

Abstract
Both genetic mutations and ultraviolet (UV) irradiation can predispose individuals to melanoma. Although BRAFV600E is the most prevalent oncogene in melanoma, the BRAFV600E mutant is not sufficient to induce tumors in vivo. Mutation at the CDKN2A locus is another melanoma-predisposing event that can disrupt the function of both p16INK4a and ARF. Numerous studies have focused on the role of p16INK4a in melanoma, but the involvement of ARF, a well-known p53 activator, is still controversial. Using a transgenic BRAFV600E mouse model previously generated in our laboratory, we report that loss of ARF is able to enhance spontaneous melanoma formation and cause profound sensitivity to neonatal UVB exposure. Mechanistically, BRAFV600E and ARF deletion synergize to inhibit nucleotide excision repair by epigenetically repressing XPC and inhibiting the E2F4/DP1 complex. We suggest that the deletion of ARF promotes melanomagenesis not by abrogating p53 activation but by acting in concert with BRAFV600E to increase the load of DNA damage caused by UV irradiation.
Keywords: BRAFV600E, ARF, UV melanomas, XPC, promoter methylation
Introduction
Derived from the professional pigment-producing melanocytes, melanoma is considered the most lethal form of skin cancer (1). Mutation in the BRAF protein, especially the V600E point mutation, is the most prevalent genetic alteration in human melanoma (2). Interestingly, the BRAFV600E mutation on its own is not sufficient to induce tumor formation in vitro and in vivo, consistent with the fact that expression of BRAFV600E in melanocytes either in the culture dish or in animals results in senescence (3–6). Therefore, additional genetic alterations are required for the progression of BRAFV600E-expressing melanocytes to melanoma, at least in part to achieve suppression of the senescence response.
In addition to BRAF mutation, the CDKN2A locus is frequently targeted in melanoma (7). Two distinct tumor suppressors, p16INK4a and p14ARF (p19ARF in mice, hereafter referred to as ARF), are encoded by this locus via alternative reading frames (8). In contrast to p16INK4a, the importance of ARF in melanomas has been debated, since germline mutations in ARF fail to associate with melanoma susceptibility (9) and CDKN2A mutations that target only p16INK4a but not ARF for inactivation have been described (10). However, the recent discovery of mutations specific to the ARF-coding region and ARF-specific deletions (11), as well as the observation that ARF ablation facilitates oncogenic induction of murine melanoma (12–15), highlight the significance of ARF in melanomagenesis. Therefore, it is increasingly accepted that p16INK4a-independent inactivation of ARF can also predispose individuals to melanoma.
The mechanistic basis of ARF’s ability to suppress melanoma may lie in ARF’s canonical role in the MDM2-p53 pathway, and indeed p53 is mostly wild-type in human melanomas (16), suggesting loss of ARF may abrogate the need for p53 mutation. However, ARF also has p53-independent functions (17, 18), which explains the fact that it can act as a melanoma suppressor in the absence of p53 (14). For example, ARF has been shown to bind to DRTF polypeptide 1 (DP1), a coactivator of E2F transcription factors, and inhibit the formation of active DP1-E2F complexes, thereby regulating the expression of E2F target genes (19–21). Therefore, the mechanism of melanoma suppression by ARF remains unclear. Understanding this mechanism may have impact on the design of future therapies for melanoma.
The ability of ARF to modulate E2F activity also extends ARF’s function to activation of the nucleotide excision repair (NER) pathway (21). NER is a DNA repair mechanism that functions to remove bulky DNA adducts such as cyclobutane pyrimidine dimers (CPD) and 6-4-photoproducts mostly caused by ultraviolet irradiation (UVR) (22). Interestingly, solar UVR is the major etiological factor for skin cancers including melanoma, although the relationship between UVR and melanoma is less evident than that of squamous and basal cell carcinomas (23). Recent studies demonstrated neonatal UVB irradiation (275–320nm) can exacerbate melanoma penetrance in various genetic mouse models (12, 13, 24); however, the mechanism leading to UV sensitivity in melanocytes is not well understood. Although BRAF mutations are not overrepresented in melanomas arising from chronic sun-exposed areas, it is unclear whether BRAFV600E alone or in combination with other genetic events increases the sensitivity to UVR.
In the present study, we used a transgenic BRAFV600E mouse model (3) to show that loss of ARF is able to shorten the latency of melanoma formation and causes profound sensitivity to neonatal UVB exposure. Oncogenic BRAFV600E instigates transient DNA damage in melanocytes which triggers senescence that cannot be bypassed by ARF deletion in vivo. However, loss of ARF cooperates with BRAF mutation to suppress the NER pathway, enhancing UVB-induced DNA damage and melanomagenesis.
Materials and Methods
Mice
All mouse experiments were performed with the approval of Tufts University/Tufts Medical Center IACUC. The generation of melanocyte-specific BRAFV600E transgenic mice was described previously (3). Founder mice were backcrossed to C57BL/6 mice for more than 10 generations. The exon 1b-specific p19ARF knockout mouse in C57BL/6 background used in this study has been described elsewhere (25) and was a generous gift from N. Rosenberg (Tufts University School of Medicine, Boston, MA). All mice were maintained in a pathogen-free mouse facility at Tufts University School of Medicine.
Cell Culture and Recombinant Vectors
All cells were cultured in a 5% CO2 humidified incubator at 37°C. Primary mouse melanocytes were isolated from neonatal mouse skins as described (26) and maintained in F12 medium supplemented with 3% fetal bovine serum (FBS, Gibco), 1% antibiotic-antimycotic (Invitrogen), 48 nM 12-O-tetradecanoyl-phorbol-13-acetate (Sigma), 0.1 mM isobutylmethyl xanthine (Sigma), 10 μg/ml bovine pituitary extract (Invitrogen) and 0.1 mM dibutyryladenosine 3′,5′-cyclic monophosphate (Sigma). Contaminating fibroblasts and keratinocytes were eliminated by treatment with 100 μg/ml geneticin (Invitrogen) for 24h. Primary mouse melanoma cells were isolated by collagenase/hyaluronidase digestion of tumor fragments for 30 min and grown in RPMI 1640 (Gibco) supplemented with 10% FBS and 1% antibiotic-antimycotic (27). Human melanoma cell line CHL1 was cultured in DMEM (Gibco) with 10% FBS and 1% penicillin-streptomycin.
The retroviral vector pBabe-puro-p19ARF was generously provided by N. Sharpless (University of North Carolina, Chapel Hill, NC), pBabe-hygro-dominant-negative-p53 was obtained from Addgene (#9058) (28). The lentiviral construct FG12-HA-BRAFE600-eGFP was a gift from D. Peeper (The Netherlands Cancer Institute, Amsterdam, The Netherlands) (5). shRNA targeting mouse p53 sequence 5′-GTACTCTCCTCCCCTCAAT-3′ was generated in the pMKO.1 retroviral vector.
UV Exposure
For irradiation of cultured cells, cells were washed with PBS and subsequently exposed to UV radiation using UVB lamps (280–314nm; UVP, Inc.) at the indicated dose. UV emittance was measured with the use of a UV photometer (model ILT1400A; UV Products).
For in vivo tumor induction, animals were irradiated under the UVB bulbs at a dose of 750mJ/cm2 at neonatal day 3.5. Following UV exposure, mice were carefully monitored for the degree of erythema and/or desquamation. Severe erythema or desquamation resulted in sacrifice of the animal but was rarely observed. The majority of animals displayed mild to moderate erythema and was monitored for tumor formation as indicated.
Histology
Tissue samples were fixed in 10% buffered formalin overnight and stored in 70% ethanol prior to paraffin embedding, sectioning and hematoxylin/eosin (H&E) staining (by the Rodent Histopathology Core, Harvard Medical School). Immunohistochemistry was performed with the following antibodies: Ki-67 (SP6, Thermo Scientific), p16 (M-156, Santa Cruz), p53 (Thermo Scientific), interleukin 6 (ab6672, Abcam), Trp2 (ab74073, Abcam), S100 (Ab-2, Thermo Scientific), Melan A (C-20, Santa Cruz) and HMB45 (ab732, Abcam). Staining was performed with Vector NovaRED Substrate Kit (Vector Laboratories). Negative control was done by replacing the primary antibody with species-matched total IgG.
Senescence-associated β-Galactosidase Staining
Skin samples were fixed in 4% buffered paraformaldehyde at room temperature for 30 min, followed by a wash with 50 mM glycine in PBS. Dehydration was performed by subsequently soaking the samples in 20% sucrose overnight and 30% sucrose until the samples settle at the bottom. Dehydrated samples were embedded in OCT, frozen and sectioned. The staining for perinuclear SA-β-gal activity was performed according to protocol described previously (29, 30).
Immunoblotting and Immunoprecipitation
For immunoblotting, tissue samples or cultured cells were homogenized and/or lysed with RIPA buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% NP-40, 1% sodium deoxycholate, 1 mM sodium pyrophosphate, 10 mM β-glycerophosphate, 0.2 mM Na3VO4, 10 mM NaF, 1 mM DTT and eComplete-Mini protease inhibitor cocktail (Roche). Primary antibodies used included p19 (ab80, Abcam), p16 (F-12, Santa Cruz), GAPDH (MAB374, Chemicon), p53 (Thermo Scientific), p21 (F-5, Santa Cruz), phosphor-p53-Ser15 (9284, Cell Signaling), pERK1/2 (4376, Cell Signaling) and ERK2 (C-14, Santa Cruz).
Co-immunoprecipitation was carried out as described by Dominguez-Brauer et al. (21). The antibodies used were p19 (ab80, Abcam), E2F4 (C-20, Santa Cruz) and DP1 (hybridoma supernatant kindly provided by J. Lees, MIT, Cambridge, MA).
Quantitative Real-time PCR
Total RNA was extracted by TRIzol (Invitrogen) and converted into cDNA using either iScript (Bio-Rad) or SMARTScribe (Clontech) cDNA synthesis kit. Gene expression was quantified using QuantiTect SYBR Green (Qiagen) or SYBR Advantage (Clontech).
Genomic DNA was extracted by phenol-chloroform, followed by methylation sensitive enzyme HpaII digestion for 3h. Quantitative PCR was carried out with 1 μl of the HpaII-digested heat-inactivated gDNA and with the XPC promoter-specific and control primers (Supplementary Table 1). The sequences between XPC forward and reverse primers harbor multiple HpaII recognition sites, thereby will be digested if not methylated; the sequences between the control primers do not have any HpaII sites, and thus served as an internal control.
CPD Immunoblot
Heat-denatured genomic DNA was dot-blotted onto a nitrocellulose membrane and was blocked with 5% milk overnight. The membrane was then incubated with anti-CPD antibody (provided by R. Cui, Boston University, Boston, MA) for 30 min at room temperature.
Growth Curve and Soft Agar Colony-formation Assay
Primary mouse melanoma cells were stably infected with dominant-negative p53 (DNp53) or empty vector and seeded in 6-well plates at a density of 1X104/well in triplicate. Cell number was counted at indicated time points.
For anchorage-independent growth assays, 2X104 cells were suspended in medium containing 0.4% agarose and plated onto a solidified layer of medium-containing 0.8% agarose in 6-well plates. Colonies were quantified in five fields in each well 3 weeks post seeding.
Chromatin Immunoprecipitation
ChIP was carried out according to the protocol described previously (31) using E2F4 antibody (C-20, Santa Cruz). Real-time PCR was performed according to Dominguez-Brauer et al.(21). ChIP data were normalized to input chromatin.
Results
ARF is a suppressor of BRAFV600E-driven murine melanoma
ARF can be induced by oncogenes such as c-Myc and Ras in cultured cells (17). To determine whether oncogenic BRAFV600E can trigger ARF expression in vivo, we performed immunoblots on skin lysates from either wild-type (WT) or melanocyte-specific BRAFV600E transgenic mice (3). ARF, but not p16INK4a, was detectable in the BRAFV600E skin from adult mice (Figure 1A), suggesting that ARF induction is a significant cellular response to chronic BRAF activation. Therefore, we introduced the germline ARF-specific deletion allele (p16INK4a is intact) into BRAFV600E mice (strain 476)(3) and monitored tumor development. Neither BRAFV600E nor BRAFV600E;ARF+/− mice developed tumors for up to 5 months, whereas BRAFV600E;ARF−/− animals formed tumors relatively rapidly, with a mean latency for survival of 85 days of age (Figure 1B). 40% of the BRAFV600E;ARF−/− mice were sacrificed because of progressive melanoma, as indicated by asterisks in the curve (Figure 1B & S1A); the remaining mice were sacrificed as a result of sarcoma and lymphoma, the prominent tumor types in germline ARF-null animals (32). Surprisingly, during the experimental period, sarcomas and lymphomas were also observed in the BRAFV600E;ARF−/− mice but not in the control (except in one BRAFV600E;ARF+/− mouse, Figure 1B), suggestive of reduced latency for these ARF-null tumors in the tyrosinase-driven BRAFV600E background. Given that expression of the BRAFV600E transgene was not readily detected in the sarcomas and lymphomas examined (Figure S1B), it is unlikely that these tumors resulted from expression of BRAFV600E in the affected tissues. Considering the existence of elevated amounts of inflammatory cytokines (Figure 3B & C), it is possible that alteration of the microenvironment by the BRAFV600E transgene facilitates the development of other tumors.
Figure 1. Loss of ARF accelerates melanoma induced by BRAFV600E in vivo.
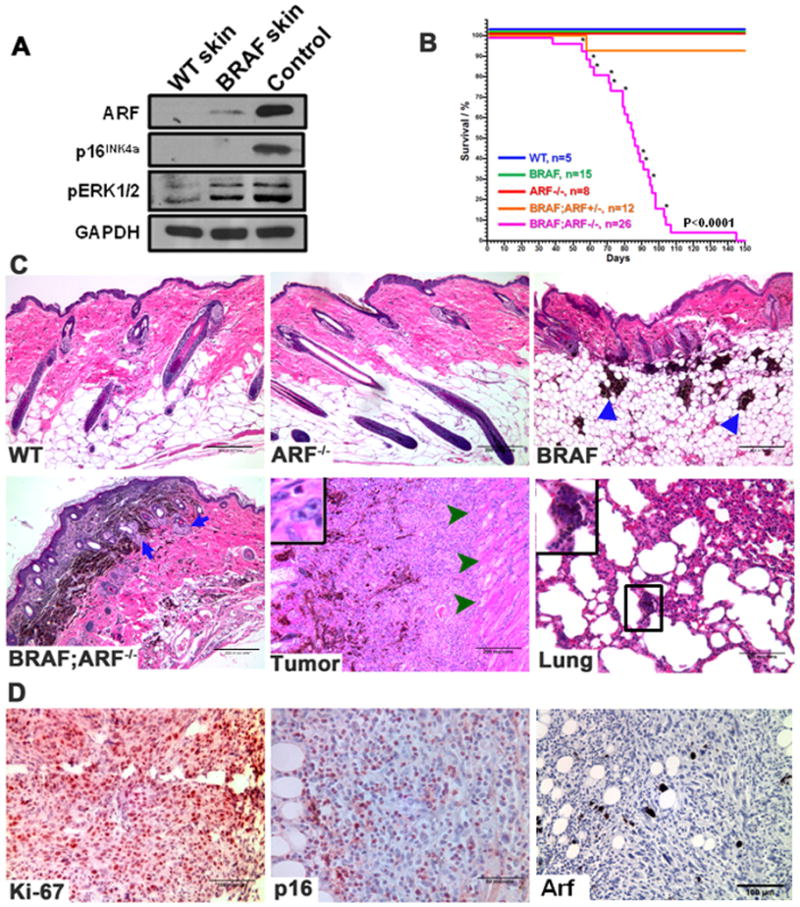
(A) Immunoblots of total skin lysate show that ARF but not p16INK4a is detected in the BRAFV600E transgenic skin. pERK1/2 was blotted to confirm the presence of active BRAF in the skin. Spontaneous melanoma derived from a BRAFV600E (470 strain) mouse was used as a positive control for both ARF and p16INK4a.
(B) Kaplan-Meier plot of total mortality of mice with different genotypes. Asterisk indicates mouse died of melanoma. The melanoma-free survival rate of the same cohort of mice is shown in Supplementary Figure 1A.
(C) H&E-stained sections from representative skins of different genotypes, primary BRAFV600E;ARF−/− tumor and lung. Triangles in BRAF skin image indicate the large nests of epithelioid cells; arrows in BRAF;ARF−/− skin image show the appearance of the Schwannian differentiated phenotype; arrowheads in BRAF;ARF−/− tumor image show tumor cells invading the muscle layer; inset in the tumor image shows mitotic figure, and inset in the lung image shows pigmented micrometastasis.
(D) Immunohistochemical (IHC) staining of Ki-67, p16INK4a and p19Arf (Arf) in representative sections from BRAFV600E;ARF−/− tumors. Arf staining in tumor section confirms loss of Arf expression. Sections were counterstained with hematoxylin.
Figure 3. Loss of ARF cannot suppress BRAFV600E-induced senescence in vivo.

(A) SA-β-gal staining on skin sections from age-matched mice of the indicated genotypes. Hematoxylin was used to counterstain the nucleus. Red boxed areas in the upper panel are enlarged in the lower panel.
(B) qRT-PCR showing the mRNA expression of selected members of the senescence-associated secretome in skins of different genotypes. Student t-test, compared to WT skin, * p<0.05, ** p<0.005.
(C) IHC of interleukin-6 (IL-6) performed on representative skin sections from BRAFV600E and BRAFV600E;ARF−/− mice. Boxed areas are enlarged as indicated and arrowheads represent the positive staining cells. Sections were counterstained with hematoxylin.
(D) IHC of p16INK4a and p53 on representative skin sections from BRAFV600E;ARF−/− mice. IgG is negative control and nucleus was counterstained by hematoxylin.
Histologically, ARF−/− skin was comparable to WT skin (Figure 1C). Expression of BRAFV600E caused melanocytic proliferation presenting as large nests of epithelioid cells in the deep dermis and subcutis (Figure 1C, triangles)(3). Knockout of ARF in BRAFV600E mice increased the apparent hyperplasia of melanocytes, with increased deposits in the dermis and subcutis and the appearance of a Schwannian differentiated phenotype (Figure 1C, arrows)(3). Melanomas that formed in the BRAFV600E;ARF−/− mice showed a transition from pigmented to unpigmented, suggesting progression from a differentiated to an undifferentiated phenotype (Figure 1C). The tumors were often very invasive, penetrating through the muscle layer underneath the skin (Figure 1C, arrowheads), and showed a high proliferation index seen in the form of mitotic figures (Figure 1C, insert) and Ki-67 staining (Figure 1D). Interestingly, this proliferation was observed despite the fact that a substantial number of cells positive for nuclear p16INK4a were detected in the tumor sections (Figure 1D). Analysis of lungs from 6 melanoma-bearing mice revealed pigmented micrometastasis in 2 cases (Figure 1C). Taken together, these data indicate that deletion of ARF accelerates invasive melanoma formation driven by BRAFV600E and thus acts as a melanoma suppressor in this context.
ARF loss does not abrogate p53 activation in BRAFV600E;ARF−/− murine melanomas
ARF is a well-characterized p53 stabilizer, therefore abrogation of ARF is expected to dampen p53 activation. Surprisingly, we detected strong p53 expression in the BRAFV600E;ARF−/− precancerous skin and tumors (Figure 2A & S2A). p53 was active based on its phosphorylation at Ser15 site and expression of the downstream target p21 (Figure 2B), suggesting that loss of ARF might not affect the function of p53 induced by chronic BRAFV600E stimulation in this model. To confirm that p53 in BRAFV600E;ARF−/− tumor cells is functional, we knocked down p53 and found that p21 level was reduced (Figure 2B). In addition, we restored ARF expression in BRAFV600E;ARF−/− tumor cells (Figure S2B), and challenged the cells with UV irradiation. Upon UV induction, p53 was rapidly phosphorylated regardless of ARF status (Figure 2D), further supporting the contention that p53 in ARF-null melanoma cells is functional.
Figure 2. p53 is functional in BRAFV600E;ARF−/− melanocytes and melanoma cells despite ARF deficiency.
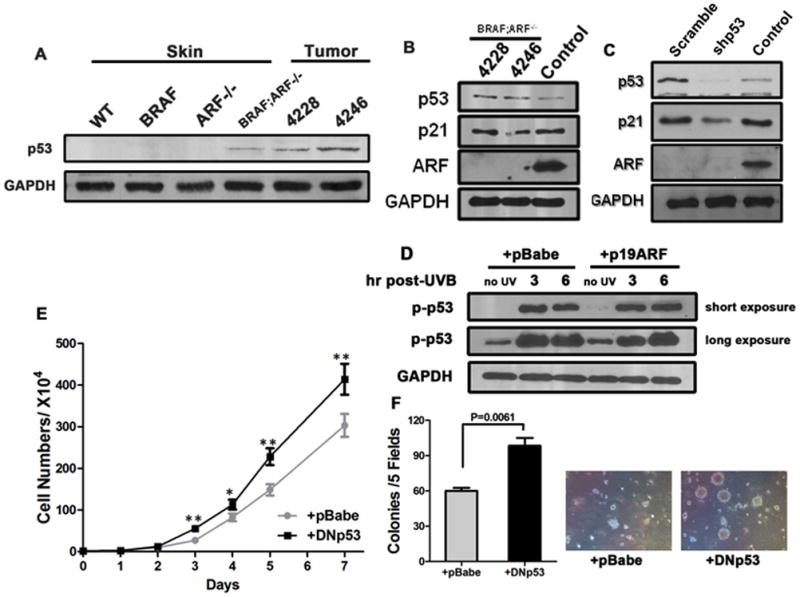
(A) Immunoblots detecting p53 from total skin and tumor lysates.
(B) Immunoblots detecting p21 and phospho-p53 at Ser 15 (p-p53) from total BRAFV600E;ARF−/− tumor lysates. Control lane was lysate from a spontaneous BRAFV600E melanoma (470 strain, the same sample with that used in Figure 1A) serving as a positive control for ARF.
(C) Immunoblots demonstrating p53 knockdown efficiency and reduced level of p21, a downstream target of p53, in isolated primary BRAFV600E;ARF−/− melanoma cells. Control sample for ARF was the same as in (B).
(D) Immunoblots demonstrating induction of phospho-p53 in primary BRAFV600E;ARF−/− melanoma cells with or without ARF restoration. p53 can be rapidly activated upon UV irradiation, regardless of ARF status, confirming that the p53 present in the cells is functional.
(E–F) Growth curve and soft agar colony-formation ability of BRAFV600E;ARF−/− primary melanoma cells. Cells (#4228) with dominant-negative p53 (DNp53) grew faster (D) and formed more colonies (E) than control. Colonies that were bigger than 30μm were quantified. Student t-test, * p<0.05, ** p<0.005.
Given that deletion of ARF does not alter p53 functionality, we reasoned that repressing the function of p53 would have an additive effect on ARF loss to enhance tumor formation. To test this, we generated BRAFV600E;ARF−/− tumor cells stably expressing either dominant-negative p53 (DNp53, Figure S2C) or mouse p53-specific shRNA. Proliferation assays showed that DNp53-expressing cells grew significantly faster than cells with vector control (Figure 2D). Soft agar colony formation assays confirmed that DNp53-expressing cells produced more and larger colonies than control cells (Figure 2E). Results using p53-shRNA were similar (Figure S2D–E). Taken together, the data suggest that loss of ARF affects targets other than p53 to increase melanoma formation, since p53 is active despite ARF deficiency.
Loss of ARF cannot suppress BRAFV600E-induced senescence in vivo
It has been shown that loss of ARF bypasses senescence in cultured primary melanocytes (14). To examine whether ARF deficiency can repress oncogene-induced senescence in vivo, we performed senescence-associated (SA) β-gal staining on skin sections from age-matched mice. This approach indicated the presence of senescence in BRAFV600E skin; however, to our surprise, strong SA-β-gal staining can also be detected in the double mutant skin (Figure 3A), suggesting senescent cells are still abundant in the ARF-null background. Quantitation of senescent cells in the skin based on SA-β-gal staining is complicated by dye retention in hair follicles and sebaceous glands. Therefore, we analyzed the expression of genes encoding members of the senescence-associated secretome as an additional marker. Quantitative RT-PCR revealed that most of the secretome factors tested were upregulated in BRAFV600E skin and considerably more highly expressed in BRAFV600E;ARF−/− skin (Figure 3B). Further, IHC staining confirmed increased expression of IL-6 adjacent to putative senescent cells (Figure 3C). The presence of an equal or increased number of senescent cells observed in the BRAFV600E;ARF−/− skins is consistent with the observation that both p16INK4a and active p53 are present in situ (Figure 3D). Therefore, the persistent expression of senescence activators and the presence of a copious number of senescent cells despite the lack of ARF strongly argue that direct evasion of senescence is not the mechanism by which loss of ARF accelerates BRAFV600E-driven melanoma formation.
Loss of ARF sensitizes BRAFV600E mice to UV-induced melanomagenesis
The data described above are consistent with the notion that increased and/or persistent cellular stress in BRAFV600E;ARF−/− melanocytes leads to genetic mutations that bypass senescence and lead to melanomagenesis. To determine if cellular changes elicited by combined BRAF and ARF mutation render melanocytes sensitive to DNA damage, we studied the response of mutant cells and animals to UV irradiation, since solar UV irradiation is the main etiological factor for melanoma. To this end, we irradiated neonatal mice of different genotypes with a single dose of UVB (750mJ/cm2, Figure 4A). During the period of study (100 days), none of the WT, BRAFV600E or ARF−/− mice succumbed to melanoma following UVB exposure (Figure 4B). Extending the observation period to 300 days also failed to reveal UV-induced tumors in WT and BRAFV600E mice, in agreement with a previous report focusing on Tyr-NRas mice (13). Significantly, every UVB-irradiated BRAFV600E;ARF−/− mouse developed multiple melanomas, with 50% of mice developing tumors by day 70 post-UV (73 days of age; Figure 4B). Thus, UVB irradiation promotes melanomagenesis in BRAFV600E;ARF−/− mice. Histopathological analysis of the tumors confirmed most of them were amelanotic melanomas, as they did not secrete melanin, but stained positive for melanoma markers Trp2, S100, Melan A and HMB45 (Figure 4C). Consistent with our observation in spontaneous melanomas, UVB-induced melanomas also preserve active p53 (Figure 4C, lower panel). Therefore, although UVB irradiation is not sufficient to induce melanomas in mice expressing melanocytic BRAFV600E, loss of ARF sensitizes BRAFV600E mice to UVB-induced melanomagenesis.
Figure 4. Loss of ARF sensitizes BRAFV600E mice to UV-induced melanoma.
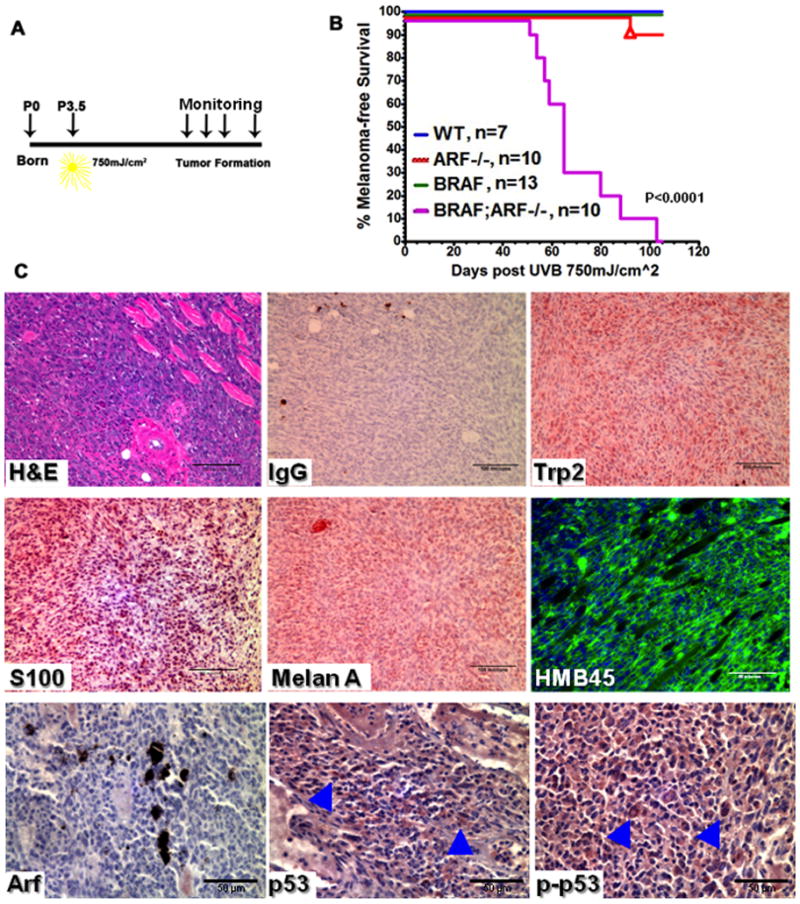
(A) Schematic representation of the neonatal UVB irradiation protocol. Mice of different genotypes were irradiated with 750mJ/cm2 UVB at postnatal day 3.5 (P3.5).
(B) Kaplan-Meier curve showing melanoma-free survival of mice receiving neonatal UVB irradiation. Triangle in the ARF−/− curve indicates mouse died of sarcoma.
(C) H&E and immunostaining of melanoma markers Trp2, S100, Melan A and HMB45 on representative sections from UVB-induced BRAFV600E;ARF−/− tumors. Arf staining confirms the absence of the protein in tumors. Arrowheads in the p53 and phospho-p53 (Ser15) images indicate the positive staining cells.
Loss of ARF impairs DNA damage repair by transcriptionally repressing XPC
UV irradiation results in bulky DNA adducts such as cyclobutane pyrimidine dimers (CPD) and 6,4-photoproducts, which can be repaired by nucleotide excision. In the absence of repair, mutagenic events may become fixed in the genome and lead to cellular transformation. Because UVB can increase melanoma development in BRAFV600E;ARF−/− mice, we wanted to know whether the combination of those two mutations has any effect on DNA repair capability. Since the xeroderma pigmentosum, complementation group C gene (XPC) is a key component in the NER pathway responsible for recognizing DNA adducts, we tested the expression of XPC in primary melanocytes with different genotypes by RT-qPCR. Single or double mutant melanocytes expressed a significantly lower level of XPC mRNA, with the lowest level seen in the double mutant cells (Figure 5A). The reduced XPC mRNA level correlated with impaired DNA repair capability as demonstrated by a CPD removal assay (Figure 5B): CPD accumulated dramatically 3h following UVB irradiation and was successfully cleared in WT melanocytes by 24h post-UVB, but persisted in cells with BRAFV600E and ARF loss. It has been reported that in mouse fibroblasts, ARF enhances XPC expression by inhibiting the transcriptional repressor activity of E2F4 via disruption of the E2F4-DP1 interaction (21). To determine whether this mechanism might be active in melanomas, we subjected BRAFV600E;ARF−/− melanoma cells with or without ARF reconstitution to UVB irradiation and measured the XPC mRNA level. Upon UVB, XPC mRNA decreased dramatically in the absence of ARF, while the presence of ARF maintained the expression of XPC mRNA (Figure 5C). Further, we confirmed that ARF is able to bind DP1, depleting the fraction of DP1 bound to E2F4 (Figure 5D). Chromatin immunoprecipitation of E2F4 confirmed the association of this transcriptional repressor with the XPC promoter in both unirradiated and irradiated melanoma cells (Figure 5E). Interestingly, E2F4 association with the XPC promoter decreased following UVB irradiation (Figure 5E) concomitant with reduction in XPC mRNA (Figure 5C), suggesting that UVB irradiation may increase the association of E2F4 with corepressive factors such as p130 (21), thus repressing promoter activity more efficiently despite reduced promoter occupancy. Importantly, reconstitution of melanoma cells with ARF further decreases the association of E2F4 with the XPC promoter and results in increased transcription, suggesting that ARF acts to counteract E2F4-mediated repression of XPC in UVB-irradiated melanoma cells, consistent with the reported role of ARF in MEFs (Figure 5E)(21).
Figure 5. Loss of ARF impairs DNA damage repair pathway by transcriptionally repressing XPC.
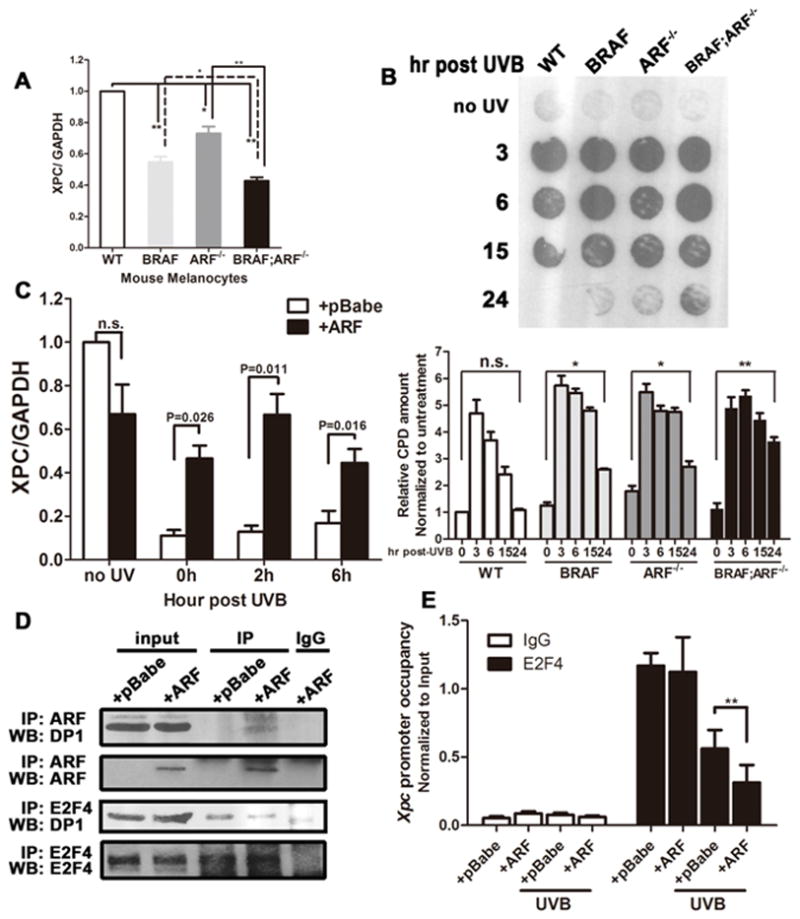
(A) mRNA expression of XPC in primary mouse melanocytes of different genotypes measured by qRT-PCR. Student t-test, * p<0.05, ** p<0.005.
(B) Immunodot blot and quantification of the level of CPD in genomic DNA at various time points after UVB irradiation. Primary mouse melanocytes of different genotypes were subjected to UVB exposure at 25mJ/cm2, followed by genomic DNA extraction at indicated time points.
(C) Expression of XPC mRNA in primary mouse melanoma cells after UVB exposure. BRAFV600E;ARF−/− melanoma cells with or without p19ARF restoration were subjected to UVB irradiation for about 3 min to reach 100 mJ/cm2, followed by RNA isolation at various time points. 0h represents RNA isolated immediately after UVB exposure.
(D) Co-immunoprecipitation showing the interaction between p19ARF/E2F4 and DP1. The binding of p19ARF and DP1 can be detected in BRAFV600E;ARF−/− melanoma cells expressing ectopic p19ARF. In the presence of p19ARF, the amount of DP1 bound to E2F4 was decreased.
(E) ChIP demonstrates XPC promoter occupancy by E2F4. Immunoprecipitation was performed using lysates of BRAFV600E;ARF−/− melanoma cells with or without p19ARF restoration receiving 25mJ/cm2 UVB; qPCR was performed using the primers described previously (22).
BRAFV600E inhibits the NER pathway by promoting methylation of the XPC promoter
The RT-qPCR result in Figure 5A shows that BRAFV600E mutation alone can reduce the level of XPC mRNA. It has been reported that in lung cancer, the promoter region of XPC is highly methylated (33). Further, perturbation of Ras signaling can regulate DNA methylation (34). Since BRAF functions downstream of Ras, it’s reasonable to postulate that activation of BRAF could also influence promoter methylation. To test the relevance of this mechanism to XPC regulation, we first analyzed the CpG island in the mouse XPC promoter that spans -1000 to -1 relative to the transcriptional start site. As is the case in the human XPC promoter, this region contains multiple CpG islands, as predicted by CpG island searcher software (35) (Figure 6A). Three HpaII restriction enzyme recognition sites are located within the XPC promoter. Therefore we performed HpaII-based quantitative PCR to determine XPC methylation in melanocytes (33). PCR products can be detected in HpaII-digested genomic DNA from BRAFV600E and BRAFV600E;ARF−/− cells, with a higher amount in the BRAFV600E;ARF−/− DNA (Figure 6B), indicating that the XPC promoter is hypermethylated in cells of these genotypes. Consistent with this, treatment of melanocytes with the DNA demethylating agent 5’-aza-2’-deoxycytidine can increase the XPC mRNA level only in cells harboring the BRAFV600E mutation (Figure 6C), suggesting a link between BRAF mutation and XPC hypermethylation.
Figure 6. Mutant BRAFV600E caused XPC promoter methylation.

(A) Diagram showing CpG islands in promoter region of the mouse XPC gene.
(B) Detection of XPC promoter methylation in mouse primary melanocytes of different genotypes. Genomic DNA was subjected to methylation-sensitive HpaII treatment, following by quantitative PCR using primers flanking the HpaII-recognition sites. Primers flanking genomic sequences that do not contain any HpaII site, thereby resistant to HpaII treatment, were used as internal control. Increased recovery of PCR product indicates increased methylation in the corresponding region. Student t-test, * p<0.05, ** p<0.005.
(C) Expression of XPC following treatment with the demethylating agent 5′-aza-2′-deoxycytidine (aza-dC). Mouse primary melanocytes of different genotypes were treated with 5μM aza-dC for 48h, followed by RNA preparation and quantitative real-time PCR.
(D–E) Acute expression of BRAFV600E increases XPC promoter methylation and inhibits XPC mRNA expression. BRAF wild-type, NRAS wild-type CHL1 melanoma cells were infected with either empty vector or HA-BRAFV600E virus, followed by treatment with mutant BRAF inhibitor PLX-4032 at indicated concentration for 48h. Protein, RNA and DNA were collected and subjected to immunoblot to confirm the activation of BRAF pathway (D), real-time quantitative PCR to determine XPC mRNA level (E) and HpaII-based qPCR to examine promoter methylation (F).
To examine whether BRAFV600E directly stimulates XPC promoter hypermethylation, we acutely expressed BRAFV600E in BRAF wild-type CHL1 melanoma cells (Figure 6D & S3A). Consistent with a previous report (36), introduction of BRAFV600E instigated senescence, presumably due to activation of the DNA damage response mainly in the form of double-strand breaks (Figure S3B). The expression of XPC was significantly inhibited in response to BRAFV600E addition (Figure 6E), correlating with a dramatic induction of methylation in the XPC promoter region (Figure 6F). Abrogating mutant BRAF activity by selective inhibition with PLX-4032 was able to not only restore XPC expression but also impaired promoter methylation (Figure 6E & F), suggesting that BRAF kinase activity is required for maintaining XPC promoter hypermethylation. Interestingly, PLX-4032 caused ERK activation in CHL1 cells with wild-type BRAF (Figure 6D) (37), which also resulted in XPC reduction and promoter methylation (Figure 6E & F), further supporting the involvement of BRAF-mediated signaling in XPC promoter hypermethylation.
Discussion
In this study, we confirmed ARF is a melanoma suppressor that can cooperate with activated BRAF and demonstrated that loss of ARF is able to sensitize BRAFV600E mice to neonatal UVB-induced melanoma. Oncogenic BRAFV600E instigates senescence in melanocytes, which cannot be bypassed by ARF deletion in vivo. Mechanistically, we showed that ARF loss did not abrogate p53 activity, but instead reduced nucleotide excision repair (NER) by elevating the inhibitory effect of E2F4-DP1 on XPC expression, as first identified in fibroblasts (21). In addition, BRAFV600E also impaired XPC expression by increasing promoter hypermethylation. Interestingly, XPC has been shown to play a role in melanoma photocarcinogenesis (38). Therefore, XPC expression appears to be a nexus for both oncogenic events studied here, and the combined effects of BRAF mutation and ARF deletion act synergistically to inhibit DNA repair and enhance melanomagenesis in the presence of UVB irradiation. We surmise that increased melanomagenesis in BRAFV600E;ARF−/− mice that have not been exposed to UV irradiation also results from decreased DNA repair capacity, rendering mutant melanocytes sensitive to the increase in double-strand breaks (DSB) that accompanies BRAFV600E expression. Interestingly, chronically reduced XPC expression has been linked to a reduced ability to repair DSBs (39), but effects of BRAFV600E and ARF loss on other forms of DNA repair may also contribute to the high rate of mutations observed in melanoma cells (40).
The efficient formation of melanomas in BRAFV600E;ARF−/− mice despite the persistence of high levels of senescence in the skin strongly suggests that the elevated mutation rate leads to selection for events that bypass senescence in individual cells that then emerge as tumor foci. The gene encoding p16INK4a is an obvious candidate, yet BRAFV600E;ARF−/− tumors retained p16INK4a expression, excluding the possibility of concurrent loss of p16INK4a as a cause. Indeed, although deletion of p16INK4a undoubtedly can predispose to melanoma formation, preservation of this protein in established melanoma is, nevertheless, not uncommon (4). Identification of mutational events that cooperate with BRAFV600E to bypass senescence and drive melanoma formation or progression is of obvious importance therapeutically, and the system described here could be useful for studying these mutations. In addition to unbiased screens for cooperating alleles, approaches to this problem include further analysis of likely candidates such as components of the Rb pathway (12), as well as several genes newly identified as susceptible to UV signature mutations in human tumors (41).
One candidate that seems surprisingly excluded from alteration in these melanomas is p53, whose UV-signature mutants are commonly linked to non-melanoma skin tumors (42). Further, although ARF has been long appreciated as a p53 stabilizer, in our model, we did not observe p53 inactivation in spite of ARF deficiency. To the contrary, persistent p53 expression and activation were detected in precancerous skins, tumor tissues and primary cell lines derived from this model. This seemingly contradictory result is actually in accord with human studies concluding that p53 is commonly preserved in melanomas (43), and at the same time undermines the argument that the lack of TP53 mutations in melanoma is compensated by functional loss of p53 activity owing to ARF deletion (7, 43). Indeed, contrary to other human malignancies, p53 in melanomas commonly remains wild-type at the genomic level (16), and also tends to be overexpressed at the protein level (43). Although p53 targeting, in combination with lineage-specific oncogenes activation, has been proven to drastically induce melanoma formation (3, 6, 13, 14), BRAFV600E;TP53−/− murine melanomas show a distinct transcript profile from that of BRAFV600E;CDKN2A−/− tumors which closely resemble human melanoma (unpublished data, FG Haluska). This in turn suggests that the TP53-null melanoma might derive from a distinct population from that giving rise to human melanoma. It is well established that ARF possesses functions independent of p53 (17), and selection against these functions of ARF appear to predominate in melanoma. However, it is likely that alterations in the p53-independent targets of ARF, the actions of hyperactivated BRAF and selected genetic and epigenetic events combine to offset the effects of activated p53 in nascent melanomas.
The presence of p53 in precancerous BRAFV600E;ARF−/− skin correlates well with our observation that senescence persists in the same tissues, although a previous report indicates ARF ablation bypasses senescence in cultured melanocytes (14). A potential explanation for this discrepancy lies in the difference of senescence triggers: oncogenic BRAFV600E stimulation in melanocytes in situ versus primary melanocytes passage in culture. Since we have not tested the stability of p53 in the context of ARF loss in this model, it’s also possible that without ARF, the turnover of p53 is augmented; however, constitutive oncogenic signaling may effectively counterbalance this, leading to the observed elevation of active p53. On the other hand, the fact that senescence persists in the skin of BRAFV600E;ARF−/− animals that eventually succumb to tumors does not challenge the tumor-suppressive effect of senescence intrinsically, but instead, supports the notion that massive senescence might be protumorigenic extrinsically. Senescent cells secrete numerous factors including inflammatory cytokines, growth factors, and extracellular matrix remodeling enzymes that are collectively called the senescence-associated secretome (44). Such secretome factors make the surrounding milieu protumorigenic, facilitating malignant progression of individual cells that have bypassed senescence due to further mutations gained spontaneously or UVB-induced. This could also potentially explain why BRAFV600E;ARF−/− mice display a decrease in the latency of sarcoma and lymphoma when compared to ARF−/− counterparts.
In addition to increased mutation events resulting from impaired XPC expression, a broader array of epigenetic changes may also lead to bypass of tumor suppressive influences such as p53 function and senescence. Indeed, epigenetic alterations have emerged as an important mechanism underlying BRAFV600E-driven tumorigenesis. For example, BRAFV600E can contribute to methylation alterations in single genes (45, 46) as well as to broader changes in methylation patterns in the genomes of melanomas and papillary thyroid cancers (47, 48). Interestingly, one report failed to associate BRAF mutations with hypermethylation of 15 cancer-linked genes in melanomas (49), suggesting that important BRAF methylation targets in melanomas remain to be discovered. Here, we provide evidence that promoter hypermethylation of XPC is such an event, but this gene is unlikely to be solely responsible for BRAFV600E-mediated changes in mutation sensitivity. Based on bioinformatic analysis, CpG islands are clearly evident in other NER genes, such as XPA and ERCC5, suggesting BRAFV600E-mediated methylation of those promoters may also occur. Additionally, the mechanism behind BRAFV600E-induced methylation remains to be elucidated. Upregulation of DNA methyltransferase 1 by BRAFV600E is a promising candidate, as has been reported (47). Interestingly, Ras-mediated methylation requires numerous factors including E2F1 (50), and this is likely to be the case for BRAFV600E as well. Because the activity of E2F1, like E2F4, requires the association of DP1, and this association can also be influenced by ARF (20), it would be intriguing to investigate whether ARF loss facilitates BRAFV600E-mediated promoter methylation, in addition to altering the expression of individual genes in an E2F-depedent manner. Clearly, the ability to reverse BRAFV600E-induced epigenetic changes that then lead to an increased incidence of genetic changes could have an important impact on both tumor progression and development of resistance to therapy. The induction of XPC observed here upon PLX4032 treatment offers a glimmer of hope that such processes remain targetable in malignant melanomas, and are therefore an important area for future study.
Supplementary Material
Acknowledgments
We thank Dr. Roderick Bronson (Harvard Medical School) for his critical analysis of mouse histology, and Drs. Naomi Rosenberg, Norman Sharpless, Daniel Peeper and Jacqueline Lees for their generosity in supplying mouse strains, constructs and antibodies as mentioned above. The insightful discussions from members of the Hinds lab and equipment support from the Kuperwasser lab are highly appreciated. This work was supported in part by National Institutes of Health grants CA095798 and AG020208 (to P.W.H.) and a Sackler Family Cancer Biology Award (to C.L.). C.L. was a Dean’s Fellow supported by funds from the Provost’s Office at Tufts University. J.S. was supported by the China Scholarship Council.
This work was supported in part by NIH grants CA095798 and AG020208 (to P.W.H.) and Sackler Family Cancer Biology Award (to C.L.). None of the materials in this manuscript has been published or is under consideration elsewhere.
Footnotes
None of the authors has a conflicting financial interest.
References
- 1.Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149–82. doi: 10.1101/gad.1437206. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.Goel VK, Ibrahim N, Jiang G, et al. Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene. 2009;28:2289–98. doi: 10.1038/onc.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhomen N, Reis-Filho JS, da Rocha Dias S, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 5.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 6.Patton EE, Widlund HR, Kutok JL, et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005;15:249–54. doi: 10.1016/j.cub.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 7.Sharpless E, Chin L. The INK4a/ARF locus and melanoma. Oncogene. 2003;22:3092–8. doi: 10.1038/sj.onc.1206461. [DOI] [PubMed] [Google Scholar]
- 8.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 9.Fargnoli MC, Chimenti S, Keller G, et al. CDKN2a/p16INK4a mutations and lack of p19ARF involvement in familial melanoma kindreds. J Invest Dermatol. 1998;111:1202–6. doi: 10.1046/j.1523-1747.1998.00412.x. [DOI] [PubMed] [Google Scholar]
- 10.Quelle DE, Cheng M, Ashmun RA, Sherr CJ. Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative reading frame protein p19ARF. Proc Natl Acad Sci U S A. 1997;94:669–73. doi: 10.1073/pnas.94.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freedberg DE, Rigas SH, Russak J, et al. Frequent p16-independent inactivation of p14ARF in human melanoma. J Natl Cancer Inst. 2008;100:784–95. doi: 10.1093/jnci/djn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kannan K, Sharpless NE, Xu J, O’Hagan RC, Bosenberg M, Chin L. Components of the Rb pathway are critical targets of UV mutagenesis in a murine melanoma model. Proc Natl Acad Sci U S A. 2003;100:1221–5. doi: 10.1073/pnas.0336397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferguson B, Konrad Muller H, Handoko HY, et al. Differential roles of the pRb and Arf/p53 pathways in murine naevus and melanoma genesis. Pigment Cell Melanoma Res. 2010;23:771–80. doi: 10.1111/j.1755-148X.2010.00752.x. [DOI] [PubMed] [Google Scholar]
- 14.Ha L, Ichikawa T, Anver M, et al. ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. Proc Natl Acad Sci U S A. 2007;104:10968–73. doi: 10.1073/pnas.0611638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharpless NE, Kannan K, Xu J, Bosenberg MW, Chin L. Both products of the mouse Ink4a/Arf locus suppress melanoma formation in vivo. Oncogene. 2003;22:5055–9. doi: 10.1038/sj.onc.1206809. [DOI] [PubMed] [Google Scholar]
- 16.Lubbe J, Reichel M, Burg G, Kleihues P. Absence of p53 gene mutations in cutaneous melanoma. J Invest Dermatol. 1994;102:819–21. doi: 10.1111/1523-1747.ep12381544. [DOI] [PubMed] [Google Scholar]
- 17.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–73. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 18.Widlund HR, Fisher DE. Potent p53-independent tumor suppressor activity of ARF in melanoma-genesis. Pigment Cell Res. 2007;20:339–40. doi: 10.1111/j.1600-0749.2007.00401.x. [DOI] [PubMed] [Google Scholar]
- 19.Datta A, Sen J, Hagen J, et al. ARF directly binds DP1: interaction with DP1 coincides with the G1 arrest function of ARF. Mol Cell Biol. 2005;25:8024–36. doi: 10.1128/MCB.25.18.8024-8036.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Datta A, Nag A, Raychaudhuri P. Differential regulation of E2F1, DP1, and the E2F1/DP1 complex by ARF. Mol Cell Biol. 2002;22:8398–408. doi: 10.1128/MCB.22.24.8398-8408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dominguez-Brauer C, Chen YJ, Brauer PM, Pimkina J, Raychaudhuri P. ARF stimulates XPC to trigger nucleotide excision repair by regulating the repressor complex of E2F4. EMBO Rep. 2009;10:1036–42. doi: 10.1038/embor.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfeifer GP, Besaratinia A. UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem Photobiol Sci. 2012;11:90–7. doi: 10.1039/c1pp05144j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdel-Malek ZA, Kadekaro AL, Swope VB. Stepping up melanocytes to the challenge of UV exposure. Pigment Cell Melanoma Res. 2010;23:171–86. doi: 10.1111/j.1755-148X.2010.00679.x. [DOI] [PubMed] [Google Scholar]
- 24.Noonan FP, Dudek J, Merlino G, De Fabo EC. Animal models of melanoma: an HGF/SF transgenic mouse model may facilitate experimental access to UV initiating events. Pigment Cell Res. 2003;16:16–25. doi: 10.1034/j.1600-0749.2003.00014.x. [DOI] [PubMed] [Google Scholar]
- 25.Kamijo T, Zindy F, Roussel MF, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–59. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 26.Cui R, Widlund HR, Feige E, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. 2007;128:853–64. doi: 10.1016/j.cell.2006.12.045. [DOI] [PubMed] [Google Scholar]
- 27.Foley CJ, Luo C, O’Callaghan K, Hinds PW, Covic L, Kuliopulos A. Matrix metalloprotease-1a promotes tumorigenesis and metastasis. J Biol Chem. 2012;287:24330–8. doi: 10.1074/jbc.M112.356303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hahn WC, Dessain SK, Brooks MW, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–23. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mao D, Hinds PW. p35 is required for CDK5 activation in cellular senescence. J Biol Chem. 2010;285:14671–80. doi: 10.1074/jbc.M109.066118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown NE, Jeselsohn R, Bihani T, et al. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-11-4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN. FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell. 2011;43:285–98. doi: 10.1016/j.molcel.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–22. [PubMed] [Google Scholar]
- 33.Wu YH, Tsai Chang JH, Cheng YW, Wu TC, Chen CY, Lee H. Xeroderma pigmentosum group C gene expression is predominantly regulated by promoter hypermethylation and contributes to p53 mutation in lung cancers. Oncogene. 2007;26:4761–73. doi: 10.1038/sj.onc.1210284. [DOI] [PubMed] [Google Scholar]
- 34.Patra SK. Ras regulation of DNA-methylation and cancer. Exp Cell Res. 2008;314:1193–201. doi: 10.1016/j.yexcr.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 35.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740–5. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maddodi N, Huang W, Havighurst T, Kim K, Longley BJ, Setaluri V. Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. J Invest Dermatol. 2010;130:1657–67. doi: 10.1038/jid.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halaban R, Zhang W, Bacchiocchi A, et al. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang G, Curley D, Bosenberg MW, Tsao H. Loss of xeroderma pigmentosum C (Xpc) enhances melanoma photocarcinogenesis in Ink4a-Arf-deficient mice. Cancer Res. 2007;67:5649–57. doi: 10.1158/0008-5472.CAN-06-3806. [DOI] [PubMed] [Google Scholar]
- 39.Despras E, Pfeiffer P, Salles B, et al. Long-term XPC silencing reduces DNA double-strand break repair. Cancer Res. 2007;67:2526–34. doi: 10.1158/0008-5472.CAN-06-3371. [DOI] [PubMed] [Google Scholar]
- 40.Walia V, Mu EW, Lin JC, Samuels Y. Delving into somatic variation in sporadic melanoma. Pigment Cell Melanoma Res. 2012;25:155–70. doi: 10.1111/j.1755-148X.2012.00976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang W, Ananthaswamy HN, Muller HK, Kripke ML. p53 protects against skin cancer induction by UV-B radiation. Oncogene. 1999;18:4247–53. doi: 10.1038/sj.onc.1202789. [DOI] [PubMed] [Google Scholar]
- 43.Ibrahim N, Haluska FG. Molecular pathogenesis of cutaneous melanocytic neoplasms. Annu Rev Pathol. 2009;4:551–79. doi: 10.1146/annurev.pathol.3.121806.151541. [DOI] [PubMed] [Google Scholar]
- 44.Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maddodi N, Bhat KM, Devi S, Zhang SC, Setaluri V. Oncogenic BRAFV600E induces expression of neuronal differentiation marker MAP2 in melanoma cells by promoter demethylation and down-regulation of transcription repressor HES1. J Biol Chem. 2010;285:242–54. doi: 10.1074/jbc.M109.068668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng C, Yang J, Scott J, Hanash S, Richardson BC. Role of the ras-MAPK signaling pathway in the DNA methyltransferase response to DNA hypomethylation. Biol Chem. 1998;379:1113–20. doi: 10.1515/bchm.1998.379.8-9.1113. [DOI] [PubMed] [Google Scholar]
- 47.Hou P, Liu D, Dong J, Xing M. The BRAF(V600E) causes widespread alterations in gene methylation in the genome of melanoma cells. Cell Cycle. 2012;11:286–95. doi: 10.4161/cc.11.2.18707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hou P, Liu D, Xing M. Genome-wide alterations in gene methylation by the BRAF V600E mutation in papillary thyroid cancer cells. Endocr Relat Cancer. 2011;18:687–97. doi: 10.1530/ERC-11-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tellez CS, Shen L, Estecio MR, Jelinek J, Gershenwald JE, Issa JP. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009;19:146–55. doi: 10.1097/cmr.0b013e32832b274e. [DOI] [PubMed] [Google Scholar]
- 50.Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449:1073–7. doi: 10.1038/nature06251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.