

Abstract
Urothelial carcinoma (UC) causes substantial morbidity and mortality worldwide. However, the molecular mechanisms underlying urothelial cancer development and tumor progression are still largely unknown. Using informatics analysis, we identified Sh3gl2 (endophilin A1) as a bladder urothelium-enriched transcript. The gene encoding Sh3gl2 is located on chromosome 9p, a region frequently altered in UC. Sh3gl2 is known to regulate endocytosis of receptor tyrosine kinases implicated in oncogenesis, such as the epidermal growth factor receptor (EGFR) and c-Met. However, its role in UC pathogenesis is unknown. Informatics analysis of expression profiles as well as immunohistochemical staining of tissue microarrays revealed Sh3gl2 expression to be decreased in UC specimens compared to nontumor tissues. Loss of Sh3gl2 was associated with increasing tumor grade and with muscle invasion, which is a reliable predictor of metastatic disease and cancer-derived mortality. Sh3gl2 expression was undetectable in 19 of 20 human UC cell lines but preserved in the low-grade cell line RT4. Stable silencing of Sh3gl2 in RT4 cells by RNA interference 1) enhanced proliferation and colony formation in vitro, 2) inhibited EGF-induced EGFR internalization and increased EGFR activation, 3) stimulated phosphorylation of Src family kinases and STAT3, and 4) promoted growth of RT4 xenografts in subrenal capsule tissue recombination experiments. Conversely, forced re-expression of Sh3gl2 in T24 cells and silenced RT4 clones attenuated oncogenic behaviors, including growth and migration. Together, these findings identify loss of Sh3gl2 as a frequent event in UC development that promotes disease progression.
Introduction
Current estimates indicate that more than 73,000 new cases of urothelial carcinoma (UC) of the bladder and upper urinary tract are diagnosed annually in the United States alone, with ∼15,000 deaths from the disease predicted annually [1]. UC affects the kidney, ureter, and bladder. UC has the highest rate of recurrence of any malignancy and, as a consequence, is the most expensive of all solid tumors to treat [2,3]. Despite this significant healthcare burden, the current standards of care for UC result in modest clinical benefit and infrequent cure. Disease management is hampered by a lack of prognostic markers capable of predicting the likely disease course and the potential for response to a given therapeutic regimen. Thus, there is an urgent need for identification and characterization of the molecular alterations that underlie lethal disease and that identify more aggressive tumors early for radical and/or novel therapies. Such findings would allow for translation of such knowledge into new clinical strategies.
A number of molecular changes have been associated with development and progression of UC. These include 1) alterations in expression and regulation of the receptor tyrosine kinases (RTKs), fibroblast growth factor receptor 3, and members of the epidermal growth factor receptor (EGFR/ERBB) family (reviewed in [4]); 2) up-regulation of signaling through the RAS and phosphatidylinositol 3-kinase/AKT pathways [5]; and 3) functional down-regulation of the tumor suppressors, p53, pRb, and p16, through deletion, mutation, and/or silencing. However, the extent to which these changes could be exploited for therapeutic benefit remains undefined. For example, ligand-mediated activation of ERBB signaling promotes multiple activities of relevance to tumorigenesis, such as cell survival, proliferation, migration, and invasion [6], suggesting that ERBB family members may be effective therapeutic targets. In spite of this, clinical trials targeting the EGFR and/or ERBB2 with tyrosine kinase inhibitors in unselected patients have been unsuccessful to date [7–10]. Thus, additional factors regulating RTK-dependent signaling are likely to influence the response to therapeutic interventions.
In this study, we employed an informatics approach to identify novel transcripts enriched in the bladder relative to other hollow organs. This analysis revealed Sh3gl2/endophilin A1, a known regulator of RTK endocytosis, to be highly expressed in normal urothelium, and lost with high frequency in human UC specimens, including in muscle-invasive disease. RNAi-mediated silencing of Sh3gl2 in well-differentiated UC cells led to increased proliferation and colony formation in vitro, increased phosphorylation of EGFR, Src family kinases (SFKs), and STAT3, and promoted growth of xenografts in vivo. Together, these findings implicate loss of Sh3gl2 in bladder cancer pathogenesis.
Materials and Methods
Bioinformatics Analysis
Sh3gl2 was identified as a transcript highly enriched in normal bladder epithelium using bioinformatics analysis of publicly available databases. Briefly, expression profiles were obtained from the Novartis SymAtlas [11] (NCBI GEO accession GSE1133) and the Genitourinary Development Molecular Anatomy Project (GUDMAP) [12,13] (NCBI GEO accession GSE11221). Both data sets were already normalized; we further removed probe sets that were Affymetrix controls or were never called present and log2-transformed the data. Between-tissue differential expression analysis was performed by computing moderated t-statistics [14] using the limma package [15] from Bioconductor [16]. We corrected for multiple hypothesis testing by using the false discovery rate [17]. With the GUDMAP data set, we compared detrusor to stroma and also detrusor to urothelium (two pairwise comparisons); in SymAtlas, we compared bladder to each of intestine, heart, kidney, liver, lung, prostate, stomach, trachea, and uterus (for a total of nine pairwise comparisons). We identified genes with a false discovery rate below 10% and fold change greater than 5 or less than -5 in every comparison within each data set. The intersection of differentially expressed genes between the two data sets (SymAtlas and GUDMAP) was determined using Entrez Gene identifiers. All analyses were performed in the statistical language R (http://www.r-project.org).
Sh3gl2 mRNA and Protein Expression in Human Subjects
Differential expression of Sh3gl2 mRNA levels in three independent cohorts of human subjects with UC [18–20] was determined using the Compendia Bioscience Oncomine bioinformatics platform (www.oncomine.com; Compendia Bioscience, Ann Arbor, MI). The reporters employed for microarray analysis were Affymetrix 205751_at [18,19] and Illumina ILMN_1661491 [20]. Expression of Sh3gl2 protein was evaluated using commercially available bladder cancer tissue microarrays from either Folio Biosciences (Columbus, OH) or US Biomax, Inc (Rockville, MD). Specific immunohistochemical (IHC) staining of Sh3gl2 in bladder tissue was verified using two independent antibodies against Sh3gl2: rabbit anti-Sh3gl2 (12345-1-AP; ProteinTech Group, Inc, Chicago, IL; not shown) or goat anti-Sh3gl2 (sc-10874; Santa Cruz Biotechnology, Santa Cruz, CA; Figure 1D, b and d). Specificity of staining was confirmed using preincubation of antibody with peptide (Figure 1D, c). Following antibody validation, tissue microarrays were stained with goat anti-Sh3gl2 antibody (1:50 dilution) using a standard immunoperoxidase approach [21]. Staining extent and intensity were scored independently by two investigators (E.M.G. and D.D.V.) and assigned values of 0, 1, 2, or 3 corresponding to absent, low, moderate, and high Sh3gl2 signal, respectively. Chi-squared analysis of Sh3gl2 expression was performed comparing benign and malignant specimens as well as between tumor grades and stages. All statistical analyses were performed with SPSS 15.0 (Chicago, IL).
Figure 1.
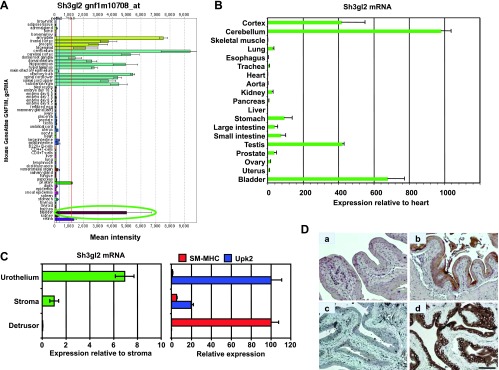
Identification of Sh3gl2 as a transcript that is highly enriched in the bladder urothelium. (A) SymAtlas screenshot of microarray hybridization analysis for Sh3gl2 in mouse organs illustrating high signal intensity in tissues from the nervous system, as well as the bladder (circled in green). (B) Semiquantitative real-time RT-PCR analysis of selected mouse tissues verified enrichment of Sh3gl2 mRNA in the nervous system tissue and bladder. Data are presented as expression level relative to that in heart (following normalization to GAPDH), which is assigned a value of 1, and represent mean ± SD of triplicate values. (C) cDNAs prepared from urothelium, stroma, and detrusor smooth muscle tissue microdissected from mouse bladders were analyzed for expression of Sh3gl2 as well as the urothelial marker uroplakin 2 and the smooth muscle marker smooth muscle myosin heavy chain by semiquantitative real-time RT-PCR. As shown in the left panel, Sh3gl2 was expressed predominantly in the urothelium, with minimal mRNA detectable in stroma or detrusor smooth muscle. Effective separation of urothelial, stromal, and smooth muscle tissue by laser capture microdissection was demonstrated by restricted expression of uroplakin 2 and smooth muscle myosin heavy chain to urothelial and smooth muscle tissues, respectively (right panel). (D) IHC staining of mouse bladder tissue revealed Sh3gl2 expression to be restricted to the urothelium (b, d), with no signal evident in submucosa or muscle. Preincubation of primary antibody with blocking peptide eliminated signal completely (c), confirming antibody specificity. No background signal was detected in sections receiving secondary antibody only (a). Scale bar, 100 µm.
Cell Culture
A panel of bladder cancer cell lines was assembled for this study as follows: RT4, T24, TCCSUP, and J82 cell lines were obtained from ATCC (Manassas, VA); 253J-P, 253J-BV, and 253J-BVR were a generous gift from Dr I. Fidler (MD Anderson Cancer Center, Houston, TX); MGH-U1, MGH-U3, and MGH-U4 were from Dr Chin-Lee Wu (Massachusetts General Hospital, Boston, MA); 5637, UM-UC3, and UM-UC9 were kindly provided by Dr Monica Liebert (University of Michigan, Ann Arbor, MI); SW780 cells were from Dr David Berman (Johns Hopkins University, Baltimore, MD); BOY cells were from Dr Enokida of Japan. TRT-HU-1 cells were generated in the laboratory of Dr Louis Liou and described by us recently [22]. Cells were cultured in McCoy's 5A medium (RT4, T24, MGH-U1, MGH-U3, MGH-U4), Dulbecco's modified Eagle's medium (TCCSUP, J82, UM-UC3, UM-UC9, 5637, TRT-HU1), minimal essential medium (253J-P, 253J-BV, 253J-BVR, BOY), or RPMI 1640 supplemented with heat-inactivated FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, and heat-inactivated FBS (Valley Biomedical, Winchester, VA). The following cell lines have been authenticated by DNA fingerprinting using short tandem repeat profiles: RT4, T24, TCCSUP, J82, 5637, UM-UC3, and UM-UC9.
Manipulation of Sh3gl2 Expression In Vitro
For stable knockdown, RT4 cells were infected with lentiviral particles encoding nontargeting shRNA or one of five independent shRNAs targeting Sh3gl2 (MISSION shRNA; all from Sigma-Aldrich, St Louis, MO). Forty-eight hours after transduction, cells were switched to medium supplemented with 0.4 µg/ml puromycin to select stable transductants. Screening of stable populations by immunoblot analysis identified sh184 and sh2103 as giving the greatest extent of silencing. Subsequent analysis of single-cell clones led to selection of sh184 C3, sh184 C9, sh2103 C8, and sh2103 C10 for initial biochemical and phenotypic analysis. To restore Sh3gl2 expression in T24 or RT4 cells, an entry clone expressing Sh3gl2 (pDONR201-Sh3gl2) was obtained from the Dana Farber/Harvard Cancer Center Plasmid Resource core and recombined into pLenti6/V5-DEST using LR Clonase (Invitrogen, Grand Island, NY). Recombinants were screened for expression in human embryonic kidney 293 (HEK293) cells and selected for packaging into viral particles using the ViraPower Lentivirus Expression System (Invitrogen). Viral supernatants from 293FT cells transfected with pLenti6/Sh3gl2 or with pLenti6/LacZ control vector were used to infect T24 or RT4 cells, and stable populations were isolated following incubation in blasticidin (6 µg/ml for T24 and 4 µg/ml for RT4). Re-expression was verified by immunoblot analysis as outlined below.
Evaluation of Sh3gl2 mRNA and Protein Levels
Levels of Sh3gl2 mRNA in cell lines or tissues were assayed by semi-quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) using gene-specific primer assays (SA Biosciences, Frederick, MD), essentially as described [23]. The relative abundance of a given transcript was estimated using the 2-ΔΔCt method, following normalization to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Sh3gl2 protein levels were evaluated by immunoblot analysis as described [23]. Antibodies used were given as follows: rabbit antiSh3gl2 (12345-1-AP; ProteinTech Group), goat anti-Sh3gl2 (sc-10874) and rabbit anti-GAPDH (sc-25778; Santa Cruz Biotechnology), rabbit anti-EGFR, rabbit anti-phospho-EGFR Y1068, rabbit anti-phospho-EGFR Y1086, mouse anti-non-phospho-Src Y416, rabbit anti-phospho-Src Y416, mouse anti-STAT3, and rabbit anti-phospho-STAT3 Y705 (all from Cell Signaling Technology, Danvers, MA).
Indirect Immunofluorescence
To assay localization of the EGFR in response to Sh3gl2 silencing, cells were seeded on coverslips in 24-well plates at 30,000 cells/well. Cells were serum-depleted in serum-free McCoy's 5A medium for 24 hours before stimulation with EGF for 10 minutes at 37°C. Following incubation, cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 minutes on ice and permeabilized with 0.2% Triton X-100 in PBS for 10 minutes. Cells were washed four times with 500 µl of PBS, and nonspecific binding sites were blocked in PBS/1% normal donkey serum/5% BSA for 1 hour at room temperature. Coverslips were rinsed once with PBS and incubated with anti-EGFR rabbit monoclonal antibody (D38B1, #4267; Cell Signaling Technology) or anti-EGFR mouse monoclonal antibody (R19/48 mix, #AHR5062; Life Technologies, Grand Island, NY), both at a dilution of 1:200 in PBS/1% normal donkey serum/5% BSA overnight at 4°C. Coverslips were washed four times with PBS for 5 minutes each wash before addition of Cy3-conjugated donkey anti-rabbit or donkey anti-mouse secondary antibody (1:500) in PBS/1% normal donkey serum for 1 hour at room temperature. Coverslips were washed four times with PBS and then mounted in Vectamount (Vector Laboratories, Inc, Burlingame, CA). Cells were visualized using an Axioplan-2 microscope (Carl Zeiss MicroImaging, Inc, Thornwood, NY). To quantify staining, cells were counted and scored qualitatively on the basis of the extent of internalization of EGFR. Three different parameters were assigned to describe localization of the EGFR. Cells with more than 90% of EGFR staining on the membrane (Memb) were counted as membrane bound. Cells with more than 90% cytoplasmic were counted as cytoplasmic (Cyto) and cells with both, i.e., less than 90% staining either on the membrane or in the cytoplasm were considered as a mixture (Memb + Cyto). The numbers obtained on the basis of the scoring method were then plotted as a percentage of the total cells scored for each population.
Proliferation Assay
To determine effects of Sh3gl2 silencing on proliferation, cells were seeded in 24-well plates at a density of 10,000 cells/well. Cell density at selected time points was assessed by crystal violet assay, as described [24], with absorbance at 570 nm determined using a microplate spectrophotometer. In some experiments, the effect of lapatinib or saracatinib on proliferation was assessed. In each case, absorbance at a given time point was expressed as a percent of absorbance on day 0 to control for variations in plating efficiency.
Clonal Growth Assay
Cells were seeded at a density of 500 cells/well in six-well tissue culture plates in complete medium. Medium was changed every 4 to 5 days during the assay. Fourteen days after plating, cells were fixed in 1% glutaraldehyde and stained with crystal violet as outlined for the proliferation assay. Plates were imaged using an Olympus SZX16 dissecting microscope with a DP71 high-resolution digital camera, and the number and size of colonies were determined using ImageJ software [National Institutes of Health (NIH), Bethesda, MD].
Phospho-kinase Proteome Array
To detect phosphorylation profiles of various intracellular signaling proteins in the control versus Sh3gl2-silenced RT4 clones, we used the Human Phospho-Kinase Array (Catalog No. ARY003; R&D Systems, Minneapolis, MN). Cells (500,000) were seeded on 6-cm dishes and allowed to grow for 48 hours. Cells were serum starved for 16 hours followed by treatment with 10 nM EGF or vehicle for 10 minutes. Cells were lysed and processed as per the manufacturer's instructions. Briefly, array membranes were incubated with 300 µg of protein lysates followed by washing as instructed. Phosphorylation status of bound proteins was subsequently detected by incubating the membranes with biotinylated detection antibodies followed by application of streptavidin-HRP and chemiluminescence detection reagents. Signals were visualized following exposure of membranes to X-ray film. Spots were measured using ImageJ software and calculated for the intensity of the average signal of the pair of duplicated spots. Background signal was subtracted from each of the spots.
Generation of Tissue Recombinant Xenografts In Vivo
To determine the significance of Sh3gl2 knockdown on tumor growth in vivo, tissue recombination experiments were performed in which control RT4 clones or clones silenced for Sh3gl2 were recombined with embryonic rat bladder mesenchyme and implanted under the renal capsule, essentially as described [25–27]. Briefly, pregnant rats (Harlan Laboratories, Tampa, FL) were sacrificed at E16 (plug day = 0) and urogenital sinus mesenchyme was isolated from embryonic bladders manually under microscopic examination. To prepare recombinants, control and Sh3gl2-silenced RT4 clones were resuspended in rat tail collagen and setting solution, and aliquots were droplet-plated in 10-cm dishes. Following the insertion of one embryonic bladder mesenchyme per aliquot, tissue recombinants were placed at 37°C for 30 minutes to promote solidification. McCoy's modified medium containing 10% FBS was then applied to solidified grafts. The following day, tissue recombinants were placed under the left kidney capsules of severe combined immunodeficient mice. Grafts were maintained in vivo for 3 weeks before euthanasia, tissue harvest, and determination of tumor burden. All animal work was conducted in accordance with institutional animal care guidelines. Tissue was subsequently embedded in optimal cutting temperature (OCT) compound (Sakura) and stored at -80°C.
Statistical Analysis
Where appropriate, comparisons between experimental groups were performed using Student's t test. P values are indicated in figure legends or on figures where relevant, with P < .05 considered statistically significant.
Results
Sh3gl2 (Endophilin A1) Is Highly Expressed in Normal Urothelium and Decreased in Bladder Cancer
Previous studies have suggested restricted expression of Sh3gl2/endophilin A1 to the brain and central nervous system [28]. However, several recent reports have identified Sh3gl2 mRNA and protein in cancers of the breast, head and neck, and ovary [29–31], indicating a broader pattern of expression than initially believed. Using an informatics-based analysis of expression profiles from normal tissues to identify novel transcripts in visceral organs, we identified the urinary bladder as a major site of expression of Sh3gl2 (Figure 1, A and B). Sh3gl2 was one of only three transcripts identified as highly enriched in our analysis of two independent publicly available data sets that used different Affymetrix microarray types (see Materials and Methods section), despite the known confounders arising from integration of independent data sets and differing Affymetrix microarray types [32,33]. As shown by laser capture microdissection coupled with semiquantitative real-time RT-PCR (Figure 1C) and IHC staining with anti-Sh3gl2 antibody (Figure 1D), expression of Sh3gl2 is largely restricted to the murine urothelium (Figure 1D, b and d). Notably, Sh3gl2 is expressed on human chromosome 9, a frequent site of genomic alterations in development of bladder cancer [34–36]. However, the association between Sh3gl2 and UC development and/or progression has not been investigated.
Interrogation of the Oncomine human tumor profiling database revealed a statistically significant decrease in Sh3gl2 mRNA levels in both superficial and invasive UC compared to normal bladder tissue in three independent studies [P < .05; Figure 2A, (i)–(iii)] [18–20]. In the study by Lee et al. [Figure 2A,(iii)], Sh3gl2 was in the top third percentile of downregulated genes. To determine whether Sh3gl2 protein was also altered in UC, we stained a human tissue microarray comprising 80 normal and cancerous specimens of bladder tissue with a validated antibody to Sh3gl2 (see Figure 1D). In agreement with data from the mouse, Sh3gl2 expression was predominantly epithelial (Figure 2B). Sh3gl2 protein levels were decreased in UC compared to noncancerous tissue, with only 19% of UC specimens versus 63% of benign tissue showing high expression of the antigen (P = .001; Figure 2B). To further determine whether loss of Sh3gl2 was associated with disease progression, we stained a bladder cancer progression array comprising 202 cases, of which 169 gave meaningful data (Table 1 and Figures 2C and W1). As shown in Figure 2D, (i), Sh3gl2 levels were low or absent at significantly higher frequency in malignant samples compared with benign samples (P < .001). Sh3gl2 levels decreased progressively with increasing tumor grade (Figure 2D, (ii), P < .001), with the majority of high-grade specimens showing little or no expression (G3 vs G1/G2, P = .001). Furthermore, Sh3gl2 expression was significantly lower in muscle-invasive compared to non-muscle-invasive bladder cancer specimens (Figure 2D, (iii), P = .03). Expression of Sh3gl2 in non-muscle-invasive specimens was also significantly lower compared to benign tissues (Figure 2C, (iii), P = .001). From these analyses, it appears that the loss of Sh3gl2 expression is a frequent and progressive event in UC pathogenesis. Differential expression of Sh3gl2 in muscle-invasive versus non-muscle-invasive specimens suggests that Sh3gl2 status in tumors may have prognostic implications.
Figure 2.

Sh3gl2 loss in bladder cancer correlates with tumor progression. (A) Analysis of three independent cohorts of bladder cancer specimens (i–iii) using the Oncomine database revealed decreased Sh3gl2 mRNA levels in both superficial and invasive lesions compared to normal bladder tissue. (B) IHC analysis of a bladder cancer tissue microarray (TMA) revealed a marked decrease in Sh3gl2 expression in cancer tissues versus noncancerous tissues, as well as a trend toward decreased expression in high-grade versus low-grade cancers. Representative images of Sh3gl2 staining in low- and high-grade lesions are presented in a and b; c represents the control receiving no primary antibody. Original magnification, x200. (C) Representative staining of (a) benign, (b) low-grade, and (c) high-grade lesions on a bladder cancer progression TMA. Original magnification, x200. (D) Quantification of IHC staining of a bladder cancer progression array revealed frequent and progressive loss of Sh3gl2. Graphs show the proportion of tissue specimens (%) in each subgroup with low (-/+) versus high (++/+++) intensity staining for Sh3gl2 protein. Sh3gl2 staining intensity was significantly lower in (i) malignant versus benign specimens (P < .001), (ii) in high-grade (G3) versus low-grade (G1/G2) specimens (P < .001), and (iii) in muscle-invasive versus non-muscle-invasive specimens (P = .03), as well as non-muscle-invasive versus benign specimens (P <.001).
Table 1.
Summary Data for Tissue Specimens on Progression Array.
| Benign | Malignant | |
| Gender | ||
| M | n = 38 (88.4%) | n = 102 (81.0%) |
| F | n = 5 (11.6%) | n = 24 (19.0%) |
| Age | 54.5 years (17–73 years) | 60.7 years (25–85 years) |
| Grade | ||
| 1 | N/A | n = 33 (26.2%) |
| 2 | N/A | n = 58 (46.0%) |
| 3 | N/A | n = 35 (27.8%) |
| Stage | ||
| T1 | N/A | n = 65 (38.5%) |
| T2 | N/A | n = 38 (22.5%) |
| T3 | N/A | n = 20 (11.8%) |
| T4 | N/A | n = 3 (1.8%) |
| Pathology | Inflammation, n = 24 (55.8%) | UC, n = 125 (99.2%) |
| Hyperplasia, n = 11 (25.6%) | Undifferentiated carcinoma, n = 1 (0.8%) | |
| Normal, n = 6 (14.0%) | ||
| Pheochromocytoma, n = 2 (5.7%) |
Silencing of Sh3gl2 Expression Enhances Proliferation
To facilitate mechanistic analysis of the role of Sh3gl2 in UC, we profiled 20 different UC cell lines representing different degrees of aggressiveness. Notably, we observed a striking absence of Sh3gl2 mRNA and protein expression in 19 of 20 cell lines, with significant expression detected only in the low-grade, well-differentiated cell line RT4 (Figure 3, A and B). To investigate the functional significance of Sh3gl2 loss in UC, we employed lentivirus-encoded shRNAs to downregulate its expression in RT4 cells. Among five independent shRNA constructs, sh184 and sh2103, which bound to seed sequences in the coding and 3′ untranslated regions of the Sh3gl2 transcript, respectively, produced >75% knockdown at the protein level in RT4 populations. Independent single-cell clones sh184 C3, sh184 C9, sh2103 C8, and sh2103 C10, along with shCtrl C2 were selected for subsequent analyses (Figure 3C). Stable silencing of Sh3gl2 led to increased proliferation of all four clones relative to control cells in which Sh3gl2 was intact (Figure 3D). Sh3gl2 silencing also promoted increased survival and colony formation in cells seeded at clonal density, as determined by colony number (Figure 3E) and colony area (Figure 3F). The extent of colony area appeared to correlate with the extent of Sh3gl2 knockdown, with greater average colony area in sh2103 clones compared to sh184 clones. To determine whether Sh3gl2 loss altered growth in vivo, we grafted recombinants comprising RT4 stably silenced with control or Sh3gl2-targeted shRNA together with rat fetal bladder mesenchyme under the renal capsule of athymic mice. As shown in Figure 3H, two of three RT4 clones lacking Sh3gl2 showed increased tumor burden compared to nontargeted controls, with a comparable increase in xenograft size observed in two independent clones targeted with sh2103. Together, these findings suggest that Sh3gl2 normally regulates survival and growth-promoting signals in urothelial cells.
Figure 3.

Sh3gl2 is lost with high frequency in bladder cancer cell lines and regulates cell growth in vitro and in vivo. Sh3gl2 mRNA (A) and protein (B) levels were analyzed in a panel of 20 bladder epithelial cell lines by semiquantitative real-time RT-PCR or immunoblot analysis, respectively; data from 14 cell lines are presented. Only the low-grade papilloma-derived cell line RT4 expressed detectable levels of Sh3gl2. (C) RT4 cells infected with lentivirus encoding shRNAs directed against five distinct regions of the Sh3gl2 transcript (sh184, sh646, sh989, sh1430, and sh2103) or with nontargeting control shRNA (shCtrl) were analyzed for Sh3gl2 expression by immunoblot analysis with two independent anti-Sh3gl2 antibodies and identified sh184 and sh2103 as yielding efficient knockdown of Sh3gl2 (left panel). Single-cell clones isolated from shCtrl (C2), sh184 (C3 and C9), and sh2103 (C8 and C10) were selected as candidate clones for functional analysis (right panel). (D) Proliferation of Sh3gl2-silenced cells, as determined in a biomass assay, was increased in all four clones compared to RT4 control cells at all time points tested (P < .05). Data are representative of at least three independent experiments. Control and Sh3gl2-targeted clones seeded at clonal density were evaluated for (E) colony number and (F) colony area 14 days after seeding. Loss of Sh3gl2 significantly increased cell survival (number) in three of four clones (*P < .01) and proliferation (area) in all four Sh3gl2-silenced clones compared to controls (*P < .01). (G) Representative images of clonal growth assays for four Sh3gl2-silenced clones and the control clone. (H) Tissue recombinants comprising RT4 cells stably silenced with control or Sh3gl2-targeted shRNA combined with rat fetal bladder mesenchyme were implanted under the renal capsule of severe combined immunodeficient mice and harvested 3 weeks after implantation. Representative images indicating grafts in situ are indicated. The graph indicates wet weight of grafts generated from three to four grafts per indicated clone and data are presented as means ± SD.
Sh3gl2 Silencing Enhances EGFR-Mediated Signaling
Sh3gl2 is known to regulate signal transduction with RTKs, such as the EGFR/ErbB1 and c-Met/HGF-R, through its ability to control endocytosis [37,38]. In agreement with this activity, stable silencing of Sh3gl2 attenuated EGF-induced EGFR internalization compared to that observed in cells with intact Sh3gl2 expression. Specifically, RT4 cells lacking Sh3gl2 retained EGFR on the plasma membrane to a greater extent than control RT4 cells in which Sh3gl2 was intact (Figure 4, A and B). Similar results were obtained with two independent anti-EGFR antibodies. Moreover, Sh3gl2 silencing consistently enhanced phosphorylation of the EGFR at multiple activating residues (Figures 4C and W2). In agreement with EGF-dependent signaling in other tumor types, HER2 was also phosphorylated in RT4 cells exposed to EGF (data not shown), suggesting that a proportion of the activated EGFR exists in a heterodimer with HER2. Sh3gl2 knockdown also decreased sensitivity to the dual specificity EGFR/HER2 inhibitor, lapatinib (Figure 4D). In the presence of vehicle (DMSO), growth of sh184 C9 and sh2103 C8 clones was increased relative to shCtrl C2 (P < .001), in a manner consistent with the extent of knockdown, with sh2103 C8 showing higher proliferation than sh184 C9. A similar pattern was observed in the presence of lapatinib, with both sh184 C9 and sh2103 C8 reaching higher densities than shCtrl C2 (P < .001). Notably, sh2103 C8 cells were essentially insensitive to the inhibitory effects of lapatinib (P = .654, sh2103 C8 DMSO vs sh2103 C8 lapatinib).
Figure 4.
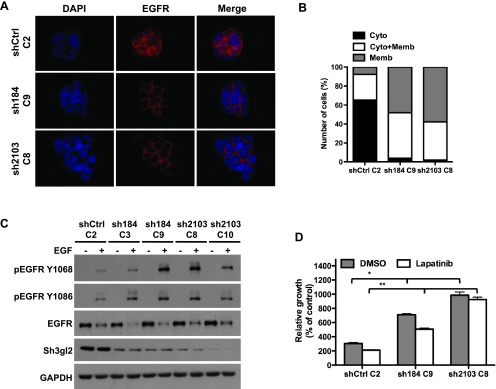
Stable Sh3gl2 silencing attenuates EGFR internalization and enhances receptor activation. (A) Control and Sh3gl2-silenced RT4 cells were seeded on coverslips, serum-depleted for 24 hours, and treated without or with 10 nM EGF for 10 minutes at 37°C. Localization of the EGFR was visualized by indirect immunofluorescence staining. Nuclei are counterstained with 4′, 6-diamidino-2-phenylindole (DAPI). Data are representative of three independent trials. Original magnification, x63. (B) The extent of EGFR internalization under the conditions indicated in A was quantified and plotted as the percentage of cells showing EGFR that was localized predominantly on the membrane (Memb), predominantly in the cytoplasm (Cyto), or present both in the cytoplasm and on the membrane (Cyto + Memb). (C) Serum-depleted control and Sh3gl2-silenced RT4 cells were treated with 10 nM EGF for 10 minutes at 37°C, and whole-cell lysates were prepared for evaluation of receptor phosphorylation. Increased EGFR phosphorylation was evident in all four clones in which Sh3gl2 was silenced compared to nontargeted control cells. Data are representative of at least four independent experiments. (D) Control and Sh3gl2-silenced RT4 cells were treated with vehicle (DMSO) or 0.3 µM lapatinib for 5 days and cell number determined by biomass assay. Data represent absorbance on day 5 expressed as a percentage of absorbance on day 0 and are the means ± SD of four values. *, shCtrl C2 DMSO versus sh184 C9 DMSO or sh2103 C8 DMSO; **, shCtrl C2 lapatinib versus sh184 C9 lapatinib or sh2103 C8 lapatinib. Data are representative of at least two independent experiments.
Sh3gl2 Silencing Increases Signaling through Src and STAT3
To explore signaling pathways activated downstream of Sh3gl2 silencing, whole-cell lysates from vehicle or EGF-treated cells were probed using a phospho-kinase array comprising 46 target phosphoproteins. From this analysis, we identified cohorts of phosphoproteins upregulated by Sh3gl2 knockdown under both basal and EGF-stimulated conditions. Increased phosphorylation of Akt (T308) and glycogen synthase kinase 3α/β (GSK3α/β), p27 (T198), and STAT4 was evident in Sh3gl2-silenced cells in the absence of growth factor treatment (Figure 5A). In EGF-treated cells, we observed increased phosphorylation of p27, Pyk2, PLCγ-1, Paxillin, c-Jun, STAT-2, -3, -5, and -6, as well as the SFKs Fgr, Fyn, Yes, Hck, and Lck (Figure 5B). Since SFKs and STAT3 have been implicated in oncogenic signaling and are known to signal within the same pathway, we proceeded to validate these targets by immunoblot analysis. As illustrated in Figure 5C, we observed increased levels of phospho-Src Y416, a site conserved in multiple SFKs, in Sh3gl2-silenced cells compared to control cells under both basal and EGF-stimulated conditions. In addition, increased phosphorylation of STAT3 Y705 was also observed in Sh3gl2-silenced cells compared to controls. To explore the functional significance of increased phosphorylation of SFKs with Sh3gl2 knockdown, we performed growth assays in the presence of the Src inhibitor saracatinib. As observed previously, cells silenced for Sh3gl2 reached higher densities than control cells with intact Sh3gl2 in the absence of inhibitor; however, growth of both control and Sh3gl2-silenced cells was inhibited significantly in the presence of saracatinib (P < .05; Figure 5D).
Figure 5.

Stable Sh3gl2 silencing increases phosphorylation of SFKs and STAT3. Lysates of control and Sh3gl2-silenced cells treated without (A) or with (B) 10 nM EGF for 10 minutes were analyzed using a phospho-kinase proteome array. Graphs and tables indicate phospho-proteins that were altered by >1.5-fold in response to Sh3gl2 silencing. Representative images of phospho-array blots from each condition are shown. (C) Independent lysates immunoblotted with the indicated antibodies revealed enhanced phosphorylation of SFKs at Y416 (or corresponding conserved residue) and STAT3 at Y705. (D) Control and Sh3gl2-silenced RT4 cells were treated with vehicle (DMSO) or 0.125 µM saracatinib for 5 days and cell number determined by biomass assay. Graphs illustrate absorbance on day 5 expressed as a percentage of absorbance on day 0, and data are the means ± SD of two values for DMSO and six values for saracatinib. Data are representative of at least two independent trials. *P < .05 compared to DMSO-treated condition.
Re-expression of Sh3gl2 Attenuates Growth and Migration
To obtain additional evidence to support a role for Sh3gl2 in regulating oncogenic behaviors, we also assessed the consequence of restoring its expression in RT4 clones silenced for Sh3gl2 and in T24 cells. Rescue of Sh3gl2 expression in the RT4 background (Figure 6A) attenuated proliferation of two independent clones, 2103C8 and 2103C10, when seeded at high density (Figure 6, B and C) and at clonal density (data not shown). In addition, forced expression of Sh3gl2 in T24 cells (Figure 6D) attenuated proliferation to a modest but statistically significant extent (Figure 6E) compared to cells expressing an irrelevant gene, LacZ. In addition, Sh3gl2 restoration inhibited EGF-induced migration in a wound healing assay by greater than 50% (Figure 6, E and F; *P < .05).
Figure 6.
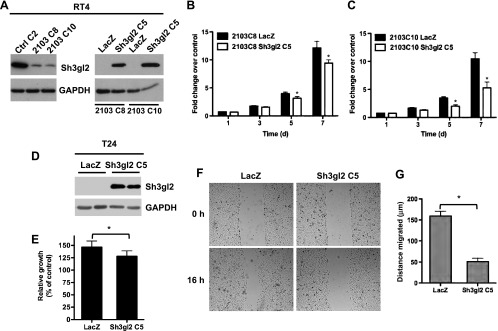
Restoration of Sh3gl2 expression inhibits proliferation and migration. (A) Rescue of Sh3gl2 expression in RT4 cells using Sh3gl2-expressing lentivirus was verified by immunoblot analysis. (B, C) Re-expression attenuated proliferation in two independent clones silenced for Sh3gl2, compared to cells expressing an irrelevant gene, LacZ. *P < .05 compared to LacZ at corresponding time point. (D) Reconstitution of Sh3gl2 expression in T24 cells was confirmed by immunoblot analysis. (E) Restoration of Sh3gl2 expression inhibited proliferation, as measured at 48 hours after seeding. *P < .05 compared to proliferation in LacZ-expressing cells. (F) Restoration of Sh3gl2 expression inhibited EGF-stimulated migration in T24 cells. (G) The graph represents average distance migrated, and data are presented as means ± SD. *P < .05 compared to distance migrated in LacZ-expressing cells.
Discussion
In this study, we describe a novel role for Sh3gl2 in regulation of urothelial cancer pathogenesis and progression. The evidence supporting our conclusions is given as follows: 1) Analysis of human tumors revealed frequent and progressive loss of Sh3gl2 mRNA and protein, as well as significantly enhanced loss of expression in muscle-invasive lesions; 2) evaluation of 20 UC cell lines revealed only one, RT4, which expressed detectable Sh3gl2 mRNA and protein; 3) RNAi-mediated silencing of Sh3gl2 in RT4 cells enhanced proliferation and colony formation, activation of ErbB receptors, SFKs and STAT3, and growth of UC xenografts in vivo; 4) reconstitution of Sh3gl2 expression in RT4 cells and T24 cells attenuated proliferation and migration, respectively. Together, these findings provide the first evidence linking alterations in Sh3gl2 to oncogenic activity in bladder cancer and implicate Sh3gl2 as an important inhibitor of tumor progression that restrains activation of kinases such as the EGFR and Src family members.
Sh3gl2 was identified initially through functional screening as an SH3 domain-containing protein [39] and, based on Northern blot analysis of a panel of human tissues, was found to be preferentially expressed in the brain [28]. In that study, however, bladder tissue was not analyzed. On the basis of both microarray hybridization data deposited in SymAtlas, as well as quantitative PCR validation by us, the level of Sh3gl2 mRNA in mouse bladder was comparable to that detected in the brain and spinal cord. Further analysis of microdissected bladder tissue confirmed restriction of Sh3gl2 mRNA and protein to the urothelial compartment. Interestingly, Sh3gl2 is expressed on human chromosome 9p (9p22), a genomic region associated with genetic and epigenetic alterations that lead to gene silencing and bladder tumor development [40]. Chromosome 9p alterations occur early in development of a number of tumor types and the biologic consequences of such changes have typically been ascribed to inactivation of CDKN2A, which encodes p16 (reviewed in [41]). However, several reports on a range of cancers describing chromosome 9p deletions distal to the CDKN2A locus [42–45] have suggested the existence of additional genes that may be targeted by genomic rearrangements. Consistent with this possibility, analyses in head and neck carcinoma as well as breast and non-small cell lung cancers have implicated Sh3gl2 as a candidate tumor suppressor gene in this region [29,30,46]. Furthermore, studies in breast as well as head and neck tumors have demonstrated hypermethylation of the Sh3gl2 promoter and suggested this as a potential mechanism of Sh3gl2 silencing [29,30]. In agreement with these findings, we found a CpG island in the Sh3gl2 proximal promoter to be hypermethylated in T24 cells that lack Sh3gl2 expression, in contrast to RT4 cells in which the Sh3gl2 promoter CpG site was completely unmethylated, as assessed by methylation-sensitive restriction analysis and PCR of genomic DNA (E.M.G. and R.M.A., unpublished observations). Thus, the absence of Sh3gl2 expression in UC tumor cells may result, at least in part, from methylation-induced gene silencing.
The biologic consequences of Sh3gl2 down-regulation are only just beginning to be delineated. Stable silencing of Sh3gl2 in the RT4 cell line, consistent with the down-regulation of Sh3gl2 mRNA and protein evident in urothelial cancer specimens, led to marked increases in cell proliferation in vitro and xenograft growth in vivo. These findings are in agreement with a recent report by Dasgupta and colleagues, in which the Sh3gl2 gene was shown by single nucleotide polymorphism (SNP) array analysis to be deleted frequently in non-small cell lung cancer. In that study, forced expression of Sh3gl2 in cell lines lacking endogenous expression attenuated oncogenic behaviors, including proliferation and colony formation [46]. In agreement with the known function of Sh3gl2 in regulating RTK endocytosis [37,38], we observed retention of the EGFR/ERBB1 on the plasma membrane in response to EGF stimulation following Sh3gl2 silencing in RT4 cells. In addition, ligand-stimulated EGFR phosphorylation at several activating residues was enhanced in Sh3gl2-silenced cells, consistent with reduced signal attenuation that typically occurs by receptor internalization. An important finding in our study was the demonstration of increased EGF-stimulated phosphorylation of multiple SFKs and STAT3 in cells lacking Sh3gl2, both of which have been implicated in urothelial cancer pathogenesis [47,48]. Sh3gl2-silenced cells were sensitive to saracatinib, an SFK inhibitor currently undergoing evaluation in phase II clinical trials for osteosarcoma and breast and pancreatic cancers [49]. Interestingly, our data suggested that Sh3gl2 silencing also decreased sensitivity to the growth-inhibitory effects of the EGFR/HER2 inhibitor lapatinib. Decreased Sh3gl2-mediated internalization of EGFR in invasive and metastatic disease may explain why EGFR-targeted therapies in UC patients have shown little or no activity in clinical studies [8]. Interestingly, a recent study by Osterberg and colleagues demonstrated that genomic loss of Sh3gl2 or CIN85, another component of the endocytosis apparatus, correlated with resistance to chemotherapy in ovarian carcinoma [31]. Notably, in that study, losses in either the Sh3gl2 or CIN85 locus were evident in 70% of resistant tumors suggesting that impaired receptor endocytosis is an important contributor to drug resistance in tumors. Thus, evaluation of Sh3gl2 status in urothelial, and possibly other tumors, may inform our understanding of which targeted therapies may be effective and the extent to which tumors are likely to respond to a given therapeutic intervention.
Loss of Sh3gl2 appears to be a frequent alteration in development of urothelial cancer, with levels continuing to decline with progression, as evidenced by the significant decrease in protein expression between non-muscle-invasive and muscle-invasive lesions. This observation supports both chromosomal deletions and methylation as contributors to significantly decreased Sh3gl2 expression in UC. Interestingly, forced expression of Sh3gl2 expression in tumor cells lacking endogenous expression attenuated invasion [46] and migration (E.M.G. and R.M.A., unpublished observations), behaviors required for cells to metastasize. Therefore, Sh3gl2 expression level, in conjunction with additional features of tumor cells, may serve as a potential marker of risk for urothelial tumor progression or recurrence and may enable determination of which patients require more aggressive intervention in the form of surgery or chemotherapy to improve prognosis. Investigation of Sh3gl2 expression with respect to clinical outcomes is currently under consideration.
In summary, we present the first evidence linking Sh3gl2 to urothelial cancer development and progression. Loss of Sh3gl2 is associated with aggressive disease and promotes oncogenic activities including up-regulation of ERBB receptors, SFKs and STAT3, and increased cell survival and proliferation. These findings provide a platform for further exploration of Sh3gl2-mediated regulation of kinase-dependent signaling in vitro and in vivo and for evaluation of the prognostic utility of Sh3gl2 status in patients with bladder cancer.
Supplementary Material
Acknowledgments
The authors thank Michael Freeman, Joshua Mauney, and Carlos Estrada, as well as members of the Urological Diseases Research Center for helpful discussions. The authors also thank Aruna Ramachandran and Michelle Mulone for technical assistance, as well as Tom Case, Simon Hayward and Douglas Strand at Vanderbilt University for their assistance with the tissue recombination experiments.
Abbreviations
- UC
urothelial carcinoma
- RTK
receptor tyrosine kinase
- SFK
Src family kinase
Footnotes
The authors acknowledge the financial support from the Children's Urological Foundation. D.J.D. was supported by the American Cancer Society Great Lakes Division- Michigan Cancer Research Fund Postdoctoral Fellowship.
This article refers to supplementary materials, which are designated by Figures W1 and W2 and are available online at www.neoplasia.com.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Riley GF, Potosky AL, Lubitz JD, Kessler LG. Medicare payments from diagnosis to death for elderly cancer patients by stage at diagnosis. Med Care. 1995;33:828–841. doi: 10.1097/00005650-199508000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Sievert KD, Amend B, Nagele U, Schilling D, Bedke J, Horstmann M, Hennenlotter J, Kruck S, Stenzl A. Economic aspects of bladder cancer: what are the benefits and costs? World J Urol. 2009;27:295–300. doi: 10.1007/s00345-009-0395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Degraff DJ, Cates JM, Mauney JR, Clark PE, Matusik RJ, Adam RM. When urothelial differentiation pathways go wrong: implications for bladder cancer development and progression. Urol Oncol. 2011 doi: 10.1016/j.urolonc.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knowles MA, Platt FM, Ross RL, Hurst CD. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305–316. doi: 10.1007/s10555-009-9198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 7.Philips GK, Halabi S, Sanford BL, Bajorin D, Small EJ. A phase II trial of cisplatin, fixed dose-rate gemcitabine and gefitinib for advanced urothelial tract carcinoma: results of the Cancer and Leukaemia Group B 90102. BJU Int. 2008;101:20–25. doi: 10.1111/j.1464-410X.2007.07226.x. [DOI] [PubMed] [Google Scholar]
- 8.Philips GK, Halabi S, Sanford BL, Bajorin D, Small EJ. A phase II trial of cisplatin (C), gemcitabine (G) and gefitinib for advanced urothelial tract carcinoma: results of Cancer and Leukemia Group B (CALGB) 90102. Ann Oncol. 2009;20:1074–1079. doi: 10.1093/annonc/mdn749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wulfing C, Machiels JP, Richel DJ, Grimm MO, Treiber U, De Groot MR, Beuzeboc P, Parikh R, Petavy F, El-Hariry IA. A single-arm, multicenter, open-label phase 2 study of lapatinib as the second-line treatment of patients with locally advanced or metastatic transitional cell carcinoma. Cancer. 2009;115:2881–2890. doi: 10.1002/cncr.24337. [DOI] [PubMed] [Google Scholar]
- 10.Petrylak DP, Tangen CM, Van Veldhuizen PJ, Jr, Goodwin JW, Twardowski PW, Atkins JN, Kakhil SR, Lange MK, Mansukhani M, Crawford ED. Results of the Southwest Oncology Group phase II evaluation (study S0031) of ZD1839 for advanced transitional cell carcinoma of the urothelium. BJU Int. 2010;105:317–321. doi: 10.1111/j.1464-410X.2009.08799.x. [DOI] [PubMed] [Google Scholar]
- 11.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Little MH, Brennan J, Georgas K, Davies JA, Davidson DR, Baldock RA, Beverdam A, Bertram JF, Capel B, Chiu HS, et al. A high-resolution anatomical ontology of the developing murine genitourinary tract. Gene Expr Patterns. 2007;7:680–699. doi: 10.1016/j.modgep.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMahon AP, Aronow BJ, Davidson DR, Davies JA, Gaido KW, Grimmond S, Lessard JL, Little MH, Potter SS, Wilder EL, et al. GUDMAP: the genitourinary developmental molecular anatomy project. J Am Soc Nephrol. 2008;19:667–671. doi: 10.1681/ASN.2007101078. [DOI] [PubMed] [Google Scholar]
- 14.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 15.Smyth GK. Gentleman R, Carey V, Dudoit S, Izarry R, Huber W. Bioinformatics and Computational Biology Solutions using R and Bioconductor. 1st ed. New York: Springer; 2005. Limma: linear models for microarray data; p. 492. [Google Scholar]
- 16.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;57:289–300. [Google Scholar]
- 18.Dyrskjot L, Kruhoffer M, Thykjaer T, Marcussen N, Jensen JL, Moller K, Orntoft TF. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64:4040–4048. doi: 10.1158/0008-5472.CAN-03-3620. [DOI] [PubMed] [Google Scholar]
- 19.Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006;24:778–789. doi: 10.1200/JCO.2005.03.2375. [DOI] [PubMed] [Google Scholar]
- 20.Lee JS, Leem SH, Lee SY, Kim SC, Park ES, Kim SB, Kim SK, Kim YJ, Kim WJ, Chu IS. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J Clin Oncol. 2010;28:2660–2667. doi: 10.1200/JCO.2009.25.0977. [DOI] [PubMed] [Google Scholar]
- 21.Adam RM, Borer JG, Williams J, Eastham JA, Loughlin KR, Freeman MR. Amphiregulin is coordinately expressed with heparin-binding epidermal growth factor-like growth factor in the interstitial smooth muscle of the human prostate. Endocrinology. 1999;140:5866–5875. doi: 10.1210/endo.140.12.7221. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Ji M, DiDonato JA, Rackley RR, Kuang M, Sadhukhan PC, Mauney JR, Keay SK, Freeman MR, Liou LS, et al. An hTERT-immortalized human urothelial cell line that responds to anti-proliferative factor. In Vitro Cell Dev Biol Anim. 2011;47:2–9. doi: 10.1007/s11626-010-9350-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramachandran A, Ranpura SA, Gong EM, Mulone M, Cannon GM, Jr, Adam RM. An Akt- and Fra-1-dependent pathway mediates platelet-derived growth factor-induced expression of thrombomodulin, a novel regulator of smooth muscle cell migration. Am J Pathol. 2010;177:119–131. doi: 10.2353/ajpath.2010.090772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stehr M, Adam RM, Khoury J, Zhuang L, Solomon KR, Peters CA, Freeman MR. Platelet derived growth factor-BB is a potent mitogen for rat ureteral and human bladder smooth muscle cells: dependence on lipid rafts for cell signaling. J Urol. 2003;169:1165–1170. doi: 10.1097/01.ju.0000041501.01323.b9. [DOI] [PubMed] [Google Scholar]
- 25.Oottamasathien S, Wang Y, Williams K, Franco OE, Wills ML, Thomas JC, Saba K, Sharif-Afshar AR, Makari JH, Bhowmick NA, et al. Directed differentiation of embryonic stem cells into bladder tissue. Dev Biol. 2007;304:556–566. doi: 10.1016/j.ydbio.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeGraff DJ, Clark PE, Cates JM, Yamashita H, Robinson VL, Yu X, Smolkin ME, Chang SS, Cookson MS, Herrick MK, et al. Loss of the urothelial differentiation marker FOXA1 is associated with high grade, late stage bladder cancer and increased tumor proliferation. PLoS One. 2012;7:e36669. doi: 10.1371/journal.pone.0036669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strand DW, Degraff DJ, Jiang M, Sameni M, Franco OE, Love HD, Hayward W, Lin Tsai O, Wang A, Cates JM, et al. Deficiency in metabolic regulators PPARγ and PTEN cooperates to drive keratinizing squamous metaplasia in novel models of human tissue regeneration. Am J Pathol. 2013;182:449–459. doi: 10.1016/j.ajpath.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giachino C, Lantelme E, Lanzetti L, Saccone S, Bella Valle G, Migone N. A novel SH3-containing human gene family preferentially expressed in the central nervous system. Genomics. 1997;41:427–434. doi: 10.1006/geno.1997.4645. [DOI] [PubMed] [Google Scholar]
- 29.Sinha S, Chunder N, Mukherjee N, Alam N, Roy A, Roychoudhury S, Panda CK. Frequent deletion and methylation in SH3GL2 and CDKN2A loci are associated with early- and late-onset breast carcinoma. Ann Surg Oncol. 2008;15:1070–1080. doi: 10.1245/s10434-007-9790-0. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh A, Ghosh S, Maiti GP, Sabbir MG, Alam N, Sikdar N, Roy B, Roychoudhury S, Panda CK. SH3GL2 and CDKN2A/2B loci are independently altered in early dysplastic lesions of head and neck: correlation with HPV infection and tobacco habit. J Pathol. 2009;217:408–419. doi: 10.1002/path.2464. [DOI] [PubMed] [Google Scholar]
- 31.Osterberg L, Levan K, Partheen K, Delle U, Olsson B, Sundfeldt K, Horvath G. Potential predictive markers of chemotherapy resistance in stage III ovarian serous carcinomas. BMC Cancer. 2009;9:368. doi: 10.1186/1471-2407-9-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kong SW, Hwang KB, Kim RD, Zhang BT, Greenberg SA, Kohane IS, Park PJ. CrossChip: a system supporting comparative analysis of different generations of Affymetrix arrays. Bioinformatics. 2005;21:2116–2117. doi: 10.1093/bioinformatics/bti288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dreyfuss JM, Johnson MD, Park PJ. Meta-analysis of glioblastoma multiforme versus anaplastic astrocytoma identifies robust gene markers. Mol Cancer. 2009;8:71. doi: 10.1186/1476-4598-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smeets W, Pauwels R, Laarakkers L, Debruyne F, Geraedts J. Chromosomal analysis of bladder cancer. III. Nonrandom alterations. Cancer Genet Cytogenet. 1987;29:29–41. doi: 10.1016/0165-4608(87)90028-8. [DOI] [PubMed] [Google Scholar]
- 35.Ruppert JM, Tokino K, Sidransky D. Evidence for two bladder cancer suppressor loci on human chromosome 9. Cancer Res. 1993;53:5093–5095. [PubMed] [Google Scholar]
- 36.Cairns P, Shaw ME, Knowles MA. Initiation of bladder cancer may involve deletion of a tumour-suppressor gene on chromosome 9. Oncogene. 1993;8:1083–1085. [PubMed] [Google Scholar]
- 37.Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature. 2002;416:183–187. doi: 10.1038/416183a. [DOI] [PubMed] [Google Scholar]
- 38.Petrelli A, Gilestro GF, Lanzardo S, Comoglio PM, Migone N, Giordano S. The endophilin-CIN85-Cbl complex mediates ligand-dependent down-regulation of c-Met. Nature. 2002;416:187–190. doi: 10.1038/416187a. [DOI] [PubMed] [Google Scholar]
- 39.Sparks AB, Hoffman NG, McConnell SJ, Fowlkes DM, Kay BK. Cloning of ligand targets: systematic isolation of SH3 domain-containing proteins. Nat Biotechnol. 1996;14:741–744. doi: 10.1038/nbt0696-741. [DOI] [PubMed] [Google Scholar]
- 40.Knowles MA. Tumor suppressor loci in bladder cancer. Front Biosci. 2007;12:2233–2251. doi: 10.2741/2226. [DOI] [PubMed] [Google Scholar]
- 41.Pollard C, Smith SC, Theodorescu D. Molecular genesis of nonmuscle-invasive urothelial carcinoma (NMIUC) Expert Rev Mol Med. 2010;12:e10. doi: 10.1017/S1462399410001407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keen AJ, Knowles MA. Definition of two regions of deletion on chromosome 9 in carcinoma of the bladder. Oncogene. 1994;9:2083–2088. [PubMed] [Google Scholar]
- 43.Farrell WE, Simpson DJ, Bicknell JE, Talbot AJ, Bates AS, Clayton RN. Chromosome 9p deletions in invasive and noninvasive nonfunctional pituitary adenomas: the deleted region involves markers outside of the MTS1 and MTS2 genes. Cancer Res. 1997;57:2703–2709. [PubMed] [Google Scholar]
- 44.Parris CN, Harris JD, Griffin DK, Cuthbert AP, Silver AJ, Newbold RF. Functional evidence of novel tumor suppressor genes for cutaneous malignant melanoma. Cancer Res. 1999;59:516–520. [PubMed] [Google Scholar]
- 45.Pollock PM, Welch J, Hayward NK. Evidence for three tumor suppressor loci on chromosome 9p involved in melanoma development. Cancer Res. 2001;61:1154–1161. [PubMed] [Google Scholar]
- 46.Dasgupta S, Jang JS, Shao C, Mukhopadhyay ND, Sokhi UK, Das SK, Brait M, Talbot C, Yung RC, Begum S, et al. SH3GL2 is frequently deleted in non-small cell lung cancer and downregulates tumor growth by modulating EGFR signaling. J Mol Med (Berl) 2012;91:381–393. doi: 10.1007/s00109-012-0955-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiang GJ, Billmeyer BR, Canes D, Stoffel J, Moinzadeh A, Austin CA, Kosakowski M, Rieger-Christ KM, Libertino JA, Summerhayes IC. The src-family kinase inhibitor PP2 suppresses the in vitro invasive phenotype of bladder carcinoma cells via modulation of Akt. BJU Int. 2005;96:416–422. doi: 10.1111/j.1464-410X.2005.05642.x. [DOI] [PubMed] [Google Scholar]
- 48.Ho PL, Lay EJ, Jian W, Parra D, Chan KS. Stat3 activation in urothelial stem cells leads to direct progression to invasive bladder cancer. Cancer Res. 2012;72:3135–3142. doi: 10.1158/0008-5472.CAN-11-3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, Allen J, Logie A, Hargreaves J, Hickinson DM, Wilkinson RW, et al. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009;3:248–261. doi: 10.1016/j.molonc.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.