

Abstract
Nanometer-sized drug carriers including polymeric nanoparticles (NPs) have been used to increase biodistribution of a drug in tumors, thereby reducing the effective dose of chemotherapy. NPs increase drug delivery to tumors to a certain extent, but the amount reaching tumors is only a small fraction of the total administered NPs because they depend on passive accumulation via the leaky vasculature surrounding tumors. In an attempt to further increase the drug delivery to tumors, we develop a polymeric NP system that interacts with an endothelial tumor marker. The NPs are decorated with quinic acid, a synthetic mimic of sialyl lewis-x, which binds to E-selectin, overexpressed on the surface of endothelial cells surrounding solid tumors. The NPs selectively bind to endothelial cells activated with tumor necrosis factor-α, with weak affinity at a relatively high shear stress. These properties may help NPs reach tumors by increasing the encounter of NPs with the peritumoral endothelium without hindering subsequent transport of the NPs.
Keywords: Nanoparticles, drug delivery, solid tumors, E-selectin, quinic acid
INTRODUCTION
Nanometer-sized drug carriers including polymeric nanoparticles (NPs) have been used to increase the distribution of a drug in tumors, thereby reducing the effective dose and non- specific toxicity of chemotherapy.1 NP delivery to tumors is largely based on the pathological features of vasculature and lymphatic systems surrounding tumors, which lead to selective extravasation and accumulation of NPs in tumors, known as the enhanced permeability and retention (EPR) effect.2 However, the amount of NPs delivered to tumors in this way is only a small fraction (~5%) of the total administered NPs.3 The use of ligands specific to receptors overexpressed on tumor cells barely increases the amount of NPs reaching the tumors, although it may increase the retention of NPs in tumors upon their arrival.4–8 To increase the amount of a drug delivered to tumors beyond the level currently possible, a new strategy to improve the extravasation of a drug carrier at tumors is needed.
One of the potential reasons of the limited NP accumulation in tumors may have to do with the fact that the first cell layer encountered by circulating NPs is the vascular endothelium,9 which--despite the defective structure--presents an access barrier to underlying tumors, the final destination of the NPs. We envision that the amount of NPs delivered to tumors may be increased by actively overcoming the access barrier posed by the vasculature surrounding tumors (“peritumoral endothelium”). We hypothesize that a NP system that recognizes and reversibly binds to the peritumoral endothelium may have a greater chance to extravasate at tumors than conventional NPs passively do. The rationale of this hypothesis is that the endothelium supplying various metastatic tumors expresses a high level of E-selectin10–15; thus, NPs with a ligand interacting with E-selectin will selectively concentrate on the peritumoral endothelium as circulating leukocytes do the inflamed endothelium. Moreover, if endothelial binding of the NPs is reversible, the NPs collected in the peritumoral endothelium will have a good chance to extravasate through endothelial defects. Although this approach may not enhance NP binding to target cells, we expect that it will increase the amount of NPs reaching tumors in the first place. As the first step to investigate this potential, we have developed a new NP system with an affinity for E-selectin, which changes with fluctuation of the blood flow.
E-selectin (ELAM-1, CD62E) is a transmembrane glycoprotein expressed on endothelial cells, primarily induced by inflammatory stimuli.16, 17 While E-selectin is mainly known for its role in inflammation as a mediator of initial interaction of leukocytes with the inflamed endothelium,18 it is also implicated in processes related to tumor progression. Many metastatic tumor cells express ligands that bind to E-selectin.15, 19–23 During circulation these tumor cells are lodged in the endothelium at distant sites via an interaction with endothelial E-selectin.15, 24, 25 In addition, a number of studies have found a positive correlation between endothelial expression of E-selectin and angiogenesis, a critical step for tumor establishment, growth, and metastasis.11, 13, 26, 27
E-selectin binds to a variety of carbohydrate structures including sialyl Lewis-x (sLex).19 sLex is found at the terminus of O-glycans, N-glycans, and neolactosphingolipids expressed on the surface of leukocytes and tumor cells.28 sLex and its mimics, such as derivatives of β-amino acid, tartaric acid, galactose, and mannose, have been explored as selectin antagonists29–36 and ligands for macromolecular or particulate drug carriers targeting the activated endothelium.37, 38 We used a quinic acid (QA) derivative as a synthetic mimic of sLex in decorating NPs, due to the stability in blood and the availability of relatively simple synthetic schemes in the literature.39 D(−)-QA has an axial-equatorial-equatorial 1,2,3-triol pattern that mimics the fucose part of sLex,40 a structural feature critical to coordinating calcium ions in the interaction with E-selectin.39, 40 By decorating polymeric NPs with the QA ligand (Fig. 1), we aimed to establish interactions between the NPs and the E-selectin-expressing endothelium, expecting that such interactions would localize the NPs on the peritumoral endothelium and enhance the chances of NP extravasation beyond the level possible with the EPR effect.
Fig. 1.
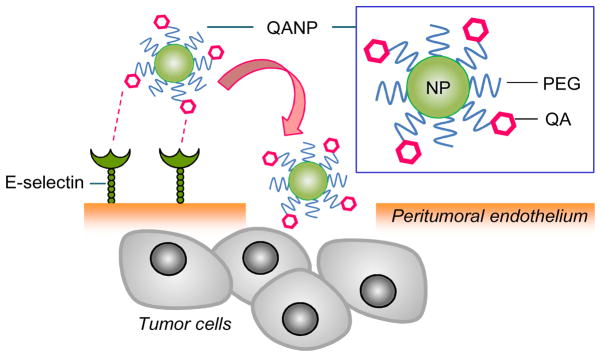
Hypothetical diagram of quinic acid conjugated NPs (QANPs) and its interaction with peritumoral endothelium, followed by extravasation.
Schematic of quinic acid conjugated NP (QANP) and its interaction with peritumoral endothelium, followed by extravasation.
EXPERIMENTAL SECTION
Synthesis and analysis of QA-NH2, QA-PEG, QA-PEG-PLGA, and PEG-PLGA (Schemes 1 and 2)
Detailed synthesis procedure and characterization are described in the supporting information. An amine-coupled QA derivative (QA-NH2, compound 4) was synthesized in an acetate-protected form (5), according to the published method with modifications.39 The amine group of protected derivative of QA-NH2 was converted to azide (PQA-Az, compound 5) for further conjugation to polymeric backbone using click chemistry.41–43 A bi-functional PEG (compound 6) was conjugated to compound 5 by click chemistry to produce a protected QA-PEG conjugate. After deprotection, the QA-PEG was conjugated to PLGA via an amide bond yielding a QA-PEG-PLGA polymer (compound 11). Structures of intermediates and the final product were elucidated using 1H-NMR, 13C-NMR, and electrospray ionization (ESI) mass spectroscopy. PEG-PLGA (compound 12 in Supporting information) was synthesized by conjugating PLGA and methoxy-PEG-amine via carbodiimide chemistry.
Preparation of PLGA NPs, PEG-PLGA NPs, and QA-conjugated NPs (QANPs)
PLGA NPs, PEG-PLGA NPs, and QANPs were prepared by the single emulsion method. Briefly, 20 mg of PLGA, PEG-PLGA, or QA-PEG-PLGA was dissolved in 0.5 mL DCM and emulsified in 3 mL of 5% polyvinyl alcohol (PVA) solution, and emulsified for 1 min using a Vibra-Cell probe sonicator (Sonics, Newtown, CT) at 80% amplitude with a 4-sec on and 2-sec off pulse mode. The polymer emulsion was stirred for 3 h to evaporate DCM, and then washed with distilled water 4 times to remove trace DCM and PVA. For preparation of fluorescently labeled NPs (denoted as *NPs), 25% of the polymer was replaced with fluoresceinamine-conjugated PLGA, produced according to a previously reported method.44 NP size was measured by the Malvern Zetasizer Nano ZS90 (Worcestershire, UK).
Preparation of PLGA NPs conjugated to E-selectin Antibody (Ab-PLGA NPs)
NPs or *NPs were created as described above and lyophilized. One milligram of dry NPs was suspended in 8 mL Tris buffer (10 mM, pH 8.5). To the suspension was added 50 μg/mL anti-E-selectin Ab dissolved in 1 mL Tris buffer (10 mM, pH 8.5) and stirred for 1 min. Freshly prepared dopamine solution (0.1 mg/mL) was added to the NP suspension so that dopamine could polymerize on the NP surface incorporating Ab. The NP suspension was stirred for 20 min, and then NPs were collected and washed 3 times to remove free dopamine and Ab.
E-selectin expression on human umbilical vein endothelial cells (HUVEC)
To evaluate the interaction of QANPs and E-selectin, HUVEC (Lonza; Basel, Switzerland) were prepared to express E-selectin. HUVEC (passage 3) were plated in 24-well plates coated with collagen type-I. The cells became confluent in 24 h. Cells were exposed to 1 mL of phenol red-free medium 199 supplemented with 2 mM L-glutamine and 0.2% BSA (serum-starved medium, SSM) overnight.45, 46 The serum-starved cells were then treated with tumor necrosis factor-α (TNF-α) (1–10 ng/mL) for 4 h according to the conditions described in the literature.45, 46 To confirm E-selectin expression, the cells were incubated with mouse anti-human E-selectin monoclonal Ab for 1 h at room temperature and washed 3 times with SSM to remove unbound Ab. Secondary Ab (anti-mouse IgG-horse radish peroxidase conjugate) was diluted in fresh SSM and added to the cells. After 1 h incubation, the secondary Ab was removed and cells were washed 3 times with SSM. 3,3′,5,5′-Tetramethylbenzidine (TMB) 200 μL was added to each well and kept protected from light for 10 min. HCl (1 M, 300 μL) was used to quench the reaction, and the absorbance was read at 450 nm.
HL-60 binding-inhibition assay
HUVEC (passage 3) were cultured in a 24-well plate and treated with TNF-α 10 ng/mL in SSM for 4 h to express E-selectin. After removing the medium, cells were treated with one of the following: QA-NH2 (20 mM), PLGA NP (0.1 mg/mL), PEG-PLGA NP (0.1 mg/mL), QANP (0.1 mg/mL, equivalent to 2 μM QA), Ab-PLGA NP (0.1 mg/mL), E-selectin Ab (5 μg/mL, positive control) and SSM (negative control), for 90 min. Meanwhile, HL-60 cells were collected by centrifugation at 1500 rpm for 5 min. Cell pellet was washed with 5 mL PBS and stained with calcein-AM (1 mM) for 45 min. After removing the treatments HUVEC cells were incubated with 4–5 × 105 HL-60 cells in 0.5 mL SSM for 60 min at 37°C. Then HUVEC cells were washed 3 times with SSM to remove unbound HL-60 cells and lysed with 100 μL of cell lysis buffer. The plates were kept at −80°C overnight, thawed at 37°C, and the fluorescence of cell lysates were read with a microplate reader (ex: 485 nm; em: 535 nm). The data was expressed as normalized to the SSM-treated negative control.
Visualization of NPs adherent to the activated HUVEC cells in a static condition
HUVEC (passage 3) were seeded at a density of 50,000 cells/cm2 in a collagen-I coated 35-mm dish with a glass window (MatTek). When HUVEC became confluent, cells were washed 4 times with SSM and incubated with either 1 mL SSM (control, non-activated HUVEC) or SSM containing 10 ng/mL TNF-α (activated HUVEC) for 4 h. The media was then replaced with SSM containing 0.1 mg/mL of fluorescently labeled NP suspension (PLGA *NPs, PEG-PLGA *NPs and QA*NPs) and incubated for 3 h at 37°C. The suspension was replaced with NP-free SSM, and the NP bound to the HUVEC layer was observed using an Olympus X81 (Olympus, Japan) confocal microscope. DRAQ-5 nuclear stain (1 μL) was added 2–3 minutes prior to the imaging. The NPs and cell nuclei were excited using a 488-nm and 633-nm laser, and their emission signals were read from 500 to 600 nm and 650 to 750 nm and expressed in green and blue, respectively.
Evaluation of NP retention on the activated HUVEC under a dynamic flow condition
HUVEC (passage 3) were cultured on a coverslip (1.77 cm2) coated with collagen-I, and stimulated with TNF-α (10 ng/mL) for 4 h prior to experiments when applicable. The coverslip with a HUVEC layer was glued to a plastic disk tip using a mounting glue and immersed in medium containing 0.1 mg/mL *NPs (PLGA *NPs, PEG-PLGA *NPs, QA*NPs, or Ab-PLGA *NPs) at room temperature. The disk tip was fixed to a rotating shaft operated by Modulated Speed Rotator (Pine Research Instrumentation, Raleigh, NC). The disk rotated at ~330, ~950 and ~1500 rpm generated average shear stresses of 2, 10, and 20 and dynes/cm2 on the cell layer, respectively, relevant to the shear stresses experienced at the wall of blood vessels, specifically in capillaries.47 The rotation continued for 60 min at room temperature. Coverslips containing cells and adherent NPs were transferred to a 24 well plate, where the cells were lysed and NPs were dissolved using 200 μL of 10% DMSO solution. Twenty five microliters of the NP-cell lysate suspension was transferred to a 96 well plate, and the cell protein content (representing cells remaining on the cover slip) was measured by the BCA assay. The remaining NP-cell lysate suspension was used to measure fluorescence to quantify the NPs remaining adherent to cells. The results were expressed as nanograms of NPs per micrograms of protein at each shear stress.
Statistical Analysis
Data were expressed as averages and standard deviations of multiple measurements. Statistical difference among groups was determined by ANOVA, and contrasts between groups were determined by the Tukey test. A value of p<0.05 was considered statistically significant.
RESULTS AND DISCUSSION
Synthesis and characterization of a QA-conjugated polymer
The QANPs consisted of three components: (i) poly(lactic-co-glycolic acid) (PLGA) that formed a NP core to encapsulate a drug, (ii) polyethylene glycol (PEG), serving as a spacer between PLGA NP core and QA ligand and a stealth layer on the NP surface to enable long-term circulation of NPs, and (iii) QA ligand to enable interactions between QANPs and E-selectin. PLGA and PEG were chosen due to the well-established in-vivo fates48, 49 and terminal structures (carboxyl and amine groups) amenable to chemical modification. Relatively small molecular weight PLGA polymers (4.2 kDa or 10 kDa) were used to accommodate more QA ligands or PEG per NP mass. PLGA polymers (lactic acid: glycolic acid = 50:50) with molecular weights less than 10 kDa were expected to degrade in a couple of weeks, as extrapolated from the literature.50 PEG with a molecular weight less than 6 kDa is known to be readily removed from body via renal excretion.49, 51 We used 3.5 kDa or 5 kDa PEG according to the literature identifying an optimal molecular weight of PEG for stealth coating in this range.52
To create the QANPs, a new polymer was synthesized by conjugating QA, PEG, and PLGA linearly. QA was first modified into an amine-coupled derivative (QA-NH2, compound 4, Scheme 1), where 3,5-diaminobeonzoic acid was conjugated to the carboxyl group of QA via an amide bond. 3,5-diaminobenzoic acid was used for three reasons: (i) to create an amine terminus, which would be used as a chemical handle to link to a polymer; (ii) to regenerate the carboxyl group, which was known to be essential for the selectin-binding39 but consumed during the derivatization; and (iii) to provide a planar benzene ring that would orient the triol and carboxyl group in the same manner as that in L-fucose in sLex.
Scheme 1.

Synthesis of a quinic acid derivative, a synthetic mimic of sLex.
The QA derivative was conjugated to PEG, which was then conjugated to PLGA (Scheme 2). The amine group of a protected form of QA-NH2 (3) was converted to an azide (pQA-Az, 5). A bi-functional PEG with an amine and carboxyl termini (6) was partially protected by converting amine to N-tert-butoxycarbonyl (NH-Boc).53, 54 Subsequently, propargyl amine was conjugated to the carboxyl terminus of PEG via carbodiimide chemistry.55 The modified PEG (8) was coupled to pQA-Az (5) by click chemistry,41–43 followed by sequential deprotection of NH-Boc in PEG and acetyls and methyl in QA part. Finally, the QA-PEG (10) was linked to PLGA via amide bond to obtain QA-PEG-PLGA polymer (11).
Scheme 2.

Synthesis of a QA-PEG-PLGA.
The QA content in the resulting polymer was difficult to quantify. UV, NMR, and mass spectroscopy were used but found inadequate. First, we attempted to measure the concentration of QA-NH2 with UV spectroscopy after overnight hydrolysis of QA-PEG-PLGA at pH 4, but its UV absorbance was below the detection limit. The number of protons belonging to QA-NH2 was far fewer than that of the PEG and PLGA parts of the polymer, making it difficult to estimate additional protons attributable to QA using 1H-NMR spectroscopy. Mass spectroscopy, in particular MALDI-TOF, could not be used since PLGA degraded into heterogeneous fragments with low signals close to the noise level. Therefore, the QA content was determined with 1H-NMR in two steps, first by calculating QA content in pQA-PEG (50 mol%: i.e., 50% of PEG molecules had QA conjugation in the carboxyl termini after the completion of reaction and purification) and then PEG content in the QA-PEG-PLGA (20 mol%: i.e., 20% of PLGA molecules had PEG or QA-PEG conjugated to its carboxyl termini after the completion of reaction and purification). The QA content in the resulting polymer was thus estimated as 10 mol% (i.e., 10% of PLGA molecules had QA in one end).
Production and characterization of QANPs
QA-PEG-PLGA was made into QANPs using the single emulsion technique. In the resulting QANPs, hydrophilic QA-PEG part was expected to face water and PLGA to form a hydrophobic core. PLGA and PEG-PLGA NPs were prepared in the same method for comparison. Additionally, PLGA NPs decorated with anti-E-selectin antibody (Ab-PLGA NPs) were prepared as a positive control using a simple and versatile conjugation method based on dopamine polymerization.56 Particle sizes of NPs ranged from 180 to 250 nm (Supporting Table 1), suitable for the systemic application. At the concentration in this study (0.1 mg/mL), QANPs did not affect the viability of HUVEC after 72 h incubation, similar to PLGA and PEG-PLGA NPs (Supporting Fig. 1). This ensures that the following results were irrelevant to cytotoxicity of NPs.
Affinity of QANPs for E-selectin
Affinity of QANPs for E-selectin was evaluated by measuring their ability to interfere with HL-60 cell binding to HUVEC expressing E-selectin (Supporting Fig. 2).45, 46 HL-60 is a maturation-arrested promyelocytic cell line, which has a sLex-bearing structure on the surface and binds to E-selectin.57 Binding of HL-60 to E-selectin is inhibited by substances that preoccupy E-selectin; thus, pre-treatment with NPs or ligands with an affinity for E-selectin results in a low number of HL-60 cells remaining on the HUVEC layer. Here, HUVEC was first activated by 4 h incubation with 10 ng/mL TNF-α, which resulted in the increase of E-selectin signal (measured by cell ELISA) by 12 times as compared to the non-treated control (Supporting Fig. 3). The activated HUVEC layer was then incubated with NPs. After removing the NPs, fluorescently stained HL-60 cells were added to the HUVEC cells, washed after 60 min incubation, and then those remaining with HUVEC were quantified by measuring their fluorescence. As shown in Fig. 2, Ab and Ab-PLGA NPs decreased HL-60 binding to the activated HUVEC by 40–50%. In contrast, treatment with 20 mM QA-NH2 as a free ligand resulted in only mild suppression of HL-60 binding, reflecting inherently low affinity of QA derivatives for selectins.39 Similarly, PLGA and PEG-PLGA NPs showed no or little inhibition of HL-60 binding to the activated HUVEC. The slight decrease by PLGA NPs as compared to PEG-PLGA NPs is likely due to their non-specific hydrophobic interaction with HUVEC. On the other hand, QANPs inhibited HL-60 binding to a comparable level as Ab or Ab-PLGA NPs, indicating that QANPs were effectively bound to the E-selectin-expressing HUVEC. Given that QANPs, PLGA, and PEG-PLGA NPs had similar zeta potentials (−19.7±1.2, −17.2±1.0, and −19.5±0.5 mV, respectively), it is unlikely that such a difference was due to the surface charge difference. Notably, QANPs inhibited HL-60 binding at a level equivalent to 2 μM of QA, four orders of magnitude lower than 20 mM, the concentration at which free QA-NH2 was not effective. This indicates that multivalent QA ligands presented by NPs were more effective than free QA-NH2 in interacting with the activated HUVEC. In addition, the presence of QANPs on HUVEC mediated by the QA-E-selectin interaction would have contributed to the inhibition of HL-60 binding.
Fig. 2.

Binding-inhibition assay. Fluorescence of HL-60 bound to HUVEC, which were pre-treated with NPs (0.1 mg/mL), QA-NH2 (20 mM), or E-selectin Ab (5 μg/mL). 0.1 mg/mL QANPs was equivalent to 2 μM of QA. The fluorescence obtained with each treatment is normalized to the fluorescence of HL-60 bound to serum-starved medium (SSM)-treated HUVEC. Therefore, NPs or ligands interfering with the HL-60-HUVEC binding results in fluorescence lower than 100% (SSM-treated HUVEC, dotted line). Data are expressed as average ± standard deviation of 3–6 independently obtained results per treatment. **: p<0.01 vs. Ab; n.s.: not significant, p>0.05 vs. Ab.
QANP binding to HUVEC in a static condition
Microscopic observation was consistent with the binding inhibition assay. The NPs were fluorescently labeled (expressed as *NPs), incubated with the activated HUVEC (E-selectin-positive) or resting HUVEC (E-selectin-negative) in tissue culture plates, and their interactions with the cells were visualized using confocal laser microscopy (Fig. 3). PLGA and PEG-PLGA *NPs did not interact with HUVEC irrespective of E-selectin expression. In contrast, QA*NPs showed selective interaction with the E-selectin-expressing HUVEC cells.
Fig. 3.

Confocal scanning microscopy images of NP interaction with HUVEC.
QANP binding to HUVEC in a dynamic flow condition
Since the ultimate destination for the QANPs is not the endothelium but the underlying tumors, it is desirable that QANP-cell interaction is not too strong so that the QANPs can ultimately extravasate through the leaky endothelium. The vessel wall experiences shear stress due to blood flow, which depends on fluid viscosity, volumetric flow rate, and vessel diameter. When all other conditions are equal, the shear stress applied on the vessel wall is directly proportional to the blood flow rate.58 Given that the blood flow in tumors fluctuates,59, 60 we envision that circulating NPs at the peritumoral endothelium will experience temporally variable shear stress. Therefore, NPs that bind to the endothelium with varying affinities at different shear stresses may have a better chance to extravasate at tumors than those stuck on the endothelial surface. To evaluate the response of QANPs to shear stress, the extents that QANPs remained bound to E-selectin/HUVEC were evaluated at various levels of shear stress. The shear stresses measured for post-capillary venules are 1–10 dynes/cm2,17 and those within arteries and veins are 10–70 dynes/cm2 and 1–6 dynes/cm2,61 respectively. To mimic the shear stresses in capillaries, the activated HUVEC layer was attached on a rotating disk system, which rotated in the *NP-containing medium at rates creating various shear stresses (from 2 to 20 dynes/cm2)62 (Supporting Fig. 4). As shown in Fig. 4, all NPs showed a minimal binding to the non-activated HUVEC at all levels of shear stress. The amounts of PLGA and PEG-PLGA *NPs remained on the activated HUVEC were minimal irrespective of shear stress, similar to microscopic observation. On the other hand, QA*NPs had significantly higher interactions with the activated HUVEC than PLGA or PEG-PLGA *NPs at all levels of shear stress. Importantly, the interaction between QA*NPs and HUVEC decreased as the shear stress increased. In contrast, the amount of Ab-PLGA *NPs, which showed similar affinity for E-selectin as QA*NPs in the absence of the applied shear stress (binding-inhibition assay), remaining at 20 dynes/cm2 was 3 times more than that of QA*NPs at the same condition. This suggests that, upon exposure to a relatively high shear stress, QANPs would be less likely to be stuck on the endothelium than Ab-PLGA NPs. The fact that QANP binding to the activated HUVEC decreased with increasing shear stress also suggests that the bound QANPs have not undergone endocytosis at least under the dynamic flow condition, although the exact fate of QANPs after the binding remains to be investigated.
Fig. 4.

NP retention on HUVEC layer at varying levels of shear stresses at tumor-associated capillaries. The retention of *NPs on the activated HUVEC reflects the balance between their affinity for E-selectin and the responsiveness to the shear stress.
CONCLUSIONS
QANPs, the NPs with QA-ligand as a synthetic mimic of sLex, were synthesized in an attempt to increase extravasation of NPs at tumors. QANPs show affinity for HUVEC cells expressing E-selectin and, thus, have the potential to concentrate on the peritumoral endothelium. QANP binding to the activated HUVEC cells is not strong, especially at a relatively high shear stress. We expect that this weak interaction between QANPs and E-selectin is more desirable than stronger ones, often obtained with ligands that target other endothelial markers,63–65 in achieving enhanced extravasation. A proof of concept remains to be obtained in a comparative biodistribution study.
Supplementary Material
Acknowledgments
This work was supported by NSF DMR-1056997, NIH R21 CA135130, American Association of Colleges of Pharmacy New Investigators Program for Pharmacy Faculty, and a grant from the Lilly Endowment, Inc. to the College of Pharmacy. The authors appreciate the support of the Ronald W. Dollens Graduate Scholarship and the Bilsland Dissertation Fellowship for ZA. We also thank the Purdue University Center for Cancer Research Interdepartmental NMR Facilities and Campus-Wide Mass Spectrometry Center (supported by NCI CCSG CA23168) for the support of materials characterization.
Footnotes
Conflict of Interest Disclosure: The authors declare no competing financial interest.
Supporting Information Available: Materials and Detailed Methods, Supporting Table and Figures are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Peer D, Karp JM, Hong S, FaroKhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer-chemotherapy - mechanism of tumoritropic accumulation of proteins and the antitumor agent Smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 3.Bae YH, Park K. Targeted drug delivery to tumors: Myths, reality and possibility. J Control Release. 2011;153:198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong KL, Nielsen UB, Marks JD, Benz CC, Park JW. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732–6740. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- 5.Mamot C, Drummond DC, Noble CO, Kallab V, Guo ZX, Hong KL, Kirpotin DB, Park JW. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res. 2005;65:11631–11638. doi: 10.1158/0008-5472.CAN-05-1093. [DOI] [PubMed] [Google Scholar]
- 6.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gabizon AA, Shmeeda H, Zalipsky S. Pros and cons of the liposome platform in cancer drug targeting. J Liposome Res. 2006;16:175–183. doi: 10.1080/08982100600848769. [DOI] [PubMed] [Google Scholar]
- 8.Gabizon A, Horowitz AT, Goren D, Tzemach D, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clinical Cancer Res. 2003;9:6551–6559. [PubMed] [Google Scholar]
- 9.Ozawa MG, Zurita AJ, Dias-Neto E, Nunes DN, Sidman RL, Gelovani JG, Arap W, Pasqualini R. Beyond Receptor Expression Levels: The Relevance of Target Accessibility in Ligand-Directed Pharmacodelivery Systems. Trends Cardiovasc Med. 2008;18:126–133. doi: 10.1016/j.tcm.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Brodt P, Fallavollita L, Bresalier RS, Meterissian S, Norton CR, Wolitzky BA. Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int J Cancer. 1997;71:612–619. doi: 10.1002/(sici)1097-0215(19970516)71:4<612::aid-ijc17>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 11.Fox SB, Turner GDH, Gatter KC, Harris AL. The Increased Expression Of Adhesion Molecules Icam-3, E-Selectin And P-Selectin On Breast-Cancer Endothelium. J Pathol. 1995;177:369–376. doi: 10.1002/path.1711770407. [DOI] [PubMed] [Google Scholar]
- 12.Shaker OG, El-Deen MAA, El-Rahim MTA, Talaat RM. Gene expression of E-selectin in tissue and its protein level in serum of breast cancer patients. Tumori. 2006;92:524–530. doi: 10.1177/030089160609200610. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen M, Corless CL, Kraling BM, Tran C, Atha T, Bischoff J, Barsky SH. Vascular expression of E-selectin is increased in estrogen-receptor-negative breast cancer - A role for tumor-cell-secreted interleukin-1 alpha. Am J Pathol. 1997;150:1307–1314. [PMC free article] [PubMed] [Google Scholar]
- 14.Bhaskar V, Law D, Ibsen E, Breinberg D, Cass K, DuBridge R, Evangelista F, Henshall S, Hevezi P, Miller J, Pong M, Powers R, Senter P, Stockett D, Sutherland R, von Freeden-Jeffry U, Willhite D, Murray R, Afar D, Ramakrishnan V. E-selectin up-regulation allows for targeted drug delivery in prostate cancer. Cancer Res. 2003;63:6387–6394. [PubMed] [Google Scholar]
- 15.Kobayashi H, Boelte KC, Lin PC. Endothelial cell adhesion molecules and cancer progression. Curr Med Chem. 2007;14:377–386. doi: 10.2174/092986707779941032. [DOI] [PubMed] [Google Scholar]
- 16.Kansas GS. Selectins and their ligands: current concepts and controversies. Blood. 1996;88:3259–3287. [PubMed] [Google Scholar]
- 17.Lawrence MB, Springer TA. Leukocytes Roll on a Selectin at Physiologic Flow Rates: Distinction from and Prerequisite for Adhesion through lntegrins. Cell. 1991;65:859–873. doi: 10.1016/0092-8674(91)90393-d. [DOI] [PubMed] [Google Scholar]
- 18.Andrian UHv, Mackay CR. T-cell function an migration: Two sides of the same coin. N Engl J Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 19.Bevilacqua MP, Nelson RM. Selectins. J Clin Invest. 1993;91:379–387. doi: 10.1172/JCI116210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takada A, Ohmori K, Yoneda T, Tsuyuoka K, Hasegawa A, Kiso M, Kannagi R. Contribution Of Carbohydrate Antigens Sialyl Lewis-A And Sialyl Lewis-X To Adhesion Of Human Cancer-Cells To Vascular Endothelium. Cancer Res. 1993;53:354–361. [PubMed] [Google Scholar]
- 21.Yu CJ, Shih JY, Lee YC, Shun CT, Yuan A, Yang PC. Sialyl Lewis antigens: association with MUC5AC protein and correlation with post-operative recurrence of non-small cell lung cancer. Lung Cancer. 2005;47:59–67. doi: 10.1016/j.lungcan.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 22.Paprocka M, Dus D, Mitterrand M, Lamerant-Fayel N, Kieda C. Flow cytometric assay for quantitative and qualitative evaluation of adhesive interactions of tumor cells with endothelial cells. Microvasc Res. 2008;76:134–138. doi: 10.1016/j.mvr.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Zen K, Liu DQ, Guo YL, Wang C, Shan J, Fang M, Zhang CY, Liu Y. CD44v4 Is a Major E-Selectin Ligand that Mediates Breast Cancer Cell Transendothelial Migration. PLoS ONE. 2008;3:e1826. doi: 10.1371/journal.pone.0001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gassmann P, Enns A, Haier J. Role of tumor cell adhesion and migration in organ-specific metastasis formation. Onkologie. 2004;27:577–582. doi: 10.1159/000081343. [DOI] [PubMed] [Google Scholar]
- 25.Rice GE, Bevilacqua MP. An Inducible Endothelial Cell Surface Glycoprotein Mediates Melanoma Adhesion. Science. 1989;246:1303–1306. doi: 10.1126/science.2588007. [DOI] [PubMed] [Google Scholar]
- 26.Koch AE, Halloran MM, Haskell CJ, Shah MR, Polverini PJ. Angiogenesis mediated by soluble forms of E-selectin and vascular cell adhesion molecule-1. Nature. 1995;376:517–519. doi: 10.1038/376517a0. [DOI] [PubMed] [Google Scholar]
- 27.Aoki M, Kanamori M, Yudoh K, Ohmori K, Yasuda T, Kimura T. Effects of vascular endothelial growth factor and E-selectin on angiogenesis in the murine metastatic RCT sarcoma. Tumor Biol. 2001;22:239–246. doi: 10.1159/000050622. [DOI] [PubMed] [Google Scholar]
- 28.Barthel SR, Gavino JD, Descheny L, Dimitroff CJ. Targeting selectins and selectin ligands in inflammation and cancer. Expert Opin Ther Targets. 2007;11:1473–1491. doi: 10.1517/14728222.11.11.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pedatella S, De Nisco M, Ernst B, Guaragna A, Wagner B, Woods RJ, Palumbo G. New sialyl Lewisx mimic containing an [alpha]-substituted [beta]3-amino acid spacer. Carbohydr Res. 2008;343:31–38. doi: 10.1016/j.carres.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robina I, Moreno-Vargas A, Carmona A, Ferrali A, Molina L. Approaches to novel sialyl Lewis X mimetics: S-Linked fucoside derivatives. Abstracts of Papers, 233rd ACS National Meeting; Chicago, IL, United States. March 25–29, 2007; 2007. CARB-067. [Google Scholar]
- 31.Huang H, Wong CH. Synthesis of Biologically Active Sialyl Lewis X Mimetics. J Org Chem. 1995;60:3100–3106. [Google Scholar]
- 32.Rao BN, Anderson MB, Musser JH, Gilbert JH, Schaefer ME, Foxall C, Brandley BK. Sialyl Lewis X mimics derived from a pharmacophore search are selectin inhibitors with anti-inflammatory activity. J Biol Chem. 1994;269:19663–19666. [PubMed] [Google Scholar]
- 33.Cappi MW, Moree WJ, Qiao L, Marron TG, Weitz-Schmidt G, Wong CH. Synthesis of Novel 6-Amido-6-deoxy-L-galactose Derivatives as Sialyl Lewis X mimetics. Bioorgan Med Chem. 1997;5:283–296. doi: 10.1016/s0968-0896(96)00236-2. [DOI] [PubMed] [Google Scholar]
- 34.DeFrees SA, Phillips L, Guo L, Zalipsky S. Sialyl Lewis x Liposomes as a Multivalent Ligand and Inhibitor of E-Selectin Mediated Cellular Adhesion. J Am Chem Soc. 1996;118:6101–6104. [Google Scholar]
- 35.Zeisig R, Stahn R, Wenzel K, Behrens D, Fichtner I. Effect of sialyl Lewis X-glycoliposomes on the inhibition of E-selectin-mediated tumour cell adhesion in vitro. Biochim Biophys Acta. 2004;1660:31–40. doi: 10.1016/j.bbamem.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 36.Marron TG, Weitz-Schmidt G, Woltering TJ, Wong CH. Sialyl Lewis X mimetics incorporating mannopeptides. 5837862 A. United States Patent. 1998
- 37.Eniola AO, Hammer DA. In vitro characterization of leukocyte mimetic for targeting therapeutics to the endothelium using two receptors. Biomaterials. 2005;26:7136–7144. doi: 10.1016/j.biomaterials.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 38.Tsuruta W, Tsurushima H, Yamamoto T, Suzuki K, Yamazaki N, Matsumura A. Application of liposomes incorporating doxorubicin with sialyl Lewis X to prevent stenosis after rat carotid artery injury. Biomaterials. 2009;30:118–125. doi: 10.1016/j.biomaterials.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Kaila N, Somers WS, Thomas BE, Thakker P, Janz K, DeBernardo S, Tam S, Moore WJ, Yang R, Wrona W, Bedard PW, Crommie D, Keith JC, Tsao DHH, Alvarez JC, Ni H, Marchese E, Patton JT, Magnani JL, Camphausen RT. Quinic Acid Derivatives as Sialyl Lewis x-Mimicking Selectin Inhibitors: Design, Synthesis, and Crystal Structure in Complex with E-Selectin. J Med Chem. 2005;48:4346–4357. doi: 10.1021/jm050049l. [DOI] [PubMed] [Google Scholar]
- 40.Girard C, Dourlat J, Savarin A, Surcin C, Leue S, Escriou V, Largeau C, Herscovici J, Scherman D. Sialyl Lewisx analogs based on a quinic acid scaffold as the fucose mimic. Bioorg Med Chem Lett. 2005;15:3224–3228. doi: 10.1016/j.bmcl.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 41.de Graaf AJ, Kooijman M, Hennink WE, Mastrobattista E. Nonnatural Amino Acids for Site-Specific Protein Conjugation. Bioconjugate Chem. 2009;20:1281–1295. doi: 10.1021/bc800294a. [DOI] [PubMed] [Google Scholar]
- 42.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 43.Barral K, Moorhouse AD, Moses JE. Efficient conversion of aromatic amines into azides: A one-pot synthesis of triazole linkages. Org Lett. 2007;9:1809–1811. doi: 10.1021/ol070527h. [DOI] [PubMed] [Google Scholar]
- 44.Xu P, Gullotti E, Tong L, Highley CB, Errabelli DR, Hasan T, Cheng JX, Kohane DS, Yeo Y. Intracellular Drug Delivery by Poly(lactic-co-glycolic acid) Nanoparticles, Revisited. Mol Pharmaceut. 2009;6:190–201. doi: 10.1021/mp800137z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, Hotta K, Nishida M, Takahashi M, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y. Novel modulator for endothelial adhesion molecules - Adipocyte-derived plasma protein adiponectin. Circulation. 1999;100:2473–2476. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 46.Makondo K, Kimura K, Kitamura T, Yamaji D, Dong Jung B, Shibata H, Saito M. Hepatocyte growth factor/scatter factor suppresses TNF-[alpha]-induced E-selectin expression in human umbilical vein endothelial cells. Biochim Biophys Acta. 2004;1644:9–15. doi: 10.1016/j.bbamcr.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 47.Sen Gupta A, Wang S, Link E, Anderson EH, Hofmann C, Lewandowski J, Kottke-Marchant K, Marchant RE. Glycocalyx-mimetic dextran-modified poly(vinyl amine) surfactant coating reduces platelet adhesion on medical-grade polycarbonate surface. Biomaterials. 2006;27:3084–3095. doi: 10.1016/j.biomaterials.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 48.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers. 2011;3:1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamaoka T, Tabata Y, Ikada Y. Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J Pharm Sci. 1994;83:601–606. doi: 10.1002/jps.2600830432. [DOI] [PubMed] [Google Scholar]
- 50.Miller RA, Brady JM, Cutright DE. Degradation rates of oral resorbable implants (polylactates and polyglycolates): rate modification with changes in PLA/PGA copolymer ratios. J Biomed Mater Res. 1977;11:711–719. doi: 10.1002/jbm.820110507. [DOI] [PubMed] [Google Scholar]
- 51.Webster R, Didier E, Harris P, Siegel N, Stadler J, Tilbury L, Smith D. PEGylated Proteins: Evaluation of their safety in the absence of definitive metabolism studies. Drug Metab Dispos. 2007;35:9–16. doi: 10.1124/dmd.106.012419. [DOI] [PubMed] [Google Scholar]
- 52.Gref R, Lück M, Quellec P, Marchand M, Dellacherie E, Harnisch S, Blunk T, Müller RH. “Stealth” corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf, B. 2000;18:301–313. doi: 10.1016/s0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 53.Salmaso S, Bersani S, Scomparin A, Mastrotto F, Scherpfer R, Tonon G, Caliceti P. Tailored PEG for rh-G-CSF Analogue Site-Specific Conjugation. Bioconjugate Chem. 2009;20:1179–1185. doi: 10.1021/bc9000432. [DOI] [PubMed] [Google Scholar]
- 54.Jarowicki K, Kocienski P. Protecting groups. J Chem Soc-Perkin Trans. 2001;1:2109–2135. [Google Scholar]
- 55.Nakajima N, Ikada Y. Mechanism of amide formation by carbodiimide for bioconjugation in aqueous-media. Bioconjugate Chem. 1995;6:123–130. doi: 10.1021/bc00031a015. [DOI] [PubMed] [Google Scholar]
- 56.Lee H, Rho J, Messersmith PB. Facile Conjugation of Biomolecules onto Surfaces via Mussel Adhesive Protein Inspired Coatings. Adv Mater. 2009;21:431–434. doi: 10.1002/adma.200801222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshida M, Chien LJ, Yasukochi Y, Numano F. Differentiation-induced transmigration of HL60 cells across activated HUVEC monolayer involves E-selectin-dependent mechanism. Ann NY Acad Sci. 2000;902:307–310. doi: 10.1111/j.1749-6632.2000.tb06328.x. [DOI] [PubMed] [Google Scholar]
- 58.Papaioannou TG, Stefanadis C. Vascular wall shear stress: basic principles and methods. Hellenic J Cardiol. 2005;46:9–15. [PubMed] [Google Scholar]
- 59.Dewhirst MW, Braun RD, Lanzen JL. Temporal changes in pO2 of R3230Ac tumors in fischer-344 rats. Int J Radiat Oncol Biol Phys. 1998;42:723–726. doi: 10.1016/s0360-3016(98)00304-6. [DOI] [PubMed] [Google Scholar]
- 60.Kimura H, Braun RD, Ong ET, Hsu R, Secomb TW, Papahadjopoulos D, Hong K, Dewhirst MW. Fluctuations in Red Cell Flux in Tumor Microvessels Can Lead to Transient Hypoxia and Reoxygenation in Tumor Parenchyma. Cancer Res. 1996;56:5522–5528. [PubMed] [Google Scholar]
- 61.Malek AM, Alper SL, Izumo S. Hemodynamic Shear Stress and Its Role in Atherosclerosis. JAMA. 1999;282:2035–2042. doi: 10.1001/jama.282.21.2035. [DOI] [PubMed] [Google Scholar]
- 62.Kao WYJ. Evaluation of leukocyte adhesion on polyurethanes: the effects of shear stress and blood proteins. Biomaterials. 2000;21:2295–2303. doi: 10.1016/s0142-9612(00)00156-3. [DOI] [PubMed] [Google Scholar]
- 63.Jubeli E, Moine L, Vergnaud-Gauduchon J, Barratt G. E-selectin as a target for drug delivery and molecular imaging. J Control Release. 2012;158:194–206. doi: 10.1016/j.jconrel.2011.09.084. [DOI] [PubMed] [Google Scholar]
- 64.Kaila N, Thomas BE, IV, Thakker P, Alvarez JC, Camphausen RT, Crommie D. Design and synthesis of sialyl lewis x mimics as e-selectin inhibitors. Bioorg Med Chem Lett. 2001;11:151–155. doi: 10.1016/s0960-894x(00)00623-5. [DOI] [PubMed] [Google Scholar]
- 65.Stahn R, Schäfer H, Kernchen F, Schreiber J. Multivalent sialyl Lewis x ligands of definite structures as inhibitors of E-selectin mediated cell adhesion. Glycobiology. 1998;8:311–319. doi: 10.1093/glycob/8.4.311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.