

Abstract
Protein interaction networks are important for the understanding of regulatory mechanisms, for the explanation of experimental data and for the prediction of protein functions. Unfortunately, most interaction data is available only for model organisms. As a possible remedy, the transfer of interactions to organisms of interest is common practice, but it is not clear when interactions can be transferred from one organism to another and, thus, the confidence in the derived interactions is low. Here, we propose to use a rich set of features to train Random Forests in order to score transferred interactions. We evaluated the transfer from a range of eukaryotic organisms to S. cerevisiae using orthologs. Directly transferred interactions to S. cerevisiae are on average only 24% consistent with the current S. cerevisiae interaction network. By using commonly applied filter approaches the transfer precision can be improved, but at the cost of a large decrease in the number of transferred interactions. Our Random Forest approach uses various features derived from both the target and the source network as well as the ortholog annotations to assign confidence values to transferred interactions. Thereby, we could increase the average transfer consistency to 85%, while still transferring almost 70% of all correctly transferable interactions. We tested our approach for the transfer of interactions to other species and showed that our approach outperforms competing methods for the transfer of interactions to species where no experimental knowledge is available. Finally, we applied our predictor to score transferred interactions to 83 targets species and we were able to extend the available interactome of B. taurus, M. musculus and G. gallus with over 40,000 interactions each. Our transferred interaction networks are publicly available via our web interface, which allows to inspect and download transferred interaction sets of different sizes, for various species, and at specified expected precision levels. Availability: http://services.bio.ifi.lmu.de/coin-db/.
Introduction
Using high-throughput screening techniques such as Yeast Two Hybrid screens, mass spectrometry and protein microarrays large amounts of protein interaction data can be obtained. A protein interaction consists of proteins which bind permanent or transient together in order to carry biological functions. Interaction data collected in various databases has for example been used to study regulatory networks, to explain experimental data or to predict the functions of proteins [1]. Researchers can query protein interactions in databases like IntAct[2] or BioGrid[3], which include interactions derived from large-scale experiments, from literature curations, from user submissions, and interactions from protein structures. Besides this, repositories exist which integrate the interactions from multiple sources. The current protein interaction networks are derived from high-throughput experiments and hypothesis-driven low-throughput experiments applied to particular gene sets of interest[4].
But still the experimental identification of interactions is a time consuming and costly process, so that high-throughput experiments have mostly been conducted on model organisms such as S. cerevisiae [5], H. sapiens [6], A. thaliana [7] and D. melanogaster [8]. Thus, the available interaction networks for other species are extremely sparse (see Table 1).
Table 1. Protein interaction networks.
| Species | Interactions | |||||
| Genes | Physical | Genetic | Other | Total | Total interactions | |
| per gene | ||||||
| S. cerevisiae | 6,328 | 55,767 | 104,926 | 17,674 | 178,367 | 28.19 |
| H. sapiens | 28,383 | 43,412 | 71 | 20,992 | 64,475 | 2.27 |
| D. melanogaster | 14,321 | 19,088 | 2,118 | 17,265 | 38,471 | 2.69 |
| S. pombe | 4,958 | 1,943 | 9,665 | 804 | 12,412 | 2.5 |
| C. elegans | 20,184 | 5,483 | 1,785 | 4,208 | 11,476 | 0.57 |
| M. musculus | 24,865 | 3,513 | 3 | 2,596 | 6,112 | 0.25 |
| A. thaliana | 26,496 | 5,048 | 67 | 937 | 6,052 | 0.23 |
| P. falciparum | 5,503 | 2,215 | 0 | 4 | 2,219 | 0.4 |
| R. norvegicus | 24,770 | 804 | 0 | 867 | 1,671 | 0.07 |
| D. rerio | 24,352 | 173 | 11 | 13 | 197 | 0.01 |
Overview of protein interaction networks extracted from the database iRefIndex [33] for the ten eukaryotic model species with the largest protein interaction networks. Besides the total number of protein interactions, the number of physical, genetic and interactions with an unknown interaction type is given. Only the interaction network of S. cerevisiae, H. sapiens, D. melanogaster and S. pombe have more than 2 interaction per gene (S. cerevisiae peaks with 28.19).
Furthermore, all experimental protein interaction detection methods have different weaknesses and biases[9]. For example false positive rates up to 50% are reported for Yeast Two Hybrid screens [10], literature curations do only agree to some extent [11], and interactions from Tandem Affinity Purification (TAP), which requires further processing in order to infer physical protein interactions[12], [13].
Numerous computational approaches have been developed to predict protein interactions. In particular, knowledge from other (model) organisms can be used to predict protein interactions for a specific target organism. But link attachments, link detachments, gene duplications and gene losses lead to (evolutionary) changes in protein interaction structures [14]. For example, single nucleotide substitutions in the coding region of a gene can lead to structure changes of the encoded protein so that new binding partners can dock or other proteins can not dock anymore to the particular protein. Gene duplications lead also to duplications of interactions and again nucleotide substitutions of the genes lead to a network rewiring. While transient protein interactions are affected by rewiring, gene gains and losses, protein complexes are mostly affected by losses and gains of subunits [15], [16].
Matthews et al. [17] introduced the term interolog (an orthologous gene pair interacting in at least one species) and many methods transfer interaction data employing interologs [18]–[22]. Matthews et al. was able to experimentally validate between 16% to 32% of transferred protein interactions from S. cerevisiae to C. elegans with different ortholog identification techniques. Several features are commonly used to increase the reliability of interaction transfers via interologs. The simplest approach is to require a certain interolog quality, e.g. a minimum bootstrap score for orthologs from the InParanoid database [18] or a minimum sequence similarity between orthologs in order to transfer an interaction. Yu et al. showed that protein interactions can be safely transferred if the joint sequence identity between the orthologs involved in the transfer is larger than 80% [22]. More advanced filter approaches use thresholds for the Gene Ontology (GO) [23] annotation similarity, domain similarity, gene expression correlation or other features of the interologs [20], [21], [24]–[26]. To achieve a specified performance, random protein pairs are compared with known protein interaction partners to define thresholds for the different features. Besides the inference of protein interaction from interologs, various other approaches try to predict interactions using structural properties [27], network topology information [28], or protein domain information [29]. The STRING database follows a different approach to score interactions by combining information from experiments, databases, text-mining and transfer information [30].
Lewis et al. showed that the transfer consistency cannot easily be improved. Furthermore, they showed that the evolutionary change of interactions is too high to allow the direct transfer of interactions for phylogenetically distant species unless a strict definition of homology is used [31]. In contrast van Dam et al. showed that protein complexes are highly conserved even between H. sapiens and S. cerevisiae [15]. All network transfer studies relay on homologies which can be identified with different ortholog detection methods like simple bidirectional BLAST best hit results, graph-based methods that cluster orthologs, or tree-based methods. Benchmarks of orthologs detection methods have shown that there is no best method for ortholog detection [32]. It is obvious that with conservative ortholog detection approaches only relatively few interactions can be transferred, but that these interactions are more likely conserved, whereas with cluster based and tree based methods groups of orthologs are produced which allow to transfer more interactions. Thus, the usage of ortholog identification approaches, the choice of experimental data (only physical interaction derived from Yeast Two Hybrid studies, or more relaxed interaction data which includes interactions from TAP or Co-Immunoprecipitation experiments, or even protein complexes), and the approaches used to deal with the incompleteness of current networks result in different estimated protein interaction conservation rates.
In this paper, new features and successfully used features in the literature are exploited to train Random-Forests-Filters (RFF) for the reliable transfer of interactions to even distant species. The RFF models are trained with interactions transferred from various eukaryotic species to S. cerevisiae using all available interactions from an integrated database and orthologs from cluster based approaches. We train the models on yeast for the only reason that the S. cerevisiae network is assumed to be the most complete one, which allows to distinguish correct and incorrect transfers in the learning phase. Another assumption we make is that the learned RFF models can be used for other species as well. This is reasonable as the models learn the important features (e.g. sequence similarity, orthology, network properties, functional similarities) and their appropriate weightings, which will hold in a species-independent way (there are no particular S. cerevisiae specific features or parameters). The transfer performance on S. cerevisiae is taken as an estimate for the expected performance on other species, especially for phylogenetically closer ones. We applied the trained RFF predictor to transfer interactions on a large scale in-between various eukaryotic species. This increases the available reliable interactions for non-model organisms manyfold without inflicting too many false positives. The transferred networks are publicly available at our web interface. Compared to competing approaches to predict protein interactions we integrate a wide range of features and, instead of using fixed thresholds, employ a systematic and conservative RFF approach with an associated performance estimate for the (distant) transfer to S. cerevisiae.
Materials and Methods
Data sources
We use an integrated database to get a complete view on the currently discovered protein interaction networks and use the interaction repository iRefIndex[33] as the source database for experimentally determined protein interactions. iRefIndex provides interaction data for multiple species in a common format from the 13 different interaction databases: BIND[34], BioGRID[3], CORUM[35], DIP[36], HPRD[37], InnateDB[38], IntAct[2], MatrixDB[39], MINT[40], MPact[41], MPIDB[42], MPPI[43] and OPHID[44]. All databases include experimental validated data extracted from different sources, besides OPHID which also makes use of transferred interactions. Therefore, we excluded interactions from OPHID for our study. Furthermore, iRefIndex includes binary interactions (physical and genetic) and few protein complexes. We transfer binary interactions from iRefIndex (physical, genetic and other interaction types including ambiguous or interactions without type annotation) to target species using publicly available ortholog mappings. Orthologs are obtained from the Orthologs Matrix Project (OMA) [45], InParanoid [46] and HomoloGene [47]. These databases are used due to the evaluation results in [32] and the coverage of ortholog mappings for various eukaryotic species. The interaction partners and orthologs are mapped to UniProt [48] as a common reference to obtain annotations including GO terms, synonyms and mappings to external databases (see Table S1 for an overview of the used data sources). We consider all eukaryotes species for which we could transfer at least one interaction given the interaction and ortholog databases. Thus, we consider 83 out of the approximate 166 (until January 2013) fully sequenced eukaryotes for the subsequent analysis (for a list of sequenced eukaryotes see EBI Genomes Pages: http://www.ebi.ac.uk/genomes/eukaryota.html).
Interaction transfer
Protein interaction networks are modeled as graphs 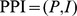 consisting of a set of proteins
consisting of a set of proteins 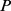 and interactions
and interactions 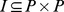 . Given an interaction network
. Given an interaction network 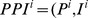 ), a target protein set
), a target protein set 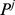 and an ortholog mapping
and an ortholog mapping 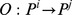 , a (directly) (interolog based) transferred interaction network consists of
, a (directly) (interolog based) transferred interaction network consists of 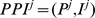 with
with 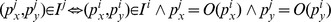 . Transferred interactions
. Transferred interactions 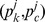 can be scored and filtered to obtain a (filtered) (interolog based) transferred interaction network. In our case, a trained Random Forest Filter (RFF) model is used and its performance for specific score thresholds is estimated via the transfer data to S. cerevisiae.
can be scored and filtered to obtain a (filtered) (interolog based) transferred interaction network. In our case, a trained Random Forest Filter (RFF) model is used and its performance for specific score thresholds is estimated via the transfer data to S. cerevisiae.
Random-Forest-Filter (RFF)
For the scoring of transferred interactions we use Random Forests (RF) from the WEKA [49] machine learning framework. Random Forests predict the outcome class (correct, incorrect) of an instance (transferred interaction) by using a voting procedure on several learned decision trees with different feature sets. Random Forests have shown good evaluation results on similar learning tasks [50] and are considered more robust against noise than other ensemble machine learning methods [51]. RF rely on two parameters, the number of trees to learn and the number of features to consider. We determine these parameters via a grid search. In addition to the output class label, the WEKA Random Forest implementation provides a score value between 0 (low confidence) and 1 (high confidence), which we use as score value for transferred interactions.
Features
As features we use the protein annotations of the interacting partners in the source and the target network and of the orthologs from which an interaction is transferred. The features can be classified into four categories: 1.) Features modeling Gene Ontology similarities (Gene Ontology), 2.) features derived from the network structure (Network), 3.) features describing the similarity between orthologs (Orthologs) and 4.) general features (General).
Gene Ontology
GO similarity
We compute the semantic GO similarity for two proteins based on Resnik[52] information content measure
| (1) |
with 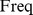 as the number of proteins annotated with a given term
as the number of proteins annotated with a given term 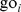 , or its descendant terms in the GO tree. For two GO terms
, or its descendant terms in the GO tree. For two GO terms  , we define the semantic GO term similarity as the
, we define the semantic GO term similarity as the 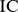 for their common ancestor. And for two proteins
for their common ancestor. And for two proteins 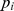 ,
, 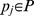 we define the semantic GO similarity as the maximum of all combination of GO annotations for the two proteins. Formally defined as
we define the semantic GO similarity as the maximum of all combination of GO annotations for the two proteins. Formally defined as
| (2) |
Given that measure, the semantic similarity is computed for the interaction partners in the source and target network and the orthologs. Besides a global semantic GO similarity, one feature is modeled for each of the GO categories cellular component, biological process and molecular function (indicated with C, B, and M behind the feature name in the following) to take the different types individually into account.
Network
Network overlap
The overlap of the neighborhood proteins for a given pair of proteins in the source and target network. For this purpose the Jaccard Index is computed for the direct neighbors of the interacting proteins with the equation
| (3) |
where 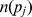 and
and 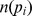 are the adjacent proteins in the protein interaction network.
are the adjacent proteins in the protein interaction network.
Network GO similarity
The average semantic GO similarity between the pair-wise neighbors of the interaction partners in the networks computed with the equation
| (4) |
General
Source interaction database
The source database from which an interaction is extracted as provided as additional information in the used integrated protein interaction database.
Edge support
The number of PubMed abstracts given as evidence for the source interaction.
Source interaction type
The source interaction type (physical, genetic or other) is used as discrete feature value. For this purpose the molecular interaction type [53] is used.
Total support
The number of times an interaction is transferred from all other networks to the target network as suggested by [54] for confidence scoring.
Gene expression correlation coefficient
Given a gene expression time series for two genes the Pearson correlation coefficient is computed for the putative interacting partners in the target network with the equations
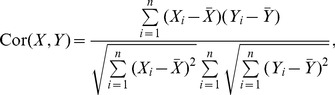 |
(5) |
where 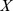 and
and 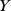 represent the expression values for the respective genes.
represent the expression values for the respective genes.
Ortholog
Sequence similarity
The sequence identity of the orthologs.
Harmonic sequence similarity
The harmonic mean of the sequence identities of the orthologs.
Synonym similarity (Token score)
From the orthologs the function of the proteins is extracted from the textual description using UniProt[48] by tokenizing, stemming and filtering stop words and to general words resulting in a set of tokens which are descriptive for the proteins. Based on these function terms we define the similarity for two proteins 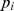 and
and 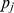 from the set of all protein
from the set of all protein 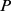 as
as
| (6) |
where 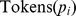 and
and 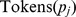 are the function terms of the proteins
are the function terms of the proteins 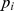 and
and 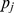 .
.
Domain/Family similarity
The InterPro and PFAM annotations from UniProt[48] are used to compute the domain/family similarity of the orthologs. For two proteins we define the domain/family similarity as
| (7) |
where 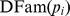 and
and 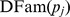 are the domain and family annotations.
are the domain and family annotations.
KEGG Pathway score
Boolean indicator whether the orthologs are involved in the same pathways or in different pathways.
Ortholog source
The database from which the orthologs used for the transfer are extracted.
Ortholog score
Given two orthologs 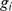 and
and 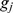 we define the ortholog score as
we define the ortholog score as
| (8) |
where 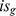 is the inparalog score and
is the inparalog score and 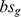 the bootstrap score provided by InParanoid for each gene
the bootstrap score provided by InParanoid for each gene 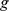 in a ortholog clusters.
in a ortholog clusters.
Ortholog support
The number of times the same ortholog relation between two genes can be found in the different ortholog databases.
Phylogenetic distance
The distance of the source and the target species in a phylogenetic tree provided by [45].
Transitive ortholog
The idea behind this feature is that more conserved orthologs can be traced from a source species to a target species along the phylogenetic tree. For this purpose a phylogenetic tree covering all species with ortholog mappings is used. Given such a tree, a path from a source to a target species is computed by:
searching the shortest path between the two species and
searching the closest leaf nodes for all inner nodes on the shortest path.
The result is a list of species which are “between” the target and the source species. An ortholog is defined as transitively consistent if a direct ortholog between the source and the target species can also be reached when going along the pairwise ortholog mappings on the estimated path.
In the case that a feature cannot be computed because of missing annotation data, the feature is replaced by a missing value indicator. Features are derived from different sources. In the rest of the article we indicate with (T), (S) and (O) after the feature name whether a feature is modeled between the protein pair in the target network, the source network, or between the orthologs, respectively.
Evaluation measures
To assess the quality of the learned models we compute the
| (9) |
| (10) |
and
 |
(11) |
for a given  value assigned by the learned model. A precision of 1.0 for a given score threshold
value assigned by the learned model. A precision of 1.0 for a given score threshold  is obtained when all transferred interactions with a score value
is obtained when all transferred interactions with a score value 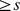 can be found in the experimentally validated network. The relative recall is 1.0 when all correctly transferable interactions using the available ortholog relations are also transferred after the filtering i.e. all transferred interactions have a score value
can be found in the experimentally validated network. The relative recall is 1.0 when all correctly transferable interactions using the available ortholog relations are also transferred after the filtering i.e. all transferred interactions have a score value 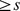 . We mostly use the relative recall instead of the regular recall in order to assess the recall with respect to a direct transfer. As overall measure for different score thresholds the area under the precision (relative) recall curve (AUPRC) and the area under the receiver operating characteristic curve (AUROC) are used [55]. Furthermore, the Information Gain (
. We mostly use the relative recall instead of the regular recall in order to assess the recall with respect to a direct transfer. As overall measure for different score thresholds the area under the precision (relative) recall curve (AUPRC) and the area under the receiver operating characteristic curve (AUROC) are used [55]. Furthermore, the Information Gain (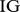 ) i.e. the reduction of entropy of the data set given information about a feature [56], is computed to estimate the impact of the different features.
) i.e. the reduction of entropy of the data set given information about a feature [56], is computed to estimate the impact of the different features.
Formally, for a data set 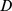 and feature
and feature 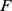 the IG is defined as
the IG is defined as
| (12) |
where  is the set of instances in
is the set of instances in 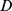 with value
with value  for feature
for feature 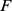 and
and 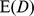 defined as
defined as
| (13) |
where 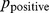 and
and 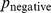 is the proportion of D belonging to the class of correctly (consistently) and incorrectly (inconsistently) transferred interactions, respectively.
is the proportion of D belonging to the class of correctly (consistently) and incorrectly (inconsistently) transferred interactions, respectively.
Results
Current Protein Interaction Networks
Table 1 gives an overview of the protein interaction networks derived from the integrated interaction database iRefIndex having the largest number of interactions.
Over 78% of the S. cerevisiae and over 90% of the D. melanogaster interactions stem from high-throughput studies where over 1,000 interactions are reported, whereas for H. sapiens only 43% of the interactions stem from high-throughput studies (see Figure S1). Furthermore, most interactions for S. cerevisiae are detected with genetic interference and affinity chromatography technology methods like Co-Immunoprecipitation or Tandem Affinity Purification, whereas for D. melanogaster most interactions are detected within one high-throughput Yeast Two Hybrid screen (see Figure S1 and S2).
The total number of interactions consists of physical interactions, genetic interactions and other protein interactions (no interaction type or ambiguous annotations).
With about 180,000 interactions the by far largest eukaryotic interaction network is available for S. cerevisiae. The majority of interactions are genetic interactions. When we only consider physical interactions the S. cerevisiae interaction network is still the largest. Especially in comparison with the second largest protein interaction network from H. sapiens it becomes clear how sparse the networks for the other species still are in current databases. The H. sapiens network has fewer physical interactions, but more than four times more genes in the network as compared to S. cerevisiae.
Furthermore, only the S. cerevisiae network consists of only one connected component. It has been estimated that the complete S. cerevisiae network has between 37,800 and 75,500 protein interactions [57]. Actually, 55,767 physical interactions are contained in iRefIndex for S. cerevisiae. Therefore, for the following, we assume that the S. cerevisiae network is almost complete and, thus, we use the S. cerevisiae network to evaluate the performance of a protein interaction transfer.
It can be expected that more complex organisms also have a more complex network. The number of genes (and maybe also the number of proteins) is not dramatically different and, thus, most likely the number of interactions is different. Therefore, the extremely small coverage of even the best investigated model organisms is apparent. For all other non-model organisms the number of available interactions are neglegible.
Interaction transfer
Experimental settings
We transfer interactions from all eukaryotic species with interaction data used in this study to S. cerevisiae to train our models. The S. cerevisiae interaction network is assumed to be almost complete and possible false negatives in the gold standard are ignored. True positives are defined as transferred interactions, which can be found in the S. cerevisiae network, and false positives as transferred interactions, which cannot be found in the network.
Three experimental settings are considered to evaluate our approach:
All interactions setting (AllI)
All interactions are transferred to S. cerevisiae and only the occurrence of the transferred interactions in the gold standard is checked.
Physical interactions setting (PhyI)
Only physical interactions are transferred to S. cerevisiae and in addition to the occurrence of the interactions also the agreement of the interaction type is checked.
Genetic interactions setting (GenI)
The same as the previous setting, but with genetic interactions.
In total 19,785 interactions from all eukaryotic species considered in this study can be transferred to S. cerevisiae. For AllI 4,745 interactions can be found in the gold standard and the other 15,040 interactions are used as negative set. The physical, PhyI, setting consists of 1,019 correctly transferred interactions and 8,174 incorrectly transferred interactions. The genetic, GenI, setting consists of 901 correctly and 5,300 incorrectly transferred interactions. The remaining 4,391 transferred interactions have an unknown, other, or an ambiguous interaction type.
The features are modeled for the protein pairs involved in the transfer. In total four proteins are considered for each transfer (two proteins from the source network and two proteins from the target network). The features are defined between the different protein pairings in the target network, in the source network and between the orthologs. In total 20 different features types are modeled where for the features used for the orthologs one feature for each of the two orthologs pairs involved in the transfer is created. E.g. for the global GO similarity one feature is modeled between the interaction partners in the source network, one feature is modeled between the interaction partners in the target network and two features are modeled between the orthologs involved in the transfer. For the gene expression feature the compiled gene expression experiment set from [58] which includes normalized intensity values from different cellular states and biological conditions is used.
Six feature sets are constructed for the training of the Random-Forest-Filter (RFF) in order to compare the performance and to estimate the feature contribution. This includes two main sets, one in which all features are considered and one setting where only features are used which can be assumed to be available for most of the species. Hence, features containing information about the network structure and the gene expression correlation are excluded in the reduced feature set. The other four feature sets (Network, Gene Ontology, General and Orthologs) consists only of the features from the respective category. In Table 2 the composition of the different feature sets and protein pairings is given.
Table 2. Feature set configuration.
| Pairing | Feature set | ||||||||
| Pairing Source network (S) | Target network (T) | Ortholog (O) | Network | GO | General | Orthologs | Reduced | Full | |
| Network overlap | x | x | x | x | |||||
| GO Network | x | x | x | x | |||||
| GO Global | x | x | x | x | x | x | |||
| GO (B) similarity (GO Biological process) | x | x | x | x | x | x | |||
| GO (C) similarity (GO Cellular component) | x | x | x | x | x | x | |||
| GO (M) similarity (GO Molecular function) | x | x | x | x | x | x | |||
| Source interaction database | x | x | x | x | |||||
| Edge support | x | x | x | x | |||||
| Source interaction type | x | x | x | x | |||||
| Total support | x | x | x | x | |||||
| Gene expression correlation | x | x | x | ||||||
| Sequence identity | x | x | x | x | |||||
| Token similarity | x | x | x | x | |||||
| Domain similarity | x | x | x | x | |||||
| KEGG pathway score | x | x | x | x | |||||
| Ortholog source | x | x | x | x | |||||
| Ortholog score | x | x | x | x | |||||
| Ortholog support | x | x | x | x | |||||
| Transitive orthology | x | x | x | x | |||||
| Phylogenetic distance | x | x | x | x | |||||
The table lists the features in the categories ”Network”, ”GO”, ”General” and ”Ortholog” and the protein pairings (proteins in the target network, proteins in the source network, orthologs). Also the configuration of the full (target network needs to be available) and the reduced feature set (used in real prediction filtering) is shown. For example the feature Global GO is modeled employing the interaction partners in the source network, in the target network and between the orthologs. Furthermore, the feature is included in the GO, the reduced and the full feature set.
Direct protein interaction transfer
Using the previously described interaction database and ortholog mappings, interactions are directly transferred to S. cerevisiae. In Figure 1 the precisions of the interaction transfers from six interaction networks using the previously introduced experimental settings are shown.
Figure 1. Precisions of a direct interaction transfer to S. cerevisiae for the different experimental settings.

We show six eukaryotic species having the largest number of interactions. For the all interaction setting (allI) only the occurrence of a transferred interaction in the S. cerevisiae network is required, whereas for the genetic (GenI) and physical (PhyI) interaction setting also the exact type of the transferred interaction is checked. In addition to the precisions, the number of total transferred interactions and consistent interactions for each species and type is shown on top of the corresponding bar. Most interactions can be transferred from S. pombe to S. cerevisiae. There the transfer precision is highest for physical and genetic interactions. For the allI setting the highest transfer precision is observed for M. musculus to S. cerevisiae. This is due to the small number of interactions, which are mostly involved in conserved biological processes like DNA replication and chromosome organization.
We use orthologs from the well established cluster based ortholog detection approaches InParanoid, OMA and HomologGene. Orthologs from these databases result in higher transfer consistencies than orthologs from tree based approaches like EnsemblCompara[59] (see Figure S3).
The overall precision of an interaction transfer from the different species to S. cerevisiae for AllI is 0.24, whereas for GenI and PhyI the transfer precision is only 0.11 and 0.15, respectively. With 4,745, 1,019 and 901 correctly transferred interactions, 3%, 2% and 1% of the S. cerevisiae network can be predicted for the respective experimental settings AllI, PhyI and GenI. The highest transfer precision of physical and genetic interactions can be achieved with a transfer from S. pombe (the phylogenetically closest species in our tree with experimentally validated interaction data).
Given complete interaction data for all species it would be expected that the highest precision would be achieved with a transfer from the phylogenetic closest species. But since the interaction data is incomplete and interologs of S. cerevisiae might be used as prior knowledge for the interaction discovery, some phylogenetically more distant species show higher interaction transfer precisions than phylogenetically closer species. Most notable is the performance of a transfer from M. musculus to S. cerevisiae with an unusually high precision of 0.36 in the AllI setting. A GO overrepresentation analysis (DAVID [60]) of the proteins involved in the transfer from M. musculus to S. cerevisiae exhibits that some highly conserved biological processes are overrepresented (like DNA-dependent DNA replication, pre-replicative complex assembly, DNA replication initiation and chromosome organization), which might explain the high precision of the interaction transfer. By looking at the transfer precisions for each biological process it can be seen that for these overrepresented biological processes the transfer precision from M. musculus to S. cerevisiae is almost the same as the transfer precision from H. sapiens to S. cerevisiae. E.g. 102 transferred interactions from H. sapiens to S. cerevisiae are associated with the biological process DNA-dependent DNA replication from which 64 are consistent, for the pre-replicative complex assembly process 30 out of 43 and for the DNA replication initiation process 31 out of 45 are consistent.
For phylogenetically distant species ortholog clusters consist of more than two genes which results in 1:n or even n:m mappings. Thus, with a direct transfer a single source interaction can be inferred between different genes in the target network. For H. sapiens and S. cerevisiae are for example on average 1.9 H. sapiens genes and 1.18 S. cerevisiae genes in one cluster, whereas for H. sapiens and M. musculus the cluster contain 1.05 and 1.01 genes, respectively.
Transfer filter
We train our Random-Forest-Filters (RFF) to score directly transferred interactions and to identify possible conservations.
In Figure 2 the precision-(relative) recall curves of the Random-Forest-Filters (RFF) trained with the full and reduced feature sets and the three experimental settings AllI, PhyI and GenI using a 10-fold cross validation are shown. A simple interaction filter using the harmonic sequence similarity between the orthologs and a filter based on the InParanoid ortholog bootstrap score are evaluated as baseline comparisons.
Figure 2. Precision - (relative) Recall curves.

Precision - (relative)Recall curves for the RFF (Random-Forest-Filter) trained with the reduced feature set, the full feature set and different experimental setting (only physical interactions (PhyI), only genetic interactions (GenI) and all interactions (AllI)) using 10-fold cross validation. Interactions are transferred from all eukaryotic species with interaction data to S. cerevisiae and filtered with the respective approaches. In addition, the precision and relative recall is given for a simple sequence similarity filter and a filter based on the InParanoid ortholog bootstrap score. The RFF for AllI trained with the full (red) and reduced (red dotted) feature set perform best. The reduced feature set performs somewhat worse than the full feature set. For the more strict PhyI and GenI settings in which also the type of an interaction is transferred, the performance drops in comparison to AllI. By comparing the different feature sets it can be seen that for physical interactions (green, green dotted) almost the same performance for the full and reduced feature set can be reached, whereas for genetic interactions (blue, blue dotted) a clear difference in the performance can be observed. But again, for these two settings a huge improvement of RFF to the baseline filters based on sequence similarity) and ortholog scores can be observed.
The RFF trained with the full feature set in the AllI setting achieves the highest 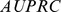 score of 0.86 and an
score of 0.86 and an 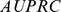 score of 0.82 with the reduced feature set. When in addition to the occurrence of an interaction also an interaction type agreement is required, the performance drops significantly. Physical interactions can be classified with an
score of 0.82 with the reduced feature set. When in addition to the occurrence of an interaction also an interaction type agreement is required, the performance drops significantly. Physical interactions can be classified with an 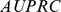 score of 0.60 and of 0.58 with the RFF trained with the full and reduced feature set, respectively. Genetic interactions can be classified with
score of 0.60 and of 0.58 with the RFF trained with the full and reduced feature set, respectively. Genetic interactions can be classified with 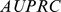 score of 0.60 and 0.48.
score of 0.60 and 0.48.
Using a maximum InParanoid ortholog bootstrap score of 1.00, a transfer precision of 0.33 for AllI can be reached resulting in an 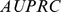 of 0.30. For physical and genetic interactions the precision of a direct transfer can barely be improved resulting in an
of 0.30. For physical and genetic interactions the precision of a direct transfer can barely be improved resulting in an 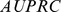 of 0.15 and 0.18, respectively.
of 0.15 and 0.18, respectively.
A high threshold has to be used for the sequence similarity filter in order to increase the transfer precision resulting in a low 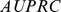 score of 0.28 for AllI. Even lower are the
score of 0.28 for AllI. Even lower are the 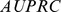 s for PhyI and GenI. This can be explained with the low sequence similarities of the orthologs used for the transfer, which ranges between 33% and 38% on average for the different species. For the full feature set the RFF for AllI yields a precision of 0.85 and a relative recall of 0.69 (regular recall of 0.02) with a typical score threshold of 0.5. With the same score threshold for PhyI a precision of 0.72 and a relative recall of 0.33 (regular recall of 0.01) can be reached, whereas for GenI a slightly lower precision of 0.68, but a higher relative recall of 0.40 is observed (0.003 regular recall).
s for PhyI and GenI. This can be explained with the low sequence similarities of the orthologs used for the transfer, which ranges between 33% and 38% on average for the different species. For the full feature set the RFF for AllI yields a precision of 0.85 and a relative recall of 0.69 (regular recall of 0.02) with a typical score threshold of 0.5. With the same score threshold for PhyI a precision of 0.72 and a relative recall of 0.33 (regular recall of 0.01) can be reached, whereas for GenI a slightly lower precision of 0.68, but a higher relative recall of 0.40 is observed (0.003 regular recall).
In general, the predictors for AllI achieve a better performance than the predictors for the more strict setting in which also the interaction type has been transferred and predicted. This is plausible as for AllI the gold standard is larger and as with a direct transfer a consistency of 25% can be reached already. For the different feature sets (full and reduced) a small drop in the AllI and PhyI setting and a large drop for the GenI setting is observed.
In the following we show examples of transferred physical interactions which receive high and low score values by RFF. On one hand, the transferred interaction between LST8 and TOR2 from WAT1 and TOR2 (in S. pombe) and also the transferred interaction between SMX3 and LSM5 from SmF and CG6610 (in D. melanogaster) get a comparable high score of  0.90. For the first, but not for the second example also an interaction is known between the orthologs in S. cerevisiae. But for the second example, both orthologs (SMX3 and LSM5) carry the Sm domain and the interaction between orthologs of SmF and CG6610 have been found in S. pombe and H. sapiens, which suggests that SMX3 and LSM5 indeed interact, but that they are not included in the S. cerevisiae gold standard. On the other hand, the transferred interaction between CRZ1 and HAT2 from Sp3 and RBBP4 (in H. sapiens, identified within a low-throughput study[61]) and the transferred interaction between ARP6 and RPS1A from Actr13E and RpS3A (in D. melanogaster which was identified in a Yeast Two Hybrid screen [8]) gets a score of
0.90. For the first, but not for the second example also an interaction is known between the orthologs in S. cerevisiae. But for the second example, both orthologs (SMX3 and LSM5) carry the Sm domain and the interaction between orthologs of SmF and CG6610 have been found in S. pombe and H. sapiens, which suggests that SMX3 and LSM5 indeed interact, but that they are not included in the S. cerevisiae gold standard. On the other hand, the transferred interaction between CRZ1 and HAT2 from Sp3 and RBBP4 (in H. sapiens, identified within a low-throughput study[61]) and the transferred interaction between ARP6 and RPS1A from Actr13E and RpS3A (in D. melanogaster which was identified in a Yeast Two Hybrid screen [8]) gets a score of  0.05. Both transferred interactions are not in the S. cerevisiae gold standard, therefore, they are filtered correctly. Due to the low-throughput experiment, which was used to discover the interaction between Sp3 and RBBP4 it can be assumed that this interaction indeed exists for H. sapiens, but not in S. cerevisiae. In contrast, the interaction between Actr13E and RpS3A could also be false positive due to the high-throughput Yeast Two Hybrid screen which was used to identify the interaction in D. melanogaster. In Figure S4 (Supporting Information) the transferred interactions together with their assigned RFF scores and their feature values in comparison to the feature distributions of correctly and incorrectly transferred interactions are shown.
0.05. Both transferred interactions are not in the S. cerevisiae gold standard, therefore, they are filtered correctly. Due to the low-throughput experiment, which was used to discover the interaction between Sp3 and RBBP4 it can be assumed that this interaction indeed exists for H. sapiens, but not in S. cerevisiae. In contrast, the interaction between Actr13E and RpS3A could also be false positive due to the high-throughput Yeast Two Hybrid screen which was used to identify the interaction in D. melanogaster. In Figure S4 (Supporting Information) the transferred interactions together with their assigned RFF scores and their feature values in comparison to the feature distributions of correctly and incorrectly transferred interactions are shown.
Feature impact
To estimate the contribution of each feature to the performance of RFF, the Information Gain (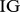 ) is computed for the different experimental settings (Figure 3 d). The
) is computed for the different experimental settings (Figure 3 d). The  for the different features differs among the experimental settings, but the sorting of the features according to their
for the different features differs among the experimental settings, but the sorting of the features according to their 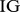 value is similar. The strongest feature is the network overlap in the target network (Network overlap (T)). But also the GO features yield a high
value is similar. The strongest feature is the network overlap in the target network (Network overlap (T)). But also the GO features yield a high 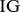 . The combined GO features have higher
. The combined GO features have higher 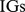 than the category-wise GO features for biological processes, cellular components and molecular functions. This can be explained by the fact that more GO terms are considered for the global semantic GO similarity, so less often a missing value indicator is assigned. From the individual GO term types, the biological processes category has the highest
than the category-wise GO features for biological processes, cellular components and molecular functions. This can be explained by the fact that more GO terms are considered for the global semantic GO similarity, so less often a missing value indicator is assigned. From the individual GO term types, the biological processes category has the highest 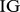 . Biological processes have also been identified by [20] as a strong feature to define thresholds for an interaction transfer filter. From the Ortholog features the synonym similarity (token score) and the ortholog score feature contributes most to the prediction.
. Biological processes have also been identified by [20] as a strong feature to define thresholds for an interaction transfer filter. From the Ortholog features the synonym similarity (token score) and the ortholog score feature contributes most to the prediction.
Figure 3. Feature impact.

Histogram of the score values for correctly (red) and incorrectly (green) transferred interactions (without interaction type) for the features: a.) Network overlap, b.) Semantic GO similarity and c.) Harmonic sequence similarity. d.) Information Gain of the individual features and experimental settings. For ortholog protein features the average Information Gain of the two orthologous partners is shown. For the features a.) Network overlap and especially for b.) Semantic GO similarity a different distribution for correctly and incorrectly transferred interactions can be observed resulting in a large Information Gain of these features. In contrast, for the harmonic sequence similarity feature only a small difference in the distributions can be observed, which explains the small Information Gain and the filter performance based only on sequence similarity.
In contrast, the gene expression correlation, which was used in other studies for the prediction of protein interaction, has a rather low 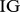 . For the two features with highest
. For the two features with highest 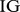 (Network overlap and GO similarity in the target network) also the score distributions of correctly and incorrectly transferred interactions for AllI are shown in Figure 3. Clearly, the fraction of correctly to incorrectly transferred interactions increases with the score value for these two features. For a feature like the harmonic sequence similarity, which has a low
(Network overlap and GO similarity in the target network) also the score distributions of correctly and incorrectly transferred interactions for AllI are shown in Figure 3. Clearly, the fraction of correctly to incorrectly transferred interactions increases with the score value for these two features. For a feature like the harmonic sequence similarity, which has a low 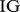 , only a small difference in the characteristics of the distribution can be observed, which explains the weak performance of filters based on sequence similarity.
, only a small difference in the characteristics of the distribution can be observed, which explains the weak performance of filters based on sequence similarity.
In Table 3 the performance of the different individual feature sets (Network, Gene Ontology, General, Ortholog, Reduced set and Full set) is summarized in addition to the filters based on the sequence similarity and the InParanoid bootstrap score. For the GO features the highest feature category-wise 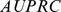 score can be reached for AllI and PhyI. For PhyI a similar
score can be reached for AllI and PhyI. For PhyI a similar 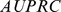 score can be achieved with the ortholog features. Using a combination of all introduced features an up to 0.08 higher
score can be achieved with the ortholog features. Using a combination of all introduced features an up to 0.08 higher 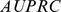 score can be obtained for the different settings. For GenI the highest category-wise
score can be obtained for the different settings. For GenI the highest category-wise 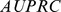 score can be reached with the network features, which is also higher than the score for the reduced feature set. This explains the performance drop for the reduced feature set for this GenI setting.
score can be reached with the network features, which is also higher than the score for the reduced feature set. This explains the performance drop for the reduced feature set for this GenI setting.
Table 3. Result details for the transfer to S. cerevisiae.
| Method | Experimental | Feature | AUPRC | AUROC |
| setting | set | |||
| RFF | All | Full | 0.86 | 0.94 |
| RFF | All | Reduced | 0.82 | 0.91 |
| RFF | All | Network | 0.79 | 0.90 |
| RFF | All | GO | 0.79 | 0.89 |
| RFF | All | Ortholog | 0.62 | 0.82 |
| RFF | All | General | 0.50 | 0.68 |
| InParanoid | All | - | 0.30 | 0.59 |
| Sequence | All | - | 0.28 | 0.55 |
| RFF | Physical | Full | 0.60 | 0.89 |
| RFF | Physical | Reduced | 0.58 | 0.88 |
| RFF | Physical | GO | 0.50 | 0.85 |
| RFF | Physical | Network | 0.42 | 0.84 |
| RFF | Physical | Ortholog | 0.48 | 0.82 |
| RFF | Physical | General | 0.19 | 0.62 |
| InParanoid | Physical | - | 0.15 | 0.61 |
| Sequence | Physical | - | 0.14 | 0.55 |
| RFF | Genetic | Full | 0.60 | 0.87 |
| RFF | Genetic | Reduced | 0.47 | 0.82 |
| RFF | Genetic | Network | 0.51 | 0.86 |
| RFF | Genetic | GO | 0.45 | 0.80 |
| RFF | Genetic | Ortholog | 0.35 | 0.75 |
| RFF | Genetic | General | 0.19 | 0.53 |
| InParanoid | Genetic | - | 0.18 | 0.60 |
| Sequence | Genetic | - | 0.15 | 0.51 |
10-fold cross validation results of the RFF (Random-Forest-Filter) trained with different feature sets, the InParanoid ortholog filter and the sequence similarity filter for different experimental settings. Interactions are transferred from all eukaryotic species with interaction data to S. cerevisiae.
For each experimental setting and feature set the area under precision recall curve (AUPRC) and the area under the receiver operating characteristic curve (AUROC) are computed. From the individual feature sets the RFF trained with the GO and Network feature set perform best for the AllI and GenI setting. Whereas for physical interactions the performance for the Network features are lower than for the GO and Ortholog feature set.
Generalizability
A general transfer approach should be able to achieve a similar performance for the interaction transfer to other species. Since the interaction networks for other species are currently too sparse (see Table 1) RFFs can not be learned and evaluated for individual species except for S. cerevisiae. Therefore, we investigate the applicability of the RFF fitted for the interaction transfer to S. cerevisiae for the transfer of interactions to other eukaryotic species. It has to be expected that:
the RFF scores transferred interactions between phylogenetically closer species higher than transferred interactions between phylogenetically distant species,
according to their importance, the ranking of features is similar for the interaction transfer to different species even though the networks are to incomplete to train a model and
that a comparable performance with competing transfer approaches should be achieved when the RFF is applied for the transfer of interactions to other species.
In the following we investigate these three points.
Transfer scores
We use the RFF with the reduced feature set trained with transferred interactions to S. cerevisiae to transfer protein interactions from the two largest interaction networks H. sapiens and S. cerevisiae to both M. musculus and B. taurus and analysed the score distributions. For physical and genetic interactions in the source network, the predictor trained with the respective interaction type (PhyI and GenI) is used and for interactions with a different type the predictor trained with all data is applied (AllI). As expected, the scores for transferred interactions from the phylogenetically closer species, in this case H. sapiens, are higher than the scores from the more distant species as shown in Figure 4. The score distribution of transferred interactions from S. cerevisiae to M. musculus and B. taurus are very similar with a median score of 0.07 for both distributions. This is comparable to the transfer of interactions to S. cerevisiae, where a median score between 0.03 and 0.09 can be observed (see Figure S5). In comparison, for the transfer of interactions between phylogenetically closer species, a median score of 0.27 and 0.25 can be observed for the transfer of interactions from H. sapiens to M. musculus and B. taurus, respectively. Thus, as expected with higher score thresholds more interactions can be transferred from H. sapiens to M. musculus as compared to H. sapiens to B. taurus. On the other hand, from S. cerevisiae almost the same number of interactions is transferred to the two species B. taurus and M. musculus with different score thresholds.
Figure 4. Score distributions.

Score distributions for transferred interactions with RFF from S. cerevisiae and H. sapiens to the two target species (a.) M. musculus and (b.) B. taurus. Transferred interactions from S. cerevisiae have significantly lower score values than transferred interactions from H. sapiens to both target species. With a low score threshold of 0.2 almost all interactions from H. sapiens will be transferred to the two target species whereas a huge fraction of the transferred S. cerevisiae interactions is filtered out.
Cross-species feature importances ranking
We transfer all available interactions to S. cerevisiae, H. sapiens, D. melanogaster, S. pombe and C. elegans and compute the Information Gain (IG) of each modeled feature given the observed consistently and inconsistently transferred interactions for the respective species. We observe that the similarity of the feature ranking decreases with the IG i.e. that those features which are important for the classifier are consistently ranked high and the ranking of those feature which are not that beneficial to our classifier differ more. For example the Network overlap feature is ranked first for all considered species expect for C. elegans. Also the feature which models the GO Similarity between the target interactions is ranked second by all considered species expect for C. elegans. In Figure S6 in the Supporting Information we show the ranking of the features according to their IG for the different species. As reference we use the ten features with highest IG for the interaction transfer to S. cerevisiae.
Comparison with other interaction transfer methods
Most protein interaction transfer methods predict interologs for H. sapiens and, in addition, quite many experimentally validated interactions are available for human. Therefore, this network is chosen to evaluate the intersections of predicted protein interactions from different data sets and a set of experimentally discovered physical protein interactions.
Transferred interactions from InteroPORC [21], the STRING database [62], InterologFinder [24], BIPS:BIANA [25] and interactions predicted with RFF are used for the comparison. In order to compare the sets, the protein identifiers are mapped to UniProt/Swissprot identifiers. The following prediction sets are constructed using the publicly available transferred networks from the considered approaches for H. sapiens:
STRING(1)
only high confidence interactions with at least one evidence of an interaction transfer from another species (interactions with a combined score below 0.7 are excluded);
STRING(2)
The combined score of STRING incorporates evidence from many sources including experimental knowledge for the respective species (direct evidence). Thus, transferred interactions with also direct evidence are scored higher, which biases the STRING set for this comparison. Therefore, an additional STRING interaction set is created where the combined score is recomputed without the scores for the direct evidence from databases, experiments and text-mining using the equation for the combined score [30]. Again for this set a combined score threshold of 0.7 is used to filter interactions.
InteroPORC
all transferred interactions;
InterologFinder
15,795 transferred interactions with highest score (the score threshold is set so that the same number of interaction as STRING(2) are predicted);
BIPS;BIANA
all transferred interactions in the online available precomputed prediction set with domain interactions or shared GO terms;
RFF
The RFF for physical source interactions (PhyI) trained with the reduced feature set and with transferred interactions to S. cerevisiae is used. All transferred interactions with a score  0.18 for the transfer to H. sapiens from all species considered in the study are used. The score value is experimentally chosen to yield roughly the same number of transferred interactions as STRING(2).
0.18 for the transfer to H. sapiens from all species considered in the study are used. The score value is experimentally chosen to yield roughly the same number of transferred interactions as STRING(2).
In the entire InteroPORC prediction set 17,111 physical interactions and in the selected BIPS:BIANA set 7,073 interactions are included. With 28,155 links between proteins the interaction set from the STRING(1) is the largest, the STRING(2) set is only slightly larger (15,795 interactions) than the set from RFF which includes 14,634 predicted physical interactions. 35,628 experimentally validated physical interactions are taken from iRefIndex (7,784 interactions are excluded because the proteins are only mappable to UniProt/TrEMBL).
In Figure 5 the consistency with experimentally validated interactions (a.) and the intersections between different H. sapiens protein interaction sets are shown (b.).
Figure 5. Method comparison.

Comparison of interaction transfer sets from various methods for H. sapiens with known H. sapiens interactions from iRefIndex. We compare interaction sets from STRING [62], InteroPORC [21], InterologFinder [24], BIPS:BIANA [25] and our Random-Forest-Filter (RFF). From the STRING database only interactions with interaction transfer information from other species and a combined score over 0.7 are included (STRING(1)). The combined score uses information from all information sources including knowledge on experimental interactions for the respective species (direct evidence). Therefore, an additional interaction set is created where the combined STRING score is recomputed excluding the scores from the direct evidence of databases, experiments and text-mining (STRING(2)). In general, the intersections between the different sets and the known interactions are small. a.) With the RFF and with STRING(1) 10% of the predicted interactions can be found in the experimental data. The modified STRING(2) interaction set is 43% smaller and only 4% of the predicted interactions are consistent with the experimental data showing a clear performance advantage of the RFF for species with no experimentally determined interactions. b.) We compare the interaction sets of RFF, STRING(1), a combined set of unique interactions from InteroPORC, InterologFinder and BIPS:BIANA and a set of known H. sapiens interactions. With the RFF 42% of predicted interactions can also be found in one of the other sets.
In general, the intersections between the sets are small. The highest consistency of 10% between the predicted interaction sets and the experimental set can be reached with the RFF and with the STRING(1) interaction set.
From the STRING(2) and BIPS:BIANA interaction set 4% and from the InteroPORC and InterologFinder around 3% of the predicted interactions are consistent with the experimental data. In total 42% of predicted interactions with the RFF can be found in at least one other set whereas for the STRING(1) set only 26%, for the BIPS:BIANA set 18%, for the InteroPORC set 17% and for the InterologFinder set 10% can be found in another interaction set. Besides BIPS:BIANA all methods transfer interactions from all available interactions in public available databases. But BIPS:BIANA explicitly excludes interactions from Tandem Affinity Purification experiments which explains the rather small interaction set. In comparison to STRING(2), BIPS:BIANA, InteroPORC and InterologFinder a clear performance gain of our RFF approach can be observed. Furthermore, RFF cannot be outperformed by STRING(1) even with the integration of experimental knowledge (which is not available for most species) via the combined score. Thus, for species without experimental knowledge but also for model organisms with experimental protein interactions a performance advantage of our approach in comparison to STRING can be expected.
Enriched protein interaction networks
As shown above via the comparison with other state-of-the-art method our RFF approach has a decent performance for the transfer of interactions to species without experimental interaction data. Therefore, we use our approach to obtain as comprehensive as possible interaction networks for various eukaryotic species. For this we use all available experimental interaction data for all 83 eukaryotic species for the transfer to all other eukaryotic species whenever ortholog mappings of appropriate quality are available. We employ three RFFs trained on S. cerevisiae: RFF, PhyI for physical source interactions, RFF, GenI for genetic source interactions and RFF, AllI for interactions for the remaining interactions including interactions without annotated interaction type. The same score threshold of 0.18 is used for all models.
With direct interaction transfer the interactome of 83 eukaryotic species can be extended from currently 321,808 interactions to 5,751,775 interactions. With the RFF 1,248,609 pair-wise interactions can be transferred (i.e. more than 78,% of transferred interactions are filtered out as possible false positive). An overview of the resulting interactomes is shown in Figure 6 using the Interactive Tree Of Life [63] (only species are shown for which at least 50% of the genes have associated GO annotations). For higher vertebrates of interest such as the farm animals B. taurus, M. musculus and G. gallus each interaction network can be enriched with over 40,000 interactions. After that, the resulting interactomes have a decent coverage of more than 2 interactions per gene on average. Still, with our method for some species only few interactions can be transferred. Examples are plants like O. sativa or V. vinifera with an average of 0.35 interactions per gene. The reason for the low coverage in these cases is the small number of available orthologs in the ortholog databases.
Figure 6. Enriched interactome.

The interactomes of 83 eukaryotic species can be increased from currently 321,808 interactions to 1,076,996 pair-wise interactions using a score threshold of 0.18 and the RFFs with reduced feature set. In the figure the enriched protein interactomes are shown for all species where at least GO annotations for half of the genes are available. Interactions are transferred from all eukaryotic species to all other species with available ortholog mappings. The color of the species nodes indicates the average number of interactions per gene and the associated bar chart indicates the fraction of physical interactions (green), genetic interactions (blue) and other interaction types (red) in the enriched interaction networks for the respective species. For species with rich annotation information including M. musculus and B. taurus over 40,000 interactions can be transferred resulting in an average number of interactions per gene larger than 2. For species with sparse annotation information and few ortholog references to UniProt only a small number of interactions can be transferred. For example for the plants O. sativa and V. vinifera only 0.35 interactions per gene on average can be obtained.
It is clear that for the large scale interaction transfer with our RFF method the limitations are the availability of ortholog relations, of mappings of the orthologs to UniProt entries and of annotations of the UniProt entries. This implies that for some species only few interactions can be transferred. Of course, RFF will profit from the expected improvements of protein annotations, ortholog mappings and further experimental protein interactions.
The transferred interaction networks for the 83 species are available on our web service and can be inspected and downloaded. The user can specify score thresholds corresponding to the expected transfer precision of our models. The database will be frequently updated to incorporate newly available experimental interactions and updates of protein annotations and orthologs for more species. In Figure 7 the web interface including the ‘transfer statistics view’ for M. musculus is outlined as an example.
Figure 7. COIN-DB web interface.

Screenshot of the web interface for the transferred and scored protein interactions. Transferred and experimentally validated interactions can be downloaded for 83 eukaryotic species for user specified score thresholds. For species of interest the transfer profiles can be inspected in detail including the number of interactions (of the different interaction types) and the number of uniquely transferred interactions, and the expected performance of the transfer.
Discussion and Conclusions
Years after high-throughput screening techniques for the identification of protein interactions were introduced most interaction data still is available for only a few model organisms, in particular for S. cerevisiae. Transferring protein interactions works best between phylogenetically close species, but already between the two yeast species S. pombe and S. cerevisiae only a consistency of 36% for transferred physical interactions can be observed. The transfer consistency between more distant species is of course much lower. The transfer consistency is also lower for genetic interactions between the two yeasts, which might be due to the incompleteness of the S. cerevisiae genetic interaction network.
We observed that for only 3% of the S. cerevisiae interactions evidence of conservation between orthologs in different species could be found. In order to improve the transfer quality and to be able to also consider interactions from phylogenetically distant species, e.g. from S. cerevisiae to M. musculus, we introduced a new method using Random Forests (Random-Forest-Filter RFF) to score and filter transferred interactions.
We trained the models with transferred interaction data from eukaryotic organisms to S. cerevisiae. We did the training on yeast, as the S. cerevisiae network is currently the largest eukaryotic interaction network and for most of the proteins in the network curated functional annotations are available. We evaluated the models with different feature sets and experimental settings and compared the models with commonly applied filter approaches e.g. using the sequence similarity and the InParanoid bootstrap score. We showed that for the task of transferring interactions to S. cerevisiae our approach performs better than commonly applied filter approaches. Based on these results we assume that the performance of the transfer to S. cerevisiae is a lower bound for the performance of the method for the transfer between phylogenetically closer species.
But still, our observed results are limited with respect to different aspects:
Possible false negatives in the S. cerevisiae network result in lower transfer consistencies, whereas false positives in the S. cerevisiae network may result in an overestimation of the consistency.
Our method makes use of interaction data from various sources like Yeast two Hybrid, or Tandem Affinity Purification and thus included measured-binary and measured-predicted binary interactions. We only address the interaction transfer on a general level and currently only consider binary-interactions. Our method will benefit from further discrimination of protein interactions e.g. discrimination between transient or permanent protein interactions, or the pre-identification of conserved protein complexes. And thus, stronger claims on the conservation rate and also a more complete interaction transfer will be possible.
Low-throughput experiments are commonly hypothesis-driven [4] and involve proteins of particular interest to the researcher performing the experiments. These low-throughput experiments can also be based on the observation that a conservation in a particular species exists, which could lead to an overestimation of the consistency and to overfitting.
The ortholog and protein annotations quality have a direct influence on our models. For example KEGG pathway information, or gene ontology and synonym annotations are themselves often inferred using homology information ( directly or indirectly). For example the KEGG databases transfers pathway information from well studied species based on manually defined ortholog groups. It is obvious that with solely transferred annotations our approach can not improve the prediction performance.
We fitted our model for the transfer to S. cerevisiae only. Due to these reasons, we can not give an accurate estimation on the performance for the protein interaction transfer to species except for S. cerevisiae.
But we could show that our approach can be applied for the transfer of interaction to species beyond S. cerevisiae as well. On one hand, we tested the generalizability of RFF with transferred interactions to H. sapiens, M. musculus and B. taurus. We showed that (as expected) transferred interactions from phylogenetically closer species get higher scores than transferred interactions from phylogenetic more distant species. Furthermore, we showed that those features which are most beneficial for the classification of interaction for the transfer to S. cerevisiae are also most beneficial for the classification of interactions for other species. On the other hand, we compared different protein interaction approaches. We showed for H. sapiens that with our approach the highest consistency of transferred interactions can be observed and that 42% of transferred interactions can be explained with high confidence relations extracted from STRING, InteroPORC, InterologFinder, BIPS:BIANA or the available experimental interactions. Furthermore, in an experimental setting where we recomputed the STRING combined edge score for H. sapiens to mimic a species without experimental knowledge, we showed that RFF predicts almost the same number of interaction as STRING, but with our approach more than twice as many interactions are consistent with the available experimental protein interaction network.
Therefore, we used RFF to transfer protein interactions to 83 eukaryotic species and we provide a web service for the download and investigation of these transferred interaction networks. Based on the above discussions, these predictions have to be used with care, but we are confident that our transferred networks are as comprehensive and also as accurate as currently possible.
Availability
Supporting Information
Fraction of interactions derived from low and high-throughput studies. Protein interactions for S. cerevisiae, H. sapiens, D. melanogaster and S. pombe from iRefIndex[33] classified into the categories: derived from low-throughput studies (detected in studies which report between 1 and 10 interactions), derived from mid-throughput studies (detected in studies which report between 10 and 100 interactions), derived from mid-high throughput (detected in studies which report between 100-1000 interactions) and derived from high-throughput studies (detected in studies which report  1000 interactions).
1000 interactions).
(PDF)
Interactions by detection method. Protein interactions by detection method for S. cerevisiae, H. sapiens, D. melanogaster and S. pombe from iRefIndex[33].
(PDF)
Direct interaction transfer to S. cerevisiae using different ortholog databases. Transfer consistencies of a protein interaction transfer from M. musculus, H. sapiens, S. pombe, C. elegans and D. melanogaster to S. cerevisiae using orthologs from the databases OMA[45], InParanoid [46], HomoloGene [47], EnsemblCompara[59], TreeFam[64] and eggNog[65] for the all interaction setting (allI).
(PDF)
Transfer examples. Examples of transferred interactions which get high and low scores by RFFs including specific feature values for these interactions and the overall feature distribution (the scores are estimated via a cross-validation setting).
(PDF)
Protein interaction scores for the transfer of interactions to S. cerevisiae . The average transfer scores for an interaction transfer from M. muscles, H. sapiens, S. pombe, A. thaliana, C. elegans and D. melanogaster to S. cerevisiae using RFFs in a cross-validation setting.
(PDF)
Information Gain feature ranking. The feature importance ranking i.e. the ranking of features, is quite similar especially for the most important features, whereas the ranking of the less important feature varies more.
(PDF)
Data sources. List of data sources used for this study.
(XLS)
Funding Statement
This work was supported by the DFG International Research Training Group (IRTG) RECESS 1563 via scholarships to RP. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Zhang A (2009) Protein Interaction Networks: Computational Analysis. Cambridge: Cambridge University Press. [Google Scholar]
- 2. Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, et al. (2012) The IntAct molecular interaction database in 2012. Nucleic Acids Res 40: D841–D846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, Boucher L, Winter A, et al. (2013) The BioGRID interaction database: 2013 update. Nucleic Acids Res 41: D816–D823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sambourg L, Thierry-Mieg N (2010) New insights into protein-protein interaction data lead to increased estimates of the s. cerevisiae interactome size. BMC Bioinformatics 11: 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gavin AC, Bösche M, Krause R, Grandi P, Marzioch M, et al. (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141–147. [DOI] [PubMed] [Google Scholar]
- 6. Ewing RM, Chu P, Elisma F, Li H, Taylor P, et al. (2007) Large-scale mapping of human proteinprotein interactions by mass spectrometry. Mol Syst Biol 3: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ehlert A, Weltmeier F, Wang X, Mayer CS, Smeekens S, et al. (2006) Two-hybrid protein-protein interaction analysis in arabidopsis protoplasts: establishment of a heterodimerization map of group c and group s bzip transcription factors. Plant J 46: 890–900. [DOI] [PubMed] [Google Scholar]
- 8. Uetz P, Pankratz MJ (2004) Protein interaction maps on the fly. Nat Biotechnol 22: 43–44. [DOI] [PubMed] [Google Scholar]
- 9. Michaut M, Kerrien S, Montecchi-Palazzi L, Chauvat F, Cassier-Chauvat C, et al. (2008) Interoporc: automated inference of highly conserved protein interaction networks. Bioinformatics 24: 1625–1631. [DOI] [PubMed] [Google Scholar]
- 10. Rhodes DR, Tomlins SA, Varambally S, Mahavisno V, Barrette T, et al. (2005) Probabilistic model of the human protein-protein interaction network. Nat Biotechnol 23: 951–959. [DOI] [PubMed] [Google Scholar]
- 11. Turinsky AL, Razick S, Turner B, Donaldson IM, Wodak SJ (2010) Literature curation of protein interactions: measuring agreement across major public databases. Database (Oxford) 2010: baq026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friedel CC, Zimmer R (2009) Identifying the topology of protein complexes from affinity purification assays. Bioinformatics 25: 2140–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berggård T, Linse S, James P (2007) Methods for the detection and analysis of protein-protein interactions. Proteomics 7: 2833–2842. [DOI] [PubMed] [Google Scholar]
- 14. Berg J, Lässig M, Wagner A (2004) Structure and evolution of protein interaction networks: a statistical model for link dynamics and gene duplications. BMC Evol Biol 4: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Dam TJP, Snel B (2008) Protein complex evolution does not involve extensive network rewiring. PLoS Comput Biol 4: e1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shou C, Bhardwaj N, Lam HYK, Yan KK, Kim PM, et al. (2011) Measuring the evolutionary rewiring of biological networks. PLoS Comput Biol 7: e1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matthews LR, Vaglio P, Reboul J, Ge H, Davis BP, et al. (2001) Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or “interologs”. Genome Res 11: 2120–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gandhi TKB, Zhong J, Mathivanan S, Karthick L, Chandrika KN, et al. (2006) Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 38: 285–293. [DOI] [PubMed] [Google Scholar]
- 19. Bork P, Jensen LJ, von Mering C, Ramani AK, Lee I, et al. (2004) Protein interaction networks from yeast to human. Curr Opin Struct Biol 14: 292–299. [DOI] [PubMed] [Google Scholar]
- 20.De Bodt S, Proost S, Vandepoele K, Rouze P, Van de Peer Y (2009) Predicting protein-protein interactions in arabidopsis thaliana through integration of orthology, gene ontology and co-expression. BMC Genomics 10: : 288+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Michaut M, Kerrien S, Montecchi-Palazzi L, Chauvat F, Cassier-Chauvat C, et al. (2008) Intero-PORC: automated inference of highly conserved protein interaction networks. Bioinformatics 24: 1625–1631. [DOI] [PubMed] [Google Scholar]
- 22. Yu H, Luscombe NM, Lu HXX, Zhu X, Xia Y, et al. (2004) Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res 14: 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene ontology: tool for the unification of biology. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wiles AM, Doderer M, Ruan J, Gu TT, Ravi D, et al. (2010) Building and analyzing protein interactome networks by cross-species comparisons. BMC Syst Biol 4: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia-Garcia J, Schleker S, Klein-Seetharaman J, Oliva B (2012) BIPS: BIANA interolog prediction server. a tool for protein-protein interaction inference. Nucleic Acids Res 40: W147–W151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gallone G, Simpson TI, Armstrong JD, Jarman AP (2011) Homology::InterologWalk–a perl module to build putative protein-protein interaction networks through interolog mapping. BMC Bioinformatics 12: 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tuncbag N, Gursoy A, Nussinov R, Keskin O (2011) Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using prism. Nat Protoc 6: 1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pao-Yang Chen GR Charlotte M Deane (2008) Predicting and validating protein interactions using network structure. PLoS Comput Biol 4(7): e1000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo Q, Pagel P, Vilne B, Frishman D (2011) DIMA 3.0: Domain interaction map. Nucleic Acids Res 39: D724–D729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, et al. (2005) String: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res 33: D433–D437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lewis ACF, Jones NS, Porter MA, Deane CM (2012) What evidence is there for the homology of protein-protein interactions? PLoS Comput Biol 8: e1002645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altenhoff AM, Dessimoz C (2009) Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput Biol 5: e1000262+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Razick S, Magklaras G, Donaldson IM (2008) iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinformatics 9: 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bader GD, Donaldson I, Wolting C, Ouellette BF, Pawson T, et al. (2001) BIND–the biomolecular interaction network database. Nucleic Acids Res 29: 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ruepp A, Waegele B, Lechner M, Brauner B, Dunger-Kaltenbach I, et al. (2010) CORUM: the comprehensive resource of mammalian protein complexes–2009. Nucleic Acids Res 38: D497–D501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xenarios I, Rice DW, Salwinski L, Baron MK, Marcotte EM, et al. (2000) DIP: the database of interacting proteins. Nucleic Acids Res 28: 289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, et al. (2009) Human protein reference database–2009 update. Nucleic Acids Res 37: D767–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lynn DJ, Winsor GL, Chan C, Richard N, Laird MR, et al. (2008) InnateDB: facilitating systemslevel analyses of the mammalian innate immune response. Mol Syst Biol 4: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chautard E, Fatoux-Ardore M, Ballut L, Thierry-Mieg N, Ricard-Blum S (2011) MatrixDB, the extracellular matrix interaction database. Nucleic Acids Res 39: D235–D240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chatr-aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, et al. (2007) MINT: the molecular interaction database. Nucleic Acids Res 35: D572–D574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Güldener U, Münsterkötter M, Oesterheld M, Pagel P, Ruepp A, et al. (2006) MPact: the MIPS protein interaction resource on yeast. Nucleic Acids Res 34: D436–D441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goll J, Rajagopala SV, Shiau SC, Wu H, Lamb BT, et al. (2008) MPIDB: the microbial protein interaction database. Bioinformatics 24: 1743–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pagel P, Kovac S, Oesterheld M, Brauner B, Dunger-Kaltenbach I, et al. (2005) The MIPS mammalian protein-protein interaction database. Bioinformatics 21: 832–834. [DOI] [PubMed] [Google Scholar]
- 44. Brown KR, Jurisica I (2005) Online predicted human interaction database. Bioinformatics 21: 2076–2082. [DOI] [PubMed] [Google Scholar]
- 45. Schneider A, Dessimoz C, Gonnet GH (2007) OMA browser–exploring orthologous relations across 352 complete genomes. Bioinformatics 23: 2180–2182. [DOI] [PubMed] [Google Scholar]
- 46. Remm M, Storm CE, Sonnhammer EL (2001) Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol 314: 1041–1052. [DOI] [PubMed] [Google Scholar]
- 47. Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, et al. (2009) Database resources of the national center for biotechnology information. Nucleic Acids Res 37: D5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Consortium TU (2011) Ongoing and future developments at the universal protein resource. Nucleic Acids Res 39: D214–D219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, et al. (2009) The WEKA data mining software: an update. SIGKDD Explorations 11: 10–18. [Google Scholar]
- 50.Caruana R, Mizil AN (2006) An empirical comparison of supervised learning algorithms. In: Proceedings of the 23rd international conference on Machine learning. pp. 161–168. [Google Scholar]
- 51. Breiman L (2001) Random forests. Machine Learning 45: 5–32. [Google Scholar]
- 52. Resnik P (1999) Semantic similarity in a taxonomy: An Information-Based measure and its application to problems of ambiguity in natural language. Journal of Artificial Intelligence Research 11: 95–130. [Google Scholar]
- 53. Côté RG, Jones P, Apweiler R, Hermjakob H (2006) The ontology lookup service, a lightweight cross-platform tool for controlled vocabulary queries. BMC Bioinformatics 7: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mika S, Rost B (2006) Protein-protein interactions more conserved within species than across species. PLoS Comput Biol 2: : e79+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis J, Goadrich M (2006) The relationship between precision-recall and roc curves. In: Proceedings of the 23rd international conference on Machine learning. New York, NY, USA: ACM, ICML '06, pp. 233–240. [Google Scholar]
- 56.Mitchell TM (1997) Machine Learning. Bosten: McGraw Hill. [Google Scholar]
- 57. Hart GT, Ramani AK, Marcotte EM (2006) How complete are current yeast and human proteininteraction networks? Genome Biol 7: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bhardwaj N, Lu H (2005) Correlation between gene expression profiles and protein-protein interactions within and across genomes. Bioinformatics 21: 2730–2738. [DOI] [PubMed] [Google Scholar]
- 59. Vilella A, Severin J, Ureta-Vidal A, Heng L, Durbin R, et al. (2009) EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19: 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang Y, Dufau ML (2002) Silencing of transcription of the human luteinizing hormone receptor gene by histone deacetylase-msin3a complex. J Biol Chem 277: 33431–33438. [DOI] [PubMed] [Google Scholar]
- 62. Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, et al. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39: D561–D568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Letunic I, Bork P (2011) Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39: W475–W478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ruan J, Li H, Chen Z, Coghlan A, Coin LJM, et al. (2008) Treefam: 2008 update. Nucleic Acids Res 36: D735–D740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, et al. (2012) eggNOG v3. 0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res 40: D284–D289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fraction of interactions derived from low and high-throughput studies. Protein interactions for S. cerevisiae, H. sapiens, D. melanogaster and S. pombe from iRefIndex[33] classified into the categories: derived from low-throughput studies (detected in studies which report between 1 and 10 interactions), derived from mid-throughput studies (detected in studies which report between 10 and 100 interactions), derived from mid-high throughput (detected in studies which report between 100-1000 interactions) and derived from high-throughput studies (detected in studies which report 1000 interactions).
(PDF)
Interactions by detection method. Protein interactions by detection method for S. cerevisiae, H. sapiens, D. melanogaster and S. pombe from iRefIndex[33].
(PDF)
Direct interaction transfer to S. cerevisiae using different ortholog databases. Transfer consistencies of a protein interaction transfer from M. musculus, H. sapiens, S. pombe, C. elegans and D. melanogaster to S. cerevisiae using orthologs from the databases OMA[45], InParanoid [46], HomoloGene [47], EnsemblCompara[59], TreeFam[64] and eggNog[65] for the all interaction setting (allI).
(PDF)
Transfer examples. Examples of transferred interactions which get high and low scores by RFFs including specific feature values for these interactions and the overall feature distribution (the scores are estimated via a cross-validation setting).
(PDF)
Protein interaction scores for the transfer of interactions to S. cerevisiae . The average transfer scores for an interaction transfer from M. muscles, H. sapiens, S. pombe, A. thaliana, C. elegans and D. melanogaster to S. cerevisiae using RFFs in a cross-validation setting.
(PDF)
Information Gain feature ranking. The feature importance ranking i.e. the ranking of features, is quite similar especially for the most important features, whereas the ranking of the less important feature varies more.
(PDF)
Data sources. List of data sources used for this study.
(XLS)