

Abstract
Peptides presented by human leukocyte antigen (HLA) molecules on the cell surface play a crucial role in adaptive immunology, mediating the communication between T cells and antigen presenting cells. Knowledge of these peptides is of pivotal importance in fundamental studies of T cell action and in cellular immunotherapy and transplantation. In this paper we present the in-depth identification and relative quantification of 14,500 peptide ligands constituting the HLA ligandome of B cells. This large number of identified ligands provides general insight into the presented peptide repertoire and antigen presentation. Our uniquely large set of HLA ligands allowed us to characterize in detail the peptides constituting the ligandome in terms of relative abundance, peptide length distribution, physicochemical properties, binding affinity to the HLA molecule, and presence of post-translational modifications. The presented B-lymphocyte ligandome is shown to be a rich source of information by the presence of minor histocompatibility antigens, virus-derived epitopes, and post-translationally modified HLA ligands, and it can be a good starting point for solving a wealth of specific immunological questions. These HLA ligands can form the basis for reversed immunology approaches to identify T cell epitopes based not on in silico predictions but on the bona fide eluted HLA ligandome.
Peptides presented by human leukocyte antigen (HLA)1 molecules on the cell surface play a crucial role in immunology and mediate the communication between T cells and antigen presenting cells. Knowledge of these peptides is of pivotal importance in fundamental studies of T cell action, the design of T-cell-mediated therapies such as tumor immunotherapy (1), and the treatment of hematological malignancies through a combination of hematopoietic stem cell transplantation and donor lymphocyte infusion (2). In addition, T cells can play an important role in organ rejection following transplantation.
The presented HLA class I ligands are the products of the intracellular processing machinery, with its continuous cycle of protein synthesis and degradation (3). Much is known about the proteins involved in antigen processing, but high fidelity ligand/epitope predictions are at present not possible. The discovery of additional involved enzymes (3, 4) and the exciting discovery of peptide splicing (5) have shown that antigen processing is even more complex than was previously thought. Moreover, gene expression studies have shown many nonstandard, unexpected protein products, including the production of antigens derived from aberrant protein fragments as a result of expression in alternative reading frames (6). Several studies report the identification of HLA ligands (7–10). Many results have been collected and discussed in a recent review on the large-scale analysis of HLA class I ligands (11). Collectively, these reports illustrate the need for in-depth elucidation of the HLA ligandome.
Elucidation of T cell epitopes has traditionally been achieved with the use of a forward immunological approach, as pioneered by Hunt and coworkers (12, 13). In this approach, the cognate peptide of T cells with the appropriate activity profile is elucidated via repeated rounds of chromatographic separation in combination with T cell recognition assays. Because T cells are not always available from the start, reverse immunological approaches (14–17) have been developed to predict T cell epitopes through a combination of bioinformatics and in vitro proteasome digests. Predicted epitopes are synthesized and tested for their capability to activate T cells. The main disadvantage of this approach is that less than 0.1% of the peptides that survive intracellular processing are presented on HLA class I molecules (3).
Therefore, we developed a large-scale peptidomics approach that is a reverse immunology approach based not on algorithms but on the bona fide eluted ligandome, which means that the identified peptides are known to have survived processing and are bona fide HLA ligands. Once the ligandome has been identified as comprehensively as possible, T cells can subsequently be selected on the basis of the immunological question at hand, as will be illustrated in a separate paper.2 The development of MHC exchange tetramers for finding relevant T cell epitopes is instrumental to this approach (18, 19).
To improve ligandome coverage, we applied and compared three off-line first dimension separation techniques, followed by on-line nano-HPLC-tandem MS.
The tandem mass spectra were interrogated by being matched against the International Protein Index (IPI) human database (20). In a second step, post-translation modifications (phosphorylation, cysteinylation) were allowed in the database search. In a third step, the tandem mass spectra were matched against a newly in-house developed database for the optimal identification of polymorphic ligands to find potential minor histocompatibility antigens (21). This led to the identification of ∼14,000 HLA class I ligands, the majority of which also were relatively quantitated. Next, we analyzed the peptides constituting our ligandome in as much detail as possible to confirm the correct identification of the vast majority of the ligands. We achieved this through a combination of several physicochemical and biological checks and comparison with existing ligand and epitope databases.
Finally, as an additional quality check, we illustrated the functional relevance of the ligandome through the identification of both previously known and new minor histocompatibility antigens, virus-derived epitopes, and post-translationally modified HLA ligands (phosphorylated ligands and cysteinylated ligands) (22–24). This is the largest ligandome reported to date, and it allows general insight into the presented peptide repertoire. This study supports the building of the “immunopeptidome” as has recently been suggested (25). A proteomics approach can be used as a starting point for contributions to immunology by providing a peptidome landscape in many immunological studies, both fundamental and applied.
EXPERIMENTAL PROCEDURES
Sample Preparation
The Epstein-Barr virus (EBV)–transformed B lymphoblastic cell lines B-LCL-HHC (typing: HLA-A*0201, B*0702, B*4402, Cw*0501, and Cw*0702) and B-LCL-JY pp65 (typing: HLA-A*0201, B*0702, and Cw*0702) were used as sources of HLA-class I molecules. The CMV-derived pp65 transduced cell line was used to introduce an internal control for the ligandome because the CMVpp65-derived T cell epitopes are known (26). Cells were expanded in roller bottles using Iscove's modified Dulbecco's medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin/streptomycin, and l-glutamine; the cells then were collected, washed with ice-cold PBS, and stored at −80 °C until use.
The hybridoma cell line was expanded in roller bottles to obtain W6/32 (anti-HLA-class I) antibody using protein-free hybridoma medium supplemented with penicillin/streptomycin and l-glutamine. Antibodies produced by the hybridoma cell lines were purified from the supernatant using Prot-A Sepharose beads and eluted from the Prot-A beads with glycine pH 2.5. The eluted antibodies were used to produce an immunoaffinity column (W6/32-Prot-A Sepharose 2.5 mg/ml). The W6/32 antibodies were covalently bound to Prot-A Sepharose beads using dimethylpimelimidate. The columns were stored in PBS pH 8.0 and 0.02% NaN3 at 4 °C.
Isolation of HLA Class I–presented Peptides
The extraction of peptides associated with HLA class I molecules was performed as described elsewhere (13, 27). Briefly, pellets from 60 × 109 B-LCL-JYpp65 cells and 40 × 109 B-LCL-HHC cells were lysed in 50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, and 0.5% Nonidet-P40 (pH 8.0) and supplemented with Complete® protease inhibitor (Sigma Aldrich). The total concentration of the cells in the lysis buffer was 0.1 × 109 cells/ml.
After 2 h incubation with tumbling of the cells in the lysis buffer at 4 °C, the preparation was centrifuged for 10 min at 2500 rpm and 4 °C. The supernatant was transferred to a new tube and centrifuged for 35 min at 11,000 rpm and 4 °C. The supernatant was pre-cleared with CL4B beads and subjected to the immunoaffinity column with a flow rate of 2.5 ml/min. After washing, bound HLA class I–peptide complexes were eluted from the column and dissociated with 10% acetic acid. Peptides were separated from the HLA class I molecules via passage through a 10 kDa membrane (Pall macrosep centrifuge devices). The filtrate was freeze dried. If an oily sample remained after freeze drying, the sample was dissolved and the peptides were further purified via solid phase extraction (C18 Oasis, 100-μl bed volume, (Waters, Milford, MA)). The peptides were eluted from the C18 Oasis column with 500 μl 50/50/0.1 water/acetonitrile (ACN)/formic acid (FA), v/v/v. The eluted peptides from B-LCL-HHC cell lines were divided into two equal portions, freeze dried, and dissolved in 95/3/0.1 water/ACN/FA, v/v/v. The eluted peptides from B-LCL-JYpp65 cell lines were divided into three equal portions, freeze dried, and dissolved in 95/3/0.1 water/ACN/FA, v/v/v.
Peptide Separation
For peptide isoelectric focusing (IEF) separations, an OFFGEL Agilent 3100 fractionator (Agilent Technologies, Waldbronn, Germany) was used. A modified method was applied involving the addition of 1 m urea to the buffer sample and rehydration buffer, instead of 5% glycerol only. Commercially available 13-cm immobilized pH gradient dry strips with a linear pH gradient range of 3–10 (GE Healthcare) were used. The strips were rehydrated with 40 μl/well rehydration solution in the assembled device for 30 min. 150 μl of the prepared samples were loaded on each well, and the cover fluid (mineral oil, Santa Clara, CA) was added onto both ends of the gel strip. The focusing method, OG12PE01, as supplied by the manufacturer, was applied for 12 well fractionations. The performance of the 3100 OFFGEL fractionator was checked under similar conditions in a separate run based on the determination of the pH using a pH indicator (pH 3–10) (Fluka Analytical, Buchs, Switzerland). Fractions were recovered and desalted via solid phase extraction, so as to avoid the presence of oil and gel pieces in the samples, using C18 Oasis columns. The column was prewetted with 10/90 water/ACN v/v and equilibrated with 95/3/0.1 water/ACN/FA v/v/v. The samples were eluted with 50/50/0.1 water/ACN/FA v/v/v, freeze dried, and dissolved in 100 μl 95/3/0.1 water/ACN/FA v/v/v. The 12 fractions generated via IEF were analyzed in triplicate and duplicate for B-LCL-HHC and B-LCL-JYpp65 cell lines, respectively, with nano-LC-MS/MS.
Next to the peptide IEF separation, two chromatographic separation techniques were applied, strong cation exchange chromatography (SCX) and RP-C18 chromatography. For SCX separations, one portion of the eluted peptides from the W6/32 column was fractionated with a homemade SCX column (320 μm inner diameter, 15 cm, polysulfoethyl A 3 μm, Poly LC) run at 4 μl/min. Gradients were run for 10 min at 100% solvent A (100% water/0.1% TFA), after which a linear gradient started to reach 100% solvent B (250 mm KCl, 35% ACN/0.1% TFA) over 15 min, followed by 100% solvent C (500 mm KCl, 35% ACN/0.1% TFA) over the next 15 min. The gradient remained at 100% solvent C for 5 min and then switched again to 100% solvent A. Twenty 4-μl fractions were collected in vials prefilled with 100 μl 95/3/0.1 water/ACN/FA v/v/v.
One portion of the eluted peptides from B-LCL-JY pp65 cell lines was fractionated on a homemade RP Reprosil-Pur C18-AQ column (200 μm inner diameter, 3 μm × 15 cm) (Dr. Maisch, GmbH, Ammerbuch, Germany). The sample was loaded in solvent A (10/90/0.1 water/ACN/FA v/v/v), and the gradient was run from 0%–50% solvent B (10/90/0.1 water/ACN/FA v/v/v) over 30 min at a flow rate of 3 μl/min. The samples were taken up in a make-up flow of 50/50/0.1 water/ACN/FA at 100 μl/min supplied via a T-piece through the annular space between the separation capillary and an auxillary capillary. In this way, 45 fractions, each half a minute wide, were collected; these were subsequently freeze dried and dissolved in solvent A for analysis via nano-LC-MS/MS.
LC-MS/MS Analysis
The dissolved fractions were analyzed via on-line nano-HPLC-MS with a system consisting of a conventional Agilent 1100 gradient HPLC system (Agilent, Waldbronn, Germany), as described by Meiring et al. (28), and an LTQ-FT Ultra mass spectrometer (Thermo, Bremen, Germany). Fractions were injected onto a homemade precolumn (100 μm × 15 mm; Reprosil-Pur C18-AQ 3 μm, Dr. Maisch, Ammerbuch, Germany) and eluted via a homemade analytical nano-HPLC column (15 cm × 50 μm; Reprosil-Pur C18-AQ 3 um). The gradient was run from 0% to 50% solvent B (10/90/0.1 water/ACN/FA v/v/v) over 90 min. The nano-HPLC column was drawn to a tip of ∼5 μm and acted as the electrospray needle of the MS source. The mass spectrometer was operated in data dependent mode, automatically switching between MS and MS/MS acquisition. Full scan mass spectra were acquired in a Fourier transform ion cyclotron resonance mass spectrometer with a resolution of 25,000 at a target value of 5,000,000. The two most intense ions were then isolated for accurate mass measurements by a selected ion monitoring scan in Fourier transform ion cyclotron resonance with a resolution of 50,000 at a target accumulation value of 50,000. The selected ions were then fragmented in the linear ion trap using collision-induced dissociation at a target value of 10,000. In a post-analysis process, raw data were converted to peak lists using Bioworks Browser software, version 3.2.0.
Data Analysis
The tandem mass spectra were matched against the IPI human database (version 3.87) using the Mascot search engine (version 2.2.04, Matrix Science, London, UK) with a precursor mass tolerance of 2 ppm, with methione oxidation as a variable modification and a product ion tolerance of 0.5 Da. For finding post-translationally modified HLA ligands, phosphorylation on serine, threonine, and tyrosine was allowed, and cysteinylation of cysteine was allowed in separate searches. Scaffold software, version 3, was subsequently used to process the Mascot output files and generate spectrum reports. Duplicates were removed, and peptides that had a best Mascot ion score of >35 and were 8 to 11 amino acids long were selected for the production of supplemental Table S1. For the immunological examples, a best Mascot score of >20 was selected, and the length was restricted to 8 to 18 amino acids. In addition to the abovementioned procedure, Proteome Discoverer 1.3 (Thermo, Bremen, Germany) was used to extract all identified peptides from the input *.RAW files, using the Mascot server mentioned above, and calculate their intensity, as reported in supplemental Table S1. False discovery rates were as determined by Proteome Discoverer for the Homo sapiens–extracted Uniprot/SWISSProt database (29) (release 2010_11) containing 20,259 protein sequences and the IPI human 3.87 database (see also supplemental Table S2). Icelogo (version 1.2) was used to generate the binding motifs as presented in Fig. 5 and supplemental Fig. S1 (30). The GRAVY index was calculated using the Protein GRAVY sequence manipulation suite. The pI values of the identified peptides were calculated using the ExPASy “Compute pI/Mw” tool. NetMHC 3.2 was used to predict the binding affinity (nm) of the identified peptides to HLA-A*0201, HLA-B*0702, and HLA-B*4402. NetMHCpan 2.4 was used to predict the binding affinity of the identified peptides to HLA-C*0501 and HLA-C*0702. Overall protein turnover values were taken from the work of Cambridge et al. (31). PhosphoSite (32) and Phospho.ELM 8.3 (33) were used to find known phosphosites in the identified peptides. NetPhos 2.0 (34) was used to predict phosphosites in identified phosphorylated ligands. For the identification of polymorphic peptides, the tandem mass spectra were matched against HSPVdb, a database optimized for finding polymorphic peptides (21). For searching CMV pp65-derived ligands/epitopes in B-LCL-JYpp65, a separate database was constructed containing only the DNA sequence of the pp65 protein (NCBI; pp65_AD169_seq with intron Human herpesvirus 5, complete genome). For searching EBV-derived epitopes, a separate database was constructed containing the DNA sequence of EBV selected from the RefSeq database (35) (>gi 82503188 ref NC_007605.1 Human herpesvirus 4 type 1, complete genome).
Fig. 5.

Binding motif of various HLA-A2 length variants. Icelogo plots of HLA-A2 binding peptides of 9-mer (n = 3302), 12-mer (n = 137), and 14-mer (n = 17) length. Peptides of different lengths clearly display similar binding motifs.
Peptide Synthesis
Peptides were synthesized according to standard fluorenylmethoxycarbonyl (Fmoc) chemistry using a SyroII peptide synthesizer (MultiSynTech, Witten, Germany). The integrity of the peptides was checked using reverse-phase HPLC and MS. Phosphopeptides were synthesized using the building blocks Fmoc-Ser(PO(OBzl)OH)-OH or Fmoc-Thr(PO(OBzl)OH)-OH. Couplings of these building blocks were identical to normal couplings, with one exception: a 3-fold excess of N-methylmorpholine was used instead of the routinely applied 2-fold excess.
RESULTS
The Peptides Constituting the HLA Ligandome
The pool of HLA peptides eluted from two EBV-transformed B lymphoblastoid cell lines (EBV-B-LCL), B-LCL-JYpp65 and B-LCL-HHC, is quite complex. From previous experiments, a simple count of the total number of peaks in the MS1 spectra in all fractions following a first dimension C18-separation resulted in a number of ∼30,000. Because of this complexity, multidimensional separations were performed to reduce the complexity before mass spectrometric analysis. Three first dimension separations were chosen, reverse-phase C18 (RP-C18) chromatography, SCX, and peptide IEF; these are based on a different separation mechanism and are the most commonly used. The second dimension separation was RP-C18 chromatography coupled on-line to the mass spectrometer in all cases. Below, the results of the three first dimension separations in combination with the second dimension on-line HPLC-MS are compared and discussed.
Peptide IEF as the First Dimension
Before we started our IEF experiments with HLA-eluted peptides, the separation performance of the OFFGEL 3100 system was tested with a trypsin-digested cell lysate using 13-cm immobilized pH gradient strips with a pH range of 3–10 (GE Healthcare). Separation efficiency was studied as a function of loading. The results were as reported in Ref. 36, with ∼83% of peptides present in a unique fraction and 96% present in one or two unique fractions, with peptide amounts up to 100 μg, confirming the high separation efficiency of the IEF process. Peptide loading below 10 μg results in considerable sample losses, which precludes the use of IEF for HLA-ligandome studies of smaller cell amounts. The application of a pH indicator (pH 3–pH 10) after IEF fractionation showed that the low-pH side of the strip was not pH 3, and was actually close to pH 4.
The 12 fractions obtained from the IEF fractionation were analyzed via on-line nano-HPLC-MS/MS, and the tandem mass spectra were matched against the IPI human protein database. The results for both B-LCLs are summarized as Venn diagrams in Figs. 1A and 1B. As can be seen, analysis of the B-LCL-HHC ligandome yielded 6878 unique 8- to 11-mer peptides with BMI > 35, and the B-LCL-JYpp65 ligandome yielded 3531 unique peptides. The large difference in the number of peptides found for B-LCL-JYpp65 and for B-LCL-HHC can be largely explained by the additional HLA molecules expressed on B-LCL-HHC relative to B-LCL-JY cells. B-LCL-JY is homozygous for the B allele (HLA-B7*0702) and C allele (HLA-Cw*0702), whereas B-LCL-HHC is heterozygous for the B allele (HLA-B*0702 and HLA-B*4402) and C allele (HLA-Cw*0501 and HLA-Cw*0702). The additional HLA molecules can present two additional sets of peptides.
Fig. 1.

Venn diagrams of identified HLA ligands per separation. The number of identified unique peptides per mass spectrometric replicate for IEF (A), SCX (C) and RP-C18 (E) for cell line B-LCL JYpp65, and for IEF (B) and SCX (D) for cell line B-LCL HHC. The area of the circles corresponds with the number of peptides. Total, fractional, and overlapping numbers of unique peptides are indicated.
SCX as the First Dimension
Twenty fractions were collected after separation on the SCX column. These fractions were analyzed, like the IEF fractions, by means of on-line nano-HPLC-MS/MS and were matched against the IPI human protein database. The results for both B-LCLs are summarized as Venn diagrams in Figs. 1C and 1D. Analysis of the B-LCL-HHC ligandome yielded 7766 unique peptides (BMI > 35), and the B-LCL-JYpp65 ligandome yielded 3410 unique peptides (BMI > 35).
RP-C18 as the First Dimension
Forty-five fractions were collected after separation on the RP-C18 column. These fractions were analyzed, like the IEF and SCX fractions, via on-line nano-HPLC-MS/MS, and the tandem mass spectra were again matched against the IPI human protein database. Analysis of the B-LCL-JYpp65 ligandome yielded 2725 unique peptides (BMI > 35). The results for B-LCL-JY are summarized as a Venn diagram in Fig. 1E. The B-LCL-HHC ligandome was not measured with the combination of RP-C18 and on-line nano-HPLC.
Combination of the Results of IEF, SCX, and RP-C18
To get an overview, a non-redundant list of all peptides obtained via the abovementioned first dimension separation techniques was produced. The results are shown as Venn diagrams in Fig. 2. The analysis of the B-LCL-HHC ligandome yielded 10,867 unique peptides (BMI > 35) (Fig. 2A), and the B-LCL-JYpp65 ligandome yielded 6493 unique peptides (BMI > 35) (Fig. 2B). Combination of the two B-LCL-derived peptide lists yields a non-redundant peptide list containing 14,065 members (Fig. 2C), of which 11,511 could be relatively quantified. This is by far the most comprehensive list of HLA ligands reported to date. A complete listing of the HLA-presented peptides, sorted by intensity, is given in supplemental Table S1, including the protein name and IPI accession number they are derived from, their intensity in B-LCL-JY and/or B-LCL-HHC, the BMI score, the binding affinity predicted by NetMHC, the GRAVY hydrophobicity value, the protein half-life, the copy number per cell calculated from the intensity and work-up yield, the peptide length, whether peptides are present in the Immune Epitope Database (IEDB) (37) and/or SYFPEITHI database (38), and the false discovery rate (FDR) calculated using the Uniprot/SWISSProt database and IPI human database. In addition, we compared our data with those reported in Refs. 7–10.
Fig. 2.

Venn diagrams of cumulative number of identified HLA ligands. Cumulative number of identified unique peptides per cell line for B-LCL HHC (A) and cell line B-LCL JYpp65 (B). C, cumulative number of identified unique peptides for B-LCLs JYpp65 and HHC. The area of the circles corresponds with the number of peptides. Total, fractional, and overlapping numbers of unique peptides are indicated.
The NetMHC algorithm was used to predict the HLA-binding affinity of the ligands. It is clear from the results that the majority of peptides are binders in particular HLA molecules (see supplemental Table S1). In supplemental Table S1, a NetMHC cut-off score of 1000 is used.
The number of peptides found in every experiment, independent of the work-up procedure, was 835, of which 770 could be quantified (Fig. 2B). Because of the shotgun nature of the experimental approach, these 835 peptides are expected to be the most abundant on average; the peptides detected in either overlap of two experiments are expected to be less abundant, and the peptides detected only in a single first dimension experiment are expected to be the lowest. This was checked for the peptides presented on B-LCL-JY, and the result is displayed in Fig. 3.
Fig. 3.

Shotgun nature of the experiment. The weighted intensity distribution of the peptides found in the various regions in the Venn diagrams of Fig. 2B. Three regions were defined (x-axis): (1) peptides found with only a unique first dimension separation technique (i.e. the “outer” regions), (2) peptides found in either of two overlapping regions, and (3) peptides always found irrespective of the first dimension separation technique. Average peptide intensity increases from region 1 to region 3, as expected in a shotgun-type of proteomics experiment. The median peptide intensity is indicated.
Peptide Length Distribution
Our large set of peptides allows a closer look at the length distribution of peptides constituting the HLA ligandome, as presented in Fig. 4. As can be seen, most ligands have a length of 9 amino acids, whereas a substantial part is either 10 or 11 amino acids long. The 8-mers and 12- to 14-mers are all below 5%. However, 708 long peptides (>11 amino acids) (amounting to 5% of the ligandome) are present that have the correct anchors for binding to the HLA-A and -B alleles. Fig. 5 shows the HLA-A2 binding motif for the 9-, 12-, and 14-mers. All different peptide lengths display the same binding motif, and a binding motif can still be discerned for 15-mers.
Fig. 4.

Peptide length distribution of the HLA ligandome. Of note are the longer peptides and the relatively high number of HLA-B7 binding peptides of 8-mer length, and the relatively wide spread of HLA-B44 binding peptides over 9 to 11 amino acids length. Number of 13-mers ∼ 80 per allele; number of 14-mers ∼ 20 per allele.
The Dynamic Range of the HLA Ligandome
In order to get an impression of the dynamic range of the presented peptide repertoire, the intensities of the presented peptides in the ligandome were determined. To this end, the *.RAW files were processed in Proteome Discoverer. A complete listing of all peptides, including their intensities, presentation on the particular B-LCL, and BMI scores, is given in supplemental Table S1. Intensity extremes, expressed as peak area values in Proteome Discoverer, range from 4E9 to 1E5, but the majority range from 1E9 to 1E6, with a range of a factor of 1000. So the dynamic range of the peptide repertoire is quite high.
The summed intensity (integration value) of all peptides eluted from 2E10 B-LCL-JY or B-LCL-HHC cells is ∼3E11, as reported by Proteome Discoverer (see supplemental Table S1). To determine to what amount of peptide this number corresponds, we followed the same analytical procedure with a known amount of a mix of 20 synthetic peptides. An integration value of 1E8 as reported by Proteome Discoverer corresponds to 1 pmol. From this we determined that the total eluted peptide amount was 3 nmol (3E11/1E8).
The composition of the ligandome can also be represented in terms of the relative intensity of the peptides, as shown in Fig. 6A. The intensities have been translated to copy numbers in Fig. 6B.
Fig. 6.
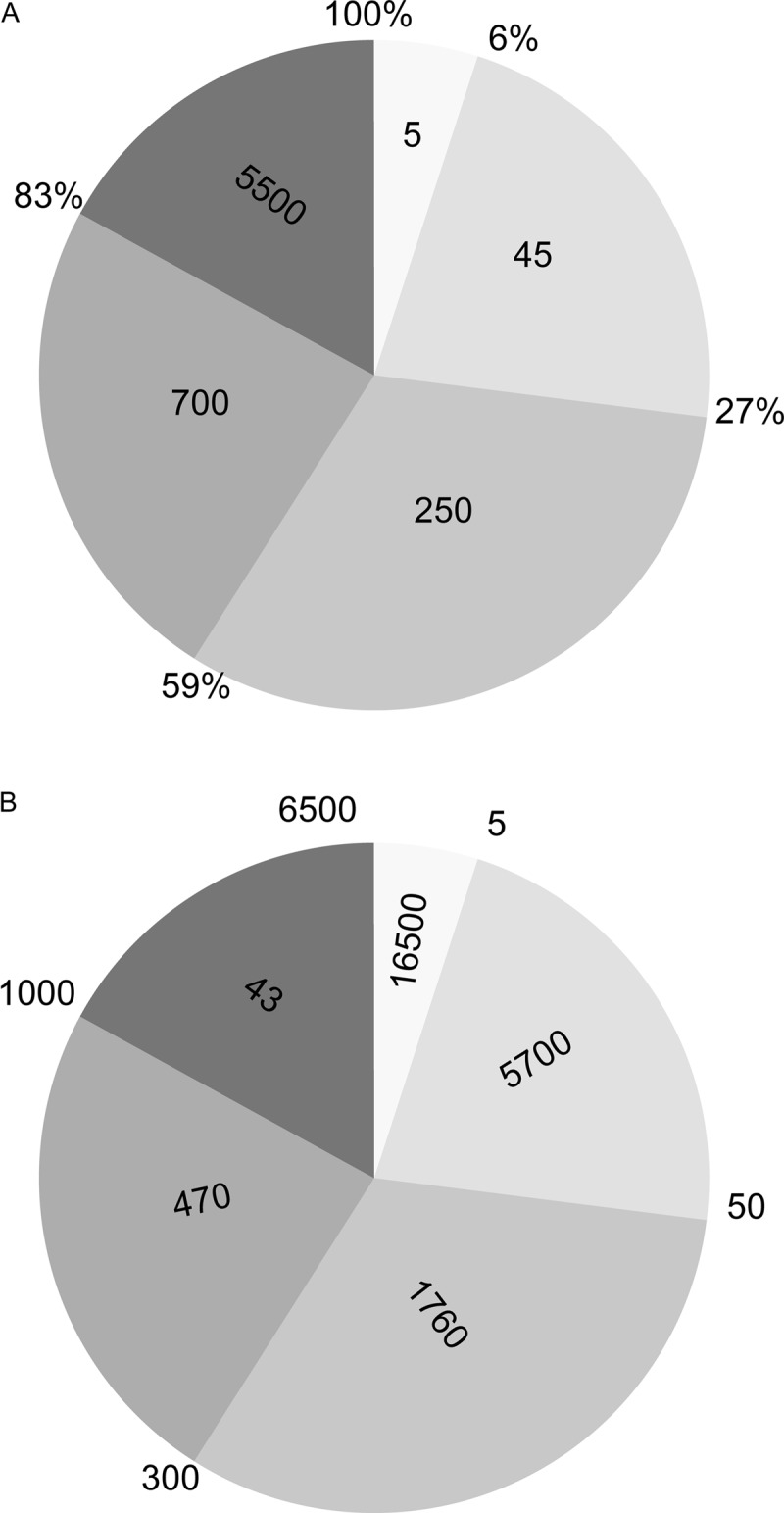
Occupation of the ligandome. The intensity distribution of peptides constituting the ligandome is shown for B-LCL JYpp65. A, the outer rim of the pie chart displays the percentage occupation of the HLA ligandome, and the number within each pie segment is the number of unique peptides in that segment. Of the total number of 6500 identified peptides, the top five abundant proteins occupy 6% of all HLA molecules, the top 50 abundant peptides occupy 27% of all HLA molecules, etc. Note that the lowest abundant 5500 peptides occupy only 17% of all HLA molecules. B, the numbers in panel A have been converted to copy numbers (i.e. the number of ligands per cell). The outer rim of the pie chart displays the number of unique peptides, and the number within each pie segment is the average copy number for the peptides in that segment. The top five abundant peptides are on average present with 16,500 copies on the cell surface, peptides 5–50 are on average present with 5700 copies, etc. The average copy number of the lower abundant peptides is 43; the very low abundant peptides are present at copy numbers close to 1 or even less.
The 14,065 HLA ligands are derived from 7059 different proteins; a graphical representation is shown Fig. 7A. In Fig. 7B, the average protein length is plotted against the number of peptides/protein. The higher the protein mass, the higher the number of derived HLA ligands. Sixty-one proteins are even represented by 10 peptides or more on the cell surface, with the exceptional case of 29 and 41 peptides/protein. The intensity of the peptides derived from the same protein varied greatly, as presented in Table II.
Fig. 7.

Representation of proteins in the HLA ligandome. A, this figure depicts the number of proteins as a function of the number of peptides identified per protein. HLA ligands derived from 7059 unique proteins are in our dataset. In our dataset, about 50% of the proteins are represented by one peptide, 20% are represented by two peptides, 10% are represented by three peptides, and so on. As can be seen, there is a gradual decrease in the number of peptides by which a protein is represented. Up to 41 peptides/protein are present in our dataset. B, the protein length is depicted as a function of the number of peptides by which the protein is represented in HLA. The number of peptides in HLA of a protein correlates well with its protein mass.
Table II. Peptide intensities derived from the same protein and intensities of different HLA-presented peptides from some selected proteins. As shown in Fig. 7, proteins can be represented by several peptides in the HLA ligandome. Here, three proteins were chosen that are represented by seven, five, and fvie peptides in HLA in B-LCL-HHC. As can be seen, these peptides can be presented by different alleles and have greatly differing hydrophobicity. Importantly, peptides derived from the same protein differ greatly in abundance. So, the abundance of an HLA ligand derived from a certain protein does not correlate with simply its protein intensity.
| Protein name | Peptide sequence | Intensity | HLA allele | BMI | NetMHC (nM) | Gravy |
|---|---|---|---|---|---|---|
| Isoform 1 of clathrin heavy chain 1 | NENSLFKSL | 7.60E+07 | B44 | 59 | 359 | −0.622 |
| NPASKVIAL | 2.64E+07 | B7 | 51 | 13 | 0.7 | |
| AEELFARKF | 2.27E+07 | B44 | 39 | 351 | −0.267 | |
| EEAFAIFRKF | 6.45E+06 | B44 | 47 | 17 | 0.11 | |
| AETGQVQKI | 5.32E+06 | B44 | 41 | 671 | −0.556 | |
| FVHDLVLYL | 3.30E+06 | A2 | 41 | 13 | 1.622 | |
| LLMVLSPRL | 1.09E+06 | A2 | 43 | 16 | 1.6 | |
| Mediator of RNA polymerase II transcription subunit 24 | SLVEQLTMV | 1.98E+07 | A2 | 70 | 12 | 1.044 |
| SPTDRSMSSSL | 1.87E+07 | B7 | 56 | 10 | −0.782 | |
| KERWSDYQW | 6.45E+06 | B44 | 36 | 123 | −2.533 | |
| AEQMHKTGF | 3.59E+06 | B44 | 53 | 594 | −0.967 | |
| SLMDPPGTAL | 1.31E+06 | A2 | 35 | 31 | 0.27 | |
| Isoform 1 of Dedicator of cytokinesis protein 2 | KMWEEAISL | 3.42E+08 | A2 | 58 | 9 | −0.067 |
| SPRLSQTFLQL | 5.26E+07 | B7 | 52 | 21 | −0.109 | |
| IPGMVGPIL | 5.20E+07 | B7 | 36 | 89 | 1.656 | |
| LEAYIQQHF | 4.28E+06 | B44 | 39 | 145 | −0.233 | |
| ILLQVAALKYI | 1.71E+06 | A2 | 49 | 63 | 1.773 |
Physicochemical Properties of the Peptides Composing the HLA Ligandome
To check whether peptide detection depended on the peptide physicochemical properties with either one of the employed first dimension separation techniques, we studied the influence of isoelectric point and hydrophobicity.
To study the influence of the isoelectric point of a peptide, we calculated the theoretical pI values of all identified ligands. The calculated pI values ranged from 3.2 to 10.4. When the peptides were assigned to pI bins as shown in Fig. 8A, it was immediately clear that ligands with pI extremes migrated out of the pI strip (these were not detected with IEF as in the first dimension separation technique, but they were detected using SCX and C18 as in the first dimension separation technique) and were therefore lost for detection when using IEF. In addition, the actual pH at the low-pH side of the strip was not 3 but almost 4, which accounts for extra losses experienced with the application of IEF. Because peptides binding to HLA-B44 are on average more acidic, they are more likely to be lost in IEF.
Fig. 8.

HLA ligandome physicochemical properties. A, distribution of HLA peptides over the pI range as a function of the first dimension separation technique used. As can be seen, IEF, SCX, and RP-C18 do not discriminate among peptides based on their theoretical pI. The only exception is that peptides with low pI values are clearly underrepresented in the IEF process. This is caused by the actual pI in the pI 3–10 strips, which in fact appeared to be pI 4–10, as also independently checked with a pH indicator after IEF. B, distribution of HLA peptides over the hydrophobicity range depending on the first dimension separation technique used. As can be seen, IEF, SCX, and RP-C18 do not discriminate among peptides based on their hydrophobicity. The plots illustrate that our HLA ligandome is a good representation of the real HLA ligandome.
Next, we studied the influence of peptide hydrophobicity on the peptide's chance of being detected. To this end we calculated the GRAVY index of all identified peptides and plotted them in hydrophobicity bins as shown in Fig. 8B. As can be seen, the distribution of the peptides is evenly spread independent of the first dimension separation technique employed. The more hydrophilic peptides are slightly disfavored by RP-C18 as the first dimension separation technique. In summary, peptides with pI extremes might be lost during IEF, but generally the majority of peptides were detected regardless of the first dimension separation technique used.
The Overall Quality of the Presented HLA Ligandome Data
To illustrate that the majority of the peptides reported here were indeed derived from the expected HLA molecules, we have shown that these peptides carry the correct anchor residues required for binding. Another way to verify the overall quality of the ligandome reported is to check the predicted binding affinity for the HLA molecule. In addition, the overall physicochemical characteristics of the peptides bound depend on the particular HLA molecule. The HLA-binding affinity can be approximated theoretically by the NetMHC algorithm. In Fig. 9, the hydrophobicity distribution for HLA-A2, HLA-B7, and HLA-B44 predicted peptides is plotted. The HLA-A2 presented peptides are centered on a GRAVY value of +1, those from HLA-B7 on 0, and those from HLA-B44 on −1.
Fig. 9.

Allele-specific distribution of HLA peptides over the hydrophobicity range. These plots show a clear average hydrophobicity shift going from HLA-B44 (most hydrophilic, median GRAVY index of −0.84) to HLA-B7 (median GRAVY index of −0.08) to HLA-A2 (median GRAVY index of 0.64). The positions of the three distributions are as expected and illustrate the overall quality of our dataset.
In addition to the overall quality check described above, we compared our data on the individual peptide level with the two main public sources of HLA ligands, which are the SYFPEITHI database and the IEDB. These databases are collections of ligands and/or epitopes that result from different immunological experiments on a variety of cell types and generally are fed not by proteomics-type experiments but by individual immunological reports. To compare our peptide lists with the SYFPEITHI database, we selected the reported HLA-A2, HLA-B7, and HLA-B44 ligands and compared them to our eluted peptide sequences. In addition, we compared our results with the peptides presented in papers by Scull et al. on B cells (7), Hillen et al. on HLA-B44 supertypes on B cells (8), Granados et al. on B cells (9), and Bade-Doeding et al. on B cells (10). The results of this comparison are shown in supplemental Table S1. We found 253 of the 784 ligands described in the SYFPEITHI database, and 72 out of 263 ligands described in the IEDB. Out of the cumulative 1179 (8- to 11-mer) selected HLA-A2 ligands recently published by Scull et al., 889 were found in our dataset (7).
Immunological and Biological Value
Below a number of immunologically relevant ligands are described. These include minor histocompatibility antigens and virus-derived epitopes.
Minor Histocompatibility Antigens
Minor histocompatibility antigens (MiHA) are peptides derived from polymorphic proteins and are relevant in allogeneic hematopoietic stem cell transplantation as targets for immunotherapy for the treatment of hematological malignancies (2, 13, 39–42).
In order to find and identify polymorphic peptides in the eluted peptides from B-LCL-HHC and B-LCL-JYpp65 cell lines, the tandem mass spectra were matched with a dedicated database, HSPVdb (21). We found 1439 polymorphic peptides of 8- to 11-mer length and a Mascot ion score of >35, accounting for 10% of the peptides in the ligandome. Six out of 16 MiHA and four allelic counterparts of known minor antigens, which could potentially be present on our cells, considering the SNP-typing of the B-LCL, were found in our eluted dataset; this was confirmed by matching the MS/MS spectra of the synthetic peptides with those of the eluted peptides. These peptides are listed in Table I. It should be noted that 3 out 10 of the peptides listed have a BMI of <35 but were correctly identified.
Table I. Listing of known MiHA and allelic counterparts of known MiHA identified in our dataset by searching the HSPV database (21), illustrating the immunological relevance of our dataset; in addition, several potential MiHA are present in our dataset that form the basis for finding new clinically relevant MiHA.
| Gene name | HLA allele | Peptide sequence | Eluted cell line | BMI |
|---|---|---|---|---|
| HA-2V | A*02:01 | YIGEVLVSV | B-LCL-JY, B-LCL-HHC | 50 |
| HA-8R | A*02:01 | RTLDKVLEV | B-LCL-JY | 34 |
| LB-SSR1-1S | A*02:01 | VLFRGGPRGSLAVA | B-LCL-HHC | 29 |
| SSR1-1L* | A*02:01 | VLFRGGPRGLLAVA | B-LCL-JY | 37 |
| LB-ERAP1-1R | B*07:02 | HPRQEQIAL | B-LCL-JY, B-LCL-HHC | 36 |
| LB-GEMIN4-1V | B*07:02 | FPALRFVEV | B-LCL-JY | 43 |
| SMCY-B7 | B*07:02 | SPSVDKARAEL | B-LCL-JY | 58 |
| EBI3-1V* | B*07:02 | RPRARYYVQV | B-LCL-JY, B-LCL-HHC | 26 |
| PDCD11-1L* | B*07:02 | GPDSSKTLLCL | B-LCL-JY | 39 |
| ACC-2G* | B*44 | KEFEDGIINW | B-LCL-HHC | 81 |
* The allelic counterpart of known MiHA.
Virus-derived Peptides
Because EBV-transformed cell lines were used in this study, our tandem mass spectra were matched with a specific database containing the genomic EBV information. Four EBV peptides were found in our data (see supplemental Table S3). The binding affinity of the newly identified peptides was predicted using NetMHC. Two peptides displayed high binding affinity for HLA-A2, and the other two showed high binding affinity for HLA-B7. The correct identification of these four peptides was confirmed by matching the tandem mass spectra of the eluted peptides with the spectra of their synthetic counterparts. These four EBV-derived peptides have not been reported before.
The B-LCL-JY cell line used in this study was transduced with the CMVpp65 protein, which gave us the opportunity to study the presentation of CMVpp65-derived ligands. To identify pp65-derived ligands, we matched the tandem mass spectra with a specific database containing the pp65 sequence. We could find two known pp65-derived epitopes (see supplemental Table S3). One epitope is presented in HLA-A2, and the other epitope is presented in HLA-B7 (26). Again, the identification of these two epitopes, with BMIs of 28 and 12, respectively, was confirmed by matching the tandem mass spectra of the eluted peptides with the spectra of their synthetic counterparts.
In summary, we identified four new EBV-derived ligand peptides and two known epitopes derived from the transduced CMVpp65 protein. The EBV peptides might be candidates for new EBV epitopes. It is important to note that low-scoring peptides should not be discarded lightly in this field of application without checking their functional relevance.
Independent immunological testing in combination with the synthesis of good candidates provides a good strategy for excluding false positive identifications without losing the immunologically relevant false negative identifications.
Together, the above findings clearly illustrate that our list of ligands harbors immunologically relevant peptides, suggesting that many more relevant peptides are in this list.
Post-translationally Modified HLA Ligands
The presentation of post-translationally modified peptides has been reported before (22, 24, 43), although not in large numbers. Therefore, we investigated our tandem mass spectra for the phosphorylation of serine, threonine, and tyrosine or the cysteinylation of cysteine residues, as both modifications have been reported to influence T cell recognition (23, 43).
Aberrant phosphorylation has been implicated in cancer. The inclusion of phosphorylation in our database-matching process yielded 451 phosphopeptide hits with lengths of 8 to 11 amino acids with a BMI > 35 (phosphorylation on anchor positions was not allowed), of which 221 were estimated to be correctly identified (see supplemental Table S4 and “Discussion”). Similarly, a cysteinylated cysteine as a post-translational modification was set as a modification in the database matching, which resulted in 1221 identified peptides with a cysteinylated cysteine (cysteinylation on anchor positions was not allowed). The peptides are listed in supplemental Table S5.
Phosphorylated HLA Ligands
The inclusion of phosphorylation in the database-matching process yielded 451 phosphopeptide hits with lengths of 8 to 11 amino acids with a best Mascot score of ≥35, as listed in supplemental Table S4. The distribution was as expected: 267 on serine, 154 on threonine, and 30 on tyrosine. Most peptides were singly phosphorylated, but 13 peptides were doubly phosphorylated.
To evaluate whether these phosphopeptides were properly assigned, we compared our results with previously reported phosphopeptides listed at PhosphoSite. This yielded 72 hits. Phospho.ELM 8.3 yielded 38 hits, all of which were also present in PhosphoSite. Next we used NetPhos 2.0 to predict phosphosites in the peptides, which resulted in 83 phosphosites, of which 28 overlapped with the 72 phosphosites detected with the two other sites mentioned above. In total, 127 out of 451 phosphopeptides with either a known or a predicted phosphosite were found.
To confirm the identification of the phosphorylated peptides in our dataset, we chose to synthesize a selection of peptides and compare the synthetic phosphopeptides with their eluted counterparts. Twenty-six peptides were selected, of which there were 9 peptides with a known or predicted phosphosite and 17 peptides without a known or predicted phosphosite, with varying phosphorylation positions in the peptide sequence and different Mascot ion scores. These were synthesized, and the MS/MS spectra of these peptides were matched with those of their eluted counterparts. The nine peptides with a known or predicted phosphosite appeared to be correctly identified. Of the other 17 peptides without known or predicted phosphosites, 29% (5 out of 17) were found to be correctly identified.
First the 127 hits found in our study, which were also either known from previously identified proteomics studies or predicted, were extrapolated to be correctly identified, leaving (451 − 127) = 324 hits. Of these 324 hits, 29% (= 94 hits) were extrapolated to be correct. Therefore, we estimate that 221 out of 451 (49%) have been correctly identified.
Cysteinylation of HLA Ligands
To find peptides with a cysteinylated cysteine as post-translational modifications in our eluted dataset, we searched the dataset obtained from B-LCL-HHC and B-LCL-JYpp65 cell lines with variable modification on cysteinylation (+119 Da). We found 1221 identified peptides with cysteinylation (see supplemental Table S5). As the binding affinity of the peptides probably does not change after cysteinylation, we used the NetMHC server to check the binding affinity of these peptides. The vast majority were predicted to bind to HLA-A2 (594), HLA-B7 (143), or HLA-B44 (294).
DISCUSSION
The Quality of the Data
The quality of the elucidated ligandome was checked on three independent levels: first, through the application of FDRs; second, by consideration of chromatographic criteria in combination with the correct distribution over physical parameters; and third, based on the predicted binding strength. Additionally, the presence of biologically relevant peptides illustrates the good quality of our dataset, but these peptides are discussed separately.
MS in HLA Ligandomics and Choice of FDRs
HLA-presented peptides arise from a complex antigen processing process involving many enzyme specificities. Therefore, enzyme restriction during the database matching process is not possible, leading to a large increase in database search space. The best way to reduce the number of falsely identified peptides is to apply a strict precursor mass tolerance of 2 ppm, which was achieved by including a selected ion monitoring scan in the Fourier transform MS measurements. Strict application of a tight FDR of 1%, or even 5%, as in trypsin-based proteomics experiments, leads to loss of a wealth of valuable peptides (false negatives), as we illustrate below. In our opinion, this is unacceptable in immunological workflows. We have chosen a BMI > 35, which we know from experience is a decent starting cut-off. Many known immunologically relevant peptide ligands are still found far below this limit. In supplemental Table S1, we list peptides with a BMI > 35, but we also indicate below what FDR percentage the peptides are, based on searches of either the smaller SWISSProt (Homo sapiens) or the larger IPI human database. We included 1%, 5%, and 10% FDRs. The results of these searches and the effects of the different search conditions are summarized in supplemental Table S2. A 10% FDR was applied by Dudek et al. in an HLA class I peptidomics-based study (44). An FDR of 37% was used in an HLA-class-II-based peptidomics study by Chornoguz et al. (45). Importantly, in targeted immunological approaches (i.e. searching for ligands/epitopes or post-translationally modified epitopes derived from a specific antigen), we advise not using a strict statistical cut-off score, be it a BMI or an FDR, during the first discovery phase. In the next step, first-round “hits” should be tested independently, be it via MS/MS analysis of the synthetic candidate epitope or a biological assay (e.g. a T cell activity assay). As illustrated by the BMI scores < 35 for 3 out of 10 validated MiHA (and 6 out of 10 with BMI < 39) and both CMV peptides, with one CMV peptide having a BMI of 12 (!), low-scoring peptides should be prevented from being assigned as false negatives. It is important to note that initially wrongly assigned peptides will immediately be filtered out by immunological follow-up experiments. Aside from to the MS methodology available, additional non-MS checkpoints were applied to estimate the value of a peptide (see below).
Chromatography and Physicochemical Properties
Sample (pre)treatment might inadvertently favor peptides with certain physicochemical properties. Therefore, we checked the distribution of our peptides over the hydrophobicity scale (GRAVY index) (see Fig. 8B) and pI scale (see Fig. 8A). The peptides were evenly distributed, so sample pretreatment did not favor peptides on the basis of their physicochemical properties. However, peptides at the pH extremes of the IEF strips were lost. It appeared that the real pH range of the strips was not 3–10 but instead closer to pH 4–9, which would explain this loss. The hydrophobicity distribution of the peptides is shown in Fig. 9 for peptides eluted from each of the three alleles used in this study. The center of the hydrophobicity distribution clearly shifted to the hydrophobic side for HLA-A2 and to the hydrophilic side for HLA-B44. The HLA-B7 peptides were at intermediate hydrophobicity. This is in perfect agreement with the general notion that HLA-A2-presented peptides are more hydrophobic (46), whereas HLA-B44-derived peptides are hydrophilic and HLA-B7 peptides have intermediate hydrophobicity (47–50). The relative positions of the hydrophobicity distributions support the correct overall assignment of the ligandome.
In addition to identifying the peptides, we determined their relative intensity. Because of the shotgun nature of the experiments, peptides found in three experiments (the 835 peptides found in Fig. 2B) are expected to have a higher intensity than those peptides present in only one experiment. The results as shown in Fig. 3 clearly illustrate that this was the case in our experiments.
Binding Prediction
Because of the large number of peptides found, peptide identities could not be verified based on MS/MS of their synthetic counterparts. As an alternative, the NetMHC algorithm was used to predict binding affinity. As can be seen in supplemental Table S1, the large majority of our reported peptides had low NetMHC scores (i.e. they were predicted to be (strong) binders). Although on the NetMHC website a score of <50 is considered indicative of a strong binder and scores between 50 and 500 denote a weak binder, from our experience with known epitopes, we know these values are too strict.
Based on the mass spectral quality, the distribution of physicochemical parameters, the expected shotgun distribution in which the overlapping regions in the Venn diagram represent the most abundant peaks, and the excellent binding affinity, we are convinced that the sample pretreatment and mass spectrometric analysis did not result in skewing of the identified ligandome as a whole. The fact that many known biologically relevant peptides were found is another strong indication of the high quality of our ligandome.
Ligandome Characteristics
Our large dataset introduces opportunities to discuss a number of properties of the HLA ligandome. It is important to realize that the peptides we report here are the survivors of the intracellular processing. Therefore, this listing is valuable for studies on antigen processing and to all in silico studies on ligand/epitope definition and the definition of antigen processing rules. In addition, it can be used to determine whether peptides from specific protein antigens can survive intracellular processing and are presented on the cell surface. Thus, it provides an indirect view of cellular processes. Many more peptides are probably presented in HLA molecules, but they might escape detection because of low abundance and/or relatively low sensitivity in the electrospray ionization process.
The Venn diagrams in Fig. 2 still display relatively large regions unique to either first dimension separation technique, which shows that many peptides are just detected or might still escape detection. Although the ligandome presented here is the most comprehensive reported to date, we interpret the occurrence of many unique peptides in a single type of “precursor dimension” as a strong indication that the HLA ligandome must contain many more peptides than we report. This notion is supported by our experience with three- and four-dimensional separations before MS analysis in the classical forward approach to find T cell epitopes. Every additional chromatographic dimension yields peptides not detected in precursor dimensions, indicating that a significant part of the presented HLA ligandome still goes undetected. Based on the current listing of 14,000 peptides and the many false negatives (i.e. correct peptides with a BMI < 35 and the peptides going undetected), we estimate that the HLA ligandome comprises at least 3 times more peptides (i.e. at least 50,000 members).
Binding Motif and Peptide Length
The ligandome contains a sizeable fraction of unusually long peptides for HLA class I binding. We found that 5% of the peptides were longer than 11 amino acids. This is in line with the results obtained by Scull et al. for a smaller set of HLA-A2 peptides (7).
In supplemental Fig. S1, the Icelogo graphs show the HLA groove can accommodate at least up to 14-mers. Peptides need to bulge out of the groove considerably to accommodate the extra amino acids between the fixed anchors at position 2 and the C terminus, as shown beautifully by crystallographic data (51). Our data on the peptide length distribution (Fig. 4) underline the need to take into account longer peptides in the search for epitopes, in both practical MS-based strategies and in silico prediction approaches.
Intensity, Dynamic Range, and Number of Peptides of the Ligandome
The total peptide intensities (sum of all identified peptide intensities) of both cell lines were similar and amounted to 3 nmol of total eluted peptide, which corresponds to ∼3 μg of peptides. Our experience with previous elutions has been that the amount of isolated HLA after immunoaffinity precipitation is 150 to 200 ng HLA/1E6 B-LCL-JY cells, which is in line with the independently reported amount of 150 ng/1E6 cells (52). Therefore, assuming 150 ng/1E6 cells, a yield of 3 mg HLA/2E10 cells, corresponding to 60 μg of peptides, was expected. Therefore, the overall work-up efficiency is about 5%. From the above, the copy number (ligands per cell) can easily be calculated. These numbers are shown in supplemental Table S1. Intensity extremes, expressed as peak area values in Proteome Discoverer, range from 4E9 to 1E5, so the dynamic range of the peptide repertoire is quite high. Considering that the lowest intensity ligands are most likely to go undetected, the actual dynamic range is probably even higher. This translates to a range from 24,000 copies/cell to (<)1 copy/cell. Clearly the chance that a protein will be represented by a peptide in the ligandome varies enormously.
With regard to cells available in limited amounts (e.g. dendritic cells), the ligandomes have been studied for only (!) a few tens of millions of cells (53, 54).The number of identified ligands, however, is low (a few hundreds). Considering the large dynamic range discussed above, it is intrinsically impossible to find the relatively low intensity ligands using small cell populations for full ligandomics experiments. An inventory of the possible ligandome on a large scale followed by multiple reaction monitoring (55) of the selected small population might be a fruitful approach. However, “more is better” definitely seems to apply to HLA ligandomics.
The composition of the ligandome in terms of the relative intensities of the peptides is shown in Fig. 6. Interestingly, it appears that in B-LCL JYpp65, the five most abundant peptides already “fill” 6% of the HLA molecules. The top 50 abundant peptides fill 27%, the top 300 fill 59%, and the top 1000 fill 83%. Importantly, this leads to the observation that all other ligands (i.e. ∼5500 peptides) fill the final 17%, as shown in Fig. 6A. The intensities have been translated to copy numbers in Fig. 6B.
The 14,065 HLA ligands are derived from 7059 different proteins; a graphical representation is shown in Fig. 7A. Half the proteins are represented by 1 peptide, and the other proteins are represented by more peptides. A gradual decline in peptide number/protein is seen. In Fig. 7B, the average protein length is plotted against the number of peptides/protein. 61 proteins are even represented by 10 peptides or more on the cell surface, with the exceptional case of 29 and 41 peptides/protein. The longer the protein, the more ligands are derived from it. It also appeared that the positions of the ligands in their “precursor” proteins were evenly distributed (data not shown). In addition, the intensities of the ligands derived from the same protein can differ greatly (see Table II for a few examples), which makes it impossible to predict the ligand copy number from the overall protein copy number. This is in line with previous work on the correlation between mRNA/protein expression and ligand density on the cell surface (56). Furthermore, no correlation could be found between peptide intensity and binding strength to HLA. The former observations might seem logical, but these imply that there is no clear selection on the basis of protein properties during antigen processing. In fact, every part of the proteome seems equally suitable for sampling by the HLA molecules. The selection of particular ligands takes place at another level, namely, the binding affinity to the available HLA molecules.
Based on our peptide listing, it is tantalizing to speculate on the origin of HLA-presented ligands and the cellular processing machinery and distill a glimpse of the cellular protein processing landscape. A courageous effort based on a rather limited dataset has been made to describe the presence of peptides in relation to their corresponding protein turnover in terms of protein stability, defective ribosomal products, and short-lived proteins (57). Studies on the scale of that presented here might be required to provide a better view of the origin of HLA-presented peptides.
Protein turnover times, as in supplemental Table S1, cannot be correlated with HLA ligand intensity. This lack of correlation might be seen as reinforcement of the defective ribosomal product hypothesis (58). Pulse-chase experiments have shown that some HLA-presented peptides make it to the cell surface within 30 min of the pulse (57). A general predictive description of the route from a protein to its presented peptides in the HLA ligandome is disturbed by the many underlying processes leading to antigen presentation, (transient) protein abundance, HLA–peptide complex stability, and reinternalization of HLA–peptide complexes. Compartmentalization and multiple sources of peptides and a subset of rapidly degraded polypeptides have been suggested to be needed in order to get a grip on the intricate antigen processing pathways (58). However, the final result of the complex cellular protein machinery is the representation of virtually all proteins on the cell membrane. In this way all cellular proteins are under the scrutiny of T cell surveillance.
The Biological Value
Our reversed immunology approach based on the bona fide eluted ligandome was started so as to explore its possibilities for finding T cell epitopes. The above were global observations and considerations concerning the ligandome. In what follows, the focus is on the immunological value of the data contained in our dataset.
Comparison with Sources of Known Epitopes
First we looked for known epitopes contained in the IEDB and the SYFPEITHI database for HLA-A2, HLA-B7, and HLA-B44. Our comparison showed that 50% and 29% listed in the IEDB and the SYFPEITHI database, respectively, were found in our ligandome, as indicated in supplemental Table S1. Considering that both the IEDB and the SYFPEITHI database are collections of ligands from many studies on a variety of cells, the biological value of our dataset is clearly demonstrated. In addition, there are probably many more epitopes in our list that are to be explored in the field of immunology.
MiHA
The first category of biologically important T cell epitopes is MiHA, which are important in the treatment of hematological malignancies. Analysis of the dataset using our in-house-developed human short peptide variation database HSPVdb (21) led to the identification of ∼1400 polymorphic ligands. Among these were known MiHA, underlining the possibilities of our proteomics approach for finding MiHA. From the set of 1400 polymorphic peptides, 80 were selected for further study to investigate their potential for medical applications. The details of the selection method and the immunological follow-up will be described in detail elsewhere.3 The total length of all proteins in Refseq release 46 is 56,113,216 base pairs. The total number of non-synonymous SNPs and indels within a coding sequence is 296,901. Thus the chance that a nucleotide is polymorphous is 0.5%. Therefore, for a 10-mer peptide (30 nucleotides), the chance of not containing an SNP is (0.995)30 is 0.86. Therefore, the chance of a 10-mer peptide containing at least one polymorphic amino acid is 14%. The number we experimentally found is 10%, on the same order as the theoretical number. From our data there is no reason to suspect that polymorphic peptides represent a “special case.” The same conclusion was drawn in a recent paper by Granados et al. on B cells (9).
Virus-derived Peptides
The antigen presenting cells in this study were EBV-transformed, and B-LCL-JY was also CMVpp65-transduced. Therefore, the presence of EBV- and CMVpp65-derived peptides was checked in our ligandome, and the results are listed in supplemental Table S3. Four EBV-derived ligands and 2 CMV-pp65-derived epitopes were found. All identifications were checked through comparison of the tandem mass spectra to those of their synthetic counterparts. All four EBV-derived ligands were newly found.
We expected that most of the previously reported EBV epitopes would not be detected, because HLA-A2 and HLA-B7 restricted T cell clones isolated from EBV-seropositive individuals and specific for the different EBV proteins BZLF-1, BRLF-1, and EBNA-3A are not reactive or are low reactive against HLA-A2- and HLA-B7-positive EBV-LCLs, whereas the T cell clones are reactive when the genes encoding these proteins are additionally introduced into EBV-LCLs. These results indicate that the published EBV epitopes are not or hardly are presented by EBV-transformed B cells, and therefore it is very well possible that these peptides were not detected in our elutions from HLA of the EBV-LCLs. The most likely reason that these epitopes are not efficiently processed and presented is that only a minority of the cells in the EBV-transformed cell lines are in a lytic cell cycle state. The four epitopes described in this paper are derived from other EBV proteins—BGLF4, RPMS1 (derived from an alternative reading frame), EBNA3B/C, and HS4ENVGP (the latter two both derived from the UTR)—and are indicated in supplemental Table S3. CMV-pp65-specific HLA-A2-restricted T cells (peptide NLVPMVATV (found)) and CMV-pp65-specific HLA-B7-restricted T cells (peptides RPHERNGFTV (found) and TRPVTGGGAM (not found)) efficiently recognize pp65-transduced EBV-LCLs, indicating that the three pp65 peptides are processed and presented in HLA-A2 and HLA-B7. Two out of the three peptides were detected in our study, and one peptide was not. The exact reason that we did not detect this particular peptide is unknown, but this peptide might have a low expression, or it might have been missed because of the shotgun nature of the experiment.
Together, the presence of the IEDB matching peptides, the MiHA, and the virus-derived peptides in our list proves the relevance of our peptide listing for immunological research. The fact that not all previously reported epitopes were found in our analysis illustrates that a considerable fraction of the ligandome still goes undetected.
Post-translationally Modified Ligands
The inclusion of phosphorylation and cysteinylation as post-translational modifications in the database-matching process yielded ∼221 phosphorylated ligands and 1221 cysteinylated ligands. Previously, 36 and 18 HLA-A2 and 8 HLA-B7 phosphorylated peptides had been reported by Zarling et al. (22, 43) and Meyer et al. (24), respectively. Fifteen of these peptides overlap with our dataset. In summary, 2 out of 150 ligands in our study are phosphorylated. Many cysteinylated ligands were identified. The total number of cysteine-containing (modified and unmodified) peptides is 1386, which represents 9% of all peptides (15,286). The frequency of cysteine in the Swissprot human database is 2%. Therefore, we calculate the chance that a peptide ligand will contain a cysteine residue as 15% and 13% for a 10-mer (1 − (0.98)8 = 0.15) and a 9-mer (1 − (0.98)7 = 0.13) ligand, respectively (we excluded the two anchor positions for this calculation because cysteine is not favorable at anchor positions). Nine percent of our peptides contained a cysteine residue. Scull et al. found a cysteine residue in 4% (50 out of 1179 unique 8- to 11-mer peptides) of their identified peptides (7). The numbers indicate that we are close to the theoretically expected number of cysteine-containing HLA ligands. Whether the cysteinylation has occurred in vivo or in vitro, the identified cysteine-containing peptides are true identifications. Because there is no suitable criterion for (de)selecting the biological relevance of these cysteinylated peptides, we do not wish to discard these true identifications. Both phosphorylation and cysteinylation have been shown to play a role in T cell recognition (23, 24, 43). The number of phosphorylated HLA ligands described here is by far the largest reported to date. Considering that we did not apply a specific phosphopeptide-directed work-up procedure, such as described in Ref. 59, we expect a considerably larger number of phosphorylated ligands to be presented. Other post-translational modifications not included in this study will certainly raise the number of post-translationally modified ligands.
In summary, our in-depth HLA ligandome study enabled a detailed look at antigen processing and provided relevant ligands. From the presented data, we estimate that the ligandome comprises at least 50,000 ligands. Considering this number and the average number of peptides per protein (between 2 and 3), the intracellular peptide pool generated by protein breakdown represents most, if not all, proteins, which seems the perfect way to present the cellular state on the outside of the cell for immune surveillance. Overall, every part of the proteome seems equally suited for sampling by the HLA molecules, although even the abundance of peptides from the same protein varies greatly. The final composition of the ligandome is determined by the enzymes involved, the transporter associated with antigen presentation, the location of a particular protein, and the relative contribution of defective ribosomal products for a given protein. The available HLA molecules select “what fits them best.” Overall, this process leads to “delegates” from many proteins (5000 found for B-LCL-HHC) in our set.
A rich and diverse repertoire of ligands is presented to the immune system, including a considerable number of post-translationally modified ligands; an additional 10% is derived from polymorphic peptides, and 5% are >11 amino acids.
We have shown that the ligandome presented here can be a good starting point for solving a wealth of specific immunological questions. Our large list of peptides presented here seamlessly fits in the efforts toward a human immunopeptidome project (25).
Supplementary Material
Acknowledgments
We dedicate this paper to Jack Leunissen, one of the first Dutch bioinformaticians, to whom we had to say good-bye in the course of this work. N. Dolezal and R. Cordfunke are thanked for providing the synthetic peptides. Drs. G. Janssen and F. Koning are thanked for critical reading of the manuscript.
Raw data have been deposited at PeptideAtlas and can be downloaded from that site.
Footnotes
* This research was made possible by the financial assistance of the Landsteiner Foundation for Blood Transfusion Research (LSBR0713).
 This article contains supplemental material.
This article contains supplemental material.
2 Discovery of T Cell Epitopes Implementing HLA-Peptidomics into a Reverse Immunology Approach. Hombrink P., Hassan C., Kester M. G., de Ru A. H., van Bergen C. A., Nijveen H., Drijfhout J. W., Falkenburg J. H., Heemskerk M. H., van Veelen P.A. J Immunol. 2013 Mar 8. [Epub ahead of print]
3 Hombrink, P., Hassan C., Kester M.G.D., Jahn L., Pont M. J., van Bergen C.A.M., Griffioen M., Falkenburg J.H.F., van Veelen P. A., Heemskerk M.H.M.
1 The abbreviations used are:
- ACN
- acetonitrile
- BMI
- best Mascot ion score
- CMV
- cytomegalovirus
- EBV
- Epstein-Barr virus
- FA
- formic acid
- FDR
- false discovery rate
- HLA
- human leukocyte antigen
- IEDB
- Immune Epitope Database
- IEF
- isoelectric focusing
- IPI
- International Protein Index
- LCL
- lymphoblastic cell line
- MHC
- major histocompatibility antigen
- MiHA
- minor histocompatibility antigen
- SCX
- strong cation exchange
- SNP
- single nucleotide polymorphism.
REFERENCES
- 1. Kenter G. G., Welters M. J., Valentijn A. R., Lowik M. J., Berends-van der Meer D. M., Vloon A. P., Essahsah F., Fathers L. M., Offringa R., Drijfhout J. W., Wafelman A. R., Oostendorp J., Fleuren G. J., van der Burg S. H., Melief C. J. (2009) Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 361, 1838–1847 [DOI] [PubMed] [Google Scholar]
- 2. Falkenburg J. H., Marijt W. A., Heemskerk M. H., Willemze R. (2002) Minor histocompatibility antigens as targets of graft-versus-leukemia reactions. Curr. Opin. Hematol. 9, 497–502 [DOI] [PubMed] [Google Scholar]
- 3. Yewdell J. W., Reits E., Neefjes J. (2003) Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 3, 952–961 [DOI] [PubMed] [Google Scholar]
- 4. Kessler J. H., Khan S., Seifert U., Le Gall S., Chow K. M., Paschen A., Bres-Vloemans S. A., de Ru A., van Montfoort N., Franken K. L., Benckhuijsen W. E., Brooks J. M., van Hall T., Ray K., Mulder A., Doxiadis I. I., van Swieten P. F., Overkleeft H. S., Prat A., Tomkinson B., Neefjes J., Kloetzel P. M., Rodgers D. W., Hersh L. B., Drijfhout J. W., van Veelen P. A., Ossendorp F., Melief C. J. (2011) Antigen processing by nardilysin and thimet oligopeptidase generates cytotoxic T cell epitopes. Nat. Immunol. 12, 45–53 [DOI] [PubMed] [Google Scholar]
- 5. Vigneron N., Stroobant V., Chapiro J., Ooms A., Degiovanni G., Morel S., van der Bruggen P., Boon T., Van den Eynde B. J. (2004) An antigenic peptide produced by peptide splicing in the proteasome. Science 304, 587–590 [DOI] [PubMed] [Google Scholar]
- 6. van Bergen C. A. M., Kester M. G. D., Jedema I., Heemskerk M. H. M., van Luxemburg-Heijs S. A. P., Kloosterboer F. M., Marijt W. A. E., de Ru A. H., Schaafsma M. R., Willemze R., van Veelen P. A., Falkenburg J. H. (2007) Multiple myeloma-reactive T cells recognize an activation-induced minor histocompatibility antigen encoded by the ATP-dependent interferon-responsive (ADIR) gene. Blood 109, 4089–4096 [DOI] [PubMed] [Google Scholar]
- 7. Scull K. E., Dudek N. L., Corbett A. J., Ramarathinam S. H., Gorasia D. G., Williamson N. A., Purcell A. W. (2012) Secreted HLA recapitulates the immunopeptidome and allows in-depth coverage of HLA A*02:01 ligands. Mol. Immunol. 51, 136–142 [DOI] [PubMed] [Google Scholar]
- 8. Hillen N., Mester G., Lemmel C., Weinzierl A. O., Muller M., Wernet D., Hennenlotter J., Stenzl A., Rammensee H. G., Stevanovic S. (2008) Essential differences in ligand presentation and T cell epitope recognition among HLA molecules of the HLA-B44 supertype. Eur. J. Immunol. 38, 2993–3003 [DOI] [PubMed] [Google Scholar]
- 9. Granados D. P., Yahyaoui W., Laumont C. M., Daouda T., Muratore-Schroeder T. L., Cote C., Laverdure J. P., Lemieux S., Thibault P., Perreault C. (2012) MHC I-associated peptides preferentially derive from transcripts bearing miRNA response elements. Blood 119, e181–e191 [DOI] [PubMed] [Google Scholar]
- 10. Bade-Doeding C., Cano P., Huyton T., Badrinath S., Eiz-Vesper B., Hiller O., Blasczyk R. (2011) Mismatches outside exons 2 and 3 do not alter the peptide motif of the allele group B*44:02P. Hum. Immunol. 72, 1039–1044 [DOI] [PubMed] [Google Scholar]
- 11. Mester G., Hoffmann V., Stevanovic S. (2011) Insights into MHC class I antigen processing gained from large-scale analysis of class I ligands. Cell. Mol. Life Sci. 68, 1521–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. den Haan J. M. M., Meadows L. M., Wang W., Pool J., Blokland E., Bishop T. L., Reinhardus C., Shabanowitz J., Offringa R., Hunt D. F., Engelhard V. H., Goulmy E. (1998) The minor histocompatibility antigen HA-1: a diallelic gene with a single amino acid polymorphism. Science 279, 1054–1057 [DOI] [PubMed] [Google Scholar]
- 13. den Haan J. M., Sherman N. E., Blokland E., Huczko E., Koning F., Drijfhout J. W., Skipper J., Shabanowitz J., Hunt D. F., Engelhard V. H., Goulmy E. (1995) Identification of a graft versus host disease-associated human minor histocompatibility antigen. Science 268, 1476–1480 [DOI] [PubMed] [Google Scholar]
- 14. Warren E. H., Vigneron N. J., Gavin M. A., Coulie P. G., Stroobant V., Dalet A., Tykodi S. S., Xuereb S. M., Mito J. K., Riddell S. R., Van den Eynde B. J. (2006) An antigen produced by splicing of noncontiguous peptides in the reverse order. Science 313, 1444–1447 [DOI] [PubMed] [Google Scholar]
- 15. Bansal A., Carlson J., Yan J. Y., Akinsiku O. T., Schaefer M., Sabbaj S., Bet A., Levy D. N., Heath S., Tang J. M., Kaslow R. A., Walker B. D., Ndung'u T., Goulder P. J., Heckerman D., Hunter E., Goepfert P. A. (2010) CD8 T cell response and evolutionary pressure to HIV-1 cryptic epitopes derived from antisense transcription. J. Exp. Med. 207, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berger C. T., Carlson J. M., Brumme C. J., Hartman K. L., Brumme Z. L., Henry L. M., Rosato P. C., Piechocka-Trocha A., Brockman M. A., Harrigan P. R., Heckerman D., Kaufmann D. E., Brander C. (2010) Viral adaptation to immune selection pressure by HLA class I-restricted CTL responses targeting epitopes in HIV frameshift sequences. J. Exp. Med. 207, 61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hombrink P., Hadrup S. R., Bakker A., Kester M. G., Falkenburg J. H., von dem Borne P. A., Schumacher T. N., Heemskerk M. H. (2011) High-throughput identification of potential minor histocompatibility antigens by MHC tetramer-based screening: feasibility and limitations. PLoS One 6, e22523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hadrup S. R., Bakker A. H., Shu C. J., Andersen R. S., van Veluw J., Hombrink P., Castermans E., Thor Straten P., Blank C., Haanen J. B., Heemskerk M. H., Schumacher T. N. (2009) Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat. Methods 6, 520–526 [DOI] [PubMed] [Google Scholar]
- 19. Hadrup S. R., Schumacher T. N. (2010) MHC-based detection of antigen-specific CD8+ T cell responses. Cancer Immunol. Immunother. 59, 1425–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kersey P. J., Duarte J., Williams A., Karavidopoulou Y., Birney E., Apweiler R. (2004) The International Protein Index: an integrated database for proteomics experiments. Proteomics 4, 1985–1988 [DOI] [PubMed] [Google Scholar]
- 21. Nijveen H., Kester M. G., Hassan C., Viars A., de Ru A. H., de Jager M., Falkenburg J. H., Leunissen J. A., van Veelen P. A. (2011) HSPVdb—the Human Short Peptide Variation Database for improved mass spectrometry-based detection of polymorphic HLA-ligands. Immunogenetics 63, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zarling A. L., Ficarro S. B., White F. M., Shabanowitz J., Hunt D. F., Engelhard V. H. (2000) Phosphorylated peptides are naturally processed and presented by major histocompatibility complex class I molecules in vivo. J. Exp. Med. 192, 1755–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meadows L., Wang W., den Haan J. M., Blokland E., Reinhardus C., Drijfhout J. W., Shabanowitz J., Pierce R., Agulnik A. I., Bishop C. E., Hunt D. F., Goulmy E., Engelhard V. H. (1997) The HLA-A*0201-restricted H-Y antigen contains a posttranslationally modified cysteine that significantly affects T cell recognition. Immunity 6, 273–281 [DOI] [PubMed] [Google Scholar]
- 24. Meyer V. S., Drews O., Gunder M., Hennenlotter J., Rammensee H. G., Stevanovic S. (2009) Identification of natural MHC class II presented phosphopeptides and tumor-derived MHC class I phospholigands. J. Proteome Res. 8, 3666–3674 [DOI] [PubMed] [Google Scholar]
- 25. Admon A., Bassani-Sternberg M. (2011) The Human Immunopeptidome Project, a suggestion for yet another postgenome next big thing. Mol. Cell. Proteomics p. 10.1074/mcp.O111.011833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kondo E., Akatsuka Y., Kuzushima K., Tsujimura K., Asakura S., Tajima K., Kagami Y., Kodera Y., Tanimoto M., Morishima Y., Takahashi T. (2004) Identification of novel CTL epitopes of CMV-pp65 presented by a variety of HLA alleles. Blood 103, 630–638 [DOI] [PubMed] [Google Scholar]
- 27. Heemskerk M. H., de Paus R. A., Lurvink E. G., Koning F., Mulder A., Willemze R., van Rood J. J., Falkenburg J. H. (2001) Dual HLA class I and class II restricted recognition of alloreactive T lymphocytes mediated by a single T cell receptor complex. Proc. Natl. Acad. Sci. U.S.A. 98, 6806–6811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meiring H. D., van der Heeft E., ten Hove G. J., de Jong A. P. J. M. (2002) Nanoscale LC-MS(n): technical design and applications to peptide and protein analysis. J. Sep. Sci. 25, 557–568 [Google Scholar]
- 29. Apweiler R., Jesus Martin M., O'onovan C., Magrane M., Alam-Faruque Y., Antunes R., Barrera Casanova E., Bely B., Bingley M., Bower L., Bursteinas B., Mun Chan W., Chavali G., Da Silva A., Dimmer E., Eberhardt R., Fazzini F., Fedotov A., Garavelli J., Castro L. G., Gardner M., Hieta R., Huntley R., Jacobsen J., Legge D., Liu W., Luo J., Orchard S., Patient S., Pichler K., Poggioli D., Pontikos N., Pundir S., Rosanoff S., Sawford T., Sehra H., Turner E., Wardell T., Watkins X., Corbett M., Donnelly M., van Rensburg P., Goujon M., McWilliam H., Lopez R., Xenarios I., Bougueleret L., Bridge A., Poux S., Redaschi N., Argoud-Puy G., Auchincloss A., Axelsen K., Baratin D., Blatter M. C., Boeckmann B., Bolleman J., Bollondi L., Boutet E., Braconi Quintaje S., Breuza L., deCastro E., Cerutti L., Coudert E., Cuche B., Cusin I., Doche M., Dornevil D., Duvaud S., Estreicher A., Famiglietti L., Feuermann M., Gehant S., Ferro S., Gasteiger E., Gerritsen V., Gos A., Gruaz-Gumowski N., Hinz U., Hulo C., Hulo N., James J., Jimenez S., Jungo F., Kappler T., Keller G., Lara V., Lemercier P., Lieberherr D., Martin X., Masson P., Moinat M., Morgat A., Paesano S., Pedruzzi I., Pilbout S., Pozzato M., Pruess M., Rivoire C., Roechert B., Schneider M., Sigrist C., Sonesson K., Staehli S., Stanley E., Stutz A., Sundaram S., Tognolli M., Verbregue L., Veuthey A. L., Wu C. H., Arighi C. N., Arminski L., Barker W. C., Chen C., Chen Y., Dubey P., Huang H., Kukreja A., Laiho K., Mazumder R., McGarvey P., Natale D. A., Natarajan T. G., Roberts N. V., Suzek B. E., Vinayaka C., Wang Q., Wang Y., Yeh L. S., Zhang J. (2012) [Google Scholar]
- 30. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 31. Cambridge S. B., Gnad F., Nguyen C., Bermejo J. L., Kruger M., Mann M. (2011) Systems-wide proteomic analysis in mammalian cells reveals conserved, functional protein turnover. J. Proteome Res. 10, 5275–5284 [DOI] [PubMed] [Google Scholar]
- 32. Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dinkel H., Chica C., Via A., Gould C. M., Jensen L. J., Gibson T. J., Diella F. (2011) Phospho.ELM: a database of phosphorylation sites—update 2011. Nucleic Acids Res. 39, D261–D267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blom N., Gammeltoft S., Brunak S. (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294, 1351–1362 [DOI] [PubMed] [Google Scholar]
- 35. Pruitt K. D., Tatusova T., Maglott D. R. (2005) NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hubner N. C., Ren S., Mann M. (2008) Peptide separation with immobilized pI strips is an attractive alternative to in-gel protein digestion for proteome analysis. Proteomics 8, 4862–4872 [DOI] [PubMed] [Google Scholar]
- 37. Vita R., Zarebski L., Greenbaum J. A., Emami H., Hoof I., Salimi N., Damle R., Sette A., Peters B. (2010) The immune epitope database 2.0. Nucleic Acids Res. 38, D854–D862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rammensee H., Bachmann J., Emmerich N. P., Bachor O. A., Stevanovic S. (1999) SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50, 213–219 [DOI] [PubMed] [Google Scholar]
- 39. Bleakley M., Riddell S. R. (2004) Molecules and mechanisms of the graft-versus-leukaemia effect. Nat. Rev. Cancer 4, 371–380 [DOI] [PubMed] [Google Scholar]
- 40. Goulmy E. (1996) Human minor histocompatibility antigens. Curr. Opin. Immunol. 8, 75–81 [DOI] [PubMed] [Google Scholar]
- 41. Mutis T., Goulmy E. (2002) Hematopoietic system-specific antigens as targets for cellular immunotherapy of hematological malignancies. Semin. Hematol. 39, 23–31 [DOI] [PubMed] [Google Scholar]
- 42. Laurin D., Hannani D., Pernollet M., Moine A., Plumas J., Bensa J. C., Cahn J. Y., Garban F. (2010) Immunomonitoring of graft-versus-host minor histocompatibility antigen correlates with graft-versus-host disease and absence of relapse after graft. Transfusion 50, 418–428 [DOI] [PubMed] [Google Scholar]
- 43. Zarling A. L., Polefrone J. M., Evans A. M., Mikesh L. M., Shabanowitz J., Lewis S. T., Engelhard V. H., Hunt D. F. (2006) Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc. Natl. Acad. Sci. U.S.A. 103, 14889–14894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dudek N. L., Tan C. T., Gorasia D. G., Croft N. P., Illing P. T., Purcell A. W. (2012) Constitutive and inflammatory immunopeptidome of pancreatic beta-cells. Diabetes 61, 3018–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chornoguz O., Gapeev A., O'Neill M. C., Ostrand-Rosenberg S. (2012) Major histocompatibility complex class II+ invariant chain negative breast cancer cells present unique peptides that activate tumor-specific T cells from breast cancer patients. Mol. Cell. Proteomics 11, 1457–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hunt D. F., Henderson R. A., Shabanowitz J., Sakaguchi K., Michel H., Sevilir N., Cox A. L., Appella E., Engelhard V. H. (1992) Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science 255, 1261–1263 [DOI] [PubMed] [Google Scholar]
- 47. Maier R., Falk K., Rotzschke O., Maier B., Gnau V., Stevanovic S., Jung G., Rammensee H. G., Meyerhans A. (1994) Peptide motifs of HLA-A3, -A24, and -B7 molecules as determined by pool sequencing. Immunogenetics 40, 306–308 [DOI] [PubMed] [Google Scholar]
- 48. Huczko E. L., Bodnar W. M., Benjamin D., Sakaguchi K., Zhu N. Z., Shabanowitz J., Henderson R. A., Appella E., Hunt D. F., Engelhard V. H. (1993) Characteristics of endogenous peptides eluted from the class I MHC molecule HLA-B7 determined by mass spectrometry and computer modeling. J. Immunol. 151, 2572–2587 [PubMed] [Google Scholar]
- 49. Macdonald W., Williams D. S., Clements C. S., Gorman J. J., Kjer-Nielsen L., Brooks A. G., McCluskey J., Rossjohn J., Purcell A. W. (2002) Identification of a dominant self-ligand bound to three HLA B44 alleles and the preliminary crystallographic analysis of recombinant forms of each complex. FEBS Lett. 527, 27–32 [DOI] [PubMed] [Google Scholar]
- 50. DiBrino M., Parker K. C., Margulies D. H., Shiloach J., Turner R. V., Biddison W. E., Coligan J. E. (1995) Identification of the peptide binding motif for HLA-B44, one of the most common HLA-B alleles in the Caucasian population. Biochemistry 34, 10130–10138 [DOI] [PubMed] [Google Scholar]
- 51. Liu Y. C., Chen Z., Burrows S. R., Purcell A. W., McCluskey J., Rossjohn J., Gras S. (2012) The energetic basis underpinning T-cell receptor recognition of a super-bulged peptide bound to a major histocompatibility complex class I molecule. J. Biol. Chem. 287, 12267–12276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Engelhard V. H., Strominger J. L., Mescher M., Burakoff S. (1978) Induction of secondary cytotoxic T lymphocytes by purified HLA-A and HLA-B antigens reconstituted into phospholipid vesicles. Proc. Natl. Acad. Sci. U.S.A. 75, 5688–5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meiring H. D., Kuipers B., van Gaans-van den Brink J. A. M., Poelen M. C. M., Timmermans H., Baart G., Brugghe H., van Schie J., Boog C. J. P., de Jong A. P. J. M., van Els C. A. (2005) Mass tag-assisted identification of naturally processed HLA class II-presented meningococcal peptides recognized by CD4+ T lymphocytes. J. Immunol. 174, 5636–5643 [DOI] [PubMed] [Google Scholar]
- 54. van Haren S. D., Herczenik E., ten Brinke A., Mertens K., Voorberg J., Meijer A. B. (2011) HLA-DR-presented peptide repertoires derived from human monocyte-derived dendritic cells pulsed with blood coagulation factor VIII. Mol. Cell. Proteomics 10, M110.002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tan C. T., Croft N. P., Dudek N. L., Williamson N. A., Purcell A. W. (2011) Direct quantitation of MHC-bound peptide epitopes by selected reaction monitoring. Proteomics 11, 2336–2340 [DOI] [PubMed] [Google Scholar]
- 56. Weinzierl A. O., Lemmel C., Schoor O., Muller M., Kruger T., Wernet D., Hennenlotter J., Stenzl A., Klingel K., Rammensee H. G., Stevanovic S. (2007) Distorted relation between mRNA copy number and corresponding major histocompatibility complex ligand density on the cell surface. Mol. Cell. Proteomics 6, 102–113 [DOI] [PubMed] [Google Scholar]
- 57. Milner E., Barnea E., Beer I., Admon A. (2006) The turnover kinetics of major histocompatibility complex peptides of human cancer cells. Mol. Cell. Proteomics 5, 357–365 [DOI] [PubMed] [Google Scholar]
- 58. Yewdell J. W. (2011) DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. Trends Immunol. 32, 548–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pinkse M. W., Mohammed S., Gouw J. W., van Breukelen B., Vos H. R., Heck A. J. (2008) Highly robust, automated, and sensitive online TiO2-based phosphoproteomics applied to study endogenous phosphorylation in Drosophila melanogaster. J. Proteome Res. 7, 687–697 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.