

Abstract
Staphylococcus aureus CstR (CsoR-like sulfur transferase repressor) is a member of the CsoR family of transition metal sensing metalloregulatory proteins. Unlike CsoR, CstR does not form a stable complex with transition metals but instead reacts with sulfite to form a mixture of di- and trisulfide species, CstR2(RS-SR′) and CstR2(RS-S-SR′)n, n = 1 or 2, respectively. Here, we investigate if CstR performs similar chemistry with related chalcogen oxyanions selenite and tellurite. In this work we show by high resolution tandem mass spectrometry that CstR is readily modified by selenite (SeO32−) or tellurite (TeO32−) to form a mixture of intersubunit disulfides and selenotrisulfides or tellurotrisulfides, respectively, between Cys31 and Cys60′. Analogous studies with S. aureus CsoR reveals no reaction with selenite and minimal reaction with tellurite. All cross-linked forms of CstR exhibit reduced DNA binding affinity. We show that Cys31 initiates the reaction with sulfite through the formation of S-sulfocysteine (RS-SO32−) and Cys60 is required to fully derivatize CstR to CstR2(RS-SR′) and CstR2(RS-S-SR′). The modification of Cys31 also drives an allosteric switch that negatively regulates DNA binding while derivatization of Cys60 alone has no effect on DNA binding. These results highlight the differences between CstRs and CsoRs in chemical reactivity and metal ion selectivity and establish Cys31 as the functionally important cysteine residue in CstRs.
Introduction
Staphylococcus aureus is an opportunistic Gram-positive non-motile cocci that is the causative agent of numerous illnesses ranging from minor skin infections to life-threatening diseases.1, 2 In contrast to other human pathogens, S. aureus lacks a functional sulfate assimilation pathway and must use other mechanisms to obtain sulfur from organic and inorganic sources.3 Sulfur acquisition is essential for the biosynthesis of cysteine, iron-sulfur clusters, and other low molecular weight sulfur-containing compounds and maintenance of cellular redox balance.4, 5 Recent work has shown that regulation of cysteine metabolism in S. aureus and its perturbation increases susceptibility to oxidative stress and decreases the ability of this microorganism to form biofilms, which is essential for survival both inside and outside of a host.5, 6 Additionally, components of sulfur metabolism and cysteine biosynthesis in Mycobacterium tuberculosis have been identified as antimicrobial and vaccine targets.7–9
Although S. aureus lacks the sulfate assimilation pathway, it is capable of growing on inorganic thiosulfate as a sole sulfur source3, 10 presumably by assimilating the sulfane sulfur and effluxing sulfite (SO32−). Thiosulfate can be generated by the mammalian host from mitochondrial hydrogen sulfide (H2S) detoxification11 via sulfur dioxygenase and sulfide quinone reductase (SQR) activities.12 Further oxidation of thiosulfate may lead to the formation of tetrathionate,13 a highly reactive sulfur compound that disrupts the redox state of the cell by converting thiols, e.g., cysteine, to disulfides.14 Although some gut bacterial pathogens use tetrathionate as a respiratory electron acceptor thereby providing a growth advantage in this niche, S. aureus appears to lack this ability.13 In any case, S. aureus sulfur metabolism and regulation in response to stress remains relatively underexplored.
We recently described a transcriptional regulator putatively involved in inorganic sulfur metabolism or assimilation, CstR (CsoR-like sulfur-transferase repressor), a paralogue to the Cu(I)-sensing CsoR (copper-sensitive operon repressor).10 Initial studies established that CstR functions via a derepression mechanism and tightly controls the expression of the divergently transcribed cst operon, which encodes five proteins likely involved in sulfur metabolism. These include a putative sulfite/sulfonate efflux pump (TauE) and CstR transcribed in one direction, and two multi-domain sulfurtransferases (CstA and CstB), a putative sulfur dioxygenase (CstB), and a sulfide quinone oxidoreductase (SQR) transcribed in the other. CstR was previously shown to be negatively regulated in vitro by sulfite through the formation of a mixed disulfide or trisulfide across the protomer interface.10
CstRs are of unknown structure but are thought to be α-helical tetrameric bundles similar to CsoRs10, 15–17 where four protomers are arranged as a dimer of dimers (Fig. 1A). In CsoRs, two metal-binding ligands are contributed from one protomer while the third lies across the dimer interface (Fig. 1B). CstRs retain two cysteines per protomer, Cys31 and Cys60, but lack a key metal-binding histidine residue. It is Cys31 and Cys60′ that reduce sulfite to a mixture of intrerprotomer disulfides, CstR2(RS-SR′), and trisulfides, CstR2(RS-S-SR′) across the dimer interface.10
Fig. 1.
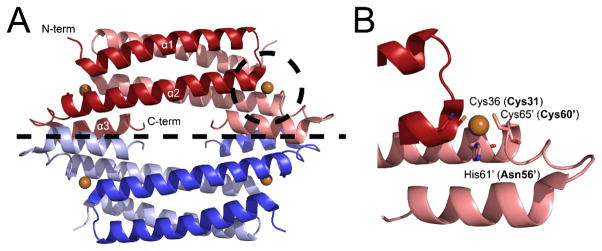
Crystal structure of Mycobacterium tuberculosis CsoR15 (A) and Cu(I) binding site (B). CsoRs feature a tetrameric bundle as a “dimer of dimers” displayed as blue and red and separated by a dashed line. There are four Cu(I) binding sites (bronze spheres), two per dimer. In CstR, Cys31 and Cys60′ form cross-links with the corresponding protomer of the dimer.7 Labelled residues are that of M. tuberculosis CsoR and those in parentheses are the analogous residues in S. aureus CstR.
It was postulated that CstR would react with related chalcogen oxyanions, selenite (SeO32−) and tellurite (TeO32−), both of which are biologically available and toxic to many microorganisms.18 Chalcogen oxyanions display increasing reactivity as one moves down the periodic table and are generally more reactive than sulfite.4, 19 Toxicity from these compounds is presumed to be from disruption of the low molecular weight thiol pool,20 e.g., glutathione, cysteine, or coenzyme A. These thiols undergo redox reactions (H2SeO3 + 4RSH → RS-Se-SR + RS-SR + 3H2O) to form a mixtures of disulfides and selenotrisulfides;21 analogous chemistry can also occur at enzyme active site or metal-chelating cysteine residues.22 This could in turn induce a cellular response similar to that of hypochlorite23 and nitrite,24 each of which strongly induce the cst operon, presumably by disrupting the redox balance of the cell. Many firmicutes and related Gram-positive bacteria, including S. aureus, exhibit strong resistance to these compounds through unknown mechanisms25 and will form pink or black colonies from the reduction of SeO32− or TeO32− to Se0 or Te0, respectively.18
In this work, we characterize by mass spectrometry the reaction products of S. aureus CstR with the chalcogen oxyanions SeO32− and TeO32−. In each case we establish that the cross-linking of the cysteine pair negatively regulates DNA binding. In addition, we explore the relative impact of the reaction of cysteine residues with sulfite and establish functional importance for Cys31 in driving negative regulation of operator DNA binding by CstR.
Materials and Methods
Plasmid Preparation, Protein Expression, and Purification
CstR cysteine mutants were prepared by introducing nucleotide substitutions with standard Quikchange™ site-directed mutagenesis techniques and appropriate primers (Table S1). All expression plasmids were verified with DNA sequencing at the Indiana University Molecular Biology Institute. CsoR, CstR and CstR cysteine mutants were purified as previously described10 with minor modifications. Throughout the purification of CstR and CstR cysteine mutants, 50 mM Tris at pH 8.0 was used in place of 50 mM HEPES at pH 7.0 and all buffers were degassed on a Schlenk line immediately prior to use. Experimental buffers were also passed over Chelex 100 resin to remove trace metal ions and degassed/backfilled with argon gas three times prior to anaerobic use. All glassware to be used anaerobically was soaked in a 2% nitric acid bath overnight and extensively rinsed with Milli-Q water. Dialysis tubing and stir bars used for anaerobic dialysis were washed with 0.5 M EDTA solution followed by thorough rinsing with Milli-Q water. Reduced thiols were confirmed by LC-ESI-MS and quantified with a DTNB assay.26
CstR and CsoR Chalcogen Oxyanion Reactions
Fifteen micromolar samples of CstR (protomer) were reacted anaerobically in a Vacuum Atmostpheres (Amesbury, MA) glovebox in 10 mM HEPES, 200 mM NaCl at pH 7.0 with a 5- fold thiol excess of modifying reagent (SO32−, SeO32−, TeO32−, or S4O62−) for 17 h at 22° C. After 17 h, samples were sealed in septa cap vials for immediate LC-ESI-MS analysis. Samples used for tryptic digest and fluorescence anisotropy were reacted for 36 h to ensure complete modification of thiols.
LC-ESI-MS Analysis of Intact CstR and CsoR
LC-ESI-MS analysis was performed at the Indiana University Mass Spectrometry Facility using a Waters/Micromass LCT Classic time of flight (TOF, Milford, MA) mass spectrometer with a CapLC inlet. Proteins were loaded onto a 50 mm Agilent BioBasic C8 reverse-phase column with a 5 μm particle size and 300 Å pore size in Solvent A (5% acetonitrile, 95% water, 0.1% formic acid) and eluted with a 20 min linear gradient from 10% Solvent A to 90% Solvent B (95% acetonitrile, 5% water, 0.1% formic acid). Elution was monitored at 215 nm. Data were collected and analyzed using MassLynx Software (Waters, Milford, MA).
LTQ-Orbitrap Tandem MS Analysis of Tryptic Peptides
Reacted and unreacted CstR samples were solution digested with proteomics-grade trypsin from porcine pancreas (Sigma-Aldrich, St. Louis, MO) at 37°C for 18 h with a 1:50 ratio of trypsin:CstR. Following digestion, samples were desalted using a C18 Zip-Tip column (Millipore, Billerica, MA), vacuum-centrifuged to dryness at 45 °C, and resuspended in water immediately prior to analysis. Parameters used for LTQ-orbitrap tandem mass spectral analysis are similar to those previously described.27 Briefly, five microliters of protein digest were loaded onto a 15 mm × 100 μm i.d. C18 reversed-phase trapping column. Peptides were eluted through a 150 mm × 75 μm internal diameter analytical column packed with 5 μm, 100 Å Magic C18AQ packing material (Microm BioResourses Inc., Auburn, CA), using a 60 min gradient from 97 % to 60 % solvent A, 97: 3: 0. 1 water/acetonitrile/formic acid (Solvent B is 0.1% formic acid in acetonitrile) at 250 nL/min on a Dionex UltiMate 3000 nanoLC (Sunnyvale, CA). Eluent from the column was ionized and electrosprayed directly into a ThermoFinnigan (San Jose, CA) LTQ-Orbitrap XL mass spectrometer which recorded mass spectra and “top five” data-dependent tandem mass spectra of the peptide ions. Data were interpreted by manual investigation to identify candidate MS/MS spectra and assign identifications of the fragment ions for confirmation of the modified CstR. In some cases, cross-linked peptides eluted within the exclusion window of a previous peak and were not selected for fragmentation. To obtain fragmentation spectra of these species, multiple reaction monitoring (MRM) was employed to ensure collection of fragmentation data.28
Fluorescence Anisotropy Titrations
Double stranded fluorescein-labeled cst OP1 DNA constructs (Table S1) were prepared as described previously.27 A typical anisotropy experiment was performed with 10 nM cst OP1 dsDNA in a buffer containing 10 mM HEPES at pH 7.0 and 200 mM NaCl at 25 °C under strictly anaerobic conditions. Injections of 1–5 μL were equilibrated for 3 min and then anisotropy was monitored using an ISS PC1 Spectrofluorometer (Champaign, IL). The fluorescein was excited at 490 nm and polarization monitored with a 515 nm cut-off filter in the L-format. For each data point, five measurements were obtained and averaged. Data collected were fit to a non-dissociable tetramer (CstR4) binding model where two tetramers sequentially bind cst OP1 operator DNA10 using Dynafit29 to calculate K1 and K2. The macroscopic binding constant, Ktet, was calculated as Ktet = (K1•K2)½ given the uncertainty in extracting unique values for Ki due to their strong inverse correlation and lack of significantly sigmoidal behavior in the binding isotherms. Previous findings10 establish a linear relationship between robs and νi, the binding density at the ith addition of protein titrant.
Results and Discussion
S. aureus CstR reacts with selenite and tellurite to form di-and trisulfide-like species
To test the reactivity of selenite and tellurite, we reacted a fivefold thiol excess of each chalcogen with fully reduced CstR under anaerobic conditions for 17 h. Products of each reaction and a control sample were analyzed with LC-ESI-MS (Fig. 2). Here, reduced CstR and cross-linked CstR are readily distinguishable due to a shift in the charge state distribution from +8–13 to +11–20, respectively (Fig. S1A–C). The shift in charge state distribution is due to doubling of the possible sites for charges and conformational changes induced by interprotomer cross-linking. Both selenite and tellurite-reacted samples produced similar overall charge state distributions for cross-linked species. The selenite-reacted sample contained a single distribution consistent with fully cross-linked protein while the tellurite-reacted sample displayed two distinct charge state distributions (Fig. S1A and B). The first is consistent with reduced and unreacted CstR and the second with cross-linked CstR.
Fig. 2.

LC-ESI-MS analysis of CstR (A–C) and CsoR (D–E) following reaction with SeO32− or TeO32− and seleno- and tellurotrisulfides have been denoted RS-X-SR′ for Se or Te cross-slinking, respectively. The CstR charge states displayed are +11 and +17 for the reduced and cross-linked forms, respectively. The CsoR charge states are +13 and +19 for reduced and cross-linked form, respectively.
Closer inspection of individual charge states in the selenite and tellurite-reacted samples reveals a series of peaks corresponding to disulfide and selenotrisulfide or tellurotrisulfide cross-linked CstR as well as several other oxygen adducted species (Fig. 2B and C). The first peak in these spectra (m/z of ~1135) is the disulfide cross-linked form of CstR and is denoted RS-SR′. The next peak is a doubly oxygen adducted peak with a +32 Da mass shift followed by +79 Da or +128 Da mass shifts that are consistent with either selenium or tellurium cross-linked CstR, respectively. In addition to RS-Se-SR′ and RS-Te-SR′, we observe prominent doubly oxygen adducted cross-linked peaks of these species. A complete listing of all observed CstR peaks is given in Table S2.
These results indicate that CstR does in fact react with heavier chalcogen oxyanions to yield products similar to those observed following reaction with sulfite.10 Here the SeO32− reaction is complete at this time point while the TeO32− sample contains a large portion of unreacted or reduced CstR (Fig. S1B and C). This indicates that despite the intrinsic greater reactivity of TeO32−, CstR reacts more readily with the smaller and less reactive SeO32−.
S. aureus CsoR does not react with selenite but reacts with tellurite to form a mixed disulfide and tellurotrisulfide
Experiments analogous to those performed with CstR were also performed with S. aureus CsoR. CsoR was previously shown to be unreactive toward sulfite10 but given the higher reactivity of related chalcogens, we hypothesized that the greater reactivity of heavier chalcogen oxyanions may yield products similar to the CstR reaction. When analyzed by LC-ESI-MS the reduced form of CsoR has a charge state distribution of +7–15 and +13–23 in the cross-linked form (Fig. S1D–F). We observe only the reduced form of CsoR when reacted with SeO32− as with sulfite10 (Fig. 2E). However, the more reactive TeO32− oxyanion forms disulfide and tellurium cross-linked CsoR (Fig. 2F) but lacks the oxygen adducts observed in the CstR TeO32− reaction (Fig. 2C). A complete listing of all observed CsoR peaks is available in Table S3.
The sum of the chalcogen oxyanion experiments with CstR and CsoR further highlights the differential selectivity of these paralogous regulatory proteins. Several recent reports have begun to explore the mechanisms by which regulatory proteins are capable of forming a regulatory disulfide bond in response to oxidative stress. Examples include HypR,30 SarZ,31 MexR,32 and BigR,33 among others. The distance between Sγ of the regulatory cysteine residues in these proteins typically ranges from 7–10 Å and is similar to that of apo-CsoR (~8 Å) from Streptomyces lividans.16 S. aureus CsoR and CstR are likely to exhibit similar distances but only the later is readily susceptible to cross-linking. Although the factors contributing to differential reactivity remain unexplored, they may include sterics, structural dynamics, charge-charge interactions, and intrinsic cysteine reactivity.
Selenite cross-linked CstR contains a selenotrisulfide as observed by LTQ-Orbitrap tandem mass spectrometry
To further investigate the selenium cross-linked peaks observed in the CstR LC-ESI-MS data, we performed a tryptic digest and analyzed the peptides using LTQ-orbitrap tandem mass spectrometry. Cross-linked peptides were initially identified by accurate mass detected in the LTQ (Table 1). The most abundant cross-linked peptides identified contained two missed cleavages (indicated by “ | ” ) on opposite sides of Cys 31, 24MMEEGK|DCK|DVITQISASK42 (denoted peptide “A”), and a fully digested peptide encompassing Cys60, 48LMGIIISENLIECVK62 (denoted peptide “B”). The most intense charge state of the cross-linked peptide was +4 charge state although less abundant charge states were also observed at +3 and +5 (data not shown). Comparison of the accurate mass of the disulfide and selenotrisulfide cross-linked peptides, 946.980 and 966.960 Da, respectively, reveals a mass shift of 79.920 Da (Table 1). This is consistent with the monoisotopic mass of 80Se, 79.91652 Da. Furthermore, the isotopic distribution from the LTQ mirrors the predicated distribution for peptide A and B with the addition of a selenium atom (Fig. 3A and C).
Table 1.
Monoisotopic masses for the parent ions of tryptic peptides of interest as observed with high-resolution tandem mass spectrometry. (−) denotes no data obtained.
| Sample | Amino Acids | Charge State | Monoisotopic Mass (Da) | |
|---|---|---|---|---|
| Calculated | Observed | |||
| Control | Cys31, RS-H | + 3 | 705.004 | 705.003 |
| Cys60, RS-H | + 2 | 704.361 | 704.352 | |
|
| ||||
| +SeO32− | RS-SR′ | + 4 | 946.978 | 946.980 |
| + O | + 4 | 950.977 | 950.978 | |
| + 2 O | + 4 | 954.976 | 954.972 | |
| + 3 O | + 4 | 958.974 | 958.972 | |
| RS-Se-SR′ | + 4 | 966.958 | 966.960 | |
| + O | + 4 | 970.965 | 970.966 | |
| + 2 O | + 4 | 974.955 | - | |
|
| ||||
| +TeO32− | RS-SR′ | + 4 | 946.978 | 946.790 |
| RS-Te-SR′ | + 4 | 979.455 | - | |
Fig. 3.

Tryptic peptides of SeO32−-reacted CstR identify a selenotrisulfide. The peptides involved in the disulfide cross-link are denoted peptide “A” (red) for the Cys31 peptide 24MMEEGKDCKDVITQISASK42 and peptide “B” (blue) for Cys60′, 48′LMGIIISENLIECVK62′. Fragmentation ions, b and y, are assigned relative to the respective peptide, “A” or “B”, and cross-linked peptides are denoted as AByn. Peptide “A” remained intact for all cross-linked ions identified and all fragmentation occurred on peptide “B” (A) Accurate mass and isotopic distribution of the RS-SR′ CstR disulfide in the +4 charge state. The monoisotopic mass is 946.980 Da (Table 1) (B) Fragmentation pattern of RS-SR′ disulfide (C) Isotopic distribution of RS-Se-SR′ selenotrisulfide in the +4 charge state. The monoisotopic mass is 966.710 Da (all 12C and 80Se) and among the preceding peaks, 76Se, 77Se and 78Se were identified. (D) Fragmentation of the RS-Se-SR′ cross-link.
We next verified the assigned accurate mass assignments with the fragmentation spectra. In all cross-linked spectra, peptide A fragmented to yield exclusively Ab7+ and Ay7+ ions as major products and remained intact in all cross-linked fragments while peptide “B” fragmented more readily. Numerous cross-linked fragments (denoted AB) were identified with +78 to +80 Da mass shifts relative to disulfide cross-linked peptides (Fig. 3B and D). and confirm the existence of a selenotrisulfide between Cys31 and Cys60′ of CstR. All cross-linked spectra were cross-referenced with the fragmentation patterns of the individual ‘A’ and ‘B’ peptides (Fig. S2). Analogous samples were prepared to verify the CstR2RS-Te-SR′ tellurotrisulfide assignment but these efforts were ultimately unsuccessful (Table 1), likely due to hydrolysis of RS-Te-SR′ to RS-H or RS-SR′ and elemental tellurium, Te0.34
In addition to selenium and tellurium cross-linked CstR, we observed prominent oxygen adducts as evidenced by a +32 Da mass shift relative to the disulfide, selenotrisulfide, or tellurotrisulfide peaks (Fig. 2B–C; Table 1). Further examination of the LTQ data allowed us to recover peaks corresponding to disulfide cross-linked CstR with singly, doubly, and triply oxygen adducted AB peptides with high mass accuracy (Table 1). The most abundant oxygen adduct contains two oxygen atoms (Fig. S3A) which is consistent with our observations from the LC-ESI-MS data (Fig. 2B–C). We initially speculated that these adducts represented trapped intermediates of SeO32− and TeO32− reduction, e.g., RS-Se(O2)-SR′,21 but could not obtain evidence for this. Rather, the principal oxygen adducts were associated with the disulfide cross-linked peptide (Fig. S3A). Analysis of the fragmentation data indicates that these oxidations are not associated with nearby methionine residues either. This therefore necessitates that both oxygen atoms remain on the cysteine Sγ atoms on either side of the disulfide bond and have been assigned as a thiosulfonate or α-disulfoxide (Fig. S3B, Fig. 4).
Fig. 4.
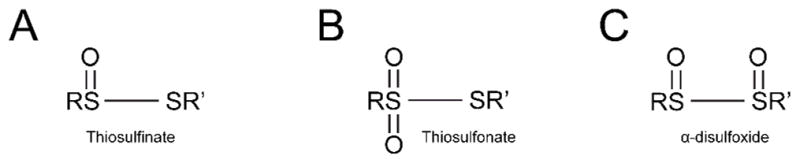
Depiction of thiosulfinate (A), thiosulfonate (B) and α-disulfoxide (C) oxygen adducts. In CstR, the oxygen adducts have been assigned to the Sγ of Cys31 and Cys60 as either thiosulfonate or α-disulfoxide (Fig. S3A). R and R′ are not defined as Cys31 or Cys60 as they can not be distinguished.
To the best of our knowledge, a protein-derived thiosulfonate (Fig. 4B) or α-disulfoxide (Fig. 4C) has not been previously observed but have been described in the organic literature in thiol-containing compounds.35 It is intriguing that a likely precursor to a thiosulfonate, specifically thiosulfinate (Fig. 4A), has been identified and characterized within an intact protein using isotopic labelling and mass spectrometry during disulfide bond formation between sulfiredoxin and peroxiredoxin.36 Other examples of protein thiol modifications are becoming increasingly recognized as functionally important including stable sulfenic acids, particularly in the context of reversible cysteine-based redox switches.37, 38 These are in contrast to sulfenic acids that are considered transient intermediates in disulfide bond formation and have recently been reviewed.39–41 With respect to the disulfide oxygen adductions observed here, it is unknown if they are functionally significant or occur as a result of over-oxidation by selenite under these conditions.
Formation of CstR cross-linked species as disulfides, selenotrisulfides, or tellurotrisulfides negatively regulates DNA binding
To test if disulfide, selenotrisulfide, or tellurotrisulfide cross-linking of CstR negatively regulates cst operator DNA binding, fluorescence anisotropy experiments were performed. Fully reduced CstR or CstR modified with tetrathionate (S4O62−) as a disulfide control42 (Table S2), SeO32− or TeO32− was titrated into a solution containing fluorescently-labeled cst OP1 under strictly anaerobic conditions (Fig. 5A). Data from these experiments were fit to a sequential tetramer binding model.10 Each modified CstR bound weakly to cst OP1 with a decrease in affinity of ≈30–60 fold relative to unmodified CstR (Table 2). These results indicate that CstR is indiscriminate with respect to the nature of the cross-link in negative regulation of DNA binding.
Fig. 5.
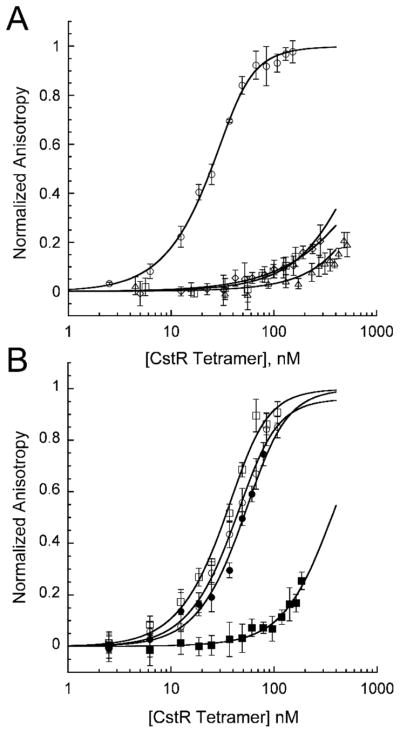
DNA binding characteristics of CstR and CstR cysteine. (A) Fluorescence anisotropy titrations of CstR (circles) and CstR reacted with SeO32− (squares), TeO32− (diamonds), or tetrathionate (S4O62−, triangle) with fluorescently-labeled cst OP1. (B) Fluorescence anisotropy titrations of C31A CstR (circles) and C60A CstR (squares). Closed circles and squares represent MMTS-derivatized thiol-modified CstRs. All data were fit to a sequential tetramer model where two tetramers bind one operator DNA in a step-wise fashion defined by K1 and K2. Ktet describes the average macroscopic binding constant (Ktet = (K1•K2)1/2). Tetrameric bundles were assumed non-dissociable.
Table 2.
Summary of binding constants obtained from fluorescence anisotropy experiments of WT CstR or CstR cysteine mutants with cst OP1
| Protein | r0 | rcomplex | Ktet (M−1) × 10−7 |
|---|---|---|---|
| CstR | 0.133 | 0.155 | 6.3 (± 0.5) |
| CstR + SeO32− | 0.132 | 0.154* | 0.20 (± 0.03) |
| CstR + TeO32− | 0.133 | 0.155* | 0.13 (± 0.14) |
| CstR + S4O62− | 0.132 | 0.154* | 0.11 (± 0.11) |
| C31A CstR | 0.135 | 0.161 | 3.3 (± 0.7) |
| C31A CstR + MMTS | 0.135 | 0.159 | 2.0 (± 0.5) |
| C60A CstR | 0.135 | 0.159 | 4.2 (± 1.0) |
| C60A CstR + MMTS | 0.136 | 0.158* | 0.31 (± 0.03) |
| C31A/C60A CstR | 0.134 | 0.157 | 12 (± 5) |
Fit to Δ anisotropy of corresponding non-derivatized CstR
CstR Cys31 initiates the reaction with sulfite and its perturbation is required to negatively regulate DNA binding
To better understand the reduction reaction of CstR with chalcogen oxyanions, we prepared single and double cysteine substitution mutants, specifically C31A, C60A, and C31A/C60A CstRs. Here, the C31A CstR mutant reports on the reactivity of Cys60 while C60A CstR reports on Cys31. Each mutant was reacted with sulfite as described previously10 to test the importance of individual cysteine residues. LC-ESI-MS analysis of sulfite-reacted C31A or C60A CstR revealed that only the C60A mutant reacted with SO32− and formed S-sulfocysteine (RS-SO32−, Fig. 6, Table S4). This result indicates that Cys31 of CstR initiates the chalcogen reduction reaction while Cys60′ is required to continue the reaction toward cross-linked products.
Fig. 6.
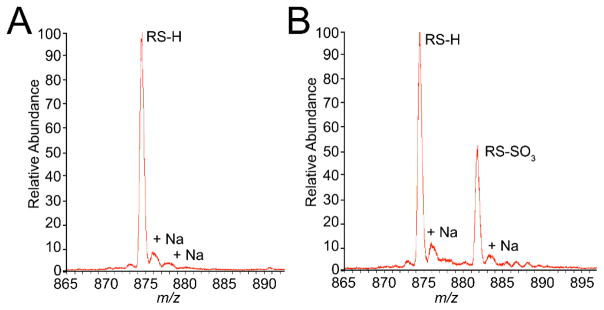
LC-ESI-MS analysis of CstR cysteine mutants following reaction with SO32−. Both sets of peaks are in the +11 charge state. C31A CstR (A) remains fully reduced while C60A CstR (B) forms an S-sulfocysteine on Cys31 as indicated by a +80 Da mass shift.
Given the importance of Cys31 in the order of reduction, we hypothesized that modification of Cys31 would be sufficient to negatively regulate DNA binding by CstR. Reduced mutant CstR was shown to bind cst OP1 with wild type-like affinity (Fig. 5B, Table 2). To test the influence of single-cysteine modifications, we reacted C31A or C60A CstR with the cysteine-modifying reagent, methylmethanethiolsulfonate (MMTS) to form CstRRS-SCH3 (Table S4). Strikingly, derivatization of Cys60 (C31A CstR) gives nearly wild type-like DNA binding affinity while derivatization of Cys31 (C60A CstR) decreased the affinity to that of derivatized WT CstR (Fig. 5B, Table 2).
These results reveal that reduction of sulfite is ordered and is not a random multi-thiol mechanism. This suggests in turn that local microenvironment near the two thiols directs these compounds to Cys31 first. Similar findings characterize the reactions of C31A and C60A CstRs with SeO32− except that both reactions form RS-SR′ and RS-Se-SR′ crosslinks. likely across the tetramer interface. C60A CstR gives a number of oxidation products including the anticipated S-selenocysteine on Cys31 (Fig. S4; Table S4). These modifications are also inhibitory to operator DNA binding (Fig. S4E). These features perhaps make CstR ideally suited to react with an wide range of cellular thiol oxidants. It is interesting to note that the formaldehyde-sensing FrmR is a CsoR-family protein that retains a single cysteine corresponding to Cys31 in CstR that is thought to react with carbon electrophiles via Michael addition.43
Conclusions
Our results show that CstR is reactive toward biologically available chalcogen oxyanions while the paralogous transcriptional regulator CsoR is weakly or not reactive under the same experimental conditions. The modification of CstR by these oxyanions takes the form of a mixed disulfide, CstR2(RS-SR′) and CstR2(RS-X-SR′)n, n = 1 or 2 where X is Se or Te. We also show that these modifications negatively regulate cst OP1 operator DNA binding. In addition, we establish that Cys31 of CstR as the thiol required to initiate the reaction between CstR and sulfite. Furthermore, derivatization of this thiol only is necessary and sufficient to negatively regulate DNA binding as modification of Cys60 does not impact DNA binding affinity. These experiments make the prediction that CstR reacts with selenite in cells thereby inducing the cst operon, although this is not yet known.
These data represent to our knowledge the first inter- or intramolecular selenotrisulfide within a native-state protein characterized by tandem mass spectrometry. Other work has been performed to identify a mixed selenotrisulfide between penicillamine-substituted glutathione and Cys-β93 of hemoglobin using MALDI-TOF.44 Penicillamine was substituted in that study due to the transient nature of the observed interaction.45 Another selenotrisulfide was analysed with ESI-MS using a model synthetic zinc-finger peptide but was not fragmented22 and thus unambiguously characterized using tandem mass spectrometry. Our identification of a stable selenotrisulfide moiety in CstR by tandem mass spectrometry provides a platform for the expansion of proteomics approaches for the detection of selenotrisulfides. There is currently no biological evidence for inter- or intramolecular selenotrisulfides in proteins in vivo but this may be due to how proteomics approaches are typically implemented. For example, it is commonplace to reduce and alkylate thiols during sample preparation46 which would reverse the adduct observed here. Approaches avoiding the reduction of thiols may provide new insights into selenium uptake, trafficking, and distribution in cells. Experiments are ongoing to further elucidate the chemical mechanism of sulfite/selenite reduction and the metabolic significance of selenite toxicity and induction of the cst operon in S. aureus Newman strain.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the NIH (GM097225 to D. P. G.). We also thank Dr. Jonathan Karty and Angela Hanson of the Indiana University Mass Spectrometry Facility for assistance with LC-ESI-MS experiments.
Footnotes
Electronic Supplementary Information (ESI) available: These include Fig. S1–S4, Tables S1–S4. See DOI: 10.1039/b000000x
Notes and References
- 1.Lowy FD. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Nizet V. J Allergy Clin Immunol. 2007;120:13–22. doi: 10.1016/j.jaci.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Lithgow JK, Hayhurst EJ, Cohen G, Aharonowitz Y, Foster SJ. J Bacteriol. 2004;186:1579–1590. doi: 10.1128/JB.186.6.1579-1590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacob C, Giles GI, Giles NM, Sies H. Angew Chem Int Ed Engl. 2003;42:4742–4758. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 5.Soutourina O, Poupel O, Coppee JY, Danchin A, Msadek T, Martin-Verstaete I. Molec Microbiol. 2009;73:194–211. doi: 10.1111/j.1365-2958.2009.06760.x. [DOI] [PubMed] [Google Scholar]
- 6.Soutourina O, Dubrac S, Poupel O, Msadek T, Martin-Verstraete I. PLoS Pathog. 2010;6:e1000894. doi: 10.1371/journal.ppat.1000894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burns KE, Baumgart S, Dorrestein PC, Zhai HL, McLafferty FW, Begley TP. J Am Chem Soc. 2005;127:11602–11603. doi: 10.1021/ja053476x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhave DP, Muse WB, 3rd, Carroll S. Infect Disord: Drug Targets. 2007;7:140–158. doi: 10.2174/187152607781001772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senaratne RH, Mougous JD, Reader JR, Williams SJ, Zhang TJ, Bertozzi CR, Riley LW. J Med Microbiol. 2007;56:454–458. doi: 10.1099/jmm.0.46983-0. [DOI] [PubMed] [Google Scholar]
- 10.Grossoehme NE, Kehl-Fie TE, Ma Z, Adams KW, Cowart DM, Scott RA, Skaar EP, Giedroc DP. J Biol Chem. 2011;286:13522–13531. doi: 10.1074/jbc.M111.220012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hildebrandt TM, Grieshaber MK. FEBS J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- 12.Jackson MR, Melideo SL, Jorns MS. Biochemistry. 2012;51:6804–6815. doi: 10.1021/bi300778t. [DOI] [PubMed] [Google Scholar]
- 13.Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, Roth JR, Baumler AJ. Nature. 2010;467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foss O. Acta Chem Scand. 1947;1:8–31. doi: 10.3891/acta.chem.scand.01-0008. [DOI] [PubMed] [Google Scholar]
- 15.Liu T, Ramesh A, Ma Z, Ward SK, Zhang L, George GN, Talaat AM, Sacchettini JC, Giedroc DP. Nat Chem Biol. 2007;3:60–68. doi: 10.1038/nchembio844. [DOI] [PubMed] [Google Scholar]
- 16.Dwarakanath S, Chaplin AK, Hough MA, Rigali S, Vijgenboom E, Worrall JAR. J Biol Chem. 2012;287:17833–17847. doi: 10.1074/jbc.M112.352740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakamoto K, Agari Y, Agari K, Kuramitsu S, Shinkai A. Microbiology. 2010;156:1993–2005. doi: 10.1099/mic.0.037382-0. [DOI] [PubMed] [Google Scholar]
- 18.Zannoni D, Borsetti F, Harrison JJ, Turner RJ. In: Adv Microb Physiol. Robert KP, editor. Vol. 53. Academic Press; 2007. pp. 1–312. [DOI] [PubMed] [Google Scholar]
- 19.Chasteen TG, Fuentes DE, Tantaleán JC, Vásquez CC. FEMS Microbiol Rev. 2009;33:820–832. doi: 10.1111/j.1574-6976.2009.00177.x. [DOI] [PubMed] [Google Scholar]
- 20.Lindemann T, Hintelmann H. Anal Chem. 2002;74:4602–4610. doi: 10.1021/ac020196g. [DOI] [PubMed] [Google Scholar]
- 21.Kice JL, Lee TWS, Pan S. J Am Chem Soc. 1980;102:4448–4455. [Google Scholar]
- 22.Larabee JL, Hocker JR, Hanas JS. J Inorg Biochem. 2009;103:419–426. doi: 10.1016/j.jinorgbio.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Chang MW, Toghrol F, Bentley WE. Environ Sci Technol. 2007;41:7570–7575. doi: 10.1021/es070929k. [DOI] [PubMed] [Google Scholar]
- 24.Schlag S, Nerz C, Birkenstock TA, Altenberend F, Götz F. J Bacteriol. 2007;189:7911–7919. doi: 10.1128/JB.00598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison JJ, Ceri H, Stremick C, Turner RJ. FEMS Microbiol Lett. 2004;235:357–362. doi: 10.1016/j.femsle.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Riddles PW, Blakeley RL, Zerner B. In: Meth Enzymol. Hirs SNTCHW, editor. Vol. 91. Academic Press; 1983. pp. 49–60. [DOI] [PubMed] [Google Scholar]
- 27.Ma Z, Cowart DM, Ward BP, Arnold RJ, DiMarchi RD, Zhang L, George GN, Scott RA, Giedroc DP. J Am Chem Soc. 2009;131:18044–18045. doi: 10.1021/ja908372b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Proc Natl Acad Sci USA. 2007;104:5860–5865. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuzmic P. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 30.Palm GJ, Khanh Chi B, Waack P, Gronau K, Becher D, Albrecht D, Hinrichs W, Read RJ, Antelmann H. Nucl Acids Res. 2012;40:4178–4192. doi: 10.1093/nar/gkr1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poor CB, Chen PR, Duguid E, Rice PA, He C. J Biol Chem. 2009;284:23517–23524. doi: 10.1074/jbc.M109.015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen H, Hu J, Chen PR, Lan L, Li Z, Hicks LM, Dinner AR, He C. Proc Natl Acad Sci U S A. 2008;105:13586–13591. doi: 10.1073/pnas.0803391105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guimarães BG, Barbosa RL, Soprano AS, Campos BM, de Souza TA, Tonoli CCC, Leme AFP, Murakami MT, Benedetti CE. J Biol Chem. 2011;286:26148–26157. doi: 10.1074/jbc.M111.234039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ba LA, Doring M, Jamier V, Jacob C. Org Biomol Chem. 2010;8:4203–4216. doi: 10.1039/c0Ob00086h. [DOI] [PubMed] [Google Scholar]
- 35.Freeman F, Angeletakis CN. J Am Chem Soc. 1983;105:4039–4049. [Google Scholar]
- 36.Jönsson TJ, Tsang AW, Lowther WT, Furdui CM. J Biol Chem. 2008;283:22890–22894. doi: 10.1074/jbc.C800124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuangthong M, Helmann JD. Proc Natl Acad Sci USA. 2002;99:6690–6695. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji Q, Zhang L, Sun F, Deng X, Liang H, Bae T, He C. J Biol Chem. 2012;287:21102–21109. doi: 10.1074/jbc.M112.359737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klomsiri C, Karplus PA, Poole LB. Antioxid Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paulsen CE, Carroll KS. ACS Chem Biol. 2009;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poole LB, Nelson KJ. Curr Opin Chem Biol. 2008;12:18–24. doi: 10.1016/j.cbpa.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milligan B, Swan JM. J Ceramic Soc. 1962;1962:683–684. [Google Scholar]
- 43.Herring CD, Blattner FR. J Bacteriol. 2004;186:6714–6720. doi: 10.1128/JB.186.20.6714-6720.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haratake M, Hongoh M, Ono M, Nakayama M. Inorg Chem. 2009;48:7805–7811. doi: 10.1021/ic900988j. [DOI] [PubMed] [Google Scholar]
- 45.Haratake M, Fujimoto K, Ono M, Nakayama M. Biochim Biophys Acta. 2005;1723:215–220. doi: 10.1016/j.bbagen.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 46.Yates JR, Ruse CI, Nakorchevsky A. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.