

Abstract
ABT has been widely used in drug development process as an irreversible inhibitor of CYP enzymes. One potential use of ABT is to potentiate pharmacological effects of rapidly-metabolized drugs on CYP expression by inhibiting their metabolism; however, ABT’s own effects on expression of CYP enzymes have been unknown. In this study, we show that ABT up-regulates expression of CYP2B6 and CYP3A4 by activating CAR. In freshly isolated human hepatocytes, ABT increased mRNA expression levels of CYP2B6 and CYP3A4 in a concentration-dependent manner. ABT also modulated CYP-inducing actions of CITCO and rifampin, the known inducers of CYP2B6 and CYP3A4. Results from luciferase reporter assays confirmed that ABT increases CYP2B6 promoter activity in CAR-expressing HepG2 cells. These results suggest that the use of ABT as a potentiator of pharmacological effects of rapidly-metabolized drugs is limited and requires precaution especially in studies examining regulation of CYP expression at the transcriptional level.
Keywords: 1-Aminobenzotriazole, CAR, CYP2B6, CYP3A4, CYP induction
INTRODUCTION
1-Aminobenzotriazole (ABT) is an irreversible CYP inhibitor that binds to the heme prosthetic group [1]. As a global CYP inhibitor, ABT has been widely used to identify CYP and non-CYP oxidative pathways in in vitro reaction phenotyping study [2] and to eliminate the concomitant drug metabolism for direct determination of transporter activity in in vitro hepatocyte systems [3]. Also, ABT is used to differentiate the pharmacological effects of parent drugs from those of metabolites in in vivo hepatotoxicity study [4].
Based on the global CYP-inhibitory effects of ABT, we have proposed a potential use of ABT as a potentiator of pharmacological effects for rapidly-metabolized drugs. For example, in examining CYP-inducing effects of potential inducers that are rapidly metabolized, ABT may be used concomitantly to slow down the CYP-mediated metabolic elimination of the inducers. 17β-Estradiol (E2), the major estrogen in human, can be one such example. E2 undergoes extensive metabolism in primary human hepatocytes, with the mean half-life of 0.6 hr (our unpublished data). In examining the effects of E2 on expression of CYP enzymes, we treated primary human hepatocytes with E2 along with ABT to inhibit the rapid metabolism of E2 by CYP enzymes and thus better simulate the physiological concentration profile (i.e., relatively constant level). E2 up-regulated expression of CYP genes such as CYP2B6 and CYP3A4 [5], and co-treatment of hepatocytes with ABT and E2 further increased the CYP expression (our unpublished data). The potentiation of E2 effects by ABT may be due to decreased metabolism of E2 as expected. However, we cannot rule out potential contribution of other factors to the results, such as effect of ABT on CYP expression or on actions of CYP inducers. In this study, we investigated such possibilities to determine whether ABT can be used as a potentiator of CYP-inducing effects for potential inducer drugs.
MATERIALS AND METHODS
Chemicals and Reagents
ABT and rifampin were purchased from Sigma (St. Louis, MO), CITCO from Biomol (Plymouth Meeting, PA).
Plasmids
Plasmid pcDNA3-PXR that expresses human PXR was previously constructed in our laboratory [6]. To construct plasmid pcDNA3-CAR that expresses human CAR, first-strand cDNA was synthesized using Superscript II reverse transcriptase (Invitrogen) using RNA isolated from HepG2 as template. With the first-strand cDNA as template, CAR was PCR-amplified using a pair of primers: forward and reverse primers of 5′-AAGGATCCGAAAACCAGCAACAGCGTGG-3′ and 5′-TTGAATTCTTCCCACTCCAGTGTATCCAG-3′ (underlined are sites for restriction enzyme digestion). The PCR product and pcDNA3 (Invitrogen) were digested by BamHI and EcoRI restriction enzymes and ligated, yielding pcDNA3-CAR. β-Galactosidase expression plasmid was previously described [6]. Two luciferase constructs harboring PXR binding elements (PXRE) or phenobarbital-responsive enhancer module (PBREM; CAR-binding elements), pGL3-UGT1A1 U2K and pGL3-CYP2B6 U2.2K, were kindly provided by Drs. Masahiko Negishi (NIEHS, NC, USA) and Hongbing Wang (University of Maryland), respectively [7, 8].
Human Hepatocyte Procurement and Drug Treatment
Freshly isolated human hepatocytes were obtained from CellzDirect (Pittsboro, NC) and Liver Tissue Cell Distribution System (Pittsburgh, PA). Briefly, hepatocytes were shipped overnight in cold preservation media. Upon receipt, the media were replaced with serum-free Williams’ E media (without phenol red) containing 0.1 μM dexamethasone, 10 μg/mL gentamicin, 15 mM HEPES, 2 mM L-glutamine, and 1% ITS (Sigma). Cells were allowed to recover from shipping for 10 hr at 37 °C in an atmosphere containing 5% CO2. After recovery, the hepatocytes were treated with media containing CITCO (100 nM), rifampin (10 μM) or vehicle (ethanol), with or without ABT (1 mM) for 72 hr.
Quantitative Real-time (qRT) PCR
Total RNAs were isolated from human hepatocytes using Trizol (Invitrogen, Carlsbad, CA) and used as template for cDNA synthesis using High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). With the cDNA as template, qRT-PCR was performed using StepOnePlus Real-Time PCR System and TaqMan® Gene expression assays (Applied Biosystems). The fold change in mRNA levels of CYP2B6 or CYP3A4 after drug treatment was determined after normalizing the gene expression levels by those of GAPDH (2−ΔΔCt method) [9]. Statistical analysis was performed by using Student’s t-test.
Cell Culture
HepG2 cells from ATCC (Manassas, VA) were cultured in complete DMEM supplemented with 10% fetal bovine serum (Gemini, Woodland, CA), 2 mM L-glutamine, 100 U penicillin/ml, 100 μg streptomycin/ml, and 1% MEM nonessential amino acids.
Luciferase Reporter Assays
HepG2 cells were seeded in 12-well plates at a density of 2.5×105 cells/ml, and on the next day, transfected with 0.3 μg of luciferase construct (pGL3-UGT1A1 U2K or pGL3-CYP2B6 U2.2K), 0.3 μg of expression plasmid (pcDNA3-PXR, pcDNA3-CAR, or pcDNA3 as a control vector), and 0.1 μg of β-galactosidase expression plasmid using Fugene 6 transfection reagent (Roche Applied Sciences) according to the manufacturer’s protocol. The transfected cells were grown for 24 hr and treated with ABT (1 mM), CITCO (100 nM), rifampin (10 μM) or ethanol vehicle (0.1%). Following 24 hr incubation, cells were harvested for determination of both luciferase and β-galactosidase activities using assay kits from Promega (Madison, WI). Luciferase activity was normalized to the β-galactosidase activity. At least two independent experiments were performed in triplicate. Statistical analysis was performed by using Student’s t-test.
RESULTS AND DISCUSSION
ABT, a well-known irreversible inhibitor of CYP enzymes, has been widely used in drug development process for various applications, but its effects on expression of CYP enzymes and actions of nuclear receptors controlling CYP expression have not been studied. To determine the effects of ABT on expression of CYP enzymes regulated by the nuclear receptors CAR and PXR, we examined mRNA levels of CYP2B6 and CYP3A4 (representative target genes of CAR and PXR, respectively) in human hepatocytes treated with ethanol (vehicle), ABT (1 mM), or known inducers (CITCO or rifampin). Also, to determine whether ABT modulates actions of gene regulatory factors during CYP induction, we examined the effects of ABT on actions of CITCO or rifampin (known CYP inducers) on CYP expression in a separate set of hepatocytes.
CITCO and rifampin, known activators of CAR and PXR in humans respectively, significantly increased the expression levels of CYP2B6 in hepatocytes from two different donors (Fig. (1)A, lanes 1, 2 and 3). These results are in agreement with previous studies in human hepatocytes that CYP2B6 expression is upregulated by activation of either CAR or PXR [10]. ABT alone also significantly increased the expression levels of CYP2B6 in both hepatocytes (7.3- and 10.8-fold, respectively) (Fig. (1)A, lanes 1 and 4), the effect being comparable to that of CITCO or rifampin. Upon co-treatment with ABT, the induction of CYP2B6 expression by CITCO or rifampin was potentiated: 12.6- and 4.0-fold for CITCO (Fig. (1)A, lanes 2 and 5) and 3.9- and 2.5-fold for rifampin (Fig. (1)A, lanes 3 and 6), indicating the additive effect of ABT on inducers of CYP2B6 expression. Interestingly, ABT had a greater potentiation effect on CITCO than on rifampin. This may be due to as yet unknown pharmacological effects of ABT on CYP expression. Alternatively, ABT-mediated inhibition of potential CITCO metabolism in hepatocytes and subsequent increase in CITCO concentration in culture media may be responsible. A previous report has indicated that induction of CYP2B6 expression by CITCO increases in a concentration-dependent manner up to 1 μM, whereas the induction by rifampin is maximal at 10 μM [10].
Fig. (1).
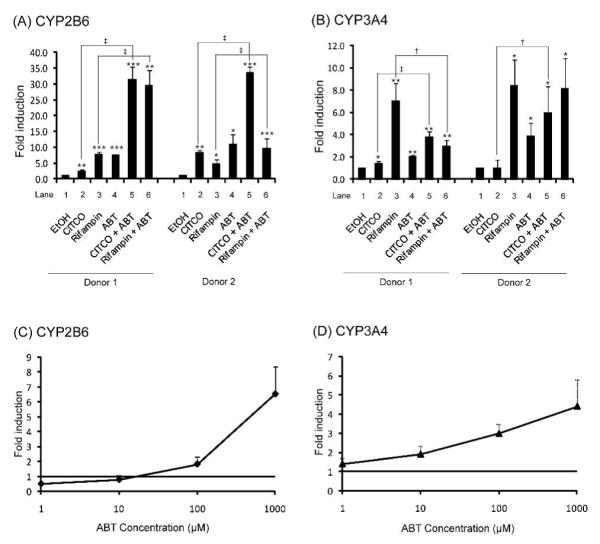
Effects of ABT on mRNA expression of CYP2B6 (A, C) and CYP3A4 (B, D). Human hepatocytes were treated with CITCO (100 nM), rifampin (10 μM), or vehicle (ethanol)-containing media in the presence or absence of ABT (1 mM) (A, B), or treated with ABT at average concentrations ranging from 1 to 1000 μM (C, D) for 72 hr. mRNA expression levels of CYP2B6 and CYP3A4 were determined by qRT-PCR. Results represent fold changes in mRNA levels of CYP2B6 and CYP3A4 relative to vehicle control (mean ± S.D.; n = 3). EC50 values were not estimated because the induction did not reach saturation. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with vehicle-treated group; †, p < 0.05; ‡, p < 0.01.
CITCO minimally affected CYP3A4 expression in human hepatocytes; CITCO increased CYP3A4 expression (by 1.4-fold) in hepatocytes from only one donor (Fig. (1)B, lanes 1 and 2). On the other hand, rifampin significantly increased the expression levels of CYP3A4 in hepatocytes from both donors (by 7.0- and 8.4- fold) (Fig. (1)B, lanes 1 and 3), confirming that upregulation of CYP3A4 expression is governed more by PXR than by CAR [10]. ABT alone increased the expression levels of CYP3A4 (by 2.0- and 3.8-fold) (Fig. (1)B, lanes 1 and 4), suggesting potential activation of PXR (or other regulatory factors) by ABT. Upon co-treatment with ABT, the effects of CITCO on CYP3A4 expression levels were potentiated by 3.8- and 6.0- fold as compared to cells treated with CITCO alone (Fig. (1)B, lanes 2 and 5). Fold induction in CYP3A4 expression by the co-treatment was greater than that by ABT alone (by 1.4- and 2.1-fold; Fig. (1)B, lanes 4 and 5). The underlying mechanism for this potentiation is unclear at this point. However, it appears unlikely that the potential increase in CITCO concentration in the culture media is responsible for the result because the effect of CITCO on CYP3A4 expression was previously shown to be independent of CITCO concentration [10]. Interestingly, ABT did not show an additive effect on rifampin-mediated induction of CYP3A4 expression; in hepatocytes from one donor, co-treatment with ABT and rifampin even decreased CYP3A4 expression as compared to cells treated with rifampin only (Fig. (1)B, lanes 3 and 6). Together, these results suggest that ABT is capable of modulating induction of CYP expression by known inducers potentially by affecting actions of certain regulatory factors during induction of CYP expression.
The inducing effects of ABT on CYP2B6 and CYP3A4 transcription were concentration-dependent (Fig. (1)C and (1)D). Typical working concentrations of ABT in in vitro hepatocyte system range from 100 μM to 1 mM. In our experimental system, ABT increased expression levels of CYP2B6 and CYP3A4 at these concentrations, indicating that ABT will probably induce CYP expression in typical experimental settings.
CYP2B6 and CYP3A4 are representative target genes of the nuclear receptors CAR and PXR. To examine the involvement of theses nuclear receptors in the induction of CYP expression by ABT, we performed luciferase reporter assays. HepG2 cells were co-transfected with an expression vector for a nuclear receptor (pcDNA3-PXR, pcDNA3-CAR, or pcDNA3 empty vector) and a luciferase construct [pGL3-UGT1A1 U2K harboring PXRE [8], or pGL3-CYP2B6-U2.2k harboring PBREM [7]] along with β-galactosidase vector (for normalization of transfection efficiency). The transfected cells were then treated with ABT (1 mM), rifampin (100 μM), CITCO (100 nM), or ethanol (vehicle), and luciferase activity was determined.
In HepG2 cells, transfection of CAR increased the CYP2B6 promoter activity by 15.8-fold (data not shown). This result is consistent with the previously reported ligand-independent activation of CAR in immortalized cells [11]. Yet, a known CAR ligand CITCO further increased CYP2B6 promoter activity by 1.6-fold in the CAR-transfected HepG2 cells, compared with vehicle-treated cells (Fig. (2)A, lanes 2 and 4), indicating that CAR can still be activated by its ligands in HepG2 cells. Similar to CITCO, treatment with ABT significantly increased the luciferase activity (by 2.4-fold) in the CAR-transfected HepG2 cells (Fig. (2)A, lanes 2 and 6). This result indicates that ABT is potentially an activator of CAR. CAR is known to be activated by either direct ligand binding or indirect mechanisms [12]. For example, CITCO is a direct activator that binds to CAR whereas phenobarbital is an indirect activator [12]. In our study, CITCO and ABT both increased the CYP2B6 promoter activity, while phenobarbital failed to increase the CYP2B6 promoter activity in the CAR-transfected HepG2 cells (data not shown). This inability of phenobarbital to activate CYP2B6 promoter activity is likely due to a lack of key transcriptional regulators required for indirect activation of CAR in HepG2 cells, and our results suggest that ABT is a direct activator of CAR similar to CITCO. Further studies such as in vitro ligand binding assays appear needed, however, because mechanism underlying indirect activation of CAR has not been fully understood. Regardless of the activation mechanism, ABT being a potential CAR activator has a significant implication in its use as a global CYP inhibitor for pharmacological studies. Because CAR is involved in various biological functions including drug metabolism and elimination, bile acid detoxification, gluconeogenesis, and carcinogenesis [13], ABT may exhibit various pharmacological effects of its own when combined with other compounds of interest.
Fig. (2).
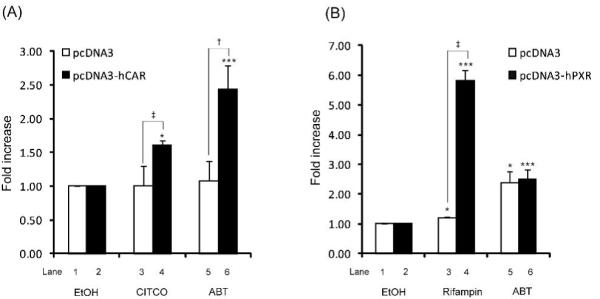
Activation of CAR (A) and PXR (B) by ABT (1 mM). HepG2 cells were transfected with luciferase constructs (pGL3-UGT1A1 U2K or pGL3-CYP2B6 U2.2K) and expression plasmids (pcDNA3, pcDNA3-PXR, or pcDNA3-CAR), along with β-galactosidase expression plasmid (for normalization of transfection efficiency). The transfected HepG2 cells were treated with ABT (1 mM), CITCO (100 nM), rifampin (10 μM), or ethanol (vehicle) for 24 hr, and luciferase assay was performed (see Materials and Methods). Results represent fold changes in luciferase activity by drug treatment relative to vehicle treatment (mean ± S.D.; n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with vehicle-treated group; †, p < 0.05; ‡, p < 0.01.
In the HepG2 cells transfected with pcDNA3-PXR, rifampin increased expression of UGT1A1 enhancer (PXRE)-driven luciferase gene by 5.8-fold as compared to those treated with vehicle (Fig. (2)B, lanes 2 and 4), confirming that rifampin is a PXR activator. Interestingly, ABT increased the luciferase activity in both control and PXR-expressing HepG2 cells (2.4- and 2.5-fold, respectively) (Fig. (2)B; lanes 1 and 5, lanes 2 and 6), suggesting that PXR is not required for activation of the promoter activity by ABT. Conceivably, ABT may activate aryl hydrocarbon receptor (AhR), which is expressed at high level in HepG2 cells [14]. Supporting this notion, AhR response element (-3319/-3304) is located close to the PXR response element (-3423/ -3409) in UGT1A1 enhancer region [15]. Furthermore, mRNA expression of CYP1A2, a representative target gene of AhR, was enhanced by ABT in human hepatocytes by 15- and 22.8-fold in our study (data not shown).
Taken together, our data suggest that ABT upregulates the expression of CYP2B6 and CYP3A4 potentially by CAR activation and it modulates actions of CYP inducers during induction of CYP expression. This limits the use of ABT as a potentiator of pharmacological effects of potential CYP inducers, especially when the effects involve regulation of CYP at the transcriptional level. Also, considering that CAR is involved in various hepatic functions, not limited to drug elimination, care should be taken when ABT is used as a global CYP inhibitor in cell-based in vitro systems.
ACKNOWLEDGEMENTS
We would like to thank Dr. Hyunwoo Lee and Dr. James H. Fischer for critical reading of this manuscript. This investigation was supported by grant HD055313 and K12HK055892 fellowship from the NICHD. Human hepatocytes were obtained through the Liver Tissue Cell Distribution System (Pittsburgh, Pennsylvania), which was funded by NIH Contract #N01-DK-7-0004 / HHSN267200700004C.
ABBREVIATIONS
- ABT
1-aminobenzotriazole
- CAR
constitutive androstane receptor
- CITCO
6-(4-Chloro phenyl)imidazo[2,1-b] [1,3]thiazole-5-carbaldehyde O-3,4-dichlorobenzyl) oxime
- CYP
cytochrome P450
- E2
17β-estradiol
- PBREM
phenobarbital-responsive enhancer module
- PXR
prengane X receptor
- PXRE
PXR-responsive element
References
- [1].de Montellano Ortiz, Mathews PR, M. J. Autocatalytic alkylation of the cytochrome P-450 prosthetic haem group by 1-aminobenzotriazole. Isolation of an NN-bridged benzyne-protoporphyrin IX adduct. Biochem J. 1981;195(3):761–764. doi: 10.1042/bj1950761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Williams JA, Hurst SI, Bauman J, Jones BC, Hyland R, Gibbs JP, Obach RS, Ball SE. Reaction phenotyping in drug discovery: moving forward with confidence? Curr Drug Metab. 2003;4(6):527–534. doi: 10.2174/1389200033489235. [DOI] [PubMed] [Google Scholar]
- [3].Hallifax D, Houston JB. Uptake and intracellular binding of lipophilic amine drugs by isolated rat hepatocytes and implications for prediction of in vivo metabolic clearance. Drug Metab Dispos. 2006;34(11):1829–1836. doi: 10.1124/dmd.106.010413. [DOI] [PubMed] [Google Scholar]
- [4].van Ravenzwaay B, Gamer AO, Leibold E, Kaufmann W. Effect of cytochrome P-450 inhibition on tetrahydrofuran-induced hepatocellular proliferation in female mice. Arch Toxicol. 2003;77(8):459–464. doi: 10.1007/s00204-003-0474-7. [DOI] [PubMed] [Google Scholar]
- [5].Koh KH, Jeong H. Effects of 17b-extradiol (E2) on expression of CYP enzymes in human hepatocytes. Drug Metab Rev. 2009;41(Suppl 3):49. [Google Scholar]
- [6].Jeong H, Choi S, Song JW, Chen H, Fischer JH. Regulation of UDP-glucuronosyltransferase (UGT) 1A1 by progesterone and its impact on labetalol elimination. Xenobiotica. 2008;38(1):62–75. doi: 10.1080/00498250701744633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang H, Faucette S, Sueyoshi T, Moore R, Ferguson S, Negishi M, LeCluyse EL. A novel distal enhancer module regulated by pregnane X receptor/constitutive androstane receptor is essential for the maximal induction of CYP2B6 gene expression. J Biol Chem. 2003;278(16):14146–14152. doi: 10.1074/jbc.M212482200. [DOI] [PubMed] [Google Scholar]
- [8].Sugatani J, Nishitani S, Yamakawa K, Yoshinari K, Sueyoshi T, Negishi M, Miwa M. Transcriptional regulation of human UGT1A1 gene expression: activated glucocorticoid receptor enhances constitutive androstane receptor/pregnane X receptor-mediated UDP-glucuronosyltransferase 1A1 regulation with glucocorticoid receptor-interacting protein 1. Mol Pharmacol. 2005;67(3):845–855. doi: 10.1124/mol.104.007161. [DOI] [PubMed] [Google Scholar]
- [9].Schmittgen T, Livak D, J. K. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- [10].Faucette SR, Sueyoshi T, Smith CM, Negishi M, Lecluyse EL, Wang H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. J Pharmacol Exp Ther. 2006;317(3):1200–1209. doi: 10.1124/jpet.105.098160. [DOI] [PubMed] [Google Scholar]
- [11].Honkakoski P, Zelko I, Sueyoshi T, Negishi M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol Cell Biol. 1998;18(10):5652–5658. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li H, Chen T, Cottrell J, Wang H. Nuclear translocation of adenoviral-enhanced yellow fluorescent protein-tagged-human constitutive androstane receptor (hCAR): a novel tool for screening hCAR activators in human primary hepatocytes. Drug Metab Dispos. 2009;37(5):1098–1106. doi: 10.1124/dmd.108.026005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kakizaki S, Yamazaki Y, Takizawa D, Negishi M. New insights on the xenobiotic-sensing nuclear receptors in liver diseases--CAR and PXR. Curr Drug Metab. 2008;9(7):614–621. doi: 10.2174/138920008785821666. [DOI] [PubMed] [Google Scholar]
- [14].Murray IA, Flaveny CA, Dinatale BC, Chairo CR, Schroeder JC, Kusnadi A, Perdew GH. Antagonism of aryl hydrocarbon receptor signaling by 6, 2′, 4′-trimethoxyflavone. J Pharmacol Exp Ther. 2009 doi: 10.1124/jpet.109.158261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yueh MF, Huang YH, Hiller A, Chen S, Nguyen N, Tukey RH. Involvement of the xenobiotic response element (XRE) in Ah receptor-mediated induction of human UDP-glucuronosyltransferase 1A1. J Biol Chem. 2003;278(17):15001–15006. doi: 10.1074/jbc.M300645200. [DOI] [PubMed] [Google Scholar]