res 31: 193–204 [DOI] [PubMed] [Google Scholar]
- Mayeda A, Krainer AR 1992. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell 68: 365–375 [DOI] [PubMed] [Google Scholar]
- McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE 1994. Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 13: 805–811 [DOI] [PubMed] [Google Scholar]
- Michlewski G, Guil S, Semple CA, Cáceres JF 2008. Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol Cell 32: 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montag D, Giese KP, Bartsch U, Martini R, Lang Y, Blüthmann H, Karthigasan J, Kirschner DA, Wintergerst ES, Nave KA 1994. Mice deficient for the myelin-associated glycoprotein show subtle abnormalities in myelin. Neuron 13: 229–246 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT 1994. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13: 757–767 [DOI] [PubMed] [Google Scholar]
- Nagy E, Maquat LE 1998. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem Sci 23: 198–199 [DOI] [PubMed] [Google Scholar]
- Okunola HL, Krainer AR 2009. Cooperative-binding and splicing-repressive properties of hnRNP A1. Mol Cell Biol 29: 5620–5631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano JM, Clingman CC, Ryder SP 2011. Quantitative approaches to monitor protein-nucleic acid interactions using fluorescent probes. RNA 17: 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor DJ, Ma H, Kierzek E, Kierzek R, Gruebele M, Bevilacqua PC 2004. Folding thermodynamics and kinetics of YNMG RNA hairpins: Specific incorporation of 8-bromoguanosine leads to stabilization by enhancement of the folding rate. Biochemistry 43: 14004–14014 [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW 2001. Molecular cloning: A laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Sheikh KA, Sun J, Liu Y, Kawai H, Crawford TO, Proia RL, Griffin JW, Schnaar RL 1999. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci 96: 7532–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu-Okabe C, Matsuda Y, Koito H, Yoshida S 2001. L-isoform but not S-isoform of myelin associated glycoprotein promotes neurite outgrowth of mouse cerebellar neurons. Neurosci Lett 311: 203–205 [DOI] [PubMed] [Google Scholar]
- Tropak MB, Johnson PW, Dunn RJ, Roder JC 1988. Differential splicing of MAG transcripts during CNS and PNS development. Brain Res 464: 143–155 [DOI] [PubMed] [Google Scholar]
- Venkatesh K, Chivatakarn O, Lee H, Joshi PS, Kantor DB, Newman BA, Mage R, Rader C, Giger RJ 2005. The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J Neurosci 25: 808–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, Diegel JV, Hippel von PH, Berglund JA 2009. The protein factors MBNL1 and U2AF65 bind alternative RNA structures to regulate splicing. Proc Natl Acad Sci 106: 9203–9208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JI, Reed RB, Grabowski PJ, Artzt K 2002. Function of quaking in myelination: Regulation of alternative splicing. Proc Natl Acad Sci 99: 4233–4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LJ, Zeller CB, Shaper NL, Kiso M, Hasegawa A, Shapiro RE, Schnaar RL 1996. Gangliosides are neuronal ligands for myelin-associated glycoprotein. Proc Natl Acad Sci 93: 814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu PH, Lee VM, Li C, Roder J, Trapp BD 1998. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci 18: 1953–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zearfoss NR, Clingman CC, Farley BM, McCoig LM, Ryder SP 2011. Quaking regulates Hnrnpa1 expression through its 3′ UTR in oligodendrocyte precursor cells. PLoS Genet 7: e1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Mandler MD, Yi H, Feng Y 2010. Quaking I controls a unique cytoplasmic pathway that regulates alternative splicing of myelin-associated glycoprotein. Proc Natl Acad Sci 107: 19061–19066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]