

Abstract
Spinal muscular atrophy (SMA) is a neurodegenerative disease that results from loss of function of the SMN1 gene, encoding the ubiquitously expressed survival of motor neuron (SMN) protein, a protein best known for its housekeeping role in the SMN–Gemin multiprotein complex involved in spliceosomal small nuclear ribonucleoprotein (snRNP) assembly. However, numerous studies reveal that SMN has many interaction partners, including mRNA binding proteins and actin regulators, suggesting its diverse role as a molecular chaperone involved in mRNA metabolism. This review focuses on studies suggesting an important role of SMN in regulating the assembly, localization, or stability of axonal messenger ribonucleoprotein (mRNP) complexes. Various animal models for SMA are discussed, and phenotypes described that indicate a predominant function for SMN in neuronal development and synapse formation. These models have begun to be used to test different therapeutic strategies that have the potential to restore SMN function. Further work to elucidate SMN mechanisms within motor neurons and other cell types involved in neuromuscular circuitry hold promise for the potential treatment of SMA.
1 Spinal Muscular Atrophy and Survival of Motor Neuron Protein
Spinal muscular atrophy (SMA) is an inherited autosomal recessive neurodegenerative disease, primarily affecting α-motor neurons of the lower spinal cord, in which proximal muscles are more severely affected then the distal muscles. SMA has an estimated incidence between 1-in-6,000 and 1-in-10,000 births, and about 1 in 35–40 people are genetic carriers (Wirth et al. 2006). SMA is classified clinically by age of onset and highest level of motor function achieved (Wirth et al. 2006; Lunn and Wang 2008; Oskoui and Kaufmann 2008). In its most severe form, known as SMA type I or Werdnig-Hoffman Disease (Online Mendelian Inheritance in Man (OMIM) #253300), SMA is the most common genetic cause of infant mortality and the second most common lethal, autosomal recessive disease after cystic fibrosis (Melki et al. 1994). Degeneration and death of the anterior horn motor neurons in the brain stem and spinal cord produce weakness in the limb muscles, as well as in muscles involved in swallowing and breathing (Nicole et al. 2002). Children with SMA present with generalized muscle weakness and hypotonia, and either do not acquire or progressively lose the ability to walk, stand, sit, and, eventually, move. Previously, SMA type I patients were predicted to die before the age of two, but in recent years, more proactive clinical care has improved survival (Oskoui et al. 2007; Oskoui and Kaufmann 2008). Intermediate SMA type II (OMIM #253550) is characterized by the onset after the age of 6 months. Patients acquire the ability to sit, but can never walk unaided. Patients with juvenile SMA type III (Kugelberg Welander disease; OMIM #253400) typically reach major milestones and can walk independently. The adult form, SMA type IV (OMIM #271150), is characterized by an age of onset beyond 30 years and relatively mild motor impairment.
SMA is caused by deletions or mutations of the survival of motor neuron gene (SMN1), which was originally cloned and characterized by Melki and colleagues (Lefebvre et al. 1995). SMN1 is an essential gene in divergent organisms, in which null mutations are lethal during early development (Schmid and DiDonato 2007). The survival of motor neuron (SMN) protein is ubiquitously expressed in all cells and tissues, with high levels in the nervous system, especially in spinal cord (Battaglia et al. 1997). Unlike other species that have only one copy of the SMN gene (e.g., mice), the SMN gene is present on human chromosome 5q13 as a single copy of the telomeric SMN1 gene, and a variable number of centromeric SMN2 genes. SMN2 appears to be unique to humans, since chimpanzees contain multiple copies of SMN1, but no SMN2 (Rochette et al. 2001). The majority of mRNAs (90%) from SMN1 encode for the full-length protein; whereas, the majority of mRNAs (90%) from SMN2 encode for a truncated and unstable protein lacking the carboxy-terminal exon-7 due to a translationally silent mutation in an exonic splicing enhancer (Lorson et al. 1999). The full-length transcripts of SMN1 and SMN2 encode proteins with an identical sequence. The most commonly inherited forms of SMA are caused by large deletions that inactivate the SMN1 gene and, while the unique presence of SMN2 in humans can protect against lethality, a neurodegenerative process occurs, leading to SMA. A major challenge is to understand how a reduction in total SMN levels results in neuronal dysfunction that leads to SMA.
The function of SMN was addressed by Dreyfuss and colleagues, who were seeking to identify proteins binding to hnRNP-U, a member of a family of heterogenous ribonucleoproteins. SMN was identified from their screen and shown to localize to nuclear structures, termed gems, based on their proximity to coiled bodies/Cajal bodies (i.e., Gemini of Cajal bodies) (Liu and Dreyfuss 1996). SMN was found tightly associated with a novel protein, SIP1, subsequently referred to as Gemin 2, and together, they form a specific complex with several spliceosomal snRNP proteins (Liu et al. 1997). The Dreyfuss lab and others subsequently identified other SMN interacting proteins (see Sect. 2.2), and extensively studied the critical role of the SMN–Gemin multiprotein complex in the assembly of spliceosomal snRNP s (Gubitz et al. 2004; Yong et al. 2004; Battle et al. 2006a; Eggert et al. 2006). The SMN–Gemin complex acts as a specificity factor to promote high-fidelity interactions between Sm core proteins and snRNAs, which prevent promiscuous interactions with other RNAs (Pellizzoni et al. 2002b). Recent data indicate that SMN deficiency alters stoichiometry of snRNAs and leads to splicing defects for numerous genes in all cells, including motor neurons (Zhang et al. 2008). In SMN-deficient mouse models of SMA, there is a preferential reduction in snRNP species that function in the minor spliceosome required for processing a rare class of introns (Gabanella et al. 2007). The function of the SMN–Gemin complex in snRNP assembly thus represents the most well understood function of SMN.
A major question needed to be addressed is how reduction of SMN in all tissues leads to a preferential impairment of motor neurons, although other neurons (e.g., sensory) and other tissues (e.g., muscle fibers) are also affected. Since the effects of SMN-deficiency on snRNP species and the stoichiometry of spliced introns appear to vary across tissues, one can also speculate that motor neurons may be more vulnerable to splicing alterations; however, to test this model, it would be necessary to demonstrate that altered splicing can have effects on protein expression and sequence composition that are deleterious to motor neurons. It is more than likely, however, that there are other functions of the SMN–Gemin complex, or even other types of SMN complexes, which contribute to the pathogenic mechanism in motor neurons. Inasmuch as SMN is localized to both the nucleus and cytoplasm (Young et al. 2000a), it becomes critical to understand whether the cytoplasmic pool of SMN is involved in activities other than that involved in the assembly of snRNPs.
2 SMN Domains and Interacting Proteins
2.1 SMN Domain Structure
The SMN1 gene contains nine exons and eight introns in a genomic region of ca. 20 kb. The ubiquitously expressed SMN1 transcript of ca. 1.7 kb encodes a highly conserved 294 amino acid protein of 38 kDa. SMN contains several functionally important regions for self assembly and protein interaction (see Fig. 1). The N-terminal part contains the binding sites that have been implicated in the interaction with Gemin2 from amino acids 13–44 (Liu et al. 1997) and 52–91 (Young et al. 2000b). The region encoded by exon2 has been shown to bind RNA directly in vitro (Lorson and Androphy 1998; Bertrandy et al. 1999). Exon 3 encodes a central Tudor domain, comprising amino acids 90–160. Tudor domains are conserved sequence motifs that were originally described for the Drosophila tudor protein. They are thought to mediate protein–protein interactions and are often found in RNA-associated proteins (Ponting 1997; Selenko et al. 2001). Tudor domain proteins have been shown to interact with proteins that contain methylated arginine and lysine residues (Brahms et al. 2001; Sprangers et al. 2003; Cote and Richard 2005). The SMN Tudor domain mediates interaction with arginine and glycine-rich motifs in several proteins (Meister et al. 2002; Paushkin et al. 2002; Gubitz et al. 2004), including the Sm core proteins. Symmetrical dimethylation of specific arginine residues enhances their affinity for SMN (Friesen et al. 2001a, b; Boisvert et al. 2002; Hebert et al. 2002; Meister and Fischer 2002). Exons 4–6 contain three stretches of poly-proline sequences that mediate the interaction with the small actin-binding protein profilin (Giesemann et al. 1999b). A conserved tyrosine/glycine-rich motif (YG box) that is found in many RNA-binding proteins is localized in exon 6, from amino acid 258–279. Regions required for dimerization or oligomerization of SMN have been mapped to self-association domains in exon 2b (52–91) and exon 6 (242–279) (Lorson et al. 1998; Young et al. 2000b).
Fig. 1.
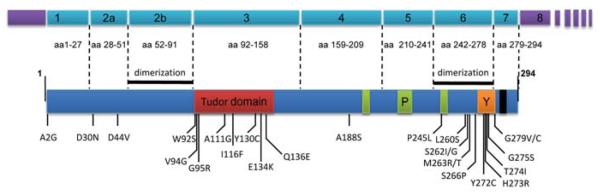
SMN1 exon boundaries and protein domain structure. Top: SMN1 is encoded by 9 exons. Coding regions are indicated in cyan and untranslated regions in purple. Bottom: Domains required for oligomerization of SMN are indicated as black bars. Other regions of interest are the Tudor domain (red), poly-proline regions (green), the YG box (orange), and the cytoplasmic targeting motif (black). Point mutations identified in SMN1 genes of SMA patients are indicated below (Wirth 2000; Alias et al. 2008)
SMA is characterized by a reduced level of full length SMN in the presence of SMN that lacks exon 7 (SMNΔ7). Although the expression of the SMNΔ7 isoform is beneficial, it cannot fully compensate for loss of full length SMN (Le et al. 2005). Therefore, the function of the protein domain encoded by exon 7 from amino acid 280–294 is of special interest. SMNΔ7 has been reported to encode an unstable and rapidly degraded protein (Le et al. 2000; Lorson and Androphy 2000) that is deficient in oligomerization activity (Lorson et al. 1999; Young et al. 2000b), binding to Sm core proteins (Pellizzoni et al. 1999), and formation of gems (Frugier et al. 2000). The SMNΔ7 protein has a twofold shorter half-life than full length SMN in cells, despite similar turnover rates, mediated by the ubiquitin-proteasome system in an in vitro assay (Burnett et al. 2008). SMNΔ7 can be stabilized by the coexpression of full length SMN, suggesting that it is stabilized by recruitment into oligomeric SMN complexes (Le et al. 2005). It is noteworthy that exon7 has also been shown to contain a cytoplasmic targeting signal that is required for the active transport of SMN into neuronal processes (Zhang et al. 2003).
Although in the large majority of cases SMA is caused by homozygous deletion of the SMN1 gene, a small percentage of SMA patients bear one SMN1 copy with small mutations. This is of considerable interest, since missense mutations may affect certain properties of SMN without causing a general loss of function. As depicted in Fig. 1, most SMA mutations have been found clustered in the tudor domain (W92S, V94G, G95R, A111G, I116F, Y130C, E134K, and Q136E) and in exon 6, within and near the Y/G box (L260S, S262G, S262I, M263R, M263T, S266P, Y272C, H273R, T274I, G275S, G279C, and G279V). This distribution of mutations suggests that the Tudor domain and the Y/G box with its flanking region are essential for SMN function. Studying these and synthetic SMN mutations may make it possible to uncouple functions required for snRNP assembly and proposed axonal functions in genetic rescue experiments (Beattie et al. 2007).
2.2 SMN Interacting Proteins
SMN is part of a well-characterized complex that facilitates assembly of Sm proteins on multiple U snRNAs to form the snRNP core (Meister et al. 2002; Paushkin et al. 2002; Gubitz et al. 2004; Kolb et al. 2007). To date, nine proteins have been identified as core components of this complex: SMN, Gemins 2–8 and unrip. SIP1 (Smn interacting protein) (Liu and Dreyfuss 1996), subsequently referred to as Gemin2, was identified as the first component of the SMN complex and shown to have a critical function in the assembly of spliceosomal snRNP s (Fischer et al. 1997; Liu et al. 1997), which has since been extensively studied by the Dreyfuss lab and others (Gubitz et al. 2004; Yong et al. 2004; Battle et al. 2006a; Eggert et al. 2006). Gemin2, Gemin3, and Gemin8 bind SMN directly (Otter et al. 2007). Gemin3 is a DEAD box RNA helicase (Charroux et al. 1999) and Gemin5 is the snRNA binding protein (Gubitz et al. 2002; Battle et al. 2006b). Gemin8 is needed for the structural organization of the SMN–Gemin complex (Carissimi et al. 2006). This SMN–Gemin core complex is found associated with spliceosomal Sm/LSm core proteins (SmB/B′, D1–3, E, F, G, LSm10, 11). It catalyzes the assembly of ring structures consisting of seven Sm core proteins onto stem loop structures of uridine-rich snRNAs (Chari et al. 2008). The high molecular weight of the SMN complex suggests that SMN and other core components are present as multimers. Oligomerization of SMN is also a prerequisite for high-affinity binding of the SMN complex to spliceosomal snRNPs (Pellizzoni et al. 1999).
In addition to the SMN complex components, SMN interacts directly or is associated with a remarkably large number of other proteins (see Table 1). This suggests that SMN may have a pleiotropic function that goes beyond the well-characterized role in snRNP assembly.
Table 1.
SMN-associated proteins, functions, and motifsa
| SMN associated protein | Function | Motifsb | References |
|---|---|---|---|
| SMN-Gemin complex | |||
| Gemin1/SMN | snRNP assembly | Tudor, Y/G box | Liu et al. (1997) |
| Gemin2/SIP1 | snRNP assembly | – | Liu et al. (1997) |
| Gemin3/DDX20/DP103 | snRNP assembly | DEXDc, HELICc | Charroux et al. (1999); Meister et al. (2000) |
| Gemin4/GIP1 | snRNP assembly | Leu zipper | Charroux et al. (2000); Meister et al. (2000) |
| Gemin5/p175 | snRNP assembly | WD40 repeats | Gubitz et al. (2002) |
| Gemin6 | snRNP assembly | – | Pellizzoni et al. (2002a) |
| Gemin7/SIP3 | snRNP assembly | RG-rich | Baccon et al. (2002) |
| Gemin8 | snRNP assembly | – | Carissimi et al. (2006) |
| unrip | snRNP assembly | WD40 repeats | Meister et al. (2001) |
| snRNP core proteins | |||
| Sm proteins/Sm B/B–, D1–3,E,F,G | snRNP assembly | Sm domain, RG rich | Liu et al. (1997); Friesen and Dreyfuss (2000) |
| Sm-like proteins/Lsm 10,11 | snRNP assembly | Sm domain, RG rich | Friesen and Dreyfuss (2000); Brahms et al. (2001) |
| snoRNP core proteins | |||
| Fibrillarin/FBL | snoRNP assembly | RGG box, RNP-2 | Liu and Dreyfuss (1996); Jones et al. (2001); Pellizzoni et al. (2001a) |
| GAR1/Nola1 | snoRNP assembly | RGG box | Pellizzoni et al. (2001a) |
| snRNP import | |||
| Snurportin and importin β | Nuclear import of snRNPs | HEAT repeat | Narayanan et al. (2002) |
| TGSI (trimethylguanosine synth. 1) | Nuclear import of snRNPs | – | Mouaikel et al. (2003) |
| Cajal body/coiled body | |||
| Coilin/p80 | Recruitment of SMN to Cajal bodies | – | Hebert et al. (2001) |
| Transcription | |||
| mSin3A | Transcriptional regulation | PAH, HDAC interact | Zou et al. (2004) |
| EWS (Ewing Sarcoma) | Transcriptional regulation | RGG box | Young et al. (2003) |
| Viral proteins | |||
| Papilloma virus E2 | Transcriptional regulation | – | Strasswimmer et al. (1999) |
| Epstein-Barr virus nuclear antigen | Transcriptional regulation | – | Barth et al. (2003) |
| Minute virus NS1 and NS2 | Viral replication and transcriptional regulation | – | Young et al. (2002b) |
| Actin metabolism | |||
| Profilin | Control of actin dynamics | – | Giesemann et al. (1999b) |
| T-Plastin/PLS3 | Actin bundling | EFh, CH | Oprea et al. (2008) |
| Apoptosis | |||
| p53 | Apoptosis | – | Young et al. (2002a) |
| Bcl-2 | Antiapoptosis | BH4, BCL | Iwahashi et al. (1997) |
| RNA metabolism | |||
| hnRNP U | RNA metabolism | SAP, SPRY, RGG | Liu and Dreyfuss (1996) |
| hnRNP Q and R | RNA metabolism | RRM, RGG | Mourelatos et al. (2001); Rossoll et al. (2002) |
| FUSE-binding protein/FBP | RNA metabolism | KH | Williams et al. (2000b) |
| KSRP/FBP2/ZBP2/MARTA1 | RNA metabolism | KH | Tadesse et al. (2008) |
| FMRP | RNA metabolism | KH, Agenet | Piazzon et al. (2008) |
| U1A | pre-mRNA splicing | RRM | Liu et al. (1997) |
| Galectin 1 and 3/LGALS1 | pre-mRNA splicing | GLECT | Park et al. (2001) |
| RNA helicase A | Transcription | DSRM, DEXDc, HELICc | Pellizzoni et al. (2001b) |
| RNA polymerase II | Transcription | – | Pellizzoni et al. (2001a) |
| Rpp20 (Ribonuclease P 20 kDa subunit) | tRNA and rRNA metabolism | – | Hua and Zhou (2004a) |
| Nucleolin and B23 | rRNA metabolism | RRM, RGG-box | Lefebvre et al. (2002) |
| ISG20 (Interferon stimul. gene 20) | Degradation of ssRNA | EXOIII | Espert et al. (2006) |
| TIAR (TIA-1-related protein) | Stress granule formation | RRM | Hua and Zhou (2004b) |
| TDP-43 (TAR DNA-binding protein) | mRNA splicing and transcription | RRM | Wang et al. (2002) |
| NFAR-1/2/nuclear factor associated with dsRNA | RNA metabolism | DZF, DSRM | Saunders et al. (2001) |
| Others | |||
| OSF (osteoclast-stimulating factor) | Src-related signaling | SH3, ANK | Kurihara et al. (2001) |
| USP9X (Ubiquitin-specific protease 9) | Deubiquitylating enzyme | – | Trinkle-Mulcahy et al. (2008) |
| PPP4 (protein phosphatase 4) | Ser/Thr protein phosphatase | PP2Ac | Carnegie et al. (2003) |
| FGF-2 (fibroblast growth factor 2) | Growth factor | FGF | Claus et al. (2003) |
| hsc70 (heat shock cognate prot. 70) | Protein folding/trafficking | – | Meister et al. (2001) |
| ZPR1 (zinc-finger protein 1) | Protein translation? | Zpr1 | Gangwani et al. (2001) |
| BAT3/HLA-B assoc. transcript 3c | Apoptosis/regulation of p53 | UBQ domain | Stelzl et al. (2005) |
| UNC119/HRG4c | Photoreceptor synaptic protein | GMP-PDE delta | Stelzl et al. (2005) |
| RIF1 (receptor interact. factor 1)c | Transcriptional repressor | – | Stelzl et al. (2005) |
| GDF9 (growth diff. factor 9)c | Growth factor | TGFB | Stelzl et al. (2005) |
| COPS6 (COP9 signalosome subunit 6)c | Regulation of ubiquitin ligases | JAB/MPN | Stelzl et al. (2005) |
| Leukocyte receptor cluster LRC member 8c | – | SAC3/GANP | Rual et al. (2005) |
| FLJ10204/C8orf32c | – | – | Rual et al. (2005) |
Based on Schultz et al. (1998)
Interaction partners identified in high throughput screens may have not been confirmed by independent methods
The largest group of SMN-associated proteins comprises components of RNP complexes that play a role in some aspects of RNA metabolism. Many of them contain domains that are enriched in arginine and glycine residues that are required for interaction with SMN (Meister et al. 2002; Paushkin et al. 2002). Arginine residues in these RG-rich domain are often methylated, which can enhance the interaction with SMN (Paushkin et al. 2002). The other interaction partners are very heterogenous and include viral and cellular transcription factors, regulators of apoptosis, and growth factors.
2.3 SMN Interacting Proteins Associated with Actin Metabolism
Interestingly, several interacting proteins have been implicated in either β-actin mRNA transport or in the regulation of actin dynamics. Examples of interacting partners that could potentially affect actin-based functions are briefly outlined as follows and further discussed below.
SMN interactions with regulators of actin dynamics. SMN was found to interact with the small actin-binding proteins profilin I and II in yeast-two-hybrid assays and to colocalize in motor neurons (Giesemann et al. 1999a). Profilin II and SMN were shown to colocalize in neurites and in growth cones of differentiating rat PC12 cells (Sharma et al. 2005). Antisense knockdown of profilin I and II isoforms inhibited neurite outgrowth of PC12 cells and caused accumulation of SMN and its associated proteins in cytoplasmic aggregates. SMN can modulate actin polymerization in vitro by reducing the inhibitory effect of profilin IIa. Therefore, reduced SMN-levels in SMA could disturb the normal regulation of microfilament growth by profilins and cause defects in axonal outgrowth. In a related study, SMN knockdown in PC12 cells altered the expression pattern of profilin II, leading to an increased formation of ROCK/profilin IIa complexes (Bowerman et al. 2007). Inappropriate activation of the RhoA/ROCK actin-remodeling pathway may result in altered cytoskeletal integrity and impaired neurite outgrowth.
Reduced actin expression in Smn-deficient distal axons. The distal distribution of β-actin mRNA and protein is defective in SMN-deficient motor neurons (see Sect. 3.1) (Rossoll et al. 2003). Treatment of primary motor neurons from Smn−/−; SMN2 embryos with a cell-permeable cAMP analog (8-CPT-cAMP) not only leads to an increased β-actin mRNA and protein level in the growth cone but also normalized the axon length and growth cone size defects to control levels (Jablonka et al. 2007). Thus, pharmacological intervention can increase distal β-actin mRNA and protein levels, and at the same time correct morphological axonal defects observed in SMA.
SMN and transport/regulation of β-actin mRNA and other transcripts. SMN interacts with several proteins thought to be involved in the transport and post-transcriptional regulation of β-actin mRNA and other transcripts: KSRP/ZBP2/MARTA1 (Tadesse et al. 2008), FBP (Williams et al. 2000b), hnRNP R, and hnRNP Q (Mourelatos et al. 2001; Rossoll et al. 2002, 2003). This is discussed in more detail in Sects. 3.1 and 3.2.
Protection from SMA by plastin 3 expression. Plastin 3/T-plastin (PLS3) has been identified as a protective modifier shown to physically interact with SMN complexes in a study of siblings discordant for SMA (Oprea et al. 2008). SMA-unaffected SMN1-deleted females exhibit significantly higher expression of PLS3 than their SMA-affected siblings. The actin-bundling protein PLS3 is important for axonogenesis through the increase in the F-actin level. Proteins of the fimbrin/plastin family share the unique property of cross-linking actin filaments into tight bundles that assist in stabilizing and rearranging the organization of the actin cytoskeleton in response to external stimuli, as well as perhaps by actin filament stabilization and anti-depolymerization activities (Oprea et al. 2008). Overexpression of PLS3 in SMN-deficient motor neurons rescued the axon length and outgrowth defects associated with SMN down-regulation.
SMN complexes with ZPR1 and eEF1A. SMN assembles into complexes with the zinc finger protein ZPR1 and eukaryotic translation elongation factor 1A (eEF1A) (Mishra et al. 2007). eEF1A binds to actin and has a noncanonical function in actin bundling (Gross and Kinzy 2005).
All considered, these data emphasize a potential role of SMN in the regulation of the axonal actin cytoskeleton, in part, by influencing β-actin mRNA transport and local translation and/or actin filament formation. Failure of locally modulated β-actin synthesis or F-actin localization in axon terminals may have a severe impact on axon growth, synapse differentiation and maintenance, the scaffolding of regulatory molecules such as synapsin, and the trafficking and release of synaptic vesicles (Shupliakov et al. 2002).
3 Localization of SMN and Links to Axonal mRNA Regulation
Previous immunocytochemical studies have localized SMN in dendrites and axons of spinal cord motor neurons in vivo (Bechade et al. 1999; Pagliardini et al. 2000). These immuno-EM analyses also depicted SMN on cytoskeletal filaments and associated with polyribosomes. Several immunofluorescence (IF) studies have detected Smn in neurites of cultured P19 cells (Fan and Simard 2002) and axons of cultured motor neurons (Rossoll et al. 2002). High-resolution imaging has revealed the presence of SMN in granules that localize to axons and growth cones of cultured neurons and align along microtubules (Zhang et al. 2003). As shown in Fig. 3, SMN-containing granules are not only abundant in the cell body and dendrites, but also along the axons and growth cones of motor neurons. SMN granules were shown by IF double labeling and fluorescence in situ hybridization (FISH) to colocalize with mRNA and ribosomes. Fluorescence imaging methods applied to living neurons showed that SMN granules are actively transported into neuronal processes and growth cones at rates over 1 μm s−1 (Zhang et al. 2003), consistent with fast axonal transport. Depolymerization of microtubules impaired the long-range transport of SMN granules, whereas disruption of F-actin impaired short-range trafficking (Zhang et al. 2003).
Fig. 3.
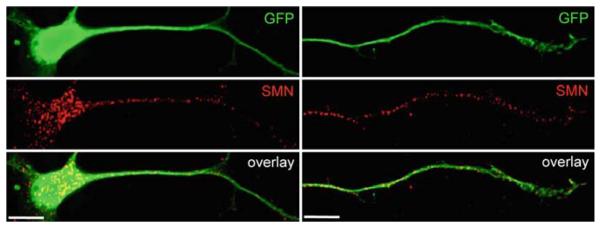
SMN-containing granules are transported along motor axons. SMN is localized in granules that are actively transported into neuronal processes and growth cones (Zhang et al. 2006). Shown above are murine primary embryonic motor neurons expressing GFP from the motor neuron-specific HB9 promoter. SMN-containing granules stained with specific antibodies are present in the cell body, dendrites, and axon, including the growth cone (red). Size bar = 10 μm (photomicrograph provided by Lei Xing)
Double label IF studies have suggested possible colocalization between SMN and Gemin proteins in neurites of PC12 cells (Sharma et al. 2005). However, significant levels of SMN appear not to colocalize with Gemin2 in axons of primary motor neurons (Jablonka et al. 2001). A later study, using high-resolution IF, digital imaging analysis, and 3D reconstruction of growth cones, showed that a population of SMN granules contained Gemin2 and Gemin3 (30–40%); however, the majority of SMN lacked Gemin proteins (Zhang et al. 2006). FRET analysis of fluorescently tagged SMN and Gemin proteins demonstrated interactions within individual granules, which were also observed to move as a complex in live neurons (Zhang et al. 2006). The QNQKE sequence from exon-7 was necessary for the sorting of the SMN–Gemin complex into the cytoplasm (Zhang et al. 2007). Of interest, the spliceosomal Sm proteins, necessary for snRNP assembly, were confined to the cell body and exhibited little colocalization with Smn–Gemin complex in neuronal processes (Zhang et al. 2006). Collectively, these studies indicate the presence of distinct SMN ribonucleoprotein complexes in neuronal processes that may play a role in mRNA regulation.
There appears to be some relationship between the neuritogenic effects of SMN and its cytoplasmic localization. The SMN Δ 7 form of SMN, which is the predominant form in SMA, was enriched within the nucleus, with much lower levels in the cytoplasm (Zhang et al. 2003). A QNQKE sequence with the carboxy-terminus of SMN was found to be necessary for cytoplasmic localization. Overexpression of SMN was neuritogenic in comparison to SMN Δ 7. This defect could be rescued by fusion of the membrane targeting sequence from GAP-43. SMN Δ 7 fused to this GAP-43 sequence now was targeted to axons and stimulated neurite growth.
A shorter splicing isoform of SMN called axonal SMN (a-Smn) has been reported to have a neuritogenic effect in NSC34 cells (Setola et al. 2007). The identified mRNA retains intron 3, which contains an in-frame stop codon. a-SMN is encoded by exons 1–3 and a small region of intron 3 that is not conserved between species. Future work is needed to address how prevalent this form of SMN is in axons. However, there has been some controversy as to whether this form of SMN could play a role in the pathogenesis of SMA (Burghes 2008). Most missense mutations found in SMA patients lie downstream of exon 3 and are clustered in exon 6. Also, there is strong evidence that SMA is caused by lack of exon7, and we know from experiments with SMA mouse models that expression of full-length murine Smn cDNA and human SMN2 can rescue the mutant phenotype. It will be interesting to assess the effects of a-SMN on rescue of axon phenotypes in animal models (discussed below).
3.1 Mechanism of β-Actin mRNA Localization in Neurons and Possible Connection to SMN and SMA
RNA localization is an essential and highly conserved biological mechanism for protein sorting that plays critical roles in neuronal polarity, axon guidance, and synaptic plasticity (Kindler et al. 2005; Bramham and Wells 2007; Lin and Holt 2008). Several mRNAs have been shown to be targeted to dendrites and/or axons, both in vivo and in cultured neurons. The molecular mechanism of mRNA localization involves recognition of cis-acting sequences within the 3′-untranslated region (UTR) by specific mRNA binding proteins and accessory factors, which are assembled into large mRNP complexes, often termed “granules” (Kiebler and Bassell 2006). mRNA granules are actively transported along microtubules and contain mRNA binding proteins, translation factors, ribosomal subunits, and accessory factors. mRNAs present within transport granules are often translationally repressed, which become derepressed in response to receptor signaling (Besse and Ephrussi 2008).
β-actin mRNA represents the most extensively studied localized mRNA, and hence provides a logical starting point to assess a possible role for SMN in the mechanism of mRNA granule localization. FISH analysis revealed that β-actin mRNAs are localized to developing axons and growth cones of primary cortical neurons in culture (Bassell et al. 1998). EM analysis showed the presence of polyribosomes within axon growth cones. A neurotrophin (NT-3) signaling pathway increased localization of β-actin mRNA from the cell body into processes and growth cones, while γ-actin mRNA remained in the cell body (Bassell et al. 1998; Zhang et al. 1999). The neurotrophin-induced localization of β-actin mRNA resulted in a protein-synthesis dependent increase in β-actin protein levels in growth cones (Zhang et al. 2001). The molecular mechanism of β-actin mRNA localization in chick forebrain neurons was shown to involve a highly conserved zipcode (54 nt) sequence, only present in the 3′UTR of β-actin, which is recognized by the mRNA binding protein, zipcode binding protein ZBP1 (Ross et al. 1997). ZBP1 has four KH-domains, which are necessary for binding to the zipcode, RNA granule formation, and association with the cytoskeleton (Farina et al. 2001).
ZBP1 also has NLS and NES and has been shown to shuttle into the nucleus where it first associates with nascent β-actin mRNA (Oleynikov and Singer 2003). Once in the cytoplasm, ZBP1 is hypothesized to function as an adapter for microfilament- and/or microtubule-dependent motors to facilitate the transport of β-actin mRNA in fibroblasts and neurons, respectively. In neurons, antisense-mediated disruption of ZBP1 binding to the zipcode impaired NT-3-induced localization of β-actin mRNA into neurites, resulting in reduced enrichment of β-actin protein and impaired growth cone dynamics (Zhang et al. 2001). In Xenopus neurons, binding of ZBP1 (VgRBP) to the β-actin zipcode was required for local β-actin synthesis and axon guidance in vitro in response to BDNF or netrin (Leung et al. 2006; Yao et al. 2006). Recent evidence indicates that ZBP1 acts to repress β-actin mRNA translation, and that in response to Src activation, ZBP1 is phosphorylated, resulting in its release from the mRNA, allowing for translation (Huttelmaier et al. 2005). Since β-actin appears to be the preferred actin isoform to induce rearrangements of the actin cytoskeleton beneath the plasma membrane (Bassell et al. 1998), it is hypothesized that local β-actin mRNA translation is necessary to promote enrichment of β-actin protein at the membrane and influence growth cone motility and guidance. ZBP1 is thus a critical factor for both β-actin mRNA transport and local protein synthesis; however, there may be other proteins within the complex that contribute to these processes, and Smn is attractive candidate based on its trafficking in the form of RNA granules.
In a study providing the first link between Smn and β-actin mRNA, Smn-deficient motor neurons, cultured from a transgenic mouse model of SMA, were shown to have reduced localization of β-actin mRNA in axons and reduced levels of β-actin protein in axonal growth cones (Rossoll et al. 2003). Of interest, Smn-deficient motor neurons displayed only defects in axonal morphology, that is, shorter axons and smaller growth cones, whereas dendrites appeared otherwise normal in morphology. These results suggest some role of SMN in the molecular mechanism of β-actin mRNA localization to axons. These results also imply that axonal defects in SMA may be due, in part, to altered localization of β-actin mRNA. Future work will be necessary to explore a possible interaction between SMN and ZBP1. However, it also possible that Smn may interact with ZBP2 to facilitate β-actin mRNA localization. ZBP2 is a KH-domain-containing protein that is predominantly localized to the nucleus, where it facilitates the hand-off of β-actin mRNA to ZBP1 that exports the mRNA into the cytoplasm (Gu et al. 2002; Pan et al. 2007). Of interest, SMN has been shown to interact with ZBP2 homologs, viz., FBP and KSRP. By yeast two-hybrid and co-immunoprecipitation (IP) analysis, SMN was shown to bind FUSE binding protein (FBP) (Williams et al. 2000a), a KH domain-containing RNA binding protein that regulates the stability of GAP-43 mRNA (Irwin et al. 1997), which is localized to axons and growth cones (Smith et al. 2004). More recent work has shown that SMN binds KSRP (Tadesse et al. 2008), a neuron-specific splicing factor that is also involved in targeting AU-rich mRNAs for degradation (Min et al. 1997). KSRP was shown to be arginine-methylated and interact with the Tudor domain of SMN. Smn and KSRP colocalized in granules within neurites and missense mutations in the Tudor domain that cause SMA abolished the colocalization with KSRP. In SMA transgenic mice, a KSRP target mRNA, p21 (cip/waf1), was increased in spinal cord, suggesting possible impairment of mRNA stability. How this relates to altered mRNA localization is unknown, but there are known links between mRNA localization and stability mechanisms; thus, it will be important for future work to assess a possible alternative or related role of SMN in regulation of mRNA stability in axons. Other studies have demonstrated a role for MARTA1, the rat KSRP ortholog, to bind the dendritic targeting element of MAP2 mRNA and facilitate its targeting in dendrites (Rehbein et al. 2002). Since Smn is localized to dendrites and axons (Bechade et al. 1999; Zhang et al. 2006), it will be important to assess a possible role of Smn in regulation of dendritic mRNA localization, which may affect postsynaptic function in spinal cord motor neurons.
3.2 SMN Interactions with hnRNP R and hnRNP Q: Another Possible Link to β-Actin mRNP Complexes
Yeast two-hybrid (Y2H) screens using SMN as a bait have identified two highly related proteins, hnRNP R (heterogenous nuclear ribonucleoprotein R) and hnRNP Q, previously discovered as glycine–arginine–tyrosine-rich RNA-binding protein (gry-rbp). hnRNP R and Q interact with wild-type SMN, but not with mutated versions of the protein found in SMA patients (Rossoll et al. 2002). In a parallel Y2H study, Mourelatos et al. have identified three human splicing isoforms of hnRNP Q as SMN interacting proteins (Mourelatos et al. 2001). hnRNP Q3/gry-rbp encodes a protein of 623 aa that is composed of an N-terminal acidic domain, followed by three consecutive RNA recognition motifs (RRM1–3) and an arginine- and glycine-rich domain (RGG box). hnRNP Q3 and hnRNP R share 82% identity and 90% similarity on the protein level. They also identified two smaller isoforms that are derived by alternative splicing of hnRNP Q3. hnRNP Q2 lacks 34 amino acids from RRM2, and in hnRNP Q1, the last 74 amino acids from the C-terminus of hnRNP Q3 are replaced by the sequence VKGVEAGPDLLQ. All yeast two-hybrid clones identified in both studies contain the C-termini, including most of the RGG box, suggesting that the RGG box of hnRNP Q is necessary and sufficient for binding to SMN. This was confirmed by GST-pull down assays that identified an RG dipeptide-rich sequence from amino acids 518–549 as the minimal SMN-binding domain.
Although hnRNP R is mainly a nuclear protein, it was also found to colocalize with Smn in axons of motor neurons (Rossoll et al. 2002). hnRNP R has been shown to associate with the 3′ UTR of β-actin mRNA and to modulate localization of actin mRNA in neurites (Rossoll et al. 2003). While hnRNP Q3 has been isolated as a predominantly nuclear protein associated with the spliceosome complex (Neubauer et al. 1998), hnRNP Q1 has been identified in several independent studies on various aspects of mRNA metabolism in the cytoplasm. Taken together, they suggest a role of hnRNP Q1 in mRNA transport, regulation of mRNA stability, and/or translation. Previously, hnRNP Q1 has been described as NSAP1, a protein interacting with the NS1 protein of minute virus of mice that is essential for viral replication in the cytoplasm (Harris et al. 1999). Regulation of mRNA stability by hnRNP Q1/NSAP1 was shown in a study on the major protein-coding-region determinant of instability (mCRD) of the c-fos proto-oncogene mRNA. hnRNP Q1/NSAP1 binds the mCRD and modulates the translationally coupled process of mRNA turnover. HnRNP Q1/NSAP overexpression stabilizes mCRD-containing mRNA by inhibiting deadenylation (Chen and Shyu 2003). In addition, hnRNP Q1/NSAP overexpression resulted in a dramatic stabilization of AU-rich element-containing mRNAs (Grosset et al. 2000). Work from several groups has demonstrated that hnRNP Q1/NSAP1 modulates cap-independent translation. It enhances IRES-dependent translation through interaction downstream of the hepatitis C virus polyprotein initiation codon (Kim et al. 2004) and it has also been shown to modulate IRES-dependent translation of cellular BiP mRNA through an RNA–protein interaction under heat stress conditions (Cho et al. 2007). Another substrate of hnRNP Q1 is mRNA related to the circadian rhythm. hnRNP Q1 has been shown to bind serotonin N-acetyltransferase (AANAT) both at its 3′ untranslated region to mediate mRNA degradation (Kim et al. 2005) and its 5′ region to regulate cap-independent translation (Kim et al. 2007). Other studies have connected hnRNP Q1 to vesicle transport and RNP transport granules. hnRNP Q1 has been described as SYNCRIP, a protein that interacts with the C2B domains of ubiquitous synaptotagmin isoforms; thereby, suggesting a potential link between vesicles and mRNA transport (Mizutani et al. 2000). hnRNP Q1/SYNCRIP has also been identified as a component of inositol 1,4,5-triphosphate receptor type I mRNA transport granules in rat hippocampal dendrites (Bannai et al. 2004). In another proteomic study, hnRNP Q1/SYNCRIP was isolated as a component of kinesin (KIF5)-containing RNA transport granules (Kanai et al. 2004). Therefore, it will be important to investigate whether SMN associates with hnRNP R/Q in neuronal processes, and whether this interaction is necessary for hnRNP R/Q mediated regulation of mRNA granules in neuronal processes.
4 A Role for SMN in Stress Granule Formation
Stress granules (SGs) are an additional type of RNA granule within neurons whose formation within the cell body and processes can be induced in response to low dose arsenate treatment (Vessey et al. 2006). In contrast to RNA transport granules, SGs have limited motility, are larger morphologically, and may assemble de-novo, or perhaps be converted from RNA transport granules (Kiebler and Bassell 2006; Anderson and Kedersha 2008). SGs serve as a depot to recruit mRNAs and translational factors (however, lacking some ribosomal components), where mRNAs are poised in translational arrest and also protected from degradation (Kedersha et al. 2005). A number of mRNA binding proteins are recruited to SGs in response to various stressors (Anderson and Kedersha 2006, 2008). The RGG box has been shown to be necessary for recruitment of some mRNA binding proteins to SGs (Yang et al. 2006). SMN was shown to target to SGs upon arsenate exposure and SMN-deficient fibroblasts from SMA patients showed impaired formation of SGs and recruitment of mRNAs (Hua and Zhou 2004b). The SG hypothesis to explain the neurodegeneration in SMA posits that impaired formation of SGs in axons, in response to various insults (i.e., oxidative stress), may lead to increased degradation of axonal mRNAs (Hua and Zhou 2004b). In that, many of the SMN-associated mRNA binding proteins are also localized to stress granules, where they play a role in mRNA stability regulation (Stohr et al. 2006), it will be important to assess how these interactions may affect the trafficking of mRNAs between various types of granules involved in mRNA transport, stability, and degradation, including stress granules and P-bodies (Kiebler and Bassell 2006). Collectively, these dynamics may have a great impact on mRNA translation in axons.
4.1 The SMN-mRNA Granule Hypothesis
As discussed earlier, a critical goal will be to continue to identify interactions between SMN and specific mRNA binding proteins that are localized to neuronal processes, and assess the function of these interactions in mRNA regulation. Although it has been shown that SMN can bind RNA in vitro, there is no evidence so far that it acts as an RNA-binding protein in vivo (Lorson and Androphy 1998; Bertrandy et al. 1999). SMN may impart binding specificity to RNA binding proteins through its role in the formation of the ribonucleoprotein complex. The past work on the SMN–Gemin complex and its role to facilitate Sm core assembly onto stem loops of snRNA may suggest a mechanism that could be used for mRNP assembly. Whether SMN interactions with mRNA binding proteins depend on Gemins will need to be addressed. Nonetheless, SMN interactions with mRNA binding proteins may modulate the binding of mRNA binding proteins to cis-acting elements and assembly of RNA transport granules. A model for the proposed role of SMN complexes in axonal RNA localization is shown in Fig. 2.
Fig. 2.

Model of a putative function of SMN complexes in axonal RNA localization. Data from different labs suggest a model whereby SMN-associated RNP complexes contribute to the localization of β-actin mRNA and probably other transcripts. SMN may facilitate the assembly of RNA cargo molecules with different RNA-binding proteins, adaptor proteins, molecular motor proteins, translational components, and auxiliary factors required for efficient transport and/or local translation (Based on a model by Lei Xing.)
5 Animal Models of SMA
SMN is present in all metazoan cells with an evolutionary conserved role in small nuclear ribonucleoprotein assembly and pre-mRNA splicing. SMN orthologs have been disrupted in different genetic model organisms, ranging from yeast to mouse, in an effort to model the disease phenotype (Schmid and DiDonato 2007). These SMA models serve as powerful research tools to elucidate the pathogenic mechanism, and also to aid in the development of therapies for the treatment of spinal muscular atrophy. Using a variety of model organisms makes it possible to exploit their respective strengths and to validate results gained from different species. Analyzing the phenotype of these model organisms suggests a unique requirement for SMN orthologs in the development and maintenance of motor neurons (Monani et al. 2000a).
5.1 Caenorhabditis elegans
The nematode Caenorhabditis elegans has been used as a valuable research tool in many fields of biological research. Establishment of a cell lineage map, sequencing of the entire genome, and a well-characterized wiring diagram of its neurons allow an in-depth investigation of the development of its nervous system (Girard et al. 2007). CeSMN is an essential protein that is required for embryonic viability. Targeting smn-1 with RNAi in all tissues (including maternal transcripts) and overexpression of the gene gives rise to locomotive defects and embryonic lethality (Miguel-Aliaga et al. 1999). Surviving hypomorphs demonstrate pleiotropic developmental defects in neuronal, muscular, and reproductive tissue that lead to locomotive defects, lack of muscle tone, vulval abnormalities, and sterility caused by a defect in germ cell maturation (Miguel-Aliaga et al. 1999).
Recently, a deletion mutant of smn-1 (ok355) has been characterized, which removes most of the gene including the translation start codon (Briese et al. 2009). Unlike the embryonic lethality caused by targeting smn-1 with RNAi, maternally contributed CeSMN in the mutant allows for normal early larval development of smn-1(ok355) mutant animals, but causes late larval arrest, sterility, decreased lifespan, as well as impaired locomotion and pharyngeal pumping activity. The smn-1(ok355) phenotype can be partially rescued by neuronal, but not muscle-directed, expression of smn-1. A C. elegans ortholog of the human Gemin2 gene and several RNA-binding proteins have also been identified as interaction partners of CeSMN (Burt et al. 2006). Knockdown of smn-1 by RNAi, as well as deletion mutants have the potential to be utilized in screens searching for phenotypic modifiers as a further step toward potential SMA therapies (Briese et al. 2009).
5.2 Drosophila melanogaster
The fruit fly Drosophila melanogaster has proven to be an excellent invertebrate model organism for studying human neurodegenerative diseases. Mutational analyses and the existence of a broad array of screening techniques allow direct observation of the effects of the loss of gene function, making it an excellent in vivo system for the identification of genetic modifiers and the testing of therapeutic compounds (Lu and Vogel 2008).
The Drosophila genome bears a single copy of the Smn gene, which encodes a highly conserved homologue of SMN (DmSMN) (Miguel-Aliaga et al. 2000). Drosophila contains a remarkably simple SMN complex that lacks most Gemin proteins (Kroiss et al. 2008). This minimal complex mediates the assembly of spliceosomal snRNPs in a manner very similar to its vertebrate counterpart, by also preventing misassembly onto nontarget RNAs (Kroiss et al. 2008). Larval-lethal Smn-null mutations do not show detectable differences in spliceosomal snRNA levels, suggesting that these animals do not die from global snRNP depletion (Rajendra et al. 2007). Ectopic expression of human SMN exerts a dominant-negative effect that leads to developmental defects and pupal lethality, presumably, by forming a nonfunctional complex with endogenous DmSMN (Miguel-Aliaga et al. 2000). Drosophila mutant strains that contain point mutations in Smn similar to those found in SMA patients show abnormal motor behavior (Chan et al. 2003). Because of maternal contribution of the smn transcript, smn zygotic mutant embryos survive early development. Mutant larvae show severe motor abnormalities, defects in late developmental stages, and late larval lethality. Defects at the neuromuscular junction (NMJ) include disorganized synaptic motor neuron boutons, reduced clustering of the neurotransmitter receptor subunit GluR IIA, and reduced excitatory post-synaptic currents. Rescue experiments show that Smn gene activity is required in both neurons and muscle to alleviate this phenotype. Taken together, these studies point to a functional role of DmSMN at the NMJ (Chan et al. 2003).
Hypomorphic mutations in Smn with a tissue specific reduction of DmSMN protein levels in the adult thorax cause flightlessness and acute muscular atrophy (Rajendra et al. 2007). Mutant flight muscle motor neurons display motor axon routing and arborization defects coupled with a loss of the flight muscle-specific actin isoform Act88F expression. The authors also observed colocalization of DmSMN with sarcomeric actin, association between SMN and the thin filament crosslinker α-actinin, and disruption of α-actinin localization in the thoracic muscles of Smn mutants. These observations suggest a specific function for DmSMN in muscle and would underline the importance of this tissue in modulating the disease phenotype (Rajendra et al. 2007). However, it is not clear whether the defects observed in the Smn hypomorphs are caused by reduced DmSMN expression in the motor neurons, the thoracic muscles, or a combination of both.
Recently, a screen for genetic modifiers of an Smn phenotype, using the Exelixis collection of transposon-induced mutations, identified 17 enhancers and 10 suppressors (Chang et al. 2008). Among these were several genes not previously known to be associated with the genetic circuitry of Smn. These include several members of the bone morphogenic protein (BMP) signaling pathway. Interestingly, the observed NMJ defects in the SMA model can be ameliorated by increased BMP signaling, suggesting that drugs that stimulate BMP activity may provide potential SMA therapeutics (Chang et al. 2008).
5.3 Danio rerio
The zebrafish Danio rerio has been developed as an excellent vertebrate model organism for studying developmental biology and genetics (Beattie et al. 2007). A very stereotyped and well characterized neuromuscular system and external embryonic development allow for easy analysis of early nervous system development. Recent advances, such as large genetic screens for mutations that disrupt development, the establishment of an effective antisense approach, and sequencing of the zebrafish genome, make them an ideal model system to study developing motor neurons and their axonal projections. Although targeted gene knockout is not yet established in the zebrafish model, most loss-of-function studies use a morpholino (MO) knockdown approach.
Beattie and colleagues have developed a zebrafish model for SMA, which demonstrate an axonal function for SMN. Using antisense morpholinos injected into zebrafish embryos at the 1–2 cell stage to reduce Smn levels lead to motor axon-specific pathfinding defects (McWhorter et al. 2003). While the number of motor neurons present at 24 h was not affected, motor axons were short or excessively branched. Importantly, specific reduction of Smn in individual motor neurons revealed that Smn is acting cell-autonomously. This study indicates an essential function of Smn in motor axon development in vivo, and suggests that these early developmental defects may contribute to later defects in synapse development that may precede motor neuron loss. To further characterize Smn functions necessary for motor axon outgrowth, smn MO were coinjected with various human SMN RNAs and assayed for their effect on motor axons (Carrel et al. 2006). Motor axon defects caused by Smn reduction were rescued by wild-type human SMN but not by SMN lacking exon-7, the predominant form in SMA, or other mutations in human SMN that also cause SMA. The identification of mutations that failed to rescue motor axon defects, but retained properties needed for snRNP assembly such as oligomerization and Sm protein binding, would indicate a critical function of SMN in motor axons that is relevant to SMA and is independent of snRNP biosynthesis (Carrel et al. 2006). SMNΔ7, SMN G279V, and SMN Y272C were defective in these properties and failed to rescue axonal defects. The human SMA mutant A111G was also unable to rescue motor axon defects while retaining Sm binding and oligomerization properties. In contrast, SMNΔ7 fused to an exon-7 sequence, containing the minimal cytoplasmic localization motif (QNQKE) (Zhang et al. 2007), was able to rescue motor axon defects, although it was deficient in oligomerization and Sm proteins binding in vitro (Carrel et al. 2006; Beattie et al. 2007). A synthetic point mutant of an exon-7 sequence (SMN Q282A) important for the cytoplasmic localization of SMN did not rescue the axon guidance defects, although this mutant retained snRNP function. Further work will be needed to show that this point mutant is fully competent for snRNP function, and it will be interesting if interactions with proteins other than Gemins are impaired. Such efforts to identify SMN domains that compromise SMN function in axon guidance, without affecting snRNP function, represent a useful approach toward identification of alternate mechanisms underlying SMA.
Another study used the morpholino knock-down approach to show an effect of SMN reduction on U snRNP metabolism (Winkler et al. 2005). Knockdown of SMN and the U snRNP assembly factors Gemin2 and pICln led to motor axon degeneration. This developmental defect was rescued by injection of purified U snRNPs into SMN- or Gemin2-deficient zebrafish embryos. These findings would imply that motor neuron degeneration in SMA patients may be a direct consequence of impaired production of U snRNPs (Winkler et al. 2005). However, these findings are contradicted by a study that did not find any aberrant motor axons caused by Gemin2 knockdown in morphologically normal embryos (McWhorter et al. 2008). Specific knockdown of Gemin2 in motor neurons by either injection into motor neurons or generation of genetic mosaics did not lead to motor axon defects. Since Gemin2 and SMN both function in snRNP biogenesis, yet, only SMN knockdown causes motor axon defects, these findings would support an axonal role for SMN independent of snRNP assembly. The authors have suggested that motor axon defects observed by Winkler et al. (2005) upon Gemin knockdown may be secondary to the overall morphological defects – including widespread defects in the nervous system – observed in these embryos.
5.4 Mouse Models of SMA
With the advent of gene targeting technology that is now routinely used to manipulate the genome, mouse models of human diseases have become the most widely used vertebrate animal models in biomedical research. Like all model organisms studied so far, mice have only a single copy of the SMN gene corresponding to human SMN1. As will be evident in the discussion below, mouse models of SMA have provided new insight into SMN function in synapse development. A list of commonly available SMA mouse models is shown in Table 2.
Table 2.
SMA mouse models available from The Jackson Laboratories
| Stock | Strain genotype | Average survival | References |
|---|---|---|---|
| 006214 | FVB.Cg-Smn1tm1Msd/J | 90–100 h | Schrank et al. (1997) |
| 005024 | FVB. Cg-Tg(SMN2)89Ahmb Smn1tm1Msd/J | 0–5 daysa | Monani et al. (2000b) |
| 006570 | STOCK Smn1tm1Msd Tg(Hlxb9-GFP) 1Tmj Tg(SMN2)89Ahmb/J | 0–5 daysa | McGovern et al. (2008) |
| 005025 | FVB.Cg-Tg(SMN2*delta7)4299Ahmb Tg(SMN2)89Ahmb Smn1tm1Msd/J | ca. 13 days | Le et al. (2005) |
| 005026 | FVB.Cg-Tg(SMN2)89Ahmb Tg(SMN1*A2G)2023Ahmb Smn1tm1Msd/J | <1 yeara | Monani et al. (2003) |
| 006146 | B6.129-Smn1tm1Jme/J | Normalb | Frugier et al. (2000) |
| 006138 | FVB.129(B6)-Smn1tm1Jme/J | Normalb | Frugier et al. (2000) |
| 005058 | FVB.Cg-Tg(SMN2)2Hung Smn1tm1Hung/J | <P10–normalc | Hsieh-Li et al. (2000) |
Difference between the survival times reported in the original publication and the survival of the strains available from The Jackson Laboratory may be due to inbreeding (http://jaxmice.jax.org/list/ra1733.html). It is the experience at The Jackson Laboratory that mice hemizygous for the SMN2 transgene do not survive
When crossed to a strain expressing Cre recombinase in a conditional manner, this mutant mouse strain will develop SMN-deficiency and associated defects in the affected tissue
Average time of survival depends on the transgene copy number
Mice that lack Smn due to homozygous insertion of a lacZ reporter gene after exon2a display massive cell death during early embryonic development and die during the morula stage prior to transition to the blastocyst stage and uterine implantation (Schrank et al. 1997). A follow-up study on Smn+/− heterozygous mice found a 46% reduction of Smn protein levels in the spinal cord and motor neuron degeneration resembling human type III SMA with normal lifespan and no reported overt phenotype (Jablonka et al. 2000). Between birth and six months of age, 40% of spinal cord motor neurons were lost relative to wild-type mice, but no further statistically significant loss occurred later. This order of pathophysiological events corresponds to an early phase of deterioration, followed by a phase of stabilization observed in SMA III patients (Crawford and Pardo 1996).
Two groups have chosen a very similar approach to model the SMA disease phenotype. These most commonly used SMA mouse models faithfully replicate the situation in human SMA patients by expressing human SMN2 transgenes in an Smn knockout background. The Burghes lab used the knockout mouse from the Sendtner lab (Schrank et al. 1997) and inserted a 35.5 kb fragment containing only the SMN2 transgene and its promoter region (Tg(SMN2)89Ahmb) (Monani et al. 2000b). The Li laboratory pursued a similar strategy by disrupting Smn exon 7 and using a larger 115 kb fragment of the human SMN2 region that also contained flanking genes (Tg(SMN2)2Hung) (Hsieh-Li et al. 2000). Smn heterozygous mice, containing the transgene, were bred to give progeny lacking endogenous Smn, but carrying the respective transgene. Aside from minor differences between these transgenic models, both had strong SMA-like characteristics. Furthermore, in both models, the human SMN2 transgene was able to complement the embryonic lethality of Smn−/− mice. The phenotype was modulated by the expression levels of SMN2 and even rescued by sufficiently high copy number of the inserted SMN2 transgene. The severely affected mice (Smn−/−, SMN2) have only one or two copies of the SMN2 transgene on an Smn null background and frequently died perinatally. Surviving mice initially appeared normal but became progressively smaller and weaker than their normal littermates and presented with decreased mobility and suckling, labored breathing, and finally, limb tremor followed by death within days after birth. These mice produce low levels of SMN protein and suffered a 35–40% loss of spinal cord and lower-brainstem motor neurons by day five, despite normal numbers at birth. Primary embryonic motor neurons cultured from these mice showed defective axon outgrowth, but normal survival (Rossoll et al. 2003). Isolated sensory neurons were less severely affected (Jablonka et al. 2006). This is in agreement with a study on SMA patients, who underwent sural nerve biopsy that showed significant sensory nerve pathology in severely affected SMA type I patients, while no sensory nerve alterations were detected in SMA type II or III patients (Rudnik-Schoneborn et al. 2003). Taken together, these results suggest that motor neuron loss is a late event, occurring subsequent to axonal denervation (discussed below), and the timing of motor neuron loss is critical for disease severity. In both mouse models, expression from several copies of the SMN2 transgene completely rescued the disease phenotype, with the exception of short, thick tails that became necrotic after about 2 weeks of age. The cause of this phenotype is currently unknown. A study with cultured primary motor neurons isolated from the Smn−/−, SMN2 mice found defective accumulation and clustering of Cav 2.2 Ca2+ channels in the axonal growth cones that was correlated with a reduction of local Ca2+ transients in distal axons and growth cones (Jablonka et al. 2007). The observed functional abnormalities of Ca2+ channels could contribute to maturation defects found in SMA.
Using the severe SMA models as a basis, several groups have inserted additional transgenes to modify the disease severity and answer specific questions about SMN function. Studies in Drosophila suggested that expression of SMN in both muscle and nervous tissue was required for complete rescue of an SMA phenotype (see above), implying that high levels of SMN expression in muscle may impact the SMA phenotype. To investigate whether this is also the case in severe mouse models of SMA, SMN was expressed in skeletal muscle fibers under the human skeletal actin (HSA) promoter and in neurons under the prion promoter (PrP) (Gavrilina et al. 2008). The results suggest that only neuronal SMN expression corrected the severe SMA phenotype in mice, whereas high SMN levels restricted to skeletal muscle fibers alone had no impact on the SMA phenotype or survival. It should be noted, however, that the transgene expressed from the PrP promoter showed “leaky” expression in skeletal muscle. Although it is possible that the timing and protein levels expressed from the HSA promoter was not sufficient to have an effect on the SMA genotype, an HSA-SMN strain that showed low levels of expression in the spinal cord and brain had significantly increased survival. Mice homozygous for PrP-SMN in an Smn−/−; SMN2 background survived for an average of 210 days, and motor neuron counts in these mice were normal. This demonstrates that increased levels of full-length SMN in neurons, but not muscle, were required to correct the SMA phenotype.
Expression of a mutant SMN transgene harboring the A2G SMA mutation in a severe SMA background (Smn−/−; SMN2) delayed the onset of motor neuron loss, resulting in mice with symptoms and neuropathology similar to patients afflicted with SMA type III. These mild SMA mice presented with motor neuron degeneration, muscle atrophy, and abnormal electromyographs, and lived to about 8 months of age (Monani et al. 2003). In the absence of the SMN2 gene, the mutant SMN A2G transgene did not rescue the embryonic lethality, suggesting that low levels of wild-type SMN are required for the formation of functional SMN complexes.
To investigate motor axon development and innervation in severe SMA mice, the HB9:GFP transgene was introduced into the Smn−/−; SMN2 +/+ strain by cross breeding to specifically label motor neurons with GFP (McGovern et al. 2008). Motor neurons innervating various muscles were examined from embryonic day 10.5 to postnatal day 2 for abnormalities in axon formation and outgrowth, but no defects were detected at any stage of development. However, a significant increase in unoccupied AChR clusters in intercostal muscles and the presence of axonal varicosities in motor axons in embryonic SMA mice indicated denervation during embryogenesis as one of the earliest detectable morphological defects in the SMA mice.
The question of whether SMNΔ7, the major splicing isoforms expressed from SMN2 locus lacking exon 7, has a beneficial or detrimental effect has been answered unequivocally by several independent studies. Transgenic mice expressing SMNΔ7 cDNA under the control of the SMN promoter were crossed onto a severe SMA background (Smn−/−; SMN2). Added expression of the SMNΔ7 transgene (Tg(SMN2*delta7) 4299Ahmb) turned out to be beneficial by extending mean survival of SMA mice homozygous for the targeted mutant Smn allele and homozygous for the two transgenic alleles from 5 to 13 days (Le et al. 2005). It is thought that expression of SMNΔ7 can rescue the SMA phenotype if a low level full-length SMN is present, probably by oligomerizing with full-length SMN and thus raising the effective concentration of SMN complexes (Le et al. 2005). In a detailed study of mice homozygous or hemizygous for the SMNΔ7 transgene, the initial effects of reduced SMN on the neuromuscular system were described as a NMJ synaptopathy (Kariya et al. 2008). The earliest structural defects appear distally prior to overt symptoms and involve the NMJ. Reduced levels of the SMN protein impair the normal maturation of acetylcholine receptor (AChR) clusters and lead to presynaptic defects, including poor terminal arborization and intermediate filament aggregation causing neurotransmission defects.
In a related study, the synaptic pathology at the neuromuscular junction in two different mouse models of SMA, the Smn−/−;SMN2 mouse model of type I SMA and the Smn−/−;SMN2;Δ7 mouse model, was analyzed in detail (Murray et al. 2008). NMJs in various muscles were disrupted in both mouse models, especially in postural muscles in the trunk. Loss of presynaptic inputs and nerve terminals with abnormal accumulations of neurofilament protein occurred even in early/mid-symptomatic animals in the most severely affected muscle groups. Skeletal muscles can be assigned to one of the two distinct classes of muscles, termed “fast synapsing” (FaSyn) and “delayed synapsing” (DeSyn) muscles, which differ in their pattern of neuromuscular synaptogenesis during embryonic development and maintenance of NMJs in adult mice (Pun et al. 2002). This study suggests that motor neurons with FaSyn-like, nonplastic characteristics may be more vulnerable to SMA-induced synapse loss than those with a DeSyn phenotype. Treatments that challenge synaptic stability result in nerve sprouting, NMJ remodeling, and ectopic synaptogenesis selectively on DeSyn muscles. Possibly, NMJs on FaSyn muscles lacking the synaptic plasticity are lost selectively and very early in SMA mice, suggesting that disease-associated motor neuron dysfunction may selectively fail to initiate maintenance processes at nonplastic FaSyn NMJs (Santos and Caroni 2003).
Electrophysiology and ultra-structure was the focus of a recent study on the function and structure of NMJ synapses in the Smn−/−;SMN2;Δ7 mouse model of severe SMA (Kong et al. 2009). While NMJ synapses remained well connected late in the disease course, decreased density of synaptic vesicles and reduced probability of vesicle release at SMA NMJs was found to be associated with impaired morphological and biochemical maturation of postsynaptic NMJ terminals and myofibers. These findings suggest that NMJ synaptic dysfunction precedes axonal degeneration in severe SMA mouse models. Similar defects were described in an earlier investigation of the NMJ ultrastructure in SMA type 1 that showed shallow subsynaptic clefts and axon terminals devoid of transmitter vesicles (Diers et al. 2005).
A different approach for the generation of SMA mouse models was chosen in the Melki lab (Frugier et al. 2000). Mice that harbor a conditional allele of Smn, in which LoxP sites flank Smn exon 7 (SmnF7), allow conditional excision of Smn exon 7 when crossed with mice expressing Cre recombinase in a tissue-specific manner. Mice carrying a homozygous deletion of Smn exon 7 directed to neurons display progressive loss of motor axons, defects in NMJs, and massive accumulation of neurofilaments in terminal axons of the remaining neuromuscular junctions (Cifuentes-Diaz et al. 2002). Selective deletion of exon7 in either skeletal muscle (Cifuentes-Diaz et al. 2001) or liver (Vitte et al. 2004) also leads to severe defects in these tissues, confirming that full length Smn is essential for all tissues and cell types. The relevance of these findings for the disease phenotype of SMA is currently not clear. The observed defects differ from those seen in human SMA patients or the other mouse models that both retain at least some expression of full length SMN. It is likely that selective reduction of SMN levels under a certain threshold level in any tissue will lead to defects due to the essential function of SMN. Future studies are needed to investigate to what extent SMN deficiency in other tissues contributes to the SMA disease phenotype.
SMA mouse models have been crossbred with other mutant mice to study the effect of neuroprotective and anti-apoptotic mutations on the SMA disease phenotype. Slow Wallerian degeneration (Wlds) mice express a fusion of nicotinamide mononucleotide adenlyl transferase 1 (Nmnat-1) and ubiquitination factor e4b (Ube4b) that protects axons from Wallerian degeneration in genetic mutants such as the pmn mouse (Ferri et al. 2003). Wlds in a SMA mouse model has no protective effect to alter the SMA phenotype, indicating that Wallerian degeneration does not directly contribute to the pathogenesis of SMA development (Rose et al. 2008; Kariya et al. 2009). Deletion of Bax, the major proapoptotic member of the Bcl-2 family, and overexpression of Bcl-xL, an anti-apoptotic of the Bcl-2 family, prevent neuronal cell death. Crossbreeding of Bax-deficient or Bcl-xL transgenic mice with SMA mice showed milder disease severity and longer life spans of the progeny as compared to their SMA littermates (Tsai et al. 2008). These results suggest that suppression of neuronal apoptosis has a potential to ameliorate the disease phenotype of SMA and may be a therapeutic target for future drug development.
Taken together, several recent studies on severe SMA mouse models emphasize early defects in NMJ synapses that include nerve terminal loss, neurofilament accumulation in presynaptic terminals, immature postsynaptic terminals, and functional abnormalities (Cifuentes-Diaz et al. 2002; Kariya et al. 2008; McGovern et al. 2008; Murray et al. 2008; Kong et al. 2009).
It will be important to compare the above phenotypes in animal models with observations from human samples. Likewise, we need to compare observations in human patients with the findings in model organisms. For example, in a recent study, SMA-I and SMA-II spinal cords showed a significant number of motor neurons that had migrated aberrantly toward the ventral spinal roots (Simic 2008; Simic et al. 2008). These heterotopic motor neurons (HMN) were located mostly in the ventral white matter and had no axon or dendrites. The authors propose that abnormal migration, differentiation, and lack of axonal outgrowth may induce cell death in displaced HMNs and contribute to the pathogenesis of SMA. It will be interesting to see whether similar phenotypes can be observed in animal models of SMA.
6 Therapeutic Strategies
SMA is the leading genetic killer of infants and toddlers with an incidence of 1/10,000 and a carrier frequency of one in 35 (Wirth et al. 2006). Currently, there is no cure for SMA or treatment to stop its progression. Treatment is symptomatic and supportive and includes treating pneumonia, curvature of the spine, and respiratory infections, if present (http://www.ninds.nih.gov/disorders/sma/sma.htm). The human toll of this disease, combined with its unique genetic profile, has focused attention from the NIH, industry, academia, and advocacy organizations on developing a treatment. SMA was selected in 2003 by the National Institute of Neurological Disorders and Stroke (NINDS) for a model translational research program aimed at accelerating drug discovery efforts against neurodegenerative diseases.
To uncover therapeutic options for SMA patients, research groups typically focus on one of the following strategies (Lunn and Stockwell 2005; Sumner 2006): (1) Elevation of endogenous full length SMN levels from the SMN2 gene by upregulating the SMN2 promoter, correcting alternative SMN2 pre-mRNA splicing, increasing read-through of SMN Δ 7 mRNA, stabilization of SMN protein, and modulating SMN2 translation; (2) Neuroprotection; (3) Delivery of SMN with viral vectors (gene therapy); and (4) Implantation of stem cell derived precursors into the spinal cord (stem cell therapy). Beyond the scope of this review, many drugs, including histone deacetylase inhibitors, such as valproic acid (VPA) or 4-phenyl-butyrate (PBA), have been shown to increase full length SMN2-derived RNA and protein levels in cell culture. None of these drugs is believed to act specifically on expression of the SMN gene, but is expected to change the expression level of hundreds of genes. Clinical trials are underway to investigate the effect of VPA, PBA, and other drugs on motor function in SMA patients (Lunn and Wang 2008; Oskoui and Kaufmann 2008). Also, beyond the scope of this review, many different approaches have been used successfully to specifically restore inclusion of exon7 in SMN2 mRNA in vitro by using small antisense RNA molecules that modulate exon7 inclusion by directly binding to SMN2 transcripts (Madocsai et al. 2005; Coady et al. 2007; Marquis et al. 2007; Singh 2007; Hua et al. 2008). While this approach holds a lot of promise, it remains to be seen whether such therapeutic oligonucleotides can be successfully delivered to spinal motor neurons.
6.1 Gene Therapy
Multiple single injections of a lentiviral vector construct that was pseudotyped with rabies virus glycoprotein in various muscles of SMA mice restored SMN expression to motor neurons, reduced motor neuron death, and increased the life expectancy by an average of 5 days, compared with untreated animals (Azzouz et al. 2004). Recently, adeno-associated virus (AAV) 9 injected intravenously has been shown to bypass the blood–brain barrier and efficiently target cells of the central nervous system (Foust et al. 2009). Intravenous injection of AAV9-GFP into neonatal mice through the facial vein resulted in extensive transduction of dorsal root ganglia and motor neurons throughout the spinal cord. These promising results suggest that lentiviral or AAV based vectors may enable the future development of gene therapies for SMA.
6.2 Stem Cells
Stem cells can be utilized as a biological tool to examine the abnormal cellular and molecular processes in these cells to gain a better understanding of the SMA pathophysiology. In a recent study, neurosphere-derived neural stem cells from the severe SMA mice (Smn−/−;SMN2) showed differences in proliferation and differentiation as compared to wild-type controls (Shafey et al. 2008). Creating a cell-culture model of human disease can also be a useful tool in developing screens for potential therapeutic agents. Human ES cell-derived neuroprogenitor cells have been generated as a new primary cell source for the purpose of developing assays for new SMA therapeutics (Wilson et al. 2007). Endogenous stem cells present in the mammalian nervous system may be manipulated to induce neural tissue repair. Alternatively, neuronal progenitor cells derived from stem cells that are committed to the motor neuron lineage can be transplanted into the injured nervous system as a therapeutic strategy. Transplanted cells may contribute to the repair of the damaged neuronal circuits, not only by directly replacing degenerated cells, but also by providing support in many other ways (Nayak et al. 2006).
In a study on paralyzed rats, the transplantation of ES cells that were treated to induce differentiation into motor neurons has shown promising results (Deshpande et al. 2006). Multipotent neural stem cells isolated from the spinal cord of GFP-expressing mice that were also primed for differentiation into motor neurons were transplanted intrathecally into SMA mice (Smn−/−; SMN2+/+; SMN Δ 7+/+) (Corti et al. 2008). Treated mice showed improved motor function and extended survival from 13 to 18 days. Transplanted cells even generated a small number of motor neurons that were properly localized to the ventral horn of the spinal cord, but secretion of trophic factors from the transplanted cells may also be responsible for the observed effect (Corti et al. 2008). Stem cell transplantation, combined with other molecular and pharmacologic approaches, may be a promising strategy in the development of SMA therapies.
An especially promising resource to develop an in vitro SMA disease model and develop novel therapies is based on two recent developments: (1) the generation of induced pluripotent stem (iPS) cells from patient dermal fibroblasts (reviewed by (Lowry and Plath 2008), and (2) the differentiation of stem cells into motor neurons (Wichterle et al. 2002). A combination of these approaches was used to generate ALS patient-specific motor neurons (Dimos et al. 2008). Recently, these two combined approaches were used in a study to generate iPS cells from skin fibroblast samples taken from a child with SMA (Ebert et al. 2008). These cells were differentiated into motor neurons that showed selective deficits compared to those derived from the child's unaffected mother. Initially, iPS-SMA cells produced similar numbers of motor neurons, but showed a decline at later time points. Diffuse synapsin staining suggested specific presynaptic maturation deficits in the iPS-SMA cultures. These results validate motor neurons derived from iPS cells as a promising new in vitro model for studying the SMA disease mechanism and drug discovery.
7 Open Questions and Perspectives
Despite recent progress in research of SMN function and the pathophysiology of SMA, many open questions remain that have important implications for SMA therapy, and are considered below.
Does a defect in the canonical housekeeping function of SMN in snRNP assembly lead to SMA?
While it is known that extremely low SMN levels affect splicing, it is not clear yet, whether a more moderate reduction, as seen in patients, cause splicing defects in motor neurons that may explain the disease phenotype. Identifying aberrantly spliced transcripts with arrays and other methods will be required to answer this question.
Does SMN play a role in the assembly of neuronal RNP complexes?
While there is evidence that SMN affects local β-actin mRNA levels in axons, it will be interesting to see whether other mRNAs are targets of SMN complexes. While SMN is transported in axons and co-localizes with interacting proteins, the composition of these putative RNA granules is not known. Proteomic studies of RNA granules to date have not identified SMN as a component. It will be important to further characterize a putative function of SMN in the assembly of axonal RNP complexes and to assess its role in the pathogenesis of SMA.
Are motor neurons the only cells affected by SMN deficiency?
The question whether SMA is caused by SMN deficiency in neurons, muscle fibers, or both appears still controversial. Most current studies suggest that low levels of SMN in motor neurons lead to SMA. It is important to clarify whether upregulation of SMN function in motor neurons is necessary and sufficient to prevent SMA. Presently, it cannot be excluded that other cells in the circuitry of the spinal cord or muscle tissue contribute to the pathophysiology of SMA. Indeed, more broadly restored SMN expression throughout the neuromuscular system, including neurons and muscle, may be necessary.
Which other genetic modifiers affect the disease phenotype of SMA?
A study in SMA discordant families with affected and asymptomatic siblings carrying the same SMN1 mutations has identified PLS3 as a modifier of SMA severity (Oprea et al. 2008). It would be interesting to see whether expression of a PLS3 transgene in spinal cord or other tissues can indeed rescue the SMA phenotype in mouse models of SMA. Comparisons across several animal models will help to further characterize these targets and assess epistatic relationships of modifiers, especially, to conduct genetic modifier screens in invertebrate animals, such as Drosophila (Chang et al. 2008) and C. elegans, which may lead to the identification of pathways that modulate the SMA phenotype and the discovery of novel therapeutic targets.
When do first neuromuscular defects develop, and can further progression be stopped?
Studies in several animal models of SMA point to early developmental defects of motor axons and NMJs and a dysfunction of motor neurons preceding cell death. The interaction of motor axons with muscle fibers in neuromuscular synapses is an important distinctive feature that distinguishes the motor neuron from all other neurons, which may underlie its specific vulnerability. It will be important to investigate how early developmental defects influence the establishment and maturation of muscle end-plate innervation patterns and may set the stage for subsequent denervation and eventual motor neuron cell death (Hausmanowa-Petrusewicz and Vrbova 2005). A question that is highly relevant for therapeutic intervention is the time window during which treatment, such as raising SMN levels, will benefit patients. The generation of conditional mouse models that allow for the induced expression of SMN at various time points (e.g., via tetracycline or tamoxifen) or conditional alleles that are engineered to revert to fully functional SMN via Cre-mediated recombination will be required to answer that question. Studies in several severe SMA models suggests that low SMN levels may lead to defects very early in development, at least in severe SMA type I, which has important implications for the development of a therapy for SMA. If an effective SMA drug treatment becomes available, intervention at a presymptomatic stage will require the establishment of neonatal screening to identify children at risk to develop SMA.
Results derived from addressing these questions will ultimately aid in the development of therapies for the treatment of SMA. Since axonal transport defects are common to many neurological disorders, including Alzheimer disease (AD), Huntington disease (HD), amyotrophic lateral sclerosis (ALS), and other forms of motor neurons disease, elucidating the cause of axonal defects in SMA may have widespread applicability to other types of neurological diseases (Duncan and Goldstein 2006).
Acknowledgment
The authors thank Claudia Fallini, Lei Xing, and Kristy Welshhans for helpful comments. Special thanks to Lei Xing for contributing to Figs. 2 and 3. The authors gratefully acknowledge funding from the Spinal Muscular Atrophy Foundation and NIH (HD055835) to GJB and Families of SMA and NIH (HD056130) to WR.
References
- Alias L, Bernal S, Fuentes-Prior P, Barcelo MJ, Also E, Martinez-Hernandez R, Rodriguez-Alvarez FJ, Martin Y, Aller E, Grau E, Pecina A, Antinolo G, Galan E, Rosa AL, Fernandez-Burriel M, Borrego S, Millan JM, Hernandez-Chico C, Baiget M, Tizzano EF. Mutation update of spinal muscular atrophy in Spain: molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum Genet. 2009;125(1):29–39. doi: 10.1007/s00439-008-0598-1. [DOI] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. RNA granules. J Cell Biol. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Azzouz M, Le T, Ralph GS, Walmsley L, Monani UR, Lee DC, Wilkes F, Mitrophanous KA, Kingsman SM, Burghes AH, Mazarakis ND. Lentivector-mediated SMN replacement in a mouse model of spinal muscular atrophy. J Clin Invest. 2004;114:1726–1731. doi: 10.1172/JCI22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccon J, Pellizzoni L, Rappsilber J, Mann M, Dreyfuss G. Identification and characterization of Gemin7, a novel component of the survival of motor neuron complex. J Biol Chem. 2002;277:31957–31962. doi: 10.1074/jbc.M203478200. [DOI] [PubMed] [Google Scholar]
- Bannai H, Fukatsu K, Mizutani A, Natsume T, Iemura S, Ikegami T, Inoue T, Mikoshiba K. An RNA-interacting protein, SYNCRIP (heterogeneous nuclear ribonuclear protein Q1/NSAP1) is a component of mRNA granule transported with inositol 1,4,5-trisphosphate receptor type 1 mRNA in neuronal dendrites. J Biol Chem. 2004;279:53427–53434. doi: 10.1074/jbc.M409732200. [DOI] [PubMed] [Google Scholar]
- Barth S, Liss M, Voss MD, Dobner T, Fischer U, Meister G, Grasser FA. Epstein-Barr virus nuclear antigen 2 binds via its methylated arginine-glycine repeat to the survival motor neuron protein. J Virol. 2003;77:5008–5013. doi: 10.1128/JVI.77.8.5008-5013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, Zhang H, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, Kosik KS. Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci. 1998;18:251–265. doi: 10.1523/JNEUROSCI.18-01-00251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia G, Princivalle A, Forti F, Lizier C, Zeviani M. Expression of the SMN gene, the spinal muscular atrophy determining gene, in the mammalian central nervous system. Hum Mol Genet. 1997;6:1961–1971. doi: 10.1093/hmg/6.11.1961. [DOI] [PubMed] [Google Scholar]
- Battle DJ, Kasim M, Yong J, Lotti F, Lau CK, Mouaikel J, Zhang Z, Han K, Wan L, Dreyfuss G. The SMN complex: an assembly machine for RNPs. Cold Spring Harb Symp Quant Biol. 2006a;71:313–320. doi: 10.1101/sqb.2006.71.001. [DOI] [PubMed] [Google Scholar]
- Battle DJ, Lau CK, Wan L, Deng H, Lotti F, Dreyfuss G. The Gemin5 protein of the SMN complex identifies snRNAs. Mol Cell. 2006b;23:273–279. doi: 10.1016/j.molcel.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Beattie CE, Carrel TL, McWhorter ML. Fishing for a mechanism: using zebrafish to understand spinal muscular atrophy. J Child Neurol. 2007;22:995–1003. doi: 10.1177/0883073807305671. [DOI] [PubMed] [Google Scholar]
- Bechade C, Rostaing P, Cisterni C, Kalisch R, La Bella V, Pettmann B, Triller A. Subcellular distribution of survival motor neuron (SMN) protein: possible involvement in nucleocytoplasmic and dendritic transport. Eur J Neurosci. 1999;11:293–304. doi: 10.1046/j.1460-9568.1999.00428.x. [DOI] [PubMed] [Google Scholar]
- Bertrandy S, Burlet P, Clermont O, Huber C, Fondrat C, Thierry-Mieg D, Munnich A, Lefebvre S. The RNA-binding properties of SMN: deletion analysis of the zebrafish orthologue defines domains conserved in evolution. Hum Mol Genet. 1999;8:775–782. doi: 10.1093/hmg/8.5.775. [DOI] [PubMed] [Google Scholar]
- Besse F, Ephrussi A. Translational control of localized mRNAs: restricting protein synthesis in space and time. Nat Rev Mol Cell Biol. 2008;9:971–980. doi: 10.1038/nrm2548. [DOI] [PubMed] [Google Scholar]
- Boisvert FM, Cote J, Boulanger MC, Cleroux P, Bachand F, Autexier C, Richard S. Symmetrical dimethylarginine methylation is required for the localization of SMN in Cajal bodies and pre-mRNA splicing. J Cell Biol. 2002;159:957–969. doi: 10.1083/jcb.200207028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman M, Shafey D, Kothary R. Smn Depletion Alters Profilin II Expression and Leads to Upregulation of the RhoA/ROCK Pathway and Defects in Neuronal Integrity. J Mol Neurosci. 2007;32:120–131. doi: 10.1007/s12031-007-0024-5. [DOI] [PubMed] [Google Scholar]
- Brahms H, Meheus L, de Brabandere V, Fischer U, Luhrmann R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B' and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA. 2001;7:1531–1542. doi: 10.1017/s135583820101442x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Wells DG. Dendritic mRNA: transport, translation and function. Nat Rev Neurosci. 2007;8:776–789. doi: 10.1038/nrn2150. [DOI] [PubMed] [Google Scholar]
- Briese M, Esmaeili B, Fraboulet S, Burt EC, Christodoulou S, Towers PR, Davies KE, Sattelle DB. Deletion of smn-1, the Caenorhabditis elegans ortholog of the spinal muscular atrophy gene, results in locomotor dysfunction and reduced lifespan. Hum Mol Genet. 2009;18:97–104. doi: 10.1093/hmg/ddn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes HM. Other forms of survival motor neuron protein and spinal muscular atrophy: an opinion. Neuromuscul Disord. 2008;18:82–83. doi: 10.1016/j.nmd.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Burnett BG, Munoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH. Regulation of SMN protein stability. Mol Cell Biol. 2008;29:1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt EC, Towers PR, Sattelle DB. Caenorhabditis elegans in the study of SMN-interacting proteins: a role for SMI-1, an orthologue of human Gemin2 and the identification of novel components of the SMN complex. Invert Neurosci. 2006;6:145–159. doi: 10.1007/s10158-006-0027-x. [DOI] [PubMed] [Google Scholar]
- Carissimi C, Saieva L, Baccon J, Chiarella P, Maiolica A, Sawyer A, Rappsilber J, Pellizzoni L. Gemin8 is a novel component of the survival motor neuron complex and functions in small nuclear ribonucleoprotein assembly. J Biol Chem. 2006;281:8126–8134. doi: 10.1074/jbc.M512243200. [DOI] [PubMed] [Google Scholar]
- Carnegie GK, Sleeman JE, Morrice N, Hastie CJ, Peggie MW, Philp A, Lamond AI, Cohen PT. Protein phosphatase 4 interacts with the Survival of Motor Neurons complex and enhances the temporal localisation of snRNPs. J Cell Sci. 2003;116:1905–1913. doi: 10.1242/jcs.00409. [DOI] [PubMed] [Google Scholar]
- Carrel TL, McWhorter ML, Workman E, Zhang H, Wolstencroft EC, Lorson C, Bassell GJ, Burghes AH, Beattie CE. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci. 2006;26:11014–11022. doi: 10.1523/JNEUROSCI.1637-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YB, Miguel-Aliaga I, Franks C, Thomas N, Trulzsch B, Sattelle DB, Davies KE, van den Heuvel M. Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet. 2003;12:1367–1376. doi: 10.1093/hmg/ddg157. [DOI] [PubMed] [Google Scholar]
- Chang HC, Dimlich DN, Yokokura T, Mukherjee A, Kankel MW, Sen A, Sridhar V, Fulga TA, Hart AC, Van Vactor D, Artavanis-Tsakonas S. Modeling spinal muscular atrophy in Drosophila. PLoS ONE. 2008;3:e3209. doi: 10.1371/journal.pone.0003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari A, Golas MM, Klingenhager M, Neuenkirchen N, Sander B, Englbrecht C, Sickmann A, Stark H, Fischer U. An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell. 2008;135:497–509. doi: 10.1016/j.cell.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Charroux B, Pellizzoni L, Perkinson RA, Shevchenko A, Mann M, Dreyfuss G. Gemin3: A novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of gems. J Cell Biol. 1999;147:1181–1194. doi: 10.1083/jcb.147.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charroux B, Pellizzoni L, Perkinson RA, Yong J, Shevchenko A, Mann M, Dreyfuss G. Gemin4. A novel component of the SMN complex that is found in both gems and nucleoli. J Cell Biol. 2000;148:1177–1186. doi: 10.1083/jcb.148.6.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Shyu AB. Rapid deadenylation triggered by a nonsense codon precedes decay of the RNA body in a mammalian cytoplasmic nonsense-mediated decay pathway. Mol Cell Biol. 2003;23:4805–4813. doi: 10.1128/MCB.23.14.4805-4813.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Park SM, Kim TD, Kim JH, Kim KT, Jang SK. BiP internal ribosomal entry site activity is controlled by heat-induced interaction of NSAP1. Mol Cell Biol. 2007;27:368–383. doi: 10.1128/MCB.00814-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Frugier T, Tiziano FD, Lacene E, Roblot N, Joshi V, Moreau MH, Melki J. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J Cell Biol. 2001;152:1107–1114. doi: 10.1083/jcb.152.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Nicole S, Velasco ME, Borra-Cebrian C, Panozzo C, Frugier T, Millet G, Roblot N, Joshi V, Melki J. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet. 2002;11:1439–1447. doi: 10.1093/hmg/11.12.1439. [DOI] [PubMed] [Google Scholar]
- Claus P, Doring F, Gringel S, Muller-Ostermeyer F, Fuhlrott J, Kraft T, Grothe C. Differential intranuclear localization of fibroblast growth factor-2 isoforms and specific interaction with the survival of motoneuron protein. J Biol Chem. 2003;278:479–485. doi: 10.1074/jbc.M206056200. [DOI] [PubMed] [Google Scholar]
- Coady TH, Shababi M, Tullis GE, Lorson CL. Restoration of SMN function: delivery of a trans-splicing RNA re-directs SMN2 pre-mRNA splicing. Mol Ther. 2007;15:1471–1478. doi: 10.1038/sj.mt.6300222. [DOI] [PubMed] [Google Scholar]
- Corti S, Nizzardo M, Nardini M, Donadoni C, Salani S, Ronchi D, Saladino F, Bordoni A, Fortunato F, Del Bo R, Papadimitriou D, Locatelli F, Menozzi G, Strazzer S, Bresolin N, Comi GP. Neural stem cell transplantation can ameliorate the phenotype of a mouse model of spinal muscular atrophy. J Clin Invest. 2008;118:3316–3330. doi: 10.1172/JCI35432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote J, Richard S. Tudor domains bind symmetrical dimethylated arginines. J Biol Chem. 2005;280:28476–28483. doi: 10.1074/jbc.M414328200. [DOI] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- Deshpande DM, Kim YS, Martinez T, Carmen J, Dike S, Shats I, Rubin LL, Drummond J, Krishnan C, Hoke A, Maragakis N, Shefner J, Rothstein JD, Kerr DA. Recovery from paralysis in adult rats using embryonic stem cells. Ann Neurol. 2006;60:32–44. doi: 10.1002/ana.20901. [DOI] [PubMed] [Google Scholar]
- Diers A, Kaczinski M, Grohmann K, Hubner C, Stoltenburg-Didinger G. The ultrastructure of peripheral nerve, motor end-plate and skeletal muscle in patients suffering from spinal muscular atrophy with respiratory distress type 1 (SMARD1) Acta Neuropathol. 2005;110:289–297. doi: 10.1007/s00401-005-1056-y. [DOI] [PubMed] [Google Scholar]
- Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Duncan JE, Goldstein LS. The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2006;2:e124. doi: 10.1371/journal.pgen.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert AD, Yu J, Rose FF, Jr, Mattis VB, Lorson CL, Thomson JA, Svendsen CN. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert C, Chari A, Laggerbauer B, Fischer U. Spinal muscular atrophy: the RNP connection. Trends Mol Med. 2006;12:113–121. doi: 10.1016/j.molmed.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Espert L, Eldin P, Gongora C, Bayard B, Harper F, Chelbi-Alix MK, Bertrand E, Degols G, Mechti N. The exonuclease ISG20 mainly localizes in the nucleolus and the Cajal (Coiled) bodies and is associated with nuclear SMN protein-containing complexes. J Cell Biochem. 2006;98:1320–1333. doi: 10.1002/jcb.20869. [DOI] [PubMed] [Google Scholar]
- Fan L, Simard LR. Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet. 2002;11:1605–1614. doi: 10.1093/hmg/11.14.1605. [DOI] [PubMed] [Google Scholar]
- Farina K, Oleynikov Y, Singer RH. Interaction of ZBP1 KH Domains with the b- actin zipcode and other RNA sequences. Molecular Biology Cell. 2001;12:1996. [Google Scholar]
- Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol. 2003;13:669–673. doi: 10.1016/s0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- Fischer U, Liu Q, Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90:1023–1029. doi: 10.1016/s0092-8674(00)80368-2. [DOI] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen WJ, Dreyfuss G. Specific sequences of the Sm and Sm-like (Lsm) proteins mediate their interaction with the spinal muscular atrophy disease gene product (SMN) J Biol Chem. 2000;275:26370–26375. doi: 10.1074/jbc.M003299200. [DOI] [PubMed] [Google Scholar]
- Friesen WJ, Massenet S, Paushkin S, Wyce A, Dreyfuss G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol Cell. 2001a;7:1111–1117. doi: 10.1016/s1097-2765(01)00244-1. [DOI] [PubMed] [Google Scholar]
- Friesen WJ, Paushkin S, Wyce A, Massenet S, Pesiridis GS, Van Duyne G, Rappsilber J, Mann M, Dreyfuss G. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol Cell Biol. 2001b;21:8289–8300. doi: 10.1128/MCB.21.24.8289-8300.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frugier T, Tiziano FD, Cifuentes-Diaz C, Miniou P, Roblot N, Dierich A, Le Meur M, Melki J. Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2000;9:849–858. doi: 10.1093/hmg/9.5.849. [DOI] [PubMed] [Google Scholar]
- Gabanella F, Butchbach ME, Saieva L, Carissimi C, Burghes AH, Pellizzoni L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE. 2007;2:e921. doi: 10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangwani L, Mikrut M, Theroux S, Sharma M, Davis RJ. Spinal muscular atrophy disrupts the interaction of ZPR1 with the SMN protein. Nat Cell Biol. 2001;3:376–383. doi: 10.1038/35070059. [DOI] [PubMed] [Google Scholar]
- Gavrilina TO, McGovern VL, Workman E, Crawford TO, Gogliotti RG, DiDonato CJ, Monani UR, Morris GE, Burghes AH. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum Mol Genet. 2008;17:1063–1075. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesemann T, Rathke-Hartlieb S, Rothkegel M, Bartsch JW, Buchmeier S, Jockusch BM, Jockusch H. A role for polyproline motifs in the spinal muscular atrophy protein SMN. Profilins bind to and colocalize with smn in nuclear gems. J Biol Chem. 1999a;274:37908–37914. doi: 10.1074/jbc.274.53.37908. [DOI] [PubMed] [Google Scholar]
- Giesemann T, Rathke-Hartlieb S, Rothkegel M, Bartsch JW, Buchmeier S, Jockusch BM, Jockusch H. A role for polyproline motifs in the spinal muscular atrophy protein SMN. Profilins bind to and colocalize with smn in nuclear gems. J Biol Chem. 1999b;274:37908–37914. doi: 10.1074/jbc.274.53.37908. [DOI] [PubMed] [Google Scholar]
- Girard LR, Fiedler TJ, Harris TW, Carvalho F, Antoshechkin I, Han M, Sternberg PW, Stein LD, Chalfie M. WormBook: the online review of Caenorhabditis elegans biology. Nucleic Acids Res. 2007;35:D472–D475. doi: 10.1093/nar/gkl894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SR, Kinzy TG. Translation elongation factor 1A is essential for regulation of the actin cytoskeleton and cell morphology. Nat Struct Mol Biol. 2005;12:772–778. doi: 10.1038/nsmb979. [DOI] [PubMed] [Google Scholar]
- Grosset C, Chen CY, Xu N, Sonenberg N, Jacquemin-Sablon H, Shyu AB. A mechanism for translationally coupled mRNA turnover: interaction between the poly(A) tail and a c-fos RNA coding determinant via a protein complex. Cell. 2000;103:29–40. doi: 10.1016/s0092-8674(00)00102-1. [DOI] [PubMed] [Google Scholar]
- Gu W, Pan F, Zhang H, Bassell GJ, Singer RH. A predominantly nuclear protein affecting cytoplasmic localization of beta-actin mRNA in fibroblasts and neurons. J Cell Biol. 2002;156:41–51. doi: 10.1083/jcb.200105133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubitz AK, Mourelatos Z, Abel L, Rappsilber J, Mann M, Dreyfuss G. Gemin5, a novel WD repeat protein component of the SMN complex that binds Sm proteins. J Biol Chem. 2002;277:5631–5636. doi: 10.1074/jbc.M109448200. [DOI] [PubMed] [Google Scholar]
- Gubitz AK, Feng W, Dreyfuss G. The SMN complex. Exp Cell Res. 2004;296:51–56. doi: 10.1016/j.yexcr.2004.03.022. [DOI] [PubMed] [Google Scholar]
- Harris CE, Boden RA, Astell CR. A novel heterogeneous nuclear ribonucleoprotein-like protein interacts with NS1 of the minute virus of mice. J Virol. 1999;73:72–80. doi: 10.1128/jvi.73.1.72-80.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmanowa-Petrusewicz I, Vrbova G. Spinal muscular atrophy: a delayed development hypothesis. Neuroreport. 2005;16:657–661. doi: 10.1097/00001756-200505120-00001. [DOI] [PubMed] [Google Scholar]
- Hebert MD, Szymczyk PW, Shpargel KB, Matera AG. Coilin forms the bridge between Cajal bodies and SMN, the spinal muscular atrophy protein. Genes Dev. 2001;15:2720–2729. doi: 10.1101/gad.908401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert MD, Shpargel KB, Ospina JK, Tucker KE, Matera AG. Coilin methylation regulates nuclear body formation. Dev Cell. 2002;3:329–337. doi: 10.1016/s1534-5807(02)00222-8. [DOI] [PubMed] [Google Scholar]
- Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Hua Y, Zhou J. Rpp20 interacts with SMN and is re-distributed into SMN granules in response to stress. Biochem Biophys Res Commun. 2004a;314:268–276. doi: 10.1016/j.bbrc.2003.12.084. [DOI] [PubMed] [Google Scholar]
- Hua Y, Zhou J. Survival motor neuron protein facilitates assembly of stress granules. FEBS Lett. 2004b;572:69–74. doi: 10.1016/j.febslet.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttelmaier S, Zenklusen D, Lederer M, Dictenberg J, Lorenz M, Meng X, Bassell GJ, Condeelis J, Singer RH. Spatial regulation of β-actin translation by Src- dependent phosphorylation of ZBP1. Nature. 2005;438:512–515. doi: 10.1038/nature04115. [DOI] [PubMed] [Google Scholar]
- Irwin N, Baekelandt V, Goritchenko L, Benowitz LI. Identification of two proteins that bind to a pyrimidine-rich sequence in the 3′-untranslated region of GAP-43 mRNA. Nucleic Acids Res. 1997;25:1281–1288. doi: 10.1093/nar/25.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahashi H, Eguchi Y, Yasuhara N, Hanafusa T, Matsuzawa Y, Tsujimoto Y. Synergistic anti-apoptotic activity between Bcl-2 and SMN implicated in spinal muscular atrophy. Nature. 1997;390:413–417. doi: 10.1038/37144. [DOI] [PubMed] [Google Scholar]
- Jablonka S, Rossoll W, Schrank B, Sendtner M. The role of SMN in spinal muscular atrophy. J Neurol. 2000;247:I37–42. doi: 10.1007/s004150050555. [DOI] [PubMed] [Google Scholar]
- Jablonka S, Bandilla M, Wiese S, Buhler D, Wirth B, Sendtner M, Fischer U. Co-regulation of survival of motor neuron (SMN) protein and its interactor SIP1 during development and in spinal muscular atrophy. Hum Mol Genet. 2001;10:497–505. doi: 10.1093/hmg/10.5.497. [DOI] [PubMed] [Google Scholar]
- Jablonka S, Karle K, Sandner B, Andreassi C, von Au K, Sendtner M. Distinct and overlapping alterations in motor and sensory neurons in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2006;15:511–518. doi: 10.1093/hmg/ddi467. [DOI] [PubMed] [Google Scholar]
- Jablonka S, Beck M, Lechner BD, Mayer C, Sendtner M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol. 2007;179:139–149. doi: 10.1083/jcb.200703187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KW, Gorzynski K, Hales CM, Fischer U, Badbanchi F, Terns RM, Terns MP. Direct interaction of the spinal muscular atrophy disease protein SMN with the small nucleolar RNA-associated protein fibrillarin. J Biol Chem. 2001;276:38645–38651. doi: 10.1074/jbc.M106161200. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43:513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya S, Mauricio R, Dai Y, Monani UR. The neuroprotective factor Wld(s) fails to mitigate distal axonal and neuromuscular junction (NMJ) defects in mouse models of spinal muscular atrophy. Neurosci Lett. 2009;449:246–251. doi: 10.1016/j.neulet.2008.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fitzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol. 2005;169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebler MA, Bassell GJ. Neuronal RNA granules: movers and makers. Neuron. 2006;51:685–690. doi: 10.1016/j.neuron.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Kim JH, Paek KY, Ha SH, Cho S, Choi K, Kim CS, Ryu SH, Jang SK. A cellular RNA-binding protein enhances internal ribosomal entry site-dependent translation through an interaction downstream of the hepatitis C virus polyprotein initiation codon. Mol Cell Biol. 2004;24:7878–7890. doi: 10.1128/MCB.24.18.7878-7890.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TD, Kim JS, Kim JH, Myung J, Chae HD, Woo KC, Jang SK, Koh DS, Kim KT. Rhythmic serotonin N-acetyltransferase mRNA degradation is essential for the maintenance of its circadian oscillation. Mol Cell Biol. 2005;25:3232–3246. doi: 10.1128/MCB.25.8.3232-3246.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TD, Woo KC, Cho S, Ha DC, Jang SK, Kim KT. Rhythmic control of AANAT translation by hnRNP Q in circadian melatonin production. Genes Dev. 2007;21:797–810. doi: 10.1101/gad.1519507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler S, Wang H, Richter D, Tiedge H. RNA transport and local control of translation. Annu Rev Cell Dev Biol. 2005;21:223–245. doi: 10.1146/annurev.cellbio.21.122303.120653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb SJ, Battle DJ, Dreyfuss G. Molecular functions of the SMN complex. J Child Neurol. 2007;22:990–994. doi: 10.1177/0883073807305666. [DOI] [PubMed] [Google Scholar]
- Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch-Marce M, Griffin JW, Rich MM, Sumner CJ. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroiss M, Schultz J, Wiesner J, Chari A, Sickmann A, Fischer U. Evolution of an RNP assembly system: a minimal SMN complex facilitates formation of UsnRNPs in Drosophila melanogaster. Proc Natl Acad Sci USA. 2008;105:10045–10050. doi: 10.1073/pnas.0802287105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara N, Menaa C, Maeda H, Haile DJ, Reddy SV. Osteoclast-stimulating factor interacts with the spinal muscular atrophy gene product to stimulate osteoclast formation. J Biol Chem. 2001;276:41035–41039. doi: 10.1074/jbc.M100233200. [DOI] [PubMed] [Google Scholar]
- Le TT, Coovert DD, Monani UR, Morris GE, Burghes AH. The survival motor neuron (SMN) protein: effect of exon loss and mutation on protein localization. Neurogenetics. 2000;3:7–16. doi: 10.1007/s100480000090. [DOI] [PubMed] [Google Scholar]
- Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burlet P, Viollet L, Bertrandy S, Huber C, Belser C, Munnich A. A novel association of the SMN protein with two major non-ribosomal nucleolar proteins and its implication in spinal muscular atrophy. Hum Mol Genet. 2002;11:1017–1027. doi: 10.1093/hmg/11.9.1017. [DOI] [PubMed] [Google Scholar]
- Leung KM, van Horck FP, Lin AC, Allison R, Standart N, Holt CE. Asymmetrical β-actin mRNA translation in growth cones mediates attractive turning to netrin-1. Nat Neurosci. 2006;9:1247–1256. doi: 10.1038/nn1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AC, Holt CE. Function and regulation of local axonal translation. Curr Opin Neurobiol. 2008;18:60–68. doi: 10.1016/j.conb.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996;15:3555–3565. [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Fischer U, Wang F, Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90:1013–1021. doi: 10.1016/s0092-8674(00)80367-0. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Androphy EJ. The domain encoded by exon 2 of the survival motor neuron protein mediates nucleic acid binding. Hum Mol Genet. 1998;7:1269–1275. doi: 10.1093/hmg/7.8.1269. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Strasswimmer J, Yao JM, Baleja JD, Hahnen E, Wirth B, Le T, Burghes AH, Androphy EJ. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- Lowry WE, Plath K. The many ways to make an iPS cell. Nat Biotechnol. 2008;26:1246–1248. doi: 10.1038/nbt1108-1246. [DOI] [PubMed] [Google Scholar]
- Lu B, Vogel H. Drosophila Models of Neurodegenerative Diseases. Annu Rev Pathol. 2008 doi: 10.1146/annurev.pathol.3.121806.151529. doi:10.1146/annurev.pathol.3.121806.151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn MR, Stockwell BR. Chemical genetics and orphan genetic diseases. Chem Biol. 2005;12:1063–1073. doi: 10.1016/j.chembiol.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- Madocsai C, Lim SR, Geib T, Lam BJ, Hertel KJ. Correction of SMN2 Pre- mRNA splicing by antisense U7 small nuclear RNAs. Mol Ther. 2005;12:1013–1022. doi: 10.1016/j.ymthe.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Marquis J, Meyer K, Angehrn L, Kampfer SS, Rothen-Rutishauser B, Schumperli D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol Ther. 2007;15:1479–1486. doi: 10.1038/sj.mt.6300200. [DOI] [PubMed] [Google Scholar]
- McGovern VL, Gavrilina TO, Beattie CE, Burghes AH. Embryonic motor axon development in the severe SMA mouse. Hum Mol Genet. 2008;17:2900–2909. doi: 10.1093/hmg/ddn189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhorter ML, Monani UR, Burghes AH, Beattie CE. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003;162:919–931. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhorter ML, Boon KL, Horan ES, Burghes AH, Beattie CE. The SMN binding protein Gemin2 is not involved in motor axon outgrowth. Dev Neurobiol. 2008;68:182–194. doi: 10.1002/dneu.20582. [DOI] [PubMed] [Google Scholar]
- Meister G, Buhler D, Laggerbauer B, Zobawa M, Lottspeich F, Fischer U. Characterization of a nuclear 20S complex containing the survival of motor neurons (SMN) protein and a specific subset of spliceosomal Sm proteins. Hum Mol Genet. 2000;9:1977–1986. doi: 10.1093/hmg/9.13.1977. [DOI] [PubMed] [Google Scholar]
- Meister G, Buhler D, Pillai R, Lottspeich F, Fischer U. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat Cell Biol. 2001;3:945–949. doi: 10.1038/ncb1101-945. [DOI] [PubMed] [Google Scholar]
- Meister G, Eggert C, Fischer U. SMN-mediated assembly of RNPs: a complex story. Trends Cell Biol. 2002;12:472–478. doi: 10.1016/s0962-8924(02)02371-1. [DOI] [PubMed] [Google Scholar]
- Meister G, Fischer U. Assisted RNP assembly: SMN and PRMT5 complexes cooperate in the formation of spliceosomal UsnRNPs. EMBO J. 2002;21:5853–5863. doi: 10.1093/emboj/cdf585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki J, Lefebvre S, Burglen L, Burlet P, Clermont O, Millasseau P, Reboullet S, Benichou B, Zeviani M, Le Paslier D, et al. De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science. 1994;264:1474–1477. doi: 10.1126/science.7910982. [DOI] [PubMed] [Google Scholar]
- Miguel-Aliaga I, Culetto E, Walker DS, Baylis HA, Sattelle DB, Davies KE. The Caenorhabditis elegans orthologue of the human gene responsible for spinal muscular atrophy is a maternal product critical for germline maturation and embryonic viability. Hum Mol Genet. 1999;8:2133–2143. doi: 10.1093/hmg/8.12.2133. [DOI] [PubMed] [Google Scholar]
- Miguel-Aliaga I, Chan YB, Davies KE, van den Heuvel M. Disruption of SMN function by ectopic expression of the human SMN gene in Drosophila. FEBS Lett. 2000;486:99–102. doi: 10.1016/s0014-5793(00)02243-2. [DOI] [PubMed] [Google Scholar]
- Min H, Turck CW, Nikolic JM, Black DL. A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev. 1997;11:1023–1036. doi: 10.1101/gad.11.8.1023. [DOI] [PubMed] [Google Scholar]
- Mishra AK, Gangwani L, Davis RJ, Lambright DG. Structural insights into the interaction of the evolutionarily conserved ZPR1 domain tandem with eukaryotic EF1A, receptors, and SMN complexes. Proc Natl Acad Sci USA. 2007;104:13930–13935. doi: 10.1073/pnas.0704915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani A, Fukuda M, Ibata K, Shiraishi Y, Mikoshiba K. SYNCRIP, a cytoplasmic counterpart of heterogeneous nuclear ribonucleoprotein R, interacts with ubiquitous synaptotagmin isoforms. J Biol Chem. 2000;275:9823–9831. doi: 10.1074/jbc.275.13.9823. [DOI] [PubMed] [Google Scholar]
- Monani UR, Coovert DD, Burghes AH. Animal models of spinal muscular atrophy. Hum Mol Genet. 2000a;9:2451–2457. doi: 10.1093/hmg/9.16.2451. [DOI] [PubMed] [Google Scholar]
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossol W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000b;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- Monani UR, Pastore MT, Gavrilina TO, Jablonka S, Le TT, Andreassi C, DiCocco JM, Lorson C, Androphy EJ, Sendtner M, Podell M, Burghes AH. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouaikel J, Narayanan U, Verheggen C, Matera AG, Bertrand E, Tazi J, Bordonne R. Interaction between the small-nuclear-RNA cap hypermethylase and the spinal muscular atrophy protein, survival of motor neuron. EMBO Rep. 2003;4:616–622. doi: 10.1038/sj.embor.embor863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Abel L, Yong J, Kataoka N, Dreyfuss G. SMN interacts with a novel family of hnRNP and spliceosomal proteins. EMBO J. 2001;20:5443–5452. doi: 10.1093/emboj/20.19.5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- Narayanan U, Ospina JK, Frey MR, Hebert MD, Matera AG. SMN, the spinal muscular atrophy protein, forms a pre-import snRNP complex with snurportin1 and importin beta. Hum Mol Genet. 2002;11:1785–1795. doi: 10.1093/hmg/11.15.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak MS, Kim YS, Goldman M, Keirstead HS, Kerr DA. Cellular therapies in motor neuron diseases. Biochim Biophys Acta. 2006;1762:1128–1138. doi: 10.1016/j.bbadis.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Neubauer G, King A, Rappsilber J, Calvio C, Watson M, Ajuh P, Sleeman J, Lamond A, Mann M. Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat Genet. 1998;20:46–50. doi: 10.1038/1700. [DOI] [PubMed] [Google Scholar]
- Nicole S, Diaz CC, Frugier T, Melki J. Spinal muscular atrophy: Recent advances and future prospects. Muscle Nerve. 2002;26:4–13. doi: 10.1002/mus.10110. [DOI] [PubMed] [Google Scholar]
- Oleynikov Y, Singer RH. Real-time visualization of ZBP1 association with β-actin mRNA during transcription and localization. Curr Biol. 2003;13:199–207. doi: 10.1016/s0960-9822(03)00044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea GE, Krober S, McWhorter ML, Rossoll W, Muller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320:524–527. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskoui M, Levy G, Garland CJ, Gray JM, O'Hagen J, De Vivo DC, Kaufmann P. The changing natural history of spinal muscular atrophy type 1. Neurology. 2007;69:1931–1936. doi: 10.1212/01.wnl.0000290830.40544.b9. [DOI] [PubMed] [Google Scholar]
- Oskoui M, Kaufmann P. Spinal muscular atrophy. Neurotherapeutics. 2008;5:499–506. doi: 10.1016/j.nurt.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otter S, Grimmler M, Neuenkirchen N, Chari A, Sickmann A, Fischer U. A comprehensive interaction map of the human survival of motor neuron (SMN) complex. J Biol Chem. 2007;282:5825–5833. doi: 10.1074/jbc.M608528200. [DOI] [PubMed] [Google Scholar]
- Pagliardini S, Giavazzi A, Setola V, Lizier C, Di Luca M, DeBiasi S, Battaglia G. Subcellular localization and axonal transport of the survival motor neuron (SMN) protein in the developing rat spinal cord. Hum Mol Genet. 2000;9:47–56. doi: 10.1093/hmg/9.1.47. [DOI] [PubMed] [Google Scholar]
- Pan F, Huttelmaier S, Singer RH, Gu W. ZBP2 facilitates binding of ZBP1 to β-actin mRNA during transcription. Mol Cell Biol. 2007;27:8340–8351. doi: 10.1128/MCB.00972-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Voss PG, Grabski S, Wang JL, Patterson RJ. Association of galectin-1 and galectin-3 with Gemin4 in complexes containing the SMN protein. Nucleic Acids Res. 2001;29:3595–3602. doi: 10.1093/nar/29.17.3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushkin S, Gubitz AK, Massenet S, Dreyfuss G. The SMN complex, an assemblyosome of ribonucleoproteins. Curr Opin Cell Biol. 2002;14:305–312. doi: 10.1016/s0955-0674(02)00332-0. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L, Charroux B, Dreyfuss G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc Natl Acad Sci USA. 1999;96:11167–11172. doi: 10.1073/pnas.96.20.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni L, Baccon J, Charroux B, Dreyfuss G. The survival of motor neurons (SMN) protein interacts with the snoRNP proteins fibrillarin and GAR1. Curr Biol. 2001a;11:1079–1088. doi: 10.1016/s0960-9822(01)00316-5. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L, Charroux B, Rappsilber J, Mann M, Dreyfuss G. A functional interaction between the survival motor neuron complex and RNA polymerase II. J Cell Biol. 2001b;152:75–85. doi: 10.1083/jcb.152.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni L, Baccon J, Rappsilber J, Mann M, Dreyfuss G. Purification of native survival of motor neurons complexes and identification of Gemin6 as a novel component. J Biol Chem. 2002a;277:7540–7545. doi: 10.1074/jbc.M110141200. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002b;298:1775–1779. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- Peri S, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–2371. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazzon N, Rage F, Schlotter F, Moine H, Branlant C, Massenet S. In vitro and in cellulo evidences for association of the survival of motor neuron complex with the fragile X mental retardation protein. J Biol Chem. 2008;283:5598–5610. doi: 10.1074/jbc.M707304200. [DOI] [PubMed] [Google Scholar]
- Ponting CP. Tudor domains in proteins that interact with RNA. Trends Biochem Sci. 1997;22:51–52. doi: 10.1016/s0968-0004(96)30049-2. [DOI] [PubMed] [Google Scholar]
- Pun S, Sigrist M, Santos AF, Ruegg MA, Sanes JR, Jessell TM, Arber S, Caroni P. An intrinsic distinction in neuromuscular junction assembly and maintenance in different skeletal muscles. Neuron. 2002;34:357–370. doi: 10.1016/s0896-6273(02)00670-0. [DOI] [PubMed] [Google Scholar]
- Rajendra TK, Gonsalvez GB, Walker MP, Shpargel KB, Salz HK, Matera AG. A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J Cell Biol. 2007;176:831–841. doi: 10.1083/jcb.200610053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehbein M, Wege K, Buck F, Schweizer M, Richter D, Kindler S. Molecular characterization of MARTA1, a protein interacting with the dendritic targeting element of MAP2 mRNAs. J Neurochem. 2002;82:1039–1046. doi: 10.1046/j.1471-4159.2002.01058.x. [DOI] [PubMed] [Google Scholar]
- Rochette CF, Gilbert N, Simard LR. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum Genet. 2001;108:255–266. doi: 10.1007/s004390100473. [DOI] [PubMed] [Google Scholar]
- Rose FF, Jr, Meehan PW, Coady TH, Garcia VB, Garcia ML, Lorson CL. The Wallerian degeneration slow (Wld(s)) gene does not attenuate disease in a mouse model of spinal muscular atrophy. Biochem Biophys Res Commun. 2008;375:119–123. doi: 10.1016/j.bbrc.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AF, Oleynikov Y, Kislauskis EH, Taneja KL, Singer RH. Characterization of a β-actin mRNA zipcode-binding protein. Mol Cell Biol. 1997;17:2158–2165. doi: 10.1128/mcb.17.4.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossoll W, Kroning AK, Ohndorf UM, Steegborn C, Jablonka S, Sendtner M. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum Mol Genet. 2002;11:93–105. doi: 10.1093/hmg/11.1.93. [DOI] [PubMed] [Google Scholar]
- Rossoll W, Jablonka S, Andreassi C, Kroning AK, Karle K, Monani UR, Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Rudnik-Schoneborn S, Goebel HH, Schlote W, Molaian S, Omran H, Ketelsen U, Korinthenberg R, Wenzel D, Lauffer H, Kreiss-Nachtsheim M, Wirth B, Zerres K. Classical infantile spinal muscular atrophy with SMN deficiency causes sensory neuronopathy. Neurology. 2003;60:983–987. doi: 10.1212/01.wnl.0000052788.39340.45. [DOI] [PubMed] [Google Scholar]
- Santos AF, Caroni P. Assembly, plasticity and selective vulnerability to disease of mouse neuromuscular junctions. J Neurocytol. 2003;32:849–862. doi: 10.1023/B:NEUR.0000020628.36013.88. [DOI] [PubMed] [Google Scholar]
- Saunders LR, Perkins DJ, Balachandran S, Michaels R, Ford R, Mayeda A, Barber GN. Characterization of two evolutionarily conserved, alternatively spliced nuclear phosphoproteins, NFAR-1 and -2, that function in mRNA processing and interact with the double-stranded RNA-dependent protein kinase, PKR. J Biol Chem. 2001;276:32300–32312. doi: 10.1074/jbc.M104207200. [DOI] [PubMed] [Google Scholar]
- Schmid A, DiDonato CJ. Animal models of spinal muscular atrophy. J Child Neurol. 2007;22:1004–1012. doi: 10.1177/0883073807305667. [DOI] [PubMed] [Google Scholar]
- Schrank B, Gotz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenko P, Sprangers R, Stier G, Buhler D, Fischer U, Sattler M. SMN tudor domain structure and its interaction with the Sm proteins. Nat Struct Biol. 2001;8:27–31. doi: 10.1038/83014. [DOI] [PubMed] [Google Scholar]
- Setola V, Terao M, Locatelli D, Bassanini S, Garattini E, Battaglia G. Axonal- SMN (a-SMN), a protein isoform of the survival motor neuron gene, is specifically involved in axonogenesis. Proc Natl Acad Sci USA. 2007;104:1959–1964. doi: 10.1073/pnas.0610660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafey D, MacKenzie AE, Kothary R. Neurodevelopmental abnormalities in neurosphere-derived neural stem cells from SMN-depleted mice. J Neurosci Res. 2008;86:2839–2847. doi: 10.1002/jnr.21743. [DOI] [PubMed] [Google Scholar]
- Sharma A, Lambrechts A, Hao le T, Le TT, Sewry CA, Ampe C, Burghes AH, Morris GE. A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp Cell Res. 2005;309:185–197. doi: 10.1016/j.yexcr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Shupliakov O, Bloom O, Gustafsson JS, Kjaerulff O, Low P, Tomilin N, Pieribone VA, Greengard P, Brodin L. Impaired recycling of synaptic vesicles after acute perturbation of the presynaptic actin cytoskeleton. Proc Natl Acad Sci USA. 2002;99:14476–14481. doi: 10.1073/pnas.212381799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simic G. Pathogenesis of proximal autosomal recessive spinal muscular atrophy. Acta Neuropathol. 2008;116:223–234. doi: 10.1007/s00401-008-0411-1. [DOI] [PubMed] [Google Scholar]
- Simic G, Mladinov M, Seso Simic D, Jovanov Milosevic N, Islam A, Pajtak A, Barisic N, Sertic J, Lucassen PJ, Hof PR, Kruslin B. Abnormal motoneuron migration, differentiation, and axon outgrowth in spinal muscular atrophy. Acta Neuropathol. 2008;115:313–326. doi: 10.1007/s00401-007-0327-1. [DOI] [PubMed] [Google Scholar]
- Singh RN. Evolving concepts on human SMN pre-mRNA splicing. RNA Biol. 2007;4:7–10. doi: 10.4161/rna.4.1.4535. [DOI] [PubMed] [Google Scholar]
- Smith CL, Afroz R, Bassell GJ, Furneaux HM, Perrone-Bizzozero NI, Burry RW. GAP-43 mRNA in growth cones is associated with HuD and ribosomes. J Neurobiol. 2004;61:222–235. doi: 10.1002/neu.20038. [DOI] [PubMed] [Google Scholar]
- Sprangers R, Groves MR, Sinning I, Sattler M. High-resolution X-ray and NMR structures of the SMN Tudor domain: conformational variation in the binding site for symmetrically dimethylated arginine residues. J Mol Biol. 2003;327:507–520. doi: 10.1016/s0022-2836(03)00148-7. [DOI] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U, et al. A human protein–protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Stohr N, Lederer M, Reinke C, Meyer S, Hatzfeld M, Singer RH, Huttelmaier S. ZBP1 regulates mRNA stability during cellular stress. J Cell Biol. 2006;175:527–534. doi: 10.1083/jcb.200608071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasswimmer J, Lorson CL, Breiding DE, Chen JJ, Le T, Burghes AH, Androphy EJ. Identification of survival motor neuron as a transcriptional activator-binding protein. Hum Mol Genet. 1999;8:1219–1226. doi: 10.1093/hmg/8.7.1219. [DOI] [PubMed] [Google Scholar]
- Sumner CJ. Therapeutics development for spinal muscular atrophy. NeuroRx. 2006;3:235–245. doi: 10.1016/j.nurx.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadesse H, Deschenes-Furry J, Boisvenue S, Cote J. KH-type splicing regulatory protein interacts with survival motor neuron protein and is misregulated in spinal muscular atrophy. Hum Mol Genet. 2008;17:506–524. doi: 10.1093/hmg/ddm327. [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Boulon S, Lam YW, Urcia R, Boisvert FM, Vandermoere F, Morrice NA, Swift S, Rothbauer U, Leonhardt H, Lamond A. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J Cell Biol. 2008;183:223–239. doi: 10.1083/jcb.200805092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LK, Tsai MS, Ting CH, Wang SH, Li H. Restoring Bcl-x(L) levels benefits a mouse model of spinal muscular atrophy. Neurobiol Dis. 2008;31:361–367. doi: 10.1016/j.nbd.2008.05.014. [DOI] [PubMed] [Google Scholar]
- Vessey JP, Vaccani A, Xie Y, Dahm R, Karra D, Kiebler MA, Macchi P. Dendritic localization of the translational repressor Pumilio 2 and its contribution to dendritic stress granules. J Neurosci. 2006;26:6496–6508. doi: 10.1523/JNEUROSCI.0649-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitte JM, Davoult B, Roblot N, Mayer M, Joshi V, Courageot S, Tronche F, Vadrot J, Moreau MH, Kemeny F, Melki J. Deletion of murine Smn exon 7 directed to liver leads to severe defect of liver development associated with iron overload. Am J Pathol. 2004;165:1731–1741. doi: 10.1016/S0002-9440(10)63428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Reddy NM, Shen CK. Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci USA. 2002;99:13583–13588. doi: 10.1073/pnas.212483099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Williams BY, Hamilton SL, Sarkar HK. The SMN protein interacts with the tranactivator FUSE Binding protein from human fetal brain. FEBBS. 2000a;470:207–210. doi: 10.1016/s0014-5793(00)01320-x. [DOI] [PubMed] [Google Scholar]
- Williams BY, Hamilton SL, Sarkar HK. The survival motor neuron protein interacts with the transactivator FUSE binding protein from human fetal brain. FEBS Lett. 2000b;470:207–210. doi: 10.1016/s0014-5793(00)01320-x. [DOI] [PubMed] [Google Scholar]
- Wilson PG, Cherry JJ, Schwamberger S, Adams AM, Zhou J, Shin S, Stice SL. An SMA project report: neural cell-based assays derived from human embryonic stem cells. Stem Cells Dev. 2007;16:1027–1041. doi: 10.1089/scd.2007.0061. [DOI] [PubMed] [Google Scholar]
- Winkler C, Eggert C, Gradl D, Meister G, Giegerich M, Wedlich D, Laggerbauer B, Fischer U. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev. 2005;19:2320–2330. doi: 10.1101/gad.342005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum Mutat. 2000;15:228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Wirth B, Brichta L, Hahnen E. Spinal muscular atrophy: from gene to therapy. Semin Pediatr Neurol. 2006;13:121–131. doi: 10.1016/j.spen.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Yang WH, Yu JH, Gulick T, Bloch KD, Bloch DB. RNA-associated protein 55 (RAP55) localizes to mRNA processing bodies and stress granules. RNA. 2006;12:547–554. doi: 10.1261/rna.2302706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Sasaki Y, Wen Z, Bassell GJ, Zheng JQ. An essential role for beta-actin mRNA localization and translation in Ca2+-dependent growth cone guidance. Nat Neurosci. 2006;9:1265–1273. doi: 10.1038/nn1773. [DOI] [PubMed] [Google Scholar]
- Yong J, Wan L, Dreyfuss G. Why do cells need an assembly machine for RNA- protein complexes? Trends Cell Biol. 2004;14:226–232. doi: 10.1016/j.tcb.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Young PJ, Le TT, thi Man N, Burghes AH, Morris GE. The relationship between SMN, the spinal muscular atrophy protein, and nuclear coiled bodies in differentiated tissues and cultured cells. Exp Cell Res. 2000a;256:365–374. doi: 10.1006/excr.2000.4858. [DOI] [PubMed] [Google Scholar]
- Young PJ, Man NT, Lorson CL, Le TT, Androphy EJ, Burghes AH, Morris GE. The exon 2b region of the spinal muscular atrophy protein, SMN, is involved in self- association and SIP1 binding. Hum Mol Genet. 2000b;9:2869–2877. doi: 10.1093/hmg/9.19.2869. [DOI] [PubMed] [Google Scholar]
- Young PJ, Day PM, Zhou J, Androphy EJ, Morris GE, Lorson CL. A direct interaction between the survival motor neuron protein and p53 and its relationship to spinal muscular atrophy. J Biol Chem. 2002a;277:2852–2859. doi: 10.1074/jbc.M108769200. [DOI] [PubMed] [Google Scholar]
- Young PJ, Jensen KT, Burger LR, Pintel DJ, Lorson CL. Minute virus of mice NS1 interacts with the SMN protein, and they colocalize in novel nuclear bodies induced by parvovirus infection. J Virol. 2002b;76:3892–3904. doi: 10.1128/JVI.76.8.3892-3904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young PJ, Francis JW, Lince D, Coon K, Androphy EJ, Lorson CL. The Ewing's sarcoma protein interacts with the Tudor domain of the survival motor neuron protein. Brain Res Mol Brain Res. 2003;119:37–49. doi: 10.1016/j.molbrainres.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Zhang H, Xing L, Rossoll W, Wichterle H, Singer RH, Bassell GJ. Multiprotein complexes of the survival of motor neuron protein SMN with Gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci. 2006;26:8622–8632. doi: 10.1523/JNEUROSCI.3967-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Xing L, Singer RH, Bassell GJ. QNQKE targeting motif for the SMN- Gemin multiprotein complexin neurons. J Neurosci Res. 2007;85:2657–2667. doi: 10.1002/jnr.21308. [DOI] [PubMed] [Google Scholar]
- Zhang HL, Singer RH, Bassell GJ. Neurotrophin regulation of beta-actin mRNA and protein localization within growth cones. J Cell Biol. 1999;147:59–70. doi: 10.1083/jcb.147.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Eom T, Oleynikov Y, Shenoy SM, Liebelt DA, Dictenberg JB, Singer RH, Bassell GJ. Neurotrophin-induced transport of a β-actin mRNP complex increases beta-actin levels and stimulates growth cone motility. Neuron. 2001;31:261–275. doi: 10.1016/s0896-6273(01)00357-9. [DOI] [PubMed] [Google Scholar]
- Zhang HL, Pan F, Hong D, Shenoy SM, Singer RH, Bassell GJ. Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J Neurosci. 2003;23:6627–6637. doi: 10.1523/JNEUROSCI.23-16-06627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Barahmand-pour F, Blackburn ML, Matsui Y, Chansky HA, Yang L. Survival motor neuron (SMN) protein interacts with transcription corepressor mSin3A. J Biol Chem. 2004;279:14922–14928. doi: 10.1074/jbc.M309218200. [DOI] [PubMed] [Google Scholar]