

Abstract
The proinflammatory activation of leukocytes in adipose tissue contributes to metabolic disease. How crosstalk between immune cells initiates and sustains adipose tissue inflammation remains an unresolved question. We have examined the hypothesis that adipose tissue macrophages (ATMs) interact with and regulate the function of T cells. Dietary obesity was shown to activate the proliferation of effector memory CD4+ T cells in adipose tissue. Our studies further demonstrate that ATMs are functional antigen-presenting cells that promote the proliferation of interferon-γ–producing CD4+ T cells in adipose tissue. ATMs from lean and obese visceral fat process and present major histocompatibility complex (MHC) class II–restricted antigens. ATMs were sufficient to promote proliferation and interferon-γ production from antigen-specific CD4+ T cells in vitro and in vivo. Diet-induced obesity increased the expression of MHC II and T-cell costimulatory molecules on ATMs in visceral fat, which correlated with an induction of T-cell proliferation in that depot. Collectively, these data indicate that ATMs provide a functional link between the innate and adaptive immune systems within visceral fat in mice.
Obesity-induced inflammation contributes to the development of type 2 diabetes, metabolic syndrome, and cardiovascular disease (1–3). Accumulation of activated leukocytes in metabolic tissues is a driving force for obesity-associated metabolic inflammation (metainflammation) and insulin resistance (3,4). In adipose tissue, a vast array of leukocytes have been identified and reported to contribute to obesity-induced metainflammation. How adipose tissue leukocytes interact to shape the inflammatory environment within fat is an important unresolved gap in our current understanding of metabolic disease.
In humans and rodent models, F4/80+ adipose tissue macrophages (ATMs) are the predominant leukocyte found in metabolically healthy and insulin-resistant fat (5). Resident (type 2) ATMs are distributed between adipocytes in healthy adipose tissue throughout development, express anti-inflammatory markers typical of “alternatively activated” or M2 polarized macrophages, and promote tissue homeostasis (6,7). Disruption of macrophage M2 polarization increases the susceptibility to insulin resistance induced by a high-fat diet (HFD) (8–10). Obesity triggers the accumulation of F4/80+ ATMs that coexpress the dendritic cell (DC) marker CD11c as well as genes typically expressed by “classically activated” or proinflammatory M1 polarized macrophages (11–13). M1 ATMs form multicellular lipid-laden clusters, known as crown-like structures (CLS), around dead adipocytes in obese fat (6,14,15) and produce inflammatory cytokines (e.g., interleukin [IL]-1β, IL-6, and tumor necrosis factor-α [TNF-α]) that can impair insulin action in adipocytes (16,17). Current models suggest that obesity promotes metainflammation in part by altering the balance between type 2 and type 1 ATMs in visceral fat (13,18).
In addition to ATMs, adipose tissue contains lymphocytes (e.g., natural killer T cells [NKTs], conventional CD4+ T cells [Tconvs], regulatory CD4+ T cells [Tregs], cytotoxic CD8+ T cells, and B cells) that are also regulated by metabolic status (19–24). Treg content in visceral fat is inversely correlated with measures of insulin resistance and inflammation (19,25,26), suggesting that Tregs are anti-inflammatory. In contrast, T helper 1 (Th1) CD4+ T cells and CD8+ adipose tissue T cells (ATTs) accumulate in fat during obesity, promoting IFN-γ and TNF-α production and insulin resistance (20,21,27). Thus, analogous to ATMs, the imbalance between anti-inflammatory Tregs and proinflammatory CD4+/CD8+ ATTs contributes to metainflammation.
The mechanisms that regulate ATTs in adipose tissue are largely unknown. Spectratyping experiments suggest that CD4+ ATTs (but not CD8+ ATTs) undergo monoclonal expansion within fat and have an effector-memory (CD44High CD62LLow) phenotype (19,21,28). This implies that ATT activation and expansion may be an adaptive immune response to an obesity-induced antigen. T-cell activation depends on an intricate relationship between T cells and antigen-presenting cells (APCs) (29). Classically, APCs (specifically, macrophages and DCs) shape CD4+ T-cell activation by three signals: 1) presentation of peptide antigens on major histocompatibility complex (MHC) class II (MHC II) molecules (signal 1), 2) expression of T-cell costimulatory molecules (e.g., CD40, CD80, and CD86) (signal 2), and 3) production of cytokines (e.g., transforming growth factor-β, IL-10, or IL-12) (signal 3). These three signals shape the differentiation of naïve CD4+ T cells into effector T-cell subsets (e.g., Th1, Th2, Th17, Treg).
The APCs that interact with ATTs in fat have not been well characterized but could include ATMs, adipose tissue DCs, adipose tissue B cells, mast cells, and neutrophils (24,30–34). Quantitative changes in ATTs can precede the accumulation of type 1 CD11c+ ATMs in visceral fat in obese mice, suggesting that APCs present in lean and obese fat could trigger an adaptive immune response. Because ATMs are the predominant leukocyte population in lean and obese fat and ATMs from obese mice and humans express MHC II molecules (35–37), we tested the hypothesis that ATMs (CD11b+ F4/80+) are capable of functioning as APCs to regulate CD4+ ATT activation and proliferation. We report that ATMs within visceral fat from mice phagocytose and process antigens for presentation, express costimulatory molecules, and induce antigen-specific CD4+ T-cell proliferation in vitro and in situ. Furthermore, we found proliferating ATTs localized with ATMs in fat-associated lymphoid clusters (FALCs) where antigen-specific T-cell activation and proliferation may be initiated. Our data indicate that ATMs meet the functional definition of APCs and suggest that MHC II-restricted antigens presented by ATMs in visceral fat regulate Tregs and Tconvs CD4+ ATTs in mice.
RESEARCH DESIGN AND METHODS
Mice.
C57BL/6J, Cx3cr1GFP/+, and OT-II mice were from The Jackson Laboratory. Male mice (6 weeks old) were fed a normal diet (ND; 4.5% fat; PMI Nutrition International) or HFD (60% fat; Research Diets). All mice procedures were approved by the University of Michigan University Committee on Use and Care of Animals and were in compliance with the Institute of Laboratory Animal Research Guide for the Care and Use of Laboratory Animals.
Quantitative real-time PCR.
Gene expression analysis was performed as described (38). Relative expression in duplicate samples was determined using the 2–ΔΔCT method after normalizing to Arbp. Sequences for PCR primers are provided in Supplementary Table 1.
Microscopy.
Fat samples and cultured cells were prepared for immunofluorescence microscopy as described (6,39). Images were captured on an Olympus inverted microscope with an Olympus DP72 camera. Confocal images were captured with a FluoView microscope and software (Olympus) or on a Zeiss LSM 700 microscope and software.
Isolation and culture of adipose tissue stromal vascular cells.
Stromal vascular cells (SVCs) were isolated as described (38). SVCs were plated at 1 × 106 cells/mL in DMEM containing 10% heat-inactivated (HI) FBS.
Flow cytometry analysis and flow-activated cell sorting.
Cells were stained for flow cytometry as described (38). Antibodies for flow cytometry are provided in Supplementary Table 2. Live/dead fixable violet staining kits (Molecular Probes) and Foxp3 staining kits (eBiosciences) were used according to the manufacturers’ instructions. Cells were analyzed on a FACSCanto II Flow Cytometer (BD Biosciences) using FlowJo 9.4 software (Treestar). SVCs (5 × 107/mL) were suspended in RPMI/2%HI-FBS containing propidium iodide (2 μg/mL) and sorted on a FACSAria (BD Biosciences). Fluorescence-activated cell sorted (FACS) cells were cultured in DMEM/10%HI-FBS or lysed in RLT buffer (Qiagen).
BrdU pulse labeling.
BrdU (0.8 mg/mL; Sigma) was added to the drinking water. Isolated cells were stained with indicated cell-surface antibodies and permeabilized using Foxp3 staining kits. Permeabilized cells were incubated in DNAseI (30 μg; Sigma) at 37°C for 1 h and stained with BrdU and Foxp3 antibodies.
Phagocytosis and antigen processing.
Fluorescein isothiocyanate (FITC)-labeled ovalbumin (OVA; 100 μg/mouse; Molecular Probes) was injected intraperitoneally into mice 1 h before fat pads were examined by immunofluorescence microscopy. SVCs were cultured on glass chamber slides (Laboratory-Tek) overnight before FITC-labeled OVA (100 μg/mL; Molecular Probes) or FITC-labeled ZymosanA (Saccharomyces cerevisiae) bioparticles (100 particles/cell; Molecular Probes) were added to the media for 1 h. Lean mice were injected intraperitoneally with DQ-OVA (100 μg/mouse; Molecular Probes), a self-quenched conjugate of OVA and boron-dipyrromethene (BODIPY) that fluoresces upon proteolytic cleavage within endolysosomes (40). Peritoneal macrophages and SVCs were isolated 1 h later and stained for flow cytometry.
Antigen-specific T-cell activation assays.
SVCs or FACS-sorted cells were grown in 96-well U bottom plates overnight, then pulsed with medium alone, OVA peptide (OVA323-339; Abbiotec), or whole OVA (ThermoScientific) for 2 h at 37°C. CD4+ T cells were isolated from the spleen and lymph nodes of OT-II mice using CD4+ T cell untouched selection kits (Miltenyi Biotec). CD4+ T cells were labeled with 2 μmol/L carboxy fluorescein succinimidyl ester (CFSE) (CellTrace kits; Molecular Probes) and added to antigen-pulsed SVCs at a 1:1 ratio in 200 μL of media. After 5 days, T cells were stained for flow cytometry, and CFSE dilution was examined in viable (Live/DeadViolet-) CD3+CD4+ lymphocytes (41). T-cell cytokines in the media were measured by ELISA.
Adoptive transfer of CD4+ T cells.
CFSE-labeled CD4+ OT-II T cells (5 × 106 cells/mouse) were adoptively transferred into lean and obese (14 weeks of HFD) mice by intraperitoneal injection. Imject Ovalbumin (ThermoScientific) or Imject BSA (100 μg/mouse) was administered by intraperitoneal injection 24 h after adoptive transfer. At the indicated times, cells from visceral fat depots, blood, and spleen were analyzed by flow cytometry.
In vivo MHC II neutralization.
Hybridoma cells secreting MHC I-A/I-E monoclonal antibody were expanded at the University of Michigan Hybridoma Core and purified on protein A/G agarose columns (Pierce). MHC I-A/I-E or rat IgG2b K isotype antibodies (200 μg/mouse) were injected intraperitoneally every 48 h for 18 days.
Metabolic evaluation.
Glucose tolerance tests were performed on mice fasted (6 h) by intraperitoneal injection of d-glucose (0.7 g/kg body weight). Blood glucose levels (mg/dL) were measured 0, 15, 30, 45, 60, 90, and 120 min after injection using a glucometer (OneTouch Ultra). Plasma insulin levels were measured by ELISA (Crystal Chem).
Statistical analysis.
Data are means ± SEM. Differences between groups were determined using two-tailed Student t tests with GraphPad Prism 5.01 software. P < 0.05 was considered significant.
RESULTS
HFD-induced obesity promotes conventional CD4+ T-cell proliferation within visceral fat in mice.
CD4+ ATTs have an activated phenotype in obesity (19,21,28), but direct evidence of T-cell proliferation in fat is lacking. Therefore, we examined CD4+ T-cell proliferation within adipose tissue in lean mice and in mice fed HFD by BrdU incorporation (Fig. 1A). HFD increased the number of CD3+ CD4+ and CD3+ CD8+ ATTs in epididymal (eWAT) and inguinal (iWAT) white adipose tissue (Supplementary Fig. 1). HFD-induced obesity increased the percentage proliferating BrdU+ CD4+ ATTs in eWAT but not in iWAT (Fig. 1B). The number of BrdU+ CD4+ T cells in spleen trended to be higher in response to HFD (Fig. 1B), but the number of BrdU+ CD4+ T cells in blood, thymus, and lymph nodes was comparable between lean and obese mice (data not shown).
FIG. 1.
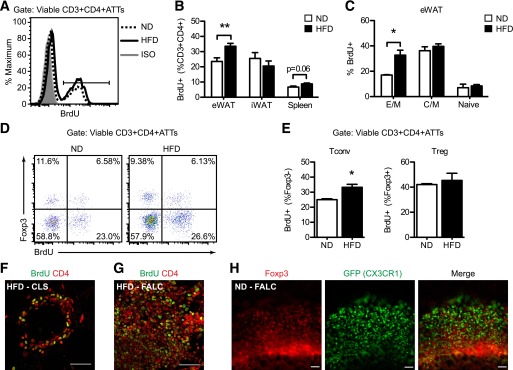
Adipose tissue CD4+ T-cell proliferation is induced in visceral fat in response to dietary obesity. Male C57BL/6 mice were fed ND or HFD (60% fat) for 14 to 17 weeks. BrdU was added to the drinking water for 5 consecutive days before splenocytes and adipose tissue stromal cells were isolated and analyzed by flow cytometry. A: Representative histogram shows BrdU incorporation in CD3+ CD4+ ATTs in eWAT from ND (dashedline) and HFD (solidline) mice. Isotype (ISO; shaded) is shown. B: Quantification of BrdU+ CD4+ T cells (expressed as percentage of viable CD3+ CD4+ lymphocytes) in eWAT, iWAT, and spleen from ND and HFD mice. Data are means ± SEM (ND eWAT: n = 5–6; HFD eWAT: n = 10; iWAT and spleen: n = 4 mice per group). C: Quantification of BrdU+ effector-memory (E/M; CD44High CD62L−), central-memory (C/M; CD44High CD62L+), and naïve (CD44Low CD62L+) CD4+ ATT subsets in eWAT from ND and HFD mice. Data are means ± SEM (ND: n = 3–4; HFD: n = 5–6 mice per group). D: Representative scatterplots (Foxp3 vs. BrdU) of viable CD3+ CD4+ ATTs demonstrating proliferation of conventional (Tconv, Foxp3−) and regulatory (Treg, Foxp3+) CD4+ T cells in eWAT from ND and HFD mice. E: Quantification of proliferating Tconvs and Tregs in eWAT from ND and HFD mice. Data are means ± SEM (ND: n = 3; HFD: n = 5 mice per group). BrdU+ (green) and CD4+ (red) lymphocytes were identified by confocal microscopy (original magnification ×60) in CLS (F) and FALC (G) within eWAT fat from HFD mice. Bar = 50 μm for F and G. H: Identification of Foxp3+ lymphocytes (red) in proximity to GFP+ ATMs (green) in FALCs in eWAT from a ND Cx3cr1GFP/+ reporter mouse. Scale bar = 100 μm. *P < 0.05, **P < 0.01 for ND vs. HFD.
Proliferation of effector-memory (CD44High CD62L–), central-memory (CD44High CD62L+), and naïve (CD44Low CD62L+) CD4+ ATT subsets was also determined. Consistent with other reports (28), E/M CD4+ ATTs were the predominant CD4+ ATTs in eWAT (Supplementary Fig. 2). HFD exposure specifically increased E/M CD4+ ATT proliferation in fat and did not alter C/M or naïve T-cell proliferation (Fig. 1C). Foxp3+ (Treg) and Foxp3– (Tconv) CD4+ T cells in eWAT both incorporated BrdU (Fig. 1D); however, HFD induced an increase in Tconv proliferation but did not alter Treg proliferation (Fig. 1E).
Within eWAT from obese mice, CLS contained BrdU+ CD4+ cells (Fig. 1F). By contrast, few CD4+ T cells were identified in contact with adipocytes in lean fat (data not shown). However, large clusters of BrdU+ CD4+ T cells were observed on the surface of visceral fat in ND and HFD mice (Fig. 1G and data not shown). These structures resembled FALCs or milky spots in mesenteric and omental fat depots that contain macrophages and lymphoid cells (42,43). In lean eWAT, FALCs contained Foxp3+ lymphocytes surrounded by CX3CR1+ macrophages (Fig. 1H).
ATMs express MHC II and costimulatory molecules that are induced by obesity and proinflammatory signals.
Increased CD4+ ATT-cell proliferation in fat suggests an interaction with MHC II+ APCs. Macrophage galactose-type C-type lectin (MGL1)+ MHC II+ ATMs were observed throughout eWAT from lean and obese mice (Fig. 2A–C). MHC II expression in ATMs was concentrated in CLS in obese eWAT (Fig. 2D–E) and in FALCs from lean and obese mice (Fig. 2F and data not shown). FALCs/milky spots in omental fat contained rare populations of MHC II+ B220+ B cells, but most of the MHC II+ cells in FALCs from eWAT were B220– by immunofluorescence (data not shown).
FIG. 2.

ATMs are the predominant MHC II+ cells in visceral fat. Male C57BL/6 mice were fed ND or HFD for 20 weeks to induce obesity. A: MHC I-A/I-E+ (green) ATMs were identified by epifluorescence microscopy dispersed between adipocytes. Bar = 100 μm. B: Identification of MGL1+ (green) and MHC I-A/I-E+ (red) ATMs in eWAT from ND mice. C and D: MHC I-A/I-E+ (green) ATMs in eWAT from HFD mice. Isolectin (red) stains blood vessels. Bar = 100 μm. E: Concentration of MHC I-A/I-E+ (red) ATMs in MGL1− (green) CLS in eWAT from HFD mice. F: MHC I-A/I-E (green) expression in a FALC in eWAT from HFD mice. Bar = 100 μm. G: H2-Ab1 gene expression in eWAT, iWAT, and spleen from ND and HFD mice. Data are means ± SEM (ND eWAT: n = 5; HFD eWAT: n = 6; iWAT: n = 5; spleen: n = 3 mice per group). H: Ciita gene expression in eWAT and spleen from ND and HFD mice (ND eWAT: n = 5; HFD eWAT: n = 6; spleen: n = 3 mice per group). I: Expression of Ciita transcripts from type I (DC), type III (B cell), and type IV (IFN-γ) promoters in eWAT from ND and HFD mice (ND: n = 5; HFD: n = 6). J: H2-Ab1 expression in FACS-sorted ATMs (F4/80+ CD11b+) and non-ATMs (F4/80− CD11b−) from ND and HFD eWAT. Data are means ± SEM (n = 4 per group). K: Surface expression of MHC I-A/I-E on ATMs (F4/80+ CD11b+ SVCs; thick line) and non-ATMs (F4/80− CD11b− SVCs; dashed line) isolated from ND and obese HFD mice. Isotype (ISO; shaded) is shown. L: Quantification of ATMs expressing MHC I-A/I-E and non-ATMs in eWAT from ND and HFD mice. Data are means ± SEM (n = 4 per group). M: MHC I-A/I-E expression (MFI, mean fluorescent intensity) on CD11c− and CD11c+ ATMs from eWAT of HFD mice as determined by flow cytometry. Data are means ± SEM (n = 4 mice). *P < 0.05, **P < 0.01, ***P < 0.001.
H2-Ab1 (MHC II) expression increased in eWAT with HFD but was reduced in iWAT and unchanged in the spleens of obese mice (Fig. 2G). The expression of the class II MHC transactivator (CIITA) was also induced in eWAT with HFD (Fig. 2H). Ciita cell-type specific promoters were assessed, and the induction in eWAT was attributed to specific activation of the DC-specific promoter (type I), which was the dominant Ciita promoter expressed in adipose tissue (Fig. 2I). The expression of the B-cell–specific Ciita promoter (type III), but not the IFN-γ inducible promoter (type IV), was modestly induced in eWAT by HFD (Fig. 2I).
SVF from ND and HFD mice were separated into F4/80+ CD11b+ ATMs and F4/80− CD11b− non-ATM fractions by FACS to further examine H2-Ab1 induction. H2-Ab1 expression was significantly higher in ATMs than in the ATM-depleted SVCs in lean and obese mice, and HFD-induced increase in H2-Ab1 expression was limited to the ATM population (Fig. 2J). Flow cytometry experiments confirmed that F4/80+ CD11b+ ATMs were the predominant MHC II+ population in eWAT (Fig. 2K). In lean mice, 55% of ATMs and less than 10% of non-ATMs expressed MHC II (Fig. 2L). HFD-induced obesity caused a significant increase in the number of MHC II+ ATMs (80%) and non-ATMs (Fig. 2L). MHC II surface expression was higher on type 1 CD11c+ ATMs than on CD11c− ATMs in HFD mice (Fig. 2M). In sum, these data demonstrate that ATMs are the predominant cell in the SVF capable of communication with T cells via MHC class II–dependent signals in lean and obese mice.
The induction of T cell costimulatory molecules (e.g., CD40, CD80, and CD86) in fat was also assessed. Cd40 and Cd80 were induced by HFD in eWAT (Fig. 3A). Cd40, Cd80, and Cd86 expression was unchanged in the spleen and suppressed in iWAT from obese mice (Fig. 3B and C). F4/80+ CD11b+ ATMs expressed higher levels of Cd40 than F4/80− CD11b− SVCs in both lean and obese eWAT, and Cd40 was only induced by HFD in ATMs (Fig. 3D).
FIG. 3.
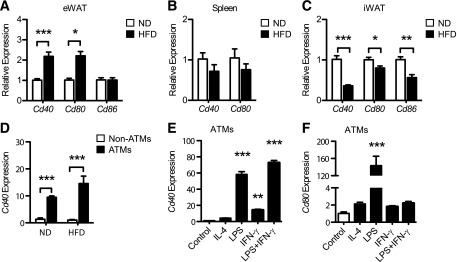
Expression of costimulatory molecules on ATMs is regulated by dietary obesity, inflammatory stimuli, and adipocyte signals. Male C57BL/6 mice were fed ND or HFD for 20 weeks. Cd40, Cd80, and Cd86 expression in eWAT (A), spleen (B), and iWAT (C) from ND and HFD mice. Data are means ± SEM (eWAT: n = 5–6; iWAT: n = 5; spleen: n = 3 mice per group). D: Cd40 expression in FACS-sorted ATMs (F4/80+ CD11b+) and non-ATMs (F4/80− CD11b−) from ND and HFD eWAT. Data are means ± SEM (n = 4 per group). Cd40 (E) and Cd80 (F) expression in cultured SVCs from HFD mice stimulated with IL-4 (20 ng/mL), LPS (100 ng/mL), IFN-γ (5 ng/mL), or LPS (100 ng/mL) plus IFN-γ (5 ng/mL) for 24 h. Data are means ± SEM (n = 3 wells) and are representative of two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
To examine the mechanisms of costimulatory molecule induction, purified ATMs were cultured in the presence of M1 and M2 macrophage-skewing stimuli. Lipopolysaccharides (LPS) induced Cd40 and Cd80 expression in ATMs, whereas IL-4 had no effect (Fig. 3D and E). IFN-γ alone or combined with LPS enhanced Cd40 but not Cd80 expression in ATMs (Fig. 3D and E).
ATMs are efficient phagocytes capable of processing antigens.
To examine the capacity of ATMs to phagocytose and process antigens, we performed experiments with OVA as a model antigen. MGL1+ ATMs throughout visceral fat and within FALCs rapidly accumulated fluorescently labeled OVA after intraperitoneal injection (Fig. 4A and B). In SVF cultures, the phagocytic capacity of cultured ATMs was heterogeneous (Fig. 4C), and OVA uptake was not an exclusive feature of MHC II+ or CD11c+ ATMs (Fig. 4D and E). F4/80+ ATMs were also capable of efficiently phagocytosing of FITC-labeled ZymosanA and Escherichia coli (Fig. 4F and data not shown).
FIG. 4.

ATMs in visceral fat are efficient phagocytes capable of processing model antigens. A: Identification of fluorescent OVA (green) in F4/80+ (red) ATMs in eWAT by confocal microscopy 1 h after intraperitoneal injection of FITC-conjugated OVA. Bar = 50 μm; original magnification ×40. B: Immunofluorescence image of fluorescent OVA (red) in a FALC in eWAT from a lean mouse. Scale bar = 100 μm. SVCs were isolated from HFD-fed (20 weeks) male mice and cultured for 18 h. Adherent cells were incubated with FITC-conjugated OVA or FITC-labeled ZymosanA particles for 1 h before immunostaining with indicated antibodies and imaged by confocal microscopy (original magnification ×63). Ex vivo uptake of FITC-OVA (green) was observed in adherent F4/80+ (red) (C), MHC I-A/I-E+ (red) (D), and CD11c+ (red) cells (E). F: Phagocytosis of ZymosanA particles (green) by cultured F4/80+ ATMs (red). Scale bar = 50 μm for C–F. Lean mice were injected intraperitoneally with DQ-OVA (100 μg/mouse). SVCs from eWAT and peritoneal macrophages were isolated 1 h later and stained for flow cytometry analysis. Representative scatterplots show DQ-OVAHigh cells in ATMs expressing MHC I-A/I-E (F4/80+ CD11b+) (G) and non-ATMs (F4/80− CD11b−) (H) from eWAT. I: DQ-OVA signals were detected in peritoneal macrophages (PMϕ; F4/80+ CD11bHigh) from the same mouse; the percentage of gated cells is indicated in each quadrant.
To determine whether ATMs in visceral fat can proteolytically process antigens, DQ-OVA was injected into mice. As expected, peritoneal macrophages (F4/80+ CD11bHigh) processed DQ-OVA (Fig. 4I), and ∼20% of F4/80+ CD11b+ MHC IIHigh ATMs from eWAT processed DQ-OVA (Fig. 4G). MHC II+ F4/80− CD11b− SVCs in eWAT, which contain B cells and nonmyeloid DCs, failed to process DQ-OVA (Fig. 4H). Approximately 4% of MHC II− F4/80− CD11b− SVCs (fibroblasts/preadipocytes) processed detectable amounts of DQ-OVA (Fig. 4H). Antigen processing in fat from obese mice could not be definitively assessed using this approach due to significant autofluorescence from obese SVCs (data not shown).
ATMs are sufficient to induce antigen-specific CD4+-cell proliferation and IFN-γ production in vitro.
We next measured the capacity of ATMS to trigger antigen-specific T-cell proliferation by culturing OVA-pulsed ATMs with CD4+ T cells from OVA-specific OT-II mice. SVCs from lean mice promoted CD4+ T-cell proliferation in an antigen dose-dependent manner (Fig. 5A and B). HFD-induced obesity significantly enhanced the ability of SVCs to induce T-cell proliferation. Antibodies blocking MHC II demonstrated that this effect was MHC II–dependent (Fig. 5C). FACs-purified (F4/80+ CD11b+) ATMs from obese mice were capable of inducing antigen-specific T-cell proliferation in vitro, but the ATM-depleted SVCs could not (Fig. 5D). SVCs pulsed with full-length OVA protein also induced OT-II CD4+ T-cell proliferation (Fig. 5E).
FIG. 5.

ATMs activate MHC II–restricted antigen-dependent CD4+ T-cell proliferation and Th1 polarization in vitro. SVCs were prepared from eWAT of lean (ND) and obese (HFD) mice and cultured overnight. Adherent cells were pulsed with the indicated amount of OVA peptide (323–339) or full OVA protein for 2 h before CFSE-labeled CD4+ cells from OT-II mice were added. After 5 days, CFSE dilution was determined by flow cytometry to quantify proliferating CD4+ cells in cocultures. Cytokines were measured by ELISA. A: Representative histograms demonstrate antigen-induced proliferation of CFSE-labeled OT-II CD4+ T cells in cocultures containing OVA peptide-pulsed SVCs from eWAT of lean (ND) and obese (HFD) mice. B: Quantitation of OT-II T-cell proliferation in cultures containing ND and HFD SVCs and increasing concentrations of OVA peptide. C: CD4+ T-cell proliferation in cultures containing OVA peptide-pulsed HFD SVCs in the presence (αMHC II) or absence (IgG) of MHC II–neutralizing antibodies. D: OT-II CD4+ T-cell proliferation induced by FACS-purified ATMs (F4/80+ CD11b+) and non-ATMs (F4/80− CD11b−) from HFD mice pulsed with OVA peptide. E: OT-II T-cell proliferation induced by ATMs from HFD mice pulsed with whole OVA protein. F: IFN-γ, IL-4, and IL-10 production in cocultures containing ND and HFD SVCs loaded with the indicated amounts of OVA peptide. G: IL-17 and IL-2 production in cocultures containing HFD SVCs pulsed with OVA peptide. nd = not detected. H: IFN-γ and IL-2 production in cocultures containing FACS-purified ATMs and non-ATMs from HFD mice loaded with OVA peptide before addition of CFSE-labeled OT-II CD4+ cells. B–H: Data are means ± SEM (n = 3–4 wells) and are representative of two to three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
To determine whether SVCs shape T-cell polarization of antigen-specific T cells, cytokines were measured in the cocultures. SVCs from lean and HFD-fed mice both promoted the accumulation of IFN-γ in the media in an antigen dose-dependent manner, but IFN-γ levels were higher when SVCs from obese mice were used as APCs (Fig. 5F). IL-10 and IL-4 production was unaltered (Fig. 5F). Notably, IL-17 production was attenuated in an antigen dose-dependent manner, whereas IL-2 was increased (Fig. 5G). IFN-γ and IL-2 production was dependent on signals from ATMs, because F4/80− CD11b− SVCs were unable to trigger a Th1 skewed T-cell response (Fig. 5H). Overall, these experiments suggest that ATMs from obese mice potently activate and skew naïve T cells toward a Th1 phenotype.
Adoptively transferred CD4+ T cells proliferate in visceral fat upon antigen exposure.
An adoptive transfer approach was used to examine antigen-specific CD4+ T-cell proliferation in visceral fat. CFSE-labeled OT-II CD4+ T cells preferentially homed to visceral fat depots and localized to FALCs 24 h after adoptive transfer (Supplementary Fig. 3). OVA, but not BSA, induced proliferation of adoptively transferred OT-II CD4+ T cells in eWAT, omental, and mesenteric fat depots, as indicated by CFSE dilution and by increased numbers of donor-derived (CD45.2+) CD4+ T cells recovered from these fat depots (Fig. 6A and B). The total number of CD4+ T cells increased in visceral fat depots, but not in spleen, 3 days after OVA exposure (Fig. 6C).
FIG. 6.

Induction of antigen-dependent CD4+ T-cell proliferation in adipose tissue in vivo. A: Proliferation of adoptively transferred CFSE-labeled OT-II CD4+ cells in eWAT from lean mice 3 days after intraperitoneal injection of OVA or BSA. Quantitation of adoptively transferred (CD45.2+) (B) and total (C) CD4+ cells in eWAT, omental (oWAT), and mesenteric (mWAT) fat depots and spleen from lean mice 3 days after intraperitoneal injection of OVA or BSA. Data are means ± SEM (n = 4 per group). Purified CD4+ OT-II T cells were adoptively transferred into obese mice (12 weeks on HFD). OVA or BSA was injected intraperitoneally 24 h after adoptive transfer. Cells from eWAT, spleen, and blood were analyzed by flow cytometry 5 days after antigen exposure. D: ATM and ATT content in eWAT from HFD mice. E: CD3+ ATTs in eWAT from HFD mice expressed as a percentage of SVCs. F: Quantification of CD4+ and CD8+ T cells in eWAT and blood from HFD mice. G: Quantification of Ki-67+ CD4+ T cells in eWAT and spleen from HFD mice. H: Quantification of ATM subtypes in eWAT from HFD mice. DN, double negative; DP, double positive. I: Random-fed blood glucose and plasma insulin levels in HFD mice. D–I: Data are means ± SEM (n = 3 mice per group). *P < 0.05, **P < 0.01, ***P < 0.001 vs. control (BSA).
OT-II CD4+ T cells were transferred into obese mice to determine the effects of antigen-specific CD4+ T-cell activation on adipose tissue inflammation in obese mice. In eWAT, OVA increased the number of CD3+ ATTs (expressed as total cells per gram or as a percentage of SVCs) but had no effect on ATM content (Fig. 6D and E). The accumulation of CD4+ T cells within fat was attributed not to increases in circulating T cells (Fig. 6F) but, rather, to localized proliferation, because OVA exposure increased the number of CD4+ ATTs expressing the cell cycle antigen Ki-67 (Fig. 6G). However, ATT proliferation alone did not alter ATM composition in eWAT (Fig. 6H) and had no significant impact on blood glucose or plasma insulin levels in obese mice (Fig. 6I).
Neutralization of MHC II reduces CD4+ ATTs but does not improve glucose homeostasis in obese mice.
We next tested whether MHC II blockade could modify T-cell inflammation and insulin resistance in mice with established obesity. MHC II immunotherapy (blocking antibody injections every 48 h for 18 days) in mice fed HFD for 14 weeks reduced the total number of CD3+ lymphocytes and the number of CD4+ ATTs in obese eWAT (Fig. 7A and B). Treg content in eWAT trended toward reduction after MHC II neutralization (Fig. 7C). Notably, there were no differences in CD4+ T-cell content in spleen or lymph nodes in obese mice after MHC II neutralization (Fig. 7D). MHC II neutralization did not attenuate the expression of inflammatory genes in eWAT (Tnfa, Il6, Il10, and Ifng; Fig. 7D). Moreover, blood glucose and insulin levels were unaffected (Fig. 7F), and glucose intolerance was similar between IgG and αMHC II–treated HFD mice (Fig. 7G). MHC II neutralization in mice fed HFD for a shorter duration (6–8 weeks) also failed to significantly improve metabolic parameters (Supplementary Fig. 4).
FIG. 7.
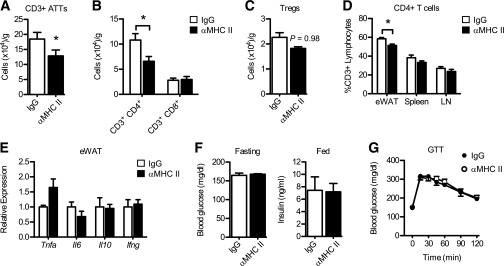
MHC II neutralization reduces CD4+ ATTs in obese mice but does not impact inflammation or glucose homeostasis. Mice were fed HFD for 14 weeks to induce obesity. Purified MHC I-A/I-E (αMHC II) or control (IgG; rat IgG2b) monoclonal antibodies were injected intraperitoneally every 48 h for 18 consecutive days. Quantification of total CD3+ ATTs (A) and total CD4+ and CD8+ ATTs (B) in eWAT of HFD mice after treatment. C: Treg content in eWAT from treated mice. D: Relative frequency of CD4+ T cells (as % total CD3+ T lymphocytes) in eWAT, spleen, and lymph nodes (LN). E: Tnfa, Il6, Il10, and Ifng expression in total eWAT from HFD mice after treatment. F: Fasting blood glucose levels (day 15) and random-fed plasma insulin levels (day 17) in HFD mice. G: Glucose tolerance test (GTT) performed in HFD mice 11 days after treatment. Data are means ± SEM (n = 3 mice per group). *P < 0.05 vs. control (IgG).
DISCUSSION
A diverse array of adipose tissue leukocytes is known to impact metabolic disease, yet we have a limited understanding of how leukocytes communicate within fat. Here, we show that ATMs from visceral fat meet the functional definition of APCs. ATMs process and present antigen on MHC II molecules, express T-cell costimulatory molecules, induce antigen-specific CD4+ T-cell proliferation, and provide accessory signals to promote the polarization of naïve CD4+ T cells into IFN-γ–producing Th1 effector cells. All of these functions were enhanced after dietary obesity due in part to the specific activation of Ciita via a DC/macrophage–specific promoter in ATMs. These data combined with the microscopic evidence of direct ATM–T-cell contact in fat support the concept that ATMs help coordinate an adaptive response to obesity by modulating T-cell activation in fat.
Our data indicate that the capacity for T-cell stimulation is an intrinsic feature of ATMs in lean mice. This provides a system whereby signals early in the course of obesity can be translated into a changed T-cell activation state, at least in part, via the ongoing interaction between CD4+ T cells and ATMs. In this way, resident ATMs may function as key sensors of the adipose tissue environment and translate signals from metabolically stressed or damaged adipocytes into an adaptive T-cell response. The potential signals that may feed into and modify this pathway are numerous and include adipokines, fatty acids, cytokines, activators of the NLRP3 inflammasome, and inflammatory CD8+ T cells. This model is consistent with the observations that ATT accumulation precedes the appearance of CD11c+ ATMs in obese visceral fat (19,21). Our data also indicate that the capacity of ATMs to induce proliferation and IFN-γ production in antigen-specific CD4+ T cells is dramatically enhanced by dietary obesity.
Because a significant fraction of ATMs expressing MHC II localize to FALCs that harbor proliferating ATTs, we believe that these atypical structures are important foci for ATM–T-cell interactions in visceral adipose tissue in mice. Notably, interactions between ATMs and ATTs redistribute more broadly with obesity, becoming centered on CLS—regions of active phagocytosis, dense CD11c+ ATM accumulation, and cytokine production. Collectively, this suggests that the APC function of ATMs may have duel roles, initially functioning to activate ATT proliferation early in response to obesity and later functioning to retain effector-memory ATTs in adipose tissue as obesity progresses. Technical limitations prevented us from definitively concluding which ATM subtype (M1 or M2) predominates in terms of APC function (data not shown). More studies are needed to define the spatial and temporal dynamics of ATM and ATT interactions in visceral fat at the onset of metabolic stress induced by overnutrition and throughout the progression of obesity.
Our group and others have primarily relied on surface markers to characterize ATMs and putative DCs in fat. Obesity induces the accumulation of myeloid cells (CD11b+) that express both macrophage (F4/80) and DC markers (CD11c) in fat, generating a confusing picture of what these cells are functionally. Given the uniqueness of the adipose tissue microenvironment, it is not surprising that myeloid cells in fat will have features that differ from classical lymphoid tissues, such as the spleen. The difficulty in distinguishing between tissue macrophages and DCs is not new to the immunology field, and many have questioned if a distinction between tissue macrophages and DCs is appropriate (44). Our studies have shifted toward clarifying the functional definition of ATMs and how this changes in obesity. A recent study described populations of F4/80− CD11c+ B220− adipose tissue DCs that are capable of promoting Th1 and Th17 T-cell responses (45). In our experience, these cells are rare in mice, because >80% of CD11c+ cells in visceral fat coexpress CD11b and F4/80. In our in vitro T-cell assays using FACS-sorted cells, F4/80− CD11c+ B220− cells were excluded from the F4/80+ ATM population but were present in the non-ATM fraction. However, these non-ATMs did not robustly stimulate T-cell proliferation or IFN-γ production in our hands. Although we cannot exclude the possibility that other SVCs may function as APCs, our data strongly imply that F4/80+ CD11b+ ATMs in visceral fat from mice are robust and efficient APCs that are significantly modified during dietary obesity.
Our initial attempts to improve insulin sensitivity in HFD-fed mice by blocking antigen presentation to CD4+ T cells with MHC II–blocking antibodies were unsuccessful in early and late obesity. MHC II neutralization decreased CD4+ ATTs specifically in the visceral fat of obese mice, supporting the notion that MHC II–restricted antigen presentation is responsible for activating and retaining CD4+ ATTs in fat. However, treating obese mice with MHC II–neutralizing antibodies alone did not improve whole-body glucose homeostasis or alter inflammatory gene expression in fat. These findings are consistent with variable results seen in other studies, and several laboratories have shown that blocking T-cell action alone is not sufficient to improve the metabolic profile in mouse models with established obesity. For example, depletion of CD4+ T cells in fat with anti-CD3 antibodies had no effect on glucose tolerance in older obese animals (28), and augmenting T-cell activation in Apoe−/− mice also had no effect on insulin resistance (46). Mice deficient for CD40 L demonstrated a significant decrease in ATTs, without any improvement in metabolic measures with obesity (47). Likewise, Treg expansion with IL-2 did not lead to significant changes in glucose or insulin tolerance in mice (19). Resolving these observations with the evidence that T cells can impact metabolism when restored in lymphopenic mice will require a more detailed view of the kinetics of T-cell activation and deactivation in fat. One explanation of why MHC II neutralization did not impact glucose metabolism may be due to the limited duration of therapeutic intervention. Two weeks of intervention may not be long enough to fully deactivate IFN-γ–producing CD4+ cells in fat. Another explanation may be related to the inability of these treatment modalities to augment the activity of proinflammatory CD11c+ ATMs, which are quantitatively the dominant proinflammatory leukocyte population in obese fat.
The identification of an antibody signature in human obesity (24) and our results suggest that protein antigens may trigger an adaptive immune response to obesity. The antigens that give rise to such responses remain elusive and will require further investigation. The limited number of total CD4+ and CD8+ ATTs in visceral fat (on the order of 1–5 × 104 cells/g) poses a significant obstacle to identifying immunogenic peptide antigens at this time. These challenges also exist in the atherosclerosis field, where T cells have been implicated for over a decade, but only a few causal antigens have been identified (48).
Beyond the identification of the individual protein antigens, the adipose tissue environment itself may significantly modify the ability of antigens to activate T cells. Antigens that are coupled to unsaturated fatty acid–based liposomes can be processed and presented on MHC I and II molecules to activate CD8+ and CD4+ T cells, respectively; by contrast, antigens coupled to saturated fatty acid–based liposomes are processed and presented via MHC II exclusively (49). In this article, we have chosen to focus on MHC II restricted CD4+ T-cell activation based on the current evidence that CD4+ T cells have a restricted T-cell receptor repertoire (19,21,28). Although we performed these experiments with non–liposomal-associated peptide antigens, whether putative obesity-induced antigens are liposomally encapsulated is unknown. This may be a potentially important node of regulation of both CD8+ and CD4+ ATTs.
In summary, we have shown that ATMs in visceral fat are efficient APCs capable of inducing the proliferative expansion of Th1 skewed effector/memory CD4+ T cells. Our observations provide a conceptual advancement to our understanding of the complexity of leukocyte networks in adipose tissue. Future studies are necessary to define how these dynamic interactions may contribute to the pathogenesis of metainflammation and metabolic diseases.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health (NIH) grants DK-090262, DK-078851, DK-087999, and DK-092873 to C.N.L. D.L.M. was supported by NIH National Research Service Award DK-091976. G.M.-S. was supported by NIH Cellular and Molecular Biology Training Grant T32-GM-007315. K.S. was supported by NIH Training Grant T32-HD-008513 and a Pediatric Endocrine Society Research Fellowship Award. This work used services at the University of Michigan and Animal Phenotyping Core, supported by NIH Grant DK-089503, and Chemistry Core(s) at the Michigan Diabetes Research and Training Center funded by P60-DK-020572 from the National Institute of Diabetes and Digestive and Kidney Diseases.
No potential conflicts of interest relevant to this article were reported.
D.L.M., K.W.C., and C.N.L. designed and performed experiments, analyzed data, and wrote the manuscript. J.L.D., K.E.O., L.M.G., G.M.-S., and K.S. performed experiments, acquired data, and performed analysis. C.N.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the analysis.
The authors thank Dr. Cheong-Hee Chang, University of Michigan, for the MHC I-A/I-E hybridoma and for providing technical expertise with the design of antigen-specific T-cell assays.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1404/-/DC1.
D.L.M. is currently affiliated with the Indiana University School of Medicine, Indianapolis, Indiana.
See accompanying commentary, p. 2656.
REFERENCES
- 1.Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res 2010;107:579–591 [DOI] [PubMed] [Google Scholar]
- 2.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol 2011;11:85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest 2011;121:2111–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest 2005;115:1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morris DL, Singer K, Lumeng CN. Adipose tissue macrophages: phenotypic plasticity and diversity in lean and obese states. Curr Opin Clin Nutr Metab Care 2011;14:341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 2007;56:16–23 [DOI] [PubMed] [Google Scholar]
- 7.Westcott DJ, Delproposto JB, Geletka LM, et al. MGL1 promotes adipose tissue inflammation and insulin resistance by regulating 7/4hi monocytes in obesity. J Exp Med 2009;206:3143–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007;447:1116–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang K, Reilly SM, Karabacak V, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab 2008;7:485–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab 2008;7:496–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007;117:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 2005;46:2347–2355 [DOI] [PubMed] [Google Scholar]
- 15.Strissel KJ, Stancheva Z, Miyoshi H, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007;56:2910–2918 [DOI] [PubMed] [Google Scholar]
- 16.Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A 1994;91:4854–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem 2003;278:45777–45784 [DOI] [PubMed] [Google Scholar]
- 18.Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol 2011;11:738–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 2009;15:930–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009;15:914–920 [DOI] [PubMed] [Google Scholar]
- 21.Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 2009;15:921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohmura K, Ishimori N, Ohmura Y, et al. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler Thromb Vasc Biol 2010;30:193–199 [DOI] [PubMed] [Google Scholar]
- 23.Zeyda M, Huber J, Prager G, Stulnig TM. Inflammation correlates with markers of T-cell subsets including regulatory T cells in adipose tissue from obese patients. Obesity (Silver Spring) 2011;19:743–780 [DOI] [PubMed]
- 24.Winer DA, Winer S, Shen L, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 2011;17:610–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ilan Y, Maron R, Tukpah AM, et al. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc Natl Acad Sci U S A 2010;107:9765–9770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deiuliis J, Shah Z, Shah N, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS ONE 2011;6:e16376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lumeng CN, Liu J, Geletka L, et al. Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J Immunol 2011;187:6208–6216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang H, Youm YH, Vandanmagsar B, et al. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol 2010;185:1836–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol 2006;6:476–483 [DOI] [PubMed] [Google Scholar]
- 30.Elgazar-Carmon V, Rudich A, Hadad N, Levy R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res 2008;49:1894–1903 [DOI] [PubMed] [Google Scholar]
- 31.Altintas MM, Azad A, Nayer B, et al. Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. J Lipid Res 2011;52:480–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertola A, Ciucci T, Rousseau D, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 2012;61:2238–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stefanovic-Racic M, Yang X, Turner MS, et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes 2012;61:2330–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talukdar S, Oh Y, Bandyopadhyay G, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 2012;18:1407–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeyda M, Farmer D, Todoric J, et al. Human adipose tissue macrophages are of an anti-inflammatory phenotype but capable of excessive pro-inflammatory mediator production. Int J Obes (Lond) 2007;31:1420–1428 [DOI] [PubMed] [Google Scholar]
- 36.Bassaganya-Riera J, Misyak S, Guri AJ, Hontecillas R. PPAR gamma is highly expressed in F4/80(hi) adipose tissue macrophages and dampens adipose-tissue inflammation. Cell Immunol 2009;258:138–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu H, Perrard XD, Wang Q, et al. CD11c expression in adipose tissue and blood and its role in diet-induced obesity. Arterioscler Thromb Vasc Biol 2010;30:186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris DL, Oatmen KE, Wang T, DelProposto JL, Lumeng CN. CX3CR1 deficiency does not influence trafficking of adipose tissue macrophages in mice with diet-induced obesity. Obesity (Silver Spring) 2012;20:1189–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008;57:3239–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daro E, Pulendran B, Brasel K, et al. Polyethylene glycol-modified GM-CSF expands CD11b(high)CD11c(high) but not CD11b(low)CD11c(high) murine dendritic cells in vivo: a comparative analysis with Flt3 ligand. J Immunol 2000;165:49–58 [DOI] [PubMed] [Google Scholar]
- 41.Quah BJ, Warren HS, Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat Protoc 2007;2:2049–2056 [DOI] [PubMed] [Google Scholar]
- 42.Moro K, Yamada T, Tanabe M, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010;463:540–544 [DOI] [PubMed] [Google Scholar]
- 43.Rangel-Moreno J, Moyron-Quiroz JE, Carragher DM, et al. Omental milky spots develop in the absence of lymphoid tissue-inducer cells and support B and T cell responses to peritoneal antigens. Immunity 2009;30:731–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol 2010;10:453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bertola A, Ciucci T, Rousseau D, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 2012;61:2238–2247 [DOI] [PMC free article] [PubMed]
- 46.Sultan A, Strodthoff D, Robertson AK, et al. T cell-mediated inflammation in adipose tissue does not cause insulin resistance in hyperlipidemic mice. Circ Res 2009;104:961–968 [DOI] [PubMed] [Google Scholar]
- 47.Wolf D, Jehle F, Ortiz Rodriguez A, et al. CD40L deficiency attenuates diet-induced adipose tissue inflammation by impairing immune cell accumulation and production of pathogenic IgG-antibodies. PLoS ONE 2012;7:e33026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Binder CJ, Chang MK, Shaw PX, et al. Innate and acquired immunity in atherogenesis. Nat Med 2002;8:1218–1226 [DOI] [PubMed] [Google Scholar]
- 49.Tanaka Y, Taneichi M, Kasai M, Kakiuchi T, Uchida T. Liposome-coupled antigens are internalized by antigen-presenting cells via pinocytosis and cross-presented to CD8 T cells. PLoS ONE 2010;5:e15225. [DOI] [PMC free article] [PubMed] [Google Scholar]