

Abstract
Cutinase is a multifunctional esterase with potential industrial applications. In the present study, a truncated version of the extracellular Thermobifida fusca cutinase without a signal peptide (referred to as cutinaseNS) was heterologously expressed in Escherichia coli BL21(DE3). The results showed that the majority of the cutinase activity was located in the culture medium. In a 3-liter fermentor, the cutinase activity in the culture medium reached 1,063.5 U/ml (2,380.8 mg/liter), and the productivity was 40.9 U/ml/h. Biochemical characterization of the purified cutinaseNS showed that it has enzymatic properties similar to those of the wild-type enzyme. In addition, E. coli cells producing inactive cutinaseNSS130A were constructed, and it was found that the majority of the inactive enzyme was located in the cytoplasm. Furthermore, T. fusca cutinase was confirmed to have hydrolytic activity toward phospholipids, an important component of the cell membrane. Compared to the cells expressing the inactive cutinaseNSS130A, the cells expressing cutinaseNS showed increased membrane permeability and irregular morphology. Based on these results, a hypothesis of “cell leakage induced by the limited phospholipid hydrolysis of cutinaseNS” was proposed to explain the underlying mechanism for the extracellular release of cutinaseNS.
INTRODUCTION
Cutinase not only catalyzes the cleavage of the ester bonds of cutin, but is also capable of hydrolyzing soluble esters, insoluble triglycerides, and a variety of polyesters (1). In addition to its hydrolytic capability, cutinase is also used in ester synthesis (2, 3) and transesterification (4). Therefore, as a multifunctional enzyme, cutinase has potential uses in the food, chemical, textile, and other industries (5).
Enzymes with cutinase activity have been found in both fungi and bacteria, such as Fusarium solani pisi and Thermobifida fusca, respectively (5–7). It has been demonstrated that cutinases from both groups belong to the α/β-hydrolase superfamily, share similar spatial structures, and display similar catalytic properties, as well as substrate specificities. In addition, consistent with their ability to accommodate large substrates like cutin, they contain an open active site, and the lid structure normally seen in most lipases is absent in cutinase. Despite these similarities, there does not appear to be significant sequence homology between fungal and bacterial cutinases; thus, it has been suggested that they be classified into prokaryotic and eukaryotic cutinase subfamilies, respectively (6).
To date, all the studied cutinases from microorganisms have been found to be secretory enzymes (6). Therefore, for heterologous expression, signal peptides are usually utilized to mediate the secretion of recombinant cutinase. Using this approach, F. solani cutinase has been expressed in a variety of host cells, such as Escherichia coli (8), Aspergillus awamori (9), Fusarium venenatum (10), Saccharomyces cerevisiae (11, 12), and Pichia pastoris (13). The highest yield of extracellular protein, 546 mg/liter, was obtained in a 5-liter bioreactor utilizing an engineered S. cerevisiae cellular system (12). Previously, our laboratory identified the open reading frame responsible for the expression of T. fusca cutinase, and secretory expression of the cutinase was performed by mediation of the PelB signal peptide in E. coli BL21(DE3) (6). Our results showed that the mature cutinase was partially located in the periplasm, while some was secreted into the culture medium (14). Secretion into the culture medium was not unexpected, since similar phenomena have been observed in the expression of other secretory proteins (15–19). After optimization of the cultivation conditions in shake flasks, the cutinase activity in the culture medium reached 149.2 U/ml (326 mg/liter) 52 h postinduction (14).
As part of our studies, an E. coli BL21(DE3) strain harboring the mature cutinase without a signal peptide (referred to as cutinaseNS) was constructed as an experimental control. Unexpectedly, significant cutinase activity was found in the culture medium. In the present study, this anomalous phenomenon of secretion was investigated, and the underlying mechanism was explored.
MATERIALS AND METHODS
Bacterial strains, vectors, and materials.
The plasmid Tfu_0883/pET-20b(+) harboring the gene for T. fusca cutinase (NCBI accession number AAZ54921) and purified wild-type cutinase from the culture medium of T. fusca were from laboratory stock (6). E. coli strain JM109 was used as the host for cloning, and E. coli strain BL21(DE3) was used for protein expression. The restriction enzymes, alkaline phosphatase, T4 DNA ligase, agarose gel DNA purification kit, and pMD18-T simple vector were obtained from TakaRa (Dalian, China). DNA sequencing and primer synthesis were performed by Shanghai Sangon Biological Engineering Technology and Services Co., Ltd. (Shanghai, China). The 4-nitrophenyl butyrate (pNPB), o-nitrophenyl-β-d-galactopyranoside (ONPG), and N-phenyl-α-naphthylamine (NPN) were obtained from Sigma (Shanghai, China). Polyvinylidene difluoride (PVDF) transfer membranes and anti-six-histidine mouse IgG, horseradish peroxidase (HRP)-labeled goat anti-mouse IgG(H+L), and DAB horseradish peroxidase color development kits were purchased from Beyotime Institute of Biotechnology (Nantong, China). The 50% egg yolk emulsion without triglyceride was purchased from Amresco (Shanghai, China). Other chemicals were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).
Plasmid construction.
The gene encoding cutinaseNS was amplified from Tfu_0883/pET-20b(+) by PCR using the primers CUT-F and CUT-R (see Table S1 in the supplemental material). The amplification product was isolated and ligated into the pMD18-T simple vector. The product of the ligation reaction was used to transform chemically competent E. coli JM109 cells. The sequence of the recombinant plasmid cutinaseNS/pMD18-T, isolated from a transformant, was confirmed by DNA sequencing. A plasmid with the correct sequence was digested with the restriction enzymes NdeI and EcoRI and then ligated into the expression vector pET-20b(+) restricted with the same restriction enzymes, as well as alkaline phosphatase. The resulting recombinant plasmid cutinaseNS/pET-20b(+), which does not contain the intrinsic PelB signal peptide, was verified by DNA sequencing.
The mutant cutinaseNSS130A/pET-20b(+) plasmid was generated from cutinaseNS/pET-20b(+), utilizing the standard QuikChange mutagenesis methodology with the complementary primers S130A-F and S130A-R (see Table S1 in the supplemental material [the lowercase letters in the primers indicate the mutated bases]). The sequence of the mutated plasmid was confirmed by DNA sequencing.
Expression of recombinant cutinases.
The recombinant plasmids for expression of cutinaseNS and cutinaseNSS130A were used to separately transform chemically competent E. coli BL21(DE3). The cells containing the desired plasmids were grown individually in TB (Terrific broth) medium containing ampicillin (100 mg/liter) at 37°C with shaking at 200 rpm. Lactose was added to a final concentration of 5 g/liter when the culture reached an optical density at 600 nm (OD600) of 1.5. After induction, the culture was grown at 25°C for an additional 24 h.
Three-liter fermentor for cutinaseNS.
E. coli BL21(DE3) harboring the cutinaseNS/pET-20b(+) plasmid was cultured in a 3-liter fermentor (BioFlo 110; New Brunswick Scientific Co., NJ, USA). The fermentation began with a fed-batch cultivation containing an initial 1.2-liter batch medium at 37°C. The pH was maintained at 7.0 by addition of ammonium hydroxide. The end of glycerol consumption was detected by a sudden increase in both the dissolved-oxygen (DO) and pH values, and continuous feeding was carried out according to the exponential-feeding method (20). When the OD600 of the culture reached 50, isopropyl-1-thio-β-d-galactopyranoside (IPTG) was added to the fermentor at a final concentration of 0.02 mM, and then lactose was fed at the rate of 0.6 g liter−1 h−1. The pH was held at 6.5 in the induction period.
Cell fractionation.
Cell fractionation was performed as follows. One milliliter of culture broth was centrifuged at 10,000 × g for 10 min, and the supernatant was collected as the extracellular fraction. To separate the periplasmic fraction, the centrifuged cells were resuspended in 1 ml 20 mM Tris-HCl buffer (pH 8.0) containing 25% (wt/vol) sucrose and 1 mM EDTA. After incubation on ice for 2 h, the cell suspension was centrifuged at 10,000 × g for 10 min. The supernatant obtained was the periplasmic fraction. The precipitate was resuspended in l ml of 20 mM Tris-HCl buffer (pH 8.0) and disrupted by sonication with a Sonifer450 (Branson, Danbury, CT, USA). After centrifugation at 10,000 × g for 10 min, the supernatant and cell debris were collected as the soluble cytoplasmic fraction and the insoluble fraction, respectively. The total cutinase activity was the sum of enzyme activity in the culture medium, periplasm, and cytoplasm.
Cutinase activity assay.
The cutinase activity was determined using pNPB as the substrate, as previously described (6). One unit of enzyme activity is defined as the production of 1 μmol of pNP per minute.
SDS-PAGE and Western blot analysis.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed using a 5% stacking gel and a 12% separating gel, and the protein bands were visualized by staining with Coomassie brilliant blue R-250. For Western blot analysis, the separated proteins were transferred electrophoretically to a PVDF transfer membrane. Since the C terminus of the target proteins was His tag labeled, the membranes were incubated with an anti-six-histidine mouse IgG (1:1,000 dilution) overnight. An HRP-labeled goat anti-mouse IgG(H+L) was used as a secondary antibody at a 1:1,000 dilution, and the bands were visualized using a DAB horseradish peroxidase color development kit.
Purification and characterization of cutinases.
The C-terminally His-tagged cutinaseNS and cutinaseNSS130A were purified first by (NH4)2SO4 fractionation, followed by purification on a nickel affinity column as previously described (6). The optimal temperature, pH optima, thermostability, and pH stability, as well as the kinetic parameters of purified cutinaseNS and wild-type cutinase, were determined as previously described (6, 21).
Phospholipase activity assay.
The phospholipase activity was measured using a modification of the egg yolk plate method as described previously (22). A certain amount of purified cutinase was added to the Oxford cup on the yolk plate (0.66% NaCl, 1.09% boric acid, 0.19% sodium tetraborate, 1.5% agar, 2% egg yolk emulsion, pH 7.2 to 7.4), and Tris-HCl buffer (20 mM, pH 8.0) was utilized as a control. The plate was incubated at 37°C overnight, and the Oxford cup was removed. The formation of a precipitation zone was considered indicative of phospholipase activity.
Assay of inner membrane permeability.
Inner membrane permeability was assessed by measuring the access of ONPG to the cytoplasm, as reported previously (23), with the following minor modifications. A sample of E. coli BL21(DE3) cells containing the target plasmid was obtained by centrifugation (3,000 × g), rinsed once, and then suspended in 10 mM sodium phosphate buffer (pH 7.4) to an OD600 of 0.16. ONPG was added to a final concentration of 100 μg/ml to a quartz cuvette containing 3 ml of the cell suspension. Substrate cleavage by β-galactosidase was monitored by light absorption measurements at 420 nm in a spectrophotometer (UV-2450; Shimadzu Co., Kyoto, Japan).
Assay of outer membrane permeability.
The permeability of the outer membrane was measured using the NPN access assay described previously (23) with minor modifications. NPN was added to a concentration of 10 μM to quartz cuvette containing 3 ml of cell suspension prepared as described above. The sample was mixed by inverting the cuvette immediately prior to fluorescence monitoring. Fluorescence was measured using a Shimadzu RF-1501 spectrofluorometer (Shimadzu Co., Kyoto, Japan) with slit widths set to 5 nm and excitation and emission wavelengths set to 350 and 420 nm, respectively.
Transmission electron microscopy (TEM).
E. coli BL21(DE3) cells containing the target plasmid were collected by centrifuging at 4,000 × g for 2 min and fixed in 2.5% glutaraldehyde for 2 to 4 h. The cell pellets were rinsed 3 times with 0.1 M sodium phosphate buffer at pH 7.2 to 7.4, postfixed in 1% osmic acid, and rinsed with the same buffer. After gradually dehydrating the cells in ethyl alcohol, they were embedded in araldite. The samples were cut into slices (70 nm thick), stained with 3% uranyl acetate and lead citrate, and observed by utilizing a JEOL JEM-7000 transmission electron microscope at 80 kV. The resulting microscopic images were photographed.
Effect of purified cutinase on intact cells.
Two different methods were utilized to investigate the effect of purified cutinase on intact cells. In the first method, E. coli BL21(DE3) cells were cultured in LB medium for 7 to 8 h. Then, 1 ml of culture broth was centrifuged at 4,000 × g for 2 min, the cells were collected, and the pellet was rinsed three times with 0.1 M sodium phosphate buffer at pH 7.2 to 7.4. The pellet was resuspended in 1 ml of the same buffer containing purified cutinaseNS to a final concentration of 1 mg/ml and incubated for 4 to 5 h. In the second method, E. coli BL21(DE3) cells were cultured in LB medium containing 0.5 mg/ml purified cutinaseNS for 7 to 8 h. The morphologies of the cells resulting from both methods were investigated. The E. coli BL21(DE3) cells treated under the same experimental conditions but without added cutinaseNS were utilized as a control.
Cell lysis determination.
Cell lysis was determined as the percentage of extracellular enzyme activity versus the sum of extracellular and intracellular activities, using β-galactosidase as the reporter protein (24, 25). Standard β-galactosidase activity was assayed according to the previously reported method (26).
Miscellaneous methods.
Cell growth was monitored by measuring the optical density of the culture broth at 600 nm using a spectrophotometer (BioPhotometer Plus; Eppendorf Co., Hamburg, Germany) after appropriate dilution. In order to determine the dry cell weight (DCW), 2 ml of culture broth was centrifuged at 10,000 × g for 10 min. The pellet was washed twice with 0.9% (wt/vol) NaCl and dried at 105°C for 16 h to a constant weight. Protein concentrations were determined using the Bio-Rad Protein Assay according to the manufacturer's protocol. Bovine serum albumin was utilized as the standard protein.
RESULTS
Cloning and expression of cutinaseNS.
The cutinaseNS gene responsible for encoding the mature form of cutinase was cloned into the T7-driven expression plasmid pET-20b(+), resulting in cutinaseNS/pET-20b(+), in which the PelB signal peptide of pET-20b(+) was eliminated. The recombinant plasmid was transformed into the E. coli BL21(DE3) strain. During cultivation in the shake flask, the cutinase activity in the cytoplasm increased during the first 8 h postinduction. After that, the activity slowly decreased to only 10.3 U/ml at 24 h postinduction. A similar profile for cutinase activity was also observed in the periplasmic fraction: first the activity increased to its maximum at 10 h and then eventually decreased to 11.9 U/ml. However, the cutinase activity in the culture medium increased rapidly from 8 h postinduction and reached a maximum of 272.8 U/ml at 24 h. This activity represented 92.5% of the sum of all the activity in the cytoplasm, periplasm, and culture medium (Fig. 1).
Fig 1.
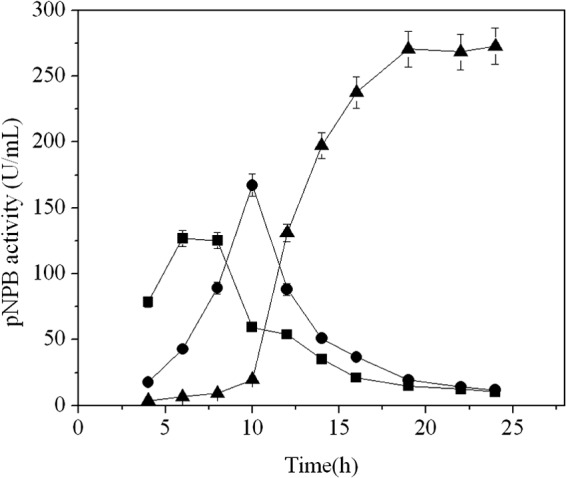
Distribution of cutinase activity postinduction. ■, intracellular fraction; ●, periplasmic fraction; ▲, culture supernatant. The error bars represent the standard deviations from three measurements.
The distribution of cutinaseNS was further investigated by SDS-PAGE (Fig. 2). The results showed that one major band around 31 kDa, which is in agreement with the calculated molecular weight of mature cutinase, was found in the culture medium. No visible band of cutinaseNS was observed in either the cytoplasm or periplasm fraction (Fig. 2). These results are consistent with that of Western blot analysis, as well as enzyme activity assay. Thus, it seems that the cutinaseNS without a signal peptide is almost completely “secreted” into the culture medium.
Fig 2.
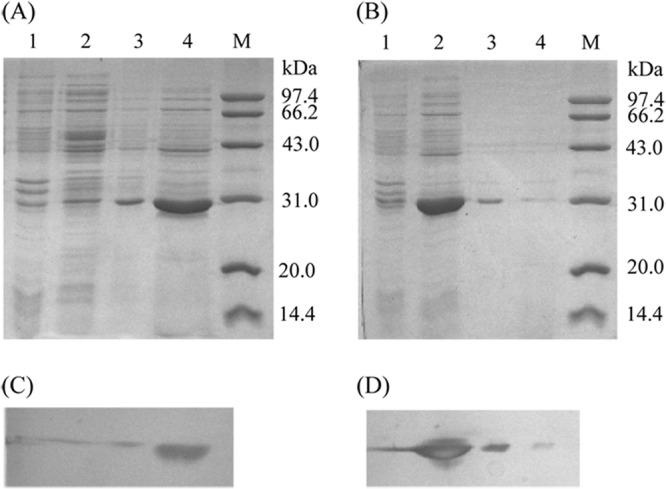
SDS-PAGE (A and B) and Western blot (C and D) analysis of the enzyme (cutinaseNS [A and C] and cutinaseNSS130A [B and D]) distribution. The primary antibody was anti-six-histidine mouse IgG, and the second was HRP-labeled goat anti-mouse IgG(H+L). Lanes: M, molecular mass standard protein; 1, intracellular insoluble fraction; 2, intracellular soluble fraction; 3, periplasmic fraction; 4, culture supernatant.
Three-liter bioreactor for production of cutinaseNS.
The extracellular “secretion” efficiency of the above-mentioned E. coli BL21(DE3) harboring cutinaseNS was further explored in a 3-liter fermentor. As shown in Fig. 3, the DCW reached 42.6 g/liter, and the extracellular cutinase activity increased to 1,063.5 U/ml 11 h postinduction, which is 3.9 times higher than that in the shake flask cultivation. The total cultivation time was about 26 h; therefore, the productivity of cutinase in the culture medium was 40.9 U/ml/h.
Fig 3.
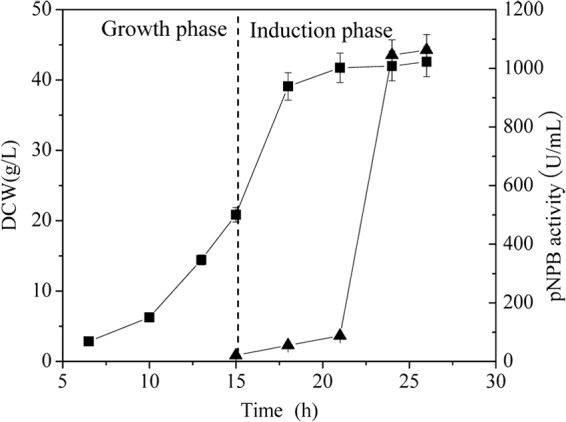
Time profiles for the production of cutinaseNS in a 3-liter fermentor. ■, DCW; ▲, cutinase activity in the culture medium. The error bars represent the standard deviations from three measurements.
Purification and characterization of cutinaseNS.
The cutinaseNS in the culture medium of the E. coli BL21(DE3) was purified first by (NH4)2SO4 fraction, dialysis to remove salt, and then nickel affinity column chromatography (Table 1). As shown in Fig. 4, the purified enzyme appears homogeneous by SDS-PAGE analysis. For comparison, the enzyme characterization was performed on both the purified cutinaseNS in this study and the previously isolated wild-type cutinase (laboratory stock). The results of these studies demonstrated that the optimum pH and pH stability, optimum temperature, and thermostability of cutinaseNS are similar to those of wild-type cutinase (see Fig. S1 in the supplemental material). Utilizing pNPB as the substrate, cutinaseNS exhibited Michaelis-Menten kinetics with a Km of 312.2 μM and a kcat of 303.8 s−1, which is similar to that obtained for wild-type cutinase (see Fig. S2 in the supplemental material).
Table 1.
Summary of purification of cutinaseNSa
| Purification step | Total protein (mg) | Total activity (U) | Sp act (U/mg) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|
| Crude extract | 243.0 | 57,344.3 | 235.9 | 100 | 1.0 |
| Ammonium sulfate fraction | 169.3 | 43,925.7 | 259.5 | 76.6 | 1.1 |
| Ni-chelating affinity chromatography | 70.4 | 31,424.7 | 446.7 | 54.8 | 1.9 |
The culture medium from 1.2 g of dried cells was used for the purification.
Fig 4.
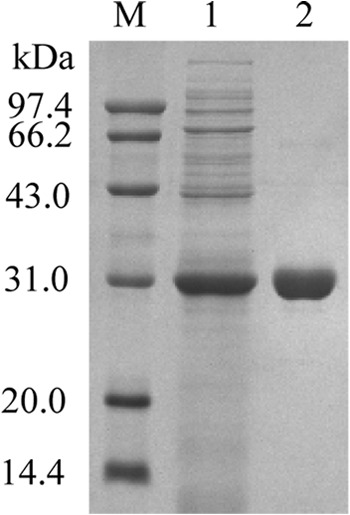
SDS-PAGE analysis of the purification of cutinaseNS. Lanes: M, molecular mass standard protein; 1, culture supernatant; 2, purified cutinaseNS.
Expression of inactive cutinaseNS.
T. fusca cutinase belongs to the α/β-hydrolase family. The examination of the cutinase structure model reveals a Ser130-His208-Asp176 catalytic triad in which Ser130 is critical to the hydrolytic activity. In addition, the mutation of Ser130 to Ala has been reported to completely eliminate activity (6).
In the present study, we performed site-directed mutagenesis to construct cutinaseNSS130A/pET-20b(+), utilizing the cutinaseNS/pET-20b(+) plasmid as the template. When the mutant was expressed in E. coli BL21(DE3), under the same conditions as for cutinaseNS, the majority of the target protein was located in the cytoplasm (Fig. 2). The mutant, cutinaseNSS130A, showed no detectable enzymatic activity.
Potential phospholipase activity of T. fusca cutinase.
The potential phospholipase activity of T. fusca cutinase was analyzed using the egg yolk plate assay, which has been reported to be a simple, straightforward method to qualitatively verify the presence of phospholipase activity (27). The results showed, compared with the control, a distinct, dense white zone of precipitation for both the purified cutinaseNS and the wild-type enzyme (Fig. 5), suggesting that T. fusca cutinase has phospholipase activity. In addition, under similar experimental conditions, the purified cutinaseNSS130A demonstrated no phospholipase activity.
Fig 5.
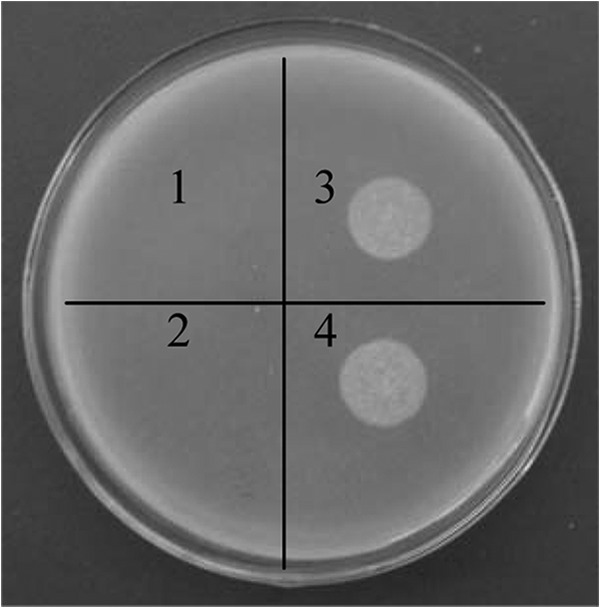
Phospholipase activity assay for T. fusca cutinase. 1, Tris-HCl buffer; 2, cutinaseNSS130A; 3, cutinaseNS; 4, wild-type cutinase.
Assay of membrane permeability.
The permeability of the inner membrane was evaluated by measuring the influx of ONPG, a substrate of cytosolic β-galactosidase. The permeability of the outer membrane was evaluated by utilizing the hydrophobic fluorescent probe NPN. As shown in Fig. 6, when E. coli cells harboring cutinaseNS/pET-20b(+) were induced for 12 h, the membrane permeability of both the inner and outer membranes was significantly higher than that of the E. coli cells harboring either pET-20b(+) or cutinaseNSS130A/pET-20b(+).
Fig 6.
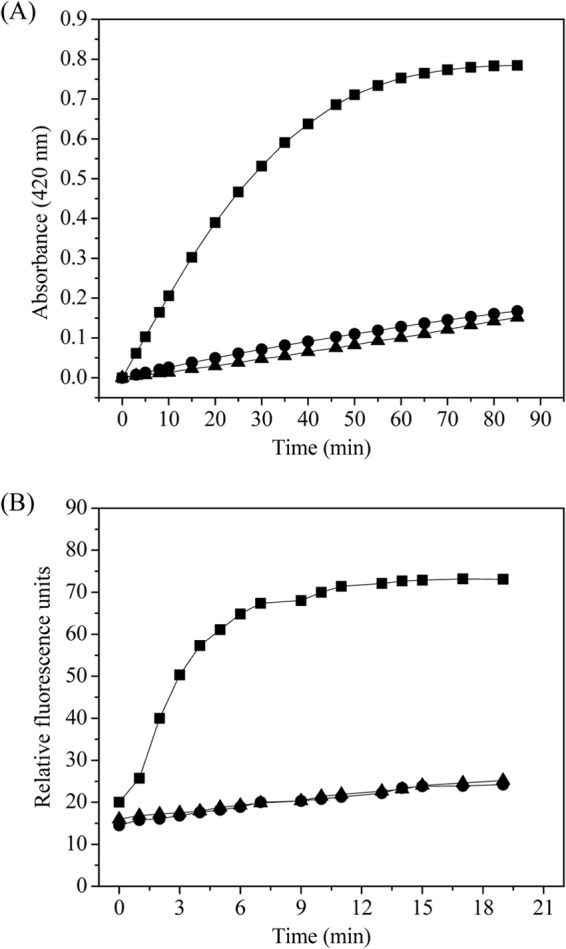
Permeability of the inner (A) and outer (B) membranes of E. coli BL21(DE3) containing target plasmid. ▲, pET-20b(+); ■, cutinaseNS/pET-20b(+); ●, cutinaseNSS130A/pET-20b(+).
TEM analysis of E. coli BL21(DE3).
The morphology of E. coli BL21(DE3) cells was evaluated using TEM. Compared to control cells harboring only the vector pET-20b(+) (Fig. 7A), the cells containing the plasmid for the expression of cutinaseNS exhibited an irregular shape with an apparently distorted cell membrane (Fig. 7B). The cells expressing the inactive cutinaseNSS130A had both a smooth membrane and a regular shape (Fig. 7C), similar to those of the control.
Fig 7.
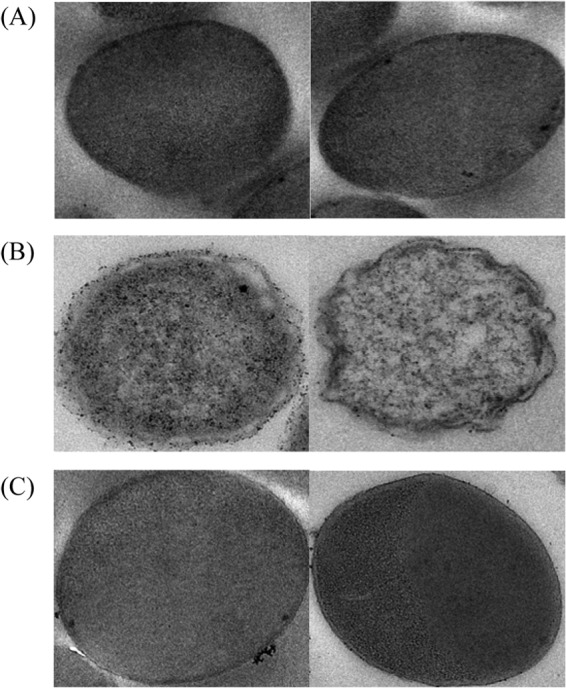
TEM images of E. coli BL21(DE3) cells containing target plasmid. (A) pET-20b(+). (B) CutinaseNS/pET-20b(+). (C) Cutinase NSS130A/pET-20b(+).
Cell lysis determination.
β-Galactosidase is a cytoplasmic protein, and under normal conditions, its level in the culture medium is very low; thus, the presence of extracellular β-galactosidase activity would be an indication of cell lysis (24, 25). In the present study, at 22 h postinduction, 90.9% of the cutinaseNS was found in the culture medium of E. coli cells harboring pET-20b(+)/cutinaseNS, while only 7.2% of the β-galactosidase activity was found in the culture medium. This amount of β-galactosidase activity is similar to that of the control E. coli cells harboring pET-20b(+)/cutinaseNSS130A or the vector pET-20b(+) without an insert. The results strongly suggest that the “secretion” of cutinaseNS was not due to cell lysis.
DISCUSSION
Cutinase can hydrolyze ester bonds. Unlike most ester hydrolases, cutinase has the capability to hydrolyze, not only soluble esters, but also insoluble triglycerides and polyester; thus, it was considered the bridge between esterases and lipases (5, 28). Since cutinase has significant potential applications in many industrial fields, the high-yield preparation of cutinase has drawn extensive attention in recent years (1).
Cutinases are natural extracellular enzymes in their native organisms. Previously, secretory expression of cutinases in different hosts was mediated by a secretion signal peptide (9–14). In the present study, cutinase activity in the culture medium from a 3-liter fermentor of T. fusca cutinase, heterologously expressed in E. coli without a signal peptide, reached 1,063.5 U/ml (2,380.8 mg/liter) after 24 h. This represents a 4.4-fold increase over the highest previously reported yield (546 mg/liter) (12). In addition, the cultivation time in the present study was much shorter than that in previous reports (12, 14). The productivity reached 40.9 U/ml/h. Higher yields and productivity should be possible upon future optimization of the culture conditions.
In general, the expression of a protein without a leader signal peptide in E. coli leads to the protein being unable to cross the cell membranes and remaining located in the cytoplasm. Protein containing a signal peptide is synthesized in the cytoplasm and then transferred across the inner membrane; in the periplasm, the signal peptide attached to the precursor protein is excised and the resulting polypeptide is folded. It has been reported that the oxidative environment in the periplasm is also very important for the correct folding of the extracellular secreted enzyme (15). In the present study, since cutinaseNS was “secreted” without the apparent mediation of a signal peptide, there was a question of whether the cutinaseNS was correctly folded. In order to clarify this issue, the cutinaseNS from the culture medium was purified to homogeneity. The enzymatic properties of both the cutinaseNS and wild-type cutinase were investigated. It was demonstrated that they exhibited similar optimum pHs, pH stability, optimum activity temperatures, and thermostability, as well as kinetic parameters (see Fig. S1 and S2 in the supplemental material). Thus, it seems that the cutinaseNS “secreted” in the culture medium was folded correctly.
The next question was, how did the cutinaseNS get into the culture medium without the mediation of a signal sequence? One possibility is that cutinaseNS itself could interact directly with transport chaperon proteins and be transferred. Unfortunately, we have not obtained experimental data to support this suggestion. A second possibility is that the cutinaseNS synthesized in the cytoplasm is partially enzymatically active and is released into the culture medium via cutinase's destruction of the cell membrane. To explore the latter option, the inactive enzyme, cutinaseNSS130A, was expressed under conditions similar to those of active cutinaseNS. It was found that the majority of the expressed inactive cutinaseNS did not cross the cell membrane and was still located in the cytoplasm (Fig. 2). Thus, it appears that the presence of extracellular cutinaseNS, intracellularly expressed without the attachment of a signal peptide, is related to cutinase's hydrolytic enzymatic activity.
E. coli, a Gram-negative bacterium, has two membranes, an inner membrane composed of phospholipids and membrane proteins and an outer asymmetric membrane, with phospholipids in the inner leaflet and lipopolysaccharide (LPS) in the outer leaflet (29, 30). Cutinase, a member of an ester hydrolase family, is capable of hydrolyzing a range of substrates. In the present study, cutinase's hydrolytic activity toward phospholipids, an important component of the cell membrane, was investigated. The results indicated that cutinase shows phospholipase activity (Fig. 5). This result is consistent with a previous report, in which the cutinase from F. solani was found to hydrolyze phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine (31).
Since T. fusca cutinase shows phospholipase activity, the “secretion” mechanism of cutinaseNS could be related to its hydrolytic function toward the phospholipid of the cell membrane. To explore this possibility, the permeability and morphology of the E. coli cells were observed. The results demonstrated that the permeability of both inner and outer membranes of the cells expressing cutinaseNS was significantly enhanced compared to that of the cells expressing inactive cutinaseNSS130A (Fig. 6). The cells expressing cutinaseNS exhibited an irregular shape under TEM analysis, while those expressing inactive cutinaseNSS130A displayed regular morphology, similar to that of control cells (Fig. 7). Further, the effect of purified cutinaseNS added to intact cells was investigated. When cutinaseNS was added in vitro either to the culture medium during the cultivation process of E. coli cells or to the suspension of harvested E. coli cells, the morphology of the cells demonstrated no obvious change under either experimental condition compared to the control (data not shown). The phenomenon might be related to the composition of the cell membrane. As described above, the outer membrane of E. coli is mainly composed of LPS, which constitutes a barrier for cutinaseNS to access the phospholipid of the cell membrane.
The increased membrane permeability and irregular morphology observed in cells expressing cutinaseNS prompted another question: did cell lysis occur? Generally, the extracellular level of β-galactosidase, a native cytoplasmic protein, is considered an indication of cell lysis (24, 25). In the present study, it was found that, at 22 h postinduction, the percentages of β-galactosidase activity in the culture medium were similar in cells expressing cutinaseNS, cells expressing inactive cutinaseNSS130A, and control cells. Additionally, it was observed by TEM analysis that the three different cell lines were intact. Furthermore, the DCW in the stationary period of cultivation was constant (Fig. 3), and at the end of cultivation, the culture solution was not viscous. Taken together, these results indicated that the cells were intact and had not thoroughly collapsed during the expression of cutinaseNS.
The above-mentioned results strongly suggest that cell leakage induced by the phospholipid hydrolysis by cutinaseNS is responsible for the presence of extracellular cutinaseNS. After cutinaseNS is synthesized, it may fold into an active form in the cytoplasm and partially hydrolyze the phospholipids in the membrane. This hydrolysis could lead to the enhanced permeability of the cell membrane, which eventually results in the release of cutinaseNS from the cytoplasm into the culture medium. It should be noted that even though cutinaseNS may hydrolyze the phospholipid of E. coli, its hydrolase activity is somewhat limited in that it does not lead to the collapse of the cells. Potentially, due to this limited hydrolyzation, the process of enzyme “secretion” was slow, so that the cutinaseNS is able to fold into the correct form.
In summary, T. fusca cutinase, expressed in E. coli BL21(DE3) without a signal peptide, is found in an active form in the culture medium. The hypothesis of cell leakage induced by the limited phospholipid hydrolysis of cutinaseNS was explored. Additional experiments are under way to further elucidate the mechanism by which cutinaseNS interacts with cell membranes and renders them permeable. This is the first report that a bacterial cutinase has hydrolytic activity toward phospholipids. In addition, we believe our study is the first successful demonstration of extracellular “secretion” of recombinant protein by the limited phospholipase activity. The yield of cutinase, 1,063.5 U/ml (2,380.8 mg/liter), obtained in the present study represents the highest production reported to date.
Supplementary Material
ACKNOWLEDGMENTS
We thank Bijun Zhu of the Biomedical Research Center of Zhongshan Hospital, Fudan University, Shanghai, China, for the TEM analysis.
Financial support for this work was provided by the Science and Technology Support Project of Jiangsu Province (no. BE2012018 and BE2012019), the 111 Project (no. 111-2-06), the High-End Foreign Experts Recruitment Program (GDW20123200116), and the Research and Innovation Project for College Graduates of Jiangsu Province (Lingqia Su).
Footnotes
Published ahead of print 19 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00239-13.
REFERENCES
- 1. Dutta K, Sen S, Veeranki VD. 2009. Production, characterization and applications of microbial cutinases. Process Biochem. 44:127–134 [Google Scholar]
- 2. De Barros DPC, Fonseca LP, Cabral JM, Weiss CK, Landfester K. 2009. Synthesis of alkyl esters by cutinase in miniemulsion and organic solvent media. Biotechnol. J. 4:674–683 [DOI] [PubMed] [Google Scholar]
- 3. De Barros DPC, Azevedo AM, Cabral JMS, Fonseca LP. 2012. Optimization of flavor esters synthesis by Fusarium solani pisi cutinase. J. Food Biochem. 36:275–284 [Google Scholar]
- 4. Borreguero I, Carvalho CML, Cabral JMS, Sinisterra JV, Alcantara AR. 2001. Enantioselective properties of Fusarium solani pisi cutinase on transesterification of acyclic diols: activity and stability evaluation. J. Mol. Catal. B 11:613–622 [Google Scholar]
- 5. Carvalho CM, Aires-Barros MR, Cabral JM. 1999. Cutinase: from molecular level to bioprocess development. Biotechnol. Bioeng. 66:17–34 [DOI] [PubMed] [Google Scholar]
- 6. Chen S, Tong X, Woodard RW, Du G, Wu J, Chen J. 2008. Identification and characterization of bacterial cutinase. J. Biol. Chem. 283:25854–25862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Egmond MR, de Vlieg J. 2000. Fusarium solani pisi cutinase. Biochimie 82:1015–1021 [DOI] [PubMed] [Google Scholar]
- 8. Griswold KE, Mahmood NA, Iverson BL, Georgiou G. 2003. Effects of codon usage versus putative 5′-mRNA structure on the expression of Fusarium solani cutinase in the Escherichia coli cytoplasm. Protein Expr. Purif. 27:134–142 [DOI] [PubMed] [Google Scholar]
- 9. van Gemeren IA, Beijersbergen A, Musters W, Gouka RJ, van den Hondel CA, Verrips CT. 1996. The effect of pre- and pro-sequences and multicopy integration on heterologous expression of the Fusarium solani pisi cutinase gene in Aspergillus awamori. Appl. Microbiol. Biotechnol. 45:755–763 [DOI] [PubMed] [Google Scholar]
- 10. Sorensen JD, Petersen EI, Wiebe MG. 2007. Production of Fusarium solani f. sp. pisi cutinase in Fusarium venenatum A3/5. Biotechnol. Lett. 29:1227–1232 [DOI] [PubMed] [Google Scholar]
- 11. Calado CR, Mannesse M, Egmond M, Cabral JM, Fonseca LP. 2002. Production of wild-type and peptide fusion cutinases by recombinant Saccharomyces cerevisiae MM01 strains. Biotechnol. Bioeng. 78:692–698 [DOI] [PubMed] [Google Scholar]
- 12. Ferreira BS, Calado CR, van Keulen F, Fonseca LP, Cabral JM, da Fonseca MM. 2003. Towards a cost effective strategy for cutinase production by a recombinant Saccharomyces cerevisiae: strain physiological aspects. Appl. Microbiol. Biotechnol. 61:69–76 [DOI] [PubMed] [Google Scholar]
- 13. Kwon MA, Kim HS, Yang TH, Song BK, Song JK. 2009. High-level expression and characterization of Fusarium solani cutinase in Pichia pastoris. Protein Expr. Purif. 68:104–109 [DOI] [PubMed] [Google Scholar]
- 14. Chen S, Liu Z, Chen J, Wu J. 2011. Study on improvement of extracellular production of recombinant Thermobifida fusca cutinase by Escherichia coli. Appl. Biochem. Biotechnol. 165:666–675 [DOI] [PubMed] [Google Scholar]
- 15. Choi JH, Lee SY. 2004. Secretory and extracellular production of recombinant proteins using Escherichia coli. Appl. Microbiol. Biotechnol. 64:625–635 [DOI] [PubMed] [Google Scholar]
- 16. Thie H, Schirrmann T, Paschke M, Dubel S, Hust M. 2008. SRP and Sec pathway leader peptides for antibody phage display and antibody fragment production in E. coli. N. Biotechnol. 25:49–54 [DOI] [PubMed] [Google Scholar]
- 17. Li Z, Li B, Gu Z, Du G, Wu J, Chen J. 2010. Extracellular expression and biochemical characterization of alpha-cyclodextrin glycosyltransferase from Paenibacillus macerans. Carbohydr. Res. 345:886–892 [DOI] [PubMed] [Google Scholar]
- 18. Yamabhai M, Emrat S, Sukasem S, Pesatcha P, Jaruseranee N, Buranabanyat B. 2008. Secretion of recombinant Bacillus hydrolytic enzymes using Escherichia coli expression systems. J. Biotechnol. 133:50–57 [DOI] [PubMed] [Google Scholar]
- 19. Takemori D, Yoshino K, Eba C, Nakano H, Iwasaki Y. 2012. Extracellular production of phospholipase A2 from Streptomyces violaceoruber by recombinant Escherichia coli. Protein Expr. Purif. 81:145–150 [DOI] [PubMed] [Google Scholar]
- 20. Cheng J, Wu D, Chen S, Chen J, Wu J. 2011. High-level extracellular production of alpha-cyclodextrin glycosyltransferase with recombinant Escherichia coli BL21(DE3). J. Agric. Food Chem. 59:3797–3802 [DOI] [PubMed] [Google Scholar]
- 21. Zhang Y, Chen S, Xu M, Cavaco-Paulo A, Wu J, Chen JA. 2010. Characterization of Thermobifida fusca cutinase-carbohydrate-binding module fusion proteins and their potential application in bioscouring. Appl. Environ. Microbiol. 76:6870–6876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Price MF, Wilkinson ID, Gentry LO. 1982. Plate method for detection of phospholipase activity in Candida albicans. Sabouraudia 20:7–14 [DOI] [PubMed] [Google Scholar]
- 23. Li Z, Gu Z, Wang M, Du G, Wu J, Chen J. 2010. Delayed supplementation of glycine enhances extracellular secretion of the recombinant alpha-cyclodextrin glycosyltransferase in Escherichia coli. Appl. Microbiol. Biotechnol. 85:553–561 [DOI] [PubMed] [Google Scholar]
- 24. Shin HD, Chen RR. 2008. Extracellular recombinant protein production from an Escherichia coli lpp deletion mutant. Biotechnol. Bioeng. 101:1288–1296 [DOI] [PubMed] [Google Scholar]
- 25. Cai Z, Xu W, Xue R, Lin Z. 2008. Facile, reagentless and in situ release of Escherichia coli intracellular enzymes by heat-inducible autolytic vector for high-throughput screening. Protein Eng. Des. Sel. 21:681–687 [DOI] [PubMed] [Google Scholar]
- 26. Martinez A, Ramirez OT, Valle F. 1997. Improvement of culture conditions to overproduce beta-galactosidase from Escherichia coli in Bacillus subtilis. Appl. Microbiol. Biotechnol. 47:40–45 [DOI] [PubMed] [Google Scholar]
- 27. Ramrakhiani L, Chand S. 2011. Recent progress on phospholipases: different sources, assay methods, industrial potential and pathogenicity. Appl. Biochem. Biotechnol. 164:991–1022 [DOI] [PubMed] [Google Scholar]
- 28. Martinez C, Nicolas A, van Tilbeurgh H, Egloff MP, Cudrey C, Verger R, Cambillau C. 1994. Cutinase, a lipolytic enzyme with a preformed oxyanion hole. Biochemistry 33:83–89 [DOI] [PubMed] [Google Scholar]
- 29. Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2:a000414. 10.1101/cshperspect.a000414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lugtenberg EJ, Peters R. 1976. Distribution of lipids in cytoplasmic and outer membranes of Escherichia coli K12. Biochim. Biophys. Acta 441:38–47 [DOI] [PubMed] [Google Scholar]
- 31. Parker SK, Curtin KM, Vasil ML. 2007. Purification and characterization of mycobacterial phospholipase A: an activity associated with mycobacterial cutinase. J. Bacteriol. 189:4153–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.