

Abstract
It has been hypothesized that before the emergence of modern DNA–RNA–protein life, biology evolved from an “RNA world.” However, synthesizing RNA and other organophosphates under plausible early Earth conditions has proved difficult, with the incorporation of phosphorus (P) causing a particular problem because phosphate, where most environmental P resides, is relatively insoluble and unreactive. Recently, it has been proposed that during the Hadean–Archean heavy bombardment by extraterrestrial impactors, meteorites would have provided reactive P in the form of the iron–nickel phosphide mineral schreibersite. This reacts in water, releasing soluble and reactive reduced P species, such as phosphite, that could then be readily incorporated into prebiotic molecules. Here, we report the occurrence of phosphite in early Archean marine carbonates at levels indicating that this was an abundant dissolved species in the ocean before 3.5 Ga. Additionally, we show that schreibersite readily reacts with an aqueous solution of glycerol to generate phosphite and the membrane biomolecule glycerol–phosphate under mild thermal conditions, with this synthesis using a mineral source of P. Phosphite derived from schreibersite was, hence, a plausible reagent in the prebiotic synthesis of phosphorylated biomolecules and was also present on the early Earth in quantities large enough to have affected the redox state of P in the ocean. Phosphorylated biomolecules like RNA may, thus, have first formed from the reaction of reduced P species with the prebiotic organic milieu on the early Earth.
Keywords: origin of life, prebiotic chemistry, phosphorylation, astrobiology, exobiology
Phosphorus (P) is the limiting nutrient in many ecosystems; its scarcity is linked to the insoluble nature of phosphate minerals (1, 2). Phosphorus species may have been important reagents during prebiotic chemical evolution, but their presumed low abundance has presented a problem in understanding the origin of key phosphorylated biomolecules, such as RNA (3, 4). This was recognized early in the study of prebiotic chemistry, and a possible role for more reduced P compounds was suggested because these are more soluble than phosphate (5, 6). However, no potential source of reduced P was known at the time, leaving phosphate as the only available P species under typical geochemical conditions (7).
Phosphide minerals such as schreibersite (Fe,Ni)3P are ubiquitous minor constituents of meteorites (8) and are potential sources of reduced P (9–12). Schreibersite reacts with water to release P compounds that are reduced relative to the geologically abundant P5+ phase phosphate PO43−, including P3+ phosphite HPO32− and P1+ hypophosphite H2PO2− (9–14). Both of these compounds are significantly more soluble than phosphate in waters of neutral pH and rich in the divalent cations Ca2+ and Mg2+, such as modern seawater (5). Schreibersite reaction rates indicate complete phosphide dissolution when submerged in water over about 10 y (12), implying rapid reaction with water after meteoritic impact into an ocean. The phosphite, thus, released could have supplied a major portion of the dissolved P in the Earth’s early oceans. This meteoritic signature should have been trapped during marine carbonate precipitation. Such a carbonate might preserve the phosphite through metamorphism if it also contained ferrous iron minerals such as magnetite Fe3O4, which buffer phosphite oxidation up to or beyond greenschist facies conditions. The oxidation rate of calcium phosphite is such that a rock at the magnetite–hematite oxygen fugacity buffer would preserve phosphite indefinitely even if moderately metamorphosed (details on these calculations are in Methods).
Other natural sources of phosphite include lightning strikes (15, 16), geothermal fluids (14, 17), and possibly microbial activity under extremely anaerobic conditions (14, 18). No other terrestrial sources of phosphite have been identified, because few geological processes are so reducing as to promote reduction of phosphate to phosphite and because under oxidizing conditions, phosphite readily converts to orthophosphate. Hence, the presence of phosphite in a rock likely has a meteoritic origin if lightning and geothermal sources can be eliminated. If phosphite is a constituent of subsurface samples from below the water table, a modern lightning origin is precluded (16). Phosphite from geothermal sources has been detected at a single terrestrial spring thus far and the amount of phosphite in this environment is minimal (0.06 μM) (17), suggesting little net impact to the oceanic P budget even if this source were more widespread.
Samples and Site Geology
A suite of 17 Archean and Phanerozoic sedimentary rocks (Table 1) was analyzed for P speciation, using 35 mM KOH as an eluent and then high-performance liquid chromatography (HPLC) using an AC-17 ion-chromatography (IC) column coupled to an inductively coupled plasma (ICP) mass spectrometer [HPLC(IC)-ICP-MS], following prior methods (14, 17). With the exception of the Coonterunah rocks, these samples were acquired during field work that was not associated with the present study but were chosen to span a range of times, locations, and lithologies.
Table 1.
Rocks examined in this study
| Rock locality | Rock type | Age, Ma |
| Coucal Fm., Coonterunah, Australia | Limestone (two sample sites) | 3,520 |
| Coucal Fm., Coonterunah, Australia | Hydrothermal chert | 3,520 |
| Coucal Fm., Coonterunah, Australia | Limestone (six core samples) | 3,520 |
| Strelley Pool Fm., Kelley Group, Australia | Chert | 3,400 |
| Goldman Meadows Fm., South Pass, WY | Banded Iron Fm. | 2,870 |
| Cheshire Fm., Belingwe, Zimbabwe | Carbonate | 2,700 |
| Brockman Iron Fm., Hamersley Basin, Australia | Banded Iron Fm. | 2,500 |
| Millboro Fm., Charles Town, WV | Shale | 400–380 |
| Green River Fm., Kemmerer, WY | Shale | 50 |
| Avon Park Fm., Citrus County, FL | Limestone | 35 |
The Coonterunah rocks come from the ∼6.5-km-thick, 3.518 Ga (19) Coonterunah Subgroup in the East Strelley Belt, the oldest supracrustal rocks in the Pilbara Craton of northwest Australia (Figs. 1 and 2). A regional unconformity to the south bounds the Coonterunah rocks from the overlying shallow marine sediments of the ∼3.35-Ga (20) Strelley Pool Formation (Fm.), whereas a regional intrusive contact with the ∼3.48-Ga (19) Carlindi Granitoid complex bounds outcrop to the north. Estimated local metamorphic conditions were in the lower greenschist facies, at temperatures between ∼350–400 °C, pressures of ∼1–2 kbar (21).
Fig. 1.
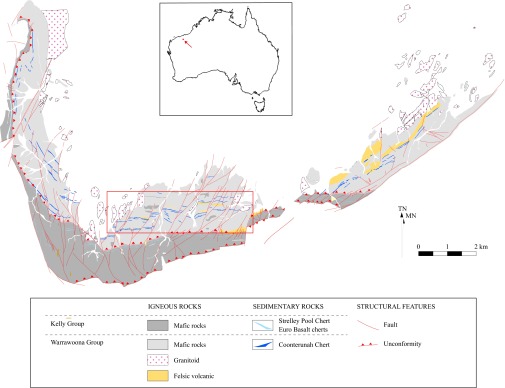
Geological map of the East Strelley Belt, with regional location shown (Inset). The box shows outline of the study area.
Fig. 2.

Stratigraphic column of tephra-carbonate sequence in the middle section of the Coucal Fm. in the Coonterunah Subgroup, Australia. Rock types are denoted by different colors, and all have been metamorphosed to at least greenschist facies. These rocks sit within the stratigraphic sequence shown in the center column, with arrows denoting the carbonate, chert, and pelitic samples investigated, and clast size shown as width of layers. This study section is a small portion of the Warrawoona group (right column), which consists of metavolcanic (V) and metasedimentary early Archean rocks, crosscut by chert dikes (x, +).
The carbonate samples were collected from the middle of the Coucal Fm., which lies conformably between ±2 km of sporadically pillowed basalt, gabbro and rare komatiites of the underlying Table Top Fm., and >2 km of pillowed tholeiitic basalt and lesser gabbros of the overlying Double Bar Fm. The most prominent and best-preserved carbonate horizon is 32 cm thick, with a lateral extent of at least 5 km (Fig. 3). Texturally similar but thinner 1- to 5-cm units also crop out. The prominent carbonate lies conformably within a ∼35-m-thick package of graded tuffaceous wackes to siltstones and chlorite–biotite–quartz metapelites, the base of which is marked by a silicified hyaloclastite. About 60 m of dolerite immediately overlies the mixed volcaniclastic-carbonate sequence, capped by an ∼8-m-thick magnetitic metachert. Associated basalts bear only rare amygdales, whereas hydrovolcanic tuff, coarse pumice, and accretionary lapilli are absent from adjacent volcaniclastic units. No coarse (more than clay size) terrigenous clastic sediments or large-scale cross-bedding are present. These igneous and sedimentary textures together indicate a low-energy depositional setting at water depths well below wave base.
Fig. 3.

Outcrop photographs of Coonterunah carbonate, with magnetitic (m1 to m4) and carbonate (c1 to c3) mesobands labeled. (A) Calcitic phase: alternating calcite and magnetite laminae. (B) Dolomitic phase: alternating dolomite/calcite and magnetite laminae. The c1 mesoband is partially silicified. (C) Slabs through dolomitic phase.
Carbonate occurs in magnetitic limestone or limestone–dolostone with 1- to 5-mm-thick planar laminae of alternating carbonate/magnetite (Fig. 3); ∼1.5- to 4.0-mm-diameter flame structures occur in both carbonate and magnetite laminae, but other sedimentary structures are absent. However, its origin is indubitably sedimentary because no secondary carbonate alteration occurs in adjacent igneous rocks, and the lateral stratigraphic continuity of layering precludes a metasomatic or metamorphic origin.
Carbonate laminae are composed of equant dolomite and calcite crystals 25–50 μm in size, with the relative proportions and chemical composition of carbonate phases exhibiting little variation within individual samples. Calcite is generally pure, with a dominant stoichiometry of Ca0.95–1.00Mg0.05–0.00(CO3), only rarely reaching Ca0.90Mg0.10(CO3). Dolomite exhibits dolomite–ankerite solid solution, varying from pure dolomite CaMg(CO3)2 to Ca1.0Mg0.8Fe0.2(CO3)2. Unlike the within-sample carbonate homogeneity, mineralogical variation between samples is pronounced, with molar Ca/Mg ratios ranging from 2.88 to 1.27, reflecting varying degrees of dolomitization. Magnetite laminae consist of pure Ni-poor magnetite, which has undergone variable weathering to maghemite. Under more extensive weathering, limonite with or without fine-grained hematite oxidation rinds and lesser goethite are also observed. Rare apatite, present as small (1- to 5-μm) anhedral crystals, is associated with the magnetite and pyrite is present in unweathered samples. The geochemistry of the carbonate further suggests a deep-sea origin (21).
Analytical Results
Of all of the samples analyzed, only the oldest, the Coonterunah carbonate samples from the early Archean of Australia, showed the presence of phosphite. These included both outcrop specimens and drill-core samples from ∼300 m subsurface (water table at ∼40 m), indicating that the phosphite is indigenous and not the product of recent or ancient lightning strikes. The Coonterunah phosphite comprised 40–67% of the total P in the rock extracts (Fig. 4), with the relative abundance of phosphite varying between the sample sites. The presence of phosphite was confirmed by spiking with a sodium phosphite standard (Methods). No phosphite was observed in the noncarbonate control samples collected from the same formation, including a magnetite-bearing chert, nor in any of the younger rocks. These results indicate that the phosphite was not introduced by diagenetic processes occurring after deposition and instead records P speciation in the ocean at ∼3.52 Ga. The high relative proportion of phosphite in these rocks indicates that it was a significant dissolved P species in the seawater, where the carbonate formed, in contrast to modern oceans, where phosphate is overwhelmingly dominant everywhere.
Fig. 4.

Chromatogram showing P speciation of two samples of the Coonterunah carbonate (ALS-A and ALS-B) and of a hydrothermal control sample (ALS-C). P3+ is phosphite. No P species appeared within the 18.2 MΩ deionized water blank, and only phosphate (P5+) appeared in extracts of the other rocks listed in Table 1. These peaks correspond to concentration ratios (P3+/P total) of 40% (ALS-A) and 67% (ALS-B). Slight shifts in peak locations are attributable to peak wander as experiments run and are constrained by comparison with standards every five samples.
Discussion
Relevance to Bombardment History.
If the Archean carbonates are representative of early Archean ocean chemistry, as might be reasonable given their deep sea setting, then these carbonates provide constraints on the bombardment history of the early Earth. Phosphides are the major P mineral in enstatite chondrites, which have recently been proposed as the source of heavy bombardment impactors (22). Corrosion of the meteoritic phosphide mineral schreibersite likely provided the phosphite detected in the present study, because few other sources of phosphite are known, and none was as widespread and voluminous as meteoritic bombardment early in Earth’s history (23). It is possible that ancient lightning strikes on Archean land surfaces could, in the absence of an oxidizing atmosphere, have provided a terrestrial fluvial flux of phosphite to the oceans via weathering of fulgurites. However, neither of our other Archean samples (Strelley Pool Fm., Goldman Meadows Fm.) dating from before the earliest evidence of oxidative weathering at ∼2.8 Ga (24) contain phosphite, indicating that any such flux was negligible and that a meteoritic source for the Coonterunah phosphite is most likely. Similarly, a significant hydrothermal phosphite source should have led to its occurrence in other Archean samples.
Phosphite and phosphate differ in solubility by a factor of about 1,000 (5, 6). If it is assumed that the phosphite to phosphate ratio has been minimally altered since deposition and is representative of the Archean ocean, then the amount of meteoritic material required to provide this quantity of phosphite to the early Earth oceans is about 1019–1020 kg, assuming an ocean volume equivalent to present day and that the average meteorite is composed of 0.1% reduced P in phosphide minerals by weight (8). This quantity is about 1% of the mass of the Earth’s crust and is comparable to other estimates of total meteorite fluxes at the time (25–27). Obviously, if other natural sources of phosphite can be identified, such as widespread production in hydrothermal systems (17), then the total meteoritic mass necessary to provide this phosphite may be reduced. More work on reduced P sources will be necessary to determine the exact contribution of separate sources of reduced P to the early Earth.
Relevance to Prebiotic Chemistry.
Phosphorus is an essential constituent in biochemical molecules such as DNA, RNA, and ATP. P may have been equally important during the evolution of life on the early Earth, including the prebiotic chemistry (28, 29) that led to the hypothesized RNA world precursor of the modern DNA–RNA–protein world (3). However, the limited availability of phosphate as a solute in water coupled to its low reactivity has led some to conclude that there was little P in the earliest biomolecules (1), with replacement of phosphate by glyoxylate (30) or peptides (31) in early replicating molecules. In contrast to calcium phosphate minerals, schreibersite is more reactive and, hence, may have provided soluble P species like phosphite necessary for the formation of organophosphates during the origin of life (9, 13).
Although aqueously corroding schreibersite has shown some reactivity toward organic compounds in previous experiments (11), it has not previously shown a significant capacity for phosphorylation. Here, we show that heating glycerol and schreibersite in water under anaerobic conditions at 65 °C produces, along with phosphite, glycerol–phosphate in 2.5% yield (Fig. 5). Glycerol is a three-carbon sugar alcohol that is one of the most common organic compounds in the Murchison meteorite (32); hence, this reaction may be similar to those that were active on the early Earth, suggesting some glycerol–phosphate may have participated in prebiotic chemistry.
Fig. 5.

31P NMR spectrum of schreibersite added to a 0.5 M aqueous solution of glycerol at neutral pH and heated to 65 °C under argon. The spectrum was acquired fully coupled to hydrogen. The peaks are identified by comparison with a glycerol phosphate standard and by their J-coupling constants. The triplet results from CH2–O–P on the terminal end of glycerol, and the doublet results from a CH–O–P interaction on the second carbon of glycerol. The presence of glycerol phosphate is also confirmed by mass spectrometry, with a peak at 171 m/z in negative ion mode.
In contrast to other experiments, this phosphorylation takes place at lower temperatures, uses a mineral source for P, occurs in water, and uses no condensing agent (1). This shows that phosphorylation can take place in a chemical system that was plausibly present on the prebiotic Earth. Although the mineral struvite has been used as a phosphorylating agent in a similar fashion (33), the evidence for this mineral being present on the early Earth is unconvincing (34). Glycerol–phosphate is a major constituent of cellular membranes, bonding with two fatty acids to make phospholipids, and is also involved in the glycerol–phosphate shuttle for transporting reducing equivalents.
Production of potential monomeric precursors to RNA involves a phosphonylation step to phosphorylate the various RNA nucleobases (29, 35). Attempts to perform this reaction using inorganic phosphate have only been successful in the presence of heat, urea, and/or formamide (36, 37). However, conceptual models of prebiotic RNA synthesis have also invoked phosphite as an important phosphorylating reagent (38). If this proves to be a worthwhile synthetic pathway, then phosphorylation by seawater phosphite potentially presents a realistic mechanism for producing prebiotic molecules for information storage, enzymatic catalysis, electron transport, and cellular segregation during the Hadean–Archean meteoritic bombardment.
Conclusion
Many approaches to prebiotic chemistry have relied on model systems without an adequate grounding in the contemporaneous geological environment (39). In large part, this is attributable to an inability to sample geological material from near the time when life is believed to have originated. Moreover, amino acids, sugars, nucleobases, and other potential prebiotic reagents rapidly degrade at the high temperatures of metamorphism, whereas P chemistry is exceptional in that it might preserve some of the unique characteristics of the early Earth that led to the development of life. If the source of the Archean phosphite reported here was indeed meteoritic schreibersite, then the conditions of the early Earth differed substantially from the modern environment, providing prebiotic reagents not readily available subsequently. Our research demonstrates the availability of reduced P species as a source of soluble reactive P on the early Earth, enabling the abiotic formation of primitive phosphorylated biomolecules such as proto-nucleic acids and lipids, thus providing a pathway to the “RNA World” and subsequent origin of life reactions.
Methods
Oxidation Rate Determination.
The oxidation rate of CaHPO3 was investigated by a series of thermal-heating experiments performed under air. CaHPO3 was synthesized by the addition of equimolar amounts of Ca(OH)2 and H3PO3 to water, and the white precipitate was separated and analyzed by 31P NMR and was shown to be CaHPO3 hydrate. A set of 0.1-g samples of this white powder was placed in an alumina crucible and then exposed to temperatures ranging from 400, 425, 450, 475, and 500 °C in an open-air oven. Portions (about 0.01 g) of each powder were taken daily from these heating experiments and were analyzed by 31P NMR to determine the ratio of P in phosphite to the oxidized species phosphate and pyrophosphate, as determined by peak integration. From these data, the oxidation rates were determined at each temperature and were pseudo–first-order reactions. These data provided an Arrhenius relationship from which the activation energy could be derived (Fig. S1).
HPLC(IC)-ICP-MS Methods.
Approximately 1 g of powders of each rock was extracted with 10 mL of a solution of Na2EDTA (0.025 M) or 0.5 M sodium acetate. These extracts were filtered through 0.45-μm in-line filters to remove large solids. The solutions were then analyzed by a Perkin-Elmer S-200 HPLC fitted with an IonPac AS17-C IC column. Total P was measured on the attached, in-line Perkin-Elmer Elan dynamic reaction cell (DRC) II ICP-MS. Initial trials were conducted in both standard and DRC mode for P at mass 31 and phosphorus oxide at mass 47 for phosphite, hypophosphite, and phosphate standards at concentrations ranging from 10−7 to 10−3 M. It was determined that 50-μL injections of P-bearing solution analyzed at a mass/charge of 31 with a dwell time of 250 ms was sufficient to resolve differences in P species concentration down to ∼10−6 M. Speciation runs were performed as a gradient with 3.5 mM KOH used as a mobile phase for the first 2 min and then increasing linearly to 35 mM over the course of 10 min. Mobile phase was held at 35 mM for 10 additional minutes. All pump rates were 1.5 mL/min.
Species Determination.
Phosphorus speciation within rock samples was determined by comparison with a standard solution consisting of sodium phosphate, sodium phosphite, and sodium hypophosphite prepared at 1 mM in 18.2 MΩ deionized water prepared in house on a Barnstead water-purification system. A series of solutions were made from this initial stock to a final concentration of 10−7 M. Two other compounds that were not studied in the current work are hypophosphate (P2O64−) and pyrophosphate (P2O74−). In prior work, these compounds always elute after phosphate (14).
Extracts performed with Na2EDTA also show the presence of phosphite in the Coonterunah rock (Fig. S2) compared with a Na2EDTA solution mixed with the standard solution (10−6 M). This method separates P species within carbonate rocks; however, the signal-to-noise ratio was weaker for these materials compared with sodium acetate extracts, likely because of overlap with 15N–O or N–17O introduced by the EDTA. Extracts done with EDTA eluted faster than those with sodium acetate.
The solutions made from extracts of the Coonterunah rocks were spiked with sodium phosphite for a molarity of 10−5 M solution of phosphite. Chromatograms of the resulting solution show an increase in the peak identified as phosphite (Fig. S3).
Reaction of Schreibersite with Glycerol.
Twenty-five milliliters of a 0.2 M solution of glycerol (C3H8O3) were added to 1 g of synthetic Fe3P powder (confirmed to be the iron analog of schreibersite by its X-ray diffractometry pattern, magnetic properties, and density). Iron sulfide (FeS) was also added to this mixture; this compound is commonly associated with schreibersite in meteorites. The solution was stirred constantly in a closed container at 65 °C for 2 d. A solution of sodium sulfide was added to the reacted material to quench the reaction (40), and the sample was filtered and centrifuged. The sample was then evaporated to dryness at 20 °C and subsequently mixed with D2O and water to give a 50% D2O:50% (vol/vol) water solution. The sample was analyzed on a 400-MHz Varian UnityInova NMR operating at 161.9 MHz in both H-coupled and H-decoupled modes, and by MS on a LC-MSD Agilent mass spectrometer in negative mode.
Supplementary Material
Acknowledgments
Drill-core samples were obtained from the Agouron Institute Drilling Program. This work was supported jointly by the National Science Foundation (NSF) and the National Aeronautics and Space Administration (NASA) Astrobiology Program, under the NSF Center for Chemical Evolution (Grant CHE-1004570), and by the NASA Exobiology and Evolutionary Biology Program (Grant NNX10AT30G).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303904110/-/DCSupplemental.
References
- 1.Keefe AD, Miller SL. Are polyphosphates or phosphate esters prebiotic reagents? J Mol Evol. 1995;41(6):693–702. doi: 10.1007/BF00173147. [DOI] [PubMed] [Google Scholar]
- 2.Oelkers EH, Valsami-Jones E. Phosphate mineral reactivity and global sustainability. Elements. 2008;4(2):83–87. [Google Scholar]
- 3.Gilbert W. Origins of life: The RNA World. Nature. 1986;319(6055):618. [Google Scholar]
- 4.Joyce GF. The antiquity of RNA-based evolution. Nature. 2002;418(6894):214–221. doi: 10.1038/418214a. [DOI] [PubMed] [Google Scholar]
- 5.Gulick A. Phosphorus as a factor in the origin of life. Am Sci. 1955;43(3):479–489. [Google Scholar]
- 6.Schwartz AW. Phosphorus in prebiotic chemistry. Philos Trans R Soc Lond B Biol Sci. 2006;361(1474):1743–1749, discussion 1749. doi: 10.1098/rstb.2006.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller SL, Urey HC. Organic compound synthesis on the primitive earth. Science. 1959;130(3370):245–251. doi: 10.1126/science.130.3370.245. [DOI] [PubMed] [Google Scholar]
- 8.Pasek MA, Lauretta DS. Extraterrestrial flux of potentially prebiotic C, N, and P to the early Earth. Orig Life Evol Biosph. 2008;38(1):5–21. doi: 10.1007/s11084-007-9110-5. [DOI] [PubMed] [Google Scholar]
- 9.Pasek MA, Lauretta DS. Aqueous corrosion of phosphide minerals from iron meteorites: A highly reactive source of prebiotic phosphorus on the surface of the early Earth. Astrobiology. 2005;5(4):515–535. doi: 10.1089/ast.2005.5.515. [DOI] [PubMed] [Google Scholar]
- 10.Bryant DE, Kee TP. Direct evidence for the availability of reactive, water soluble phosphorus on the early Earth. H-phosphinic acid from the Nantan meteorite. Chem Commun (Camb) 2006;2006(22):2344–2346. doi: 10.1039/b602651f. [DOI] [PubMed] [Google Scholar]
- 11.Pasek MA, Dworkin JP, Lauretta DS. A radical pathway for organic phosphorylation during schreibersite corrosion with implications for the origin of life. Geochim Cosmochim Acta. 2007;71(7):1721–1736. [Google Scholar]
- 12.Bryant DE, et al. Electrochemical studies of iron meteorites. Phosphorus redox chemistry on the early Earth. Int J Astrobiol. 2009;8(1):27–36. [Google Scholar]
- 13.Pasek MA. Rethinking early Earth phosphorus geochemistry. Proc Natl Acad Sci USA. 2008;105(3):853–858. doi: 10.1073/pnas.0708205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pech H, et al. Elucidating the redox cycle of environmental phosphorus using ion chromatography. J Chromatogr Sci. 2011;49(8):573–581. doi: 10.1093/chrsci/49.8.573. [DOI] [PubMed] [Google Scholar]
- 15.Essene EJ, Fisher DC. Lightning strike fusion: Extreme reduction and metal-silicate liquid immiscibility. Science. 1986;234(4773):189–193. doi: 10.1126/science.234.4773.189. [DOI] [PubMed] [Google Scholar]
- 16.Pasek MA, Block K. Lightning-induced reduction of phosphorus oxidation state. Nat Geosci. 2009;2(8):553–556. [Google Scholar]
- 17.Pech H, et al. Detection of geothermal phosphite using high-performance liquid chromatography. Environ Sci Technol. 2009;43(20):7671–7675. doi: 10.1021/es901469t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han C, et al. Determination of phosphite in a eutrophic freshwater lake by suppressed conductivity ion chromatography. Environ Sci Technol. 2012;46(19):10667–10674. doi: 10.1021/es300771a. [DOI] [PubMed] [Google Scholar]
- 19.Buick R, et al. Record of emergent continental crust ∼3.5 billion years ago in the Pilbara Craton of Australia. Nature. 1995;375(6532):574–577. [Google Scholar]
- 20.Banerjee DM, et al. Direct dating of Archean microfossil ichnofossils. Geology. 2007;35(6):487–490. [Google Scholar]
- 21.Harnmeijer JP. 2010 Squeezing Blood from a Stone: Inference into the Life and Depositional Environments. Ph.D. Thesis (University of Washington, Seattle) [Google Scholar]
- 22.Bottke WF, et al. An Archaean heavy bombardment from a destabilized extension of the asteroid belt. Nature. 2012;485(7396):78–81. doi: 10.1038/nature10967. [DOI] [PubMed] [Google Scholar]
- 23.Johnson BC, Melosh HJ. Impact spherules as a record of an ancient heavy bombardment of Earth. Nature. 2012;485(7396):75–77. doi: 10.1038/nature10982. [DOI] [PubMed] [Google Scholar]
- 24.Stüeken EE, Catling DC, Buick R. Contributions to late Archaean sulphur cycling by life on land. Nat Geosci. 2012;5(10):722–725. [Google Scholar]
- 25.Abramov O, Mojzsis SJ. Microbial habitability of the Hadean Earth during the late heavy bombardment. Nature. 2009;459(7245):419–422. doi: 10.1038/nature08015. [DOI] [PubMed] [Google Scholar]
- 26.Hartmann WK, Ryder G, Dones L, Grinspoon D. 2000. in Origin of the Earth and Moon, eds Canup R, Righter K (Univ of Arizona Press, Tucson, AZ), pp 493–512.
- 27.Ryder G, Koeberl C, Mojzsis SJ. 2000. in Origin of the Earth and Moon, eds Canup R, Righter K (Univ of Arizona Press, Tucson, AZ), pp 475–492.
- 28.Powner MW, Gerland B, Sutherland JD. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature. 2009;459(7244):239–242. doi: 10.1038/nature08013. [DOI] [PubMed] [Google Scholar]
- 29.Sutherland JD, Whitfield JN. Synthesis of potentially prebiotic RNA precursors: Cytosine and guanine derivatives. Tetrahedron Lett. 1997;38(8):1451–1454. [Google Scholar]
- 30.Bean HD, Anet FAL, Gould IR, Hud NV. Glyoxylate as a backbone linkage for a prebiotic ancestor of RNA. Orig Life Evol Biosph. 2006;36(1):39–63. doi: 10.1007/s11084-005-2082-4. [DOI] [PubMed] [Google Scholar]
- 31.Nelson KE, Levy M, Miller SL. Peptide nucleic acids rather than RNA may have been the first genetic molecule. Proc Natl Acad Sci USA. 2000;97(8):3868–3871. doi: 10.1073/pnas.97.8.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooper G, et al. Carbonaceous meteorites as a source of sugar-related organic compounds for the early Earth. Nature. 2001;414(6866):879–883. doi: 10.1038/414879a. [DOI] [PubMed] [Google Scholar]
- 33.Handschuh GJ, Orgel LE. Struvite and prebiotic phosphorylation. Science. 1973;179(4072):483–484. doi: 10.1126/science.179.4072.483. [DOI] [PubMed] [Google Scholar]
- 34.Hazen RM, et al. Mineral evolution. Am Min. 2008;93(11–12):1693–1720. [Google Scholar]
- 35.Sutherland JD, Whitfield JN. Studies on a potentially prebiotic synthesis of RNA. Tetrahedron. 1997;53(34):11595–11626. [Google Scholar]
- 36.Lohrmann R, Orgel LE. Urea-inorganic phosphate mixtures as prebiotic phosphorylating agents. Science. 1971;171(3970):490–494. doi: 10.1126/science.171.3970.490. [DOI] [PubMed] [Google Scholar]
- 37.Schoffstall AM. Prebiotic phosphorylation of nucleosides in formamide. Orig Life. 1976;7(4):399–412. doi: 10.1007/BF00927935. [DOI] [PubMed] [Google Scholar]
- 38.Benner SA, Kim H-J, Carrigan MA. Asphalt, water, and the prebiotic synthesis of ribose, ribonucleosides, and RNA. Acc Chem Res. 2012;45(12):2025–2034. doi: 10.1021/ar200332w. [DOI] [PubMed] [Google Scholar]
- 39.Shapiro R. 1986. A Skeptic’s Guide to the Creation of Life on the Earth (SummitBooks, New York), pp. 1–332.
- 40.Pasek MA, Kee TP, Bryant DE, Pavlov AA, Lunine JI. Production of potentially prebiotic condensed phosphates by phosphorus redox chemistry. Angew Chem Int Ed Engl. 2008;47(41):7918–7920. doi: 10.1002/anie.200802145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.