

Abstract
Neutrophil recruitment and survival are important control points in the development and resolution of inflammatory processes. 15-epi-lipoxin (LX)A4 interaction with formyl peptide receptor 2 (FPR2)/ALX receptor is suggested to enhance anti-inflammatory neutrophil functions and mediate resolution of airway inflammation. However, it has been reported that 15-epi-LXA4 analogues can also bind to cysteinyl leukotriene receptor 1 (CysLT1) and that the CysLT1 antagonist MK-571 binds to FPR2/ALX, so cross-reactivity between FPR2/ALX and CysLT1 ligands cannot be discarded. It is not well established whether the resolution properties reported for 15-epi-LXA4 are mediated through FPR2/ALX, or if other receptors such as CysLT1 may also be involved. Evaluation of specific FPR2/ALX ligands and CysLT1 antagonists in functional biochemical and cellular assays were performed to establish a role for both receptors in 15-epi-LXA4-mediated signalling and function. In our study, a FPR2/ALX synthetic peptide (WKYMVm) and a small molecule FPR2/ALX agonist (compound 43) induced FPR2/ALX-mediated signalling, enhancing guanosine triphosphate-gamma (GTPγ) binding and decreasing cyclic adenosine monophosphate (cAMP) levels, whereas 15-epi-LXA4 was inactive. Furthermore, 15-epi-LXA4 showed neither binding affinity nor signalling towards CysLT1. In neutrophils, 15-epi-LXA4 showed a moderate reduction of interleukin (IL)-8-mediated neutrophil chemotaxis but no effect on neutrophil survival was observed. In addition, CysLT1 antagonists were inactive in FPR2/ALX signalling or neutrophil assays. In conclusion, 15-epi-LXA4 is not a functional agonist or an antagonist of FPR2/ALX or CysLT1, shows no effect on IL-8-induced neutrophil survival and produces only moderate inhibition in IL-8-mediated neutrophil migration. Our data do not support an anti-inflammatory role of 15-epi-LXA4- FPR2/ALX interaction in IL-8-induced neutrophil inflammation.
Keywords: 15-epi-LXA4, FPR2/ALX, apoptosis, interleukin-8, neutrophils
Introduction
Neutrophils play a central role in innate immunity and are recruited rapidly to sites of infection and injury. These polymorphonuclear leucocytes are able to migrate into the inflamed lung along a gradient of increasing concentrations of chemoattractant released by other inflammatory cells, such as alveolar macrophages and epithelial cells 1. Among chemotactic factors generated during the progression of inflammation, N-formyl-Methionyl-Leucyl-Phenylalanine (fMLF), interleukin (IL)-8, complement C5a and leukotriene B4 (LTB4) are considered the crucial mediators of leucocyte recruitment and activation 1. The survival of neutrophils at the site of inflammation is influenced profoundly by signals from the inflammatory microenvironment, including bacteria, proinflammatory cytokines, chemokines and pro-apoptotic stimuli. Once the neutrophils have carried out their role, the most desirable fate for successful resolution and efficient clearance of these cells is apoptosis, followed by phagocytosis by macrophages 2. It is clear that programmed cell death has a fundamental role in almost all biological processes, and there is increasing evidence to indicate that dysregulated apoptosis driving to an excessive accumulation of neutrophils in the inflamed tissue contribute to the pathogenesis and progression of chronic inflammatory diseases such as severe asthma and chronic obstructive pulmonary disease (COPD) 2,3.
Smokers and COPD patients present increased numbers of neutrophils in sputum that correlate with disease severity 4–6 and decrease in lung function 7. The Glu-Leu-Arg (ELR+) CXC-chemokine IL-8 is one of the most relevant chemokines in COPD; its levels are increased in the sputum and plasma of COPD patients and correlate with the number of neutrophils 8. In normal conditions basal levels of IL-8, among other immune mediators, promote neutrophil migration and enhance anti-microbial host defense mechanisms, including neutrophil release of granule enzymes (MPO, neutrophil elastase) and generation of reactive oxygen species (ROS) by binding to two G-protein-coupled receptors (GPCR), CXC chemokine receptor 1 (CXCR1) and CXC chemokine receptor 2 (CXCR2) 9. However, in pathological conditions such as COPD an exaggerated production of IL-8 promotes an uncontrolled release of ROS and proteases that increase oxidative stress, tissue damage and extracellular matrix digestion that contribute to the development of emphysema. Modulation of IL-8-mediated neutrophil functions is clue to control the progression of airway inflammatory diseases.
The natural resolution of inflammation occurs via local biosynthesis of endogenous lipid mediators, such as lipoxins (LXs) and 15-epi-LXs at sites of inflamed tissue 10. 15-epi-LXs are produced locally via cell–cell interactions between leucocytes and resident cells during multi-cellular host responses to injury, inflammation and microbial invasion (reviewed in 10). Neutrophils are the more relevant cell type with specific recognition binding sites for LXA4 and 15-epi-LXA4 11, and the signalling evoked by LXs in these cells has been suggested to be through phospholipase D (PLD) activation, arachidonic acid release, presqualene diphosphate (PSDP) increase and phosphorylation of lymphocyte-specific protein 1 (LSP-1) (reviewed in 12). LXA4 and 15-epi-LXA4, as well as their stable analogues, bind with high affinity to the GPCR formyl peptide receptor 2/LXA4 receptor (FPR2/ALX) (also known as formyl peptide receptor-like 1 (FPRL1) 13. Several reports have shown the role of FPR2/ALX receptor in triggering the anti-inflammatory and pro-resolution properties associated with LXs. Deficiency in the FPR2/ALX receptor in mice decreases the ability of LXA4 to dampen inflammation in vivo 14,15, whereas over-expression of the human LX receptor in mice enhances LX-mediated resolution of inflammation 16. Of interest, in a heterodimer model using BLT1/FPR2/ALX chimera, the activation of each GPCR is mediated by the individual agonist binding to each subunit discarding transactivation mechanisms 17. In humans, up-regulation of neutrophil FPR2/ALX expression has been observed after low-dose aspirin administration in acute inflammation 18; most recently the promoter for FPR2/ALX has been identified, and LXA4 has shown to enhance both promoter activity and receptor expression in vitro 19. Besides the anti-inflammatory properties described for FPR2/ALX, the receptor can also mediate proinflammatory actions, depending on the ligand characteristics (reviewed in 12). Bioactive lipid mediators as well as specific small peptides/proteins, such as major histocompatibility complex (MHC) binding peptide and its surrogate MMK-1, and a photolytic product of the acute phase response, serum amyloid protein A (SAA), interact in vitro with the same FPR2/ALX receptor. Opposite to lipid ligands (e.g. LXs and 15-epi-LXs) that function as anti-inflammatory mediators, peptides are reported to stimulate calcium mobilization and neutrophil migration in vitro (reviewed in 12).
In addition to FPR2/ALX, 15-epi-LXA4 has also been described to bind to cysteinyl leukotriene receptor 1 (CysLT1) and competes for this receptor with equal affinity as the natural CysLT1 ligand leukotriene D4 (LTD)4 20, suggesting a double role for 15-epi-LXA4 on CysLT1 signalling as well as on FPR2/ALX-regulated neutrophil migration and function. Of interest, the MK-571 leukotriene modifier drug with a related structure to montelukast (MK-476), a potent and selective CysLT1 antagonist used widely as an oral treatment of persistent asthma 21, has been described to bind to both FPR2/ALX and CysLT1 20, suggesting the potential double function on both receptors.
It has been shown broadly that LXA4 and 15-epi-LXA4 as well as their stable analogues inhibit LTB4 and fMLF-induced neutrophil migration 22, reverse SAA and myeloperoxidase (MPO)-induced neutrophil apoptosis arrest 23,24, and act as key mediators of resolution in a wide number of inflammatory preclinical models in mice 25,26. Although LXs have been identified as crucial in resolving acute inflammation in in-vivo systems, clearer evidence in the signalling cascades triggered by FPR2/ALX and CysLT1 receptors has not been well established.
The aim of the current study was to determine whether the anti-inflammatory and resolution properties reported for 15-epi-LXA4 are mediated through FPR2/ALX or if other receptors, such as CysLT1, could also be involved. Surprisingly, using specific modulators of FPR2/ALX and CysLT1 receptors we found that the natural FPR2/ALX ligand 15-epi-LXA4 does not induce FPR2/ALX or CysLT1-mediated signalling, has no effect on neutrophil survival induced by IL-8 and exerts only minor effects on IL-8-mediated neutrophil migration. In contrast, the FPR2/ALX proinflammatory peptide (WKYMVm) and the FPR2/ALX small-molecule agonist (compound 43) induce FPR2/ALX signalling, although acting as proinflammatory mediators in neutrophils, as described previously 27,28.
Material and methods
Materials and reference compounds
Reference compounds were selected according to the reported agonist or antagonist behaviour described in the literature. 15-epi-LXA4 is described as a FPR2/ALX binding ligand with anti-inflammatory properties in in-vitro and in-vivo models 10,12; compound 43 is a small molecular weight FPR2/ALX agonist described by Amgen 29,30; the hexapeptide Trp-Lys-Tyr-Met-Val-D-Met-NH(2) (WKYMVm) is a synthetic peptide described as a proinflammatory FPR2/ALX agonist in neutrophils 12,27; montelukast and MK-571 are CysLT1 antagonists presenting bronchodilation and anti-inflammatory properties in preclinical models 21. Chemical structures of the reference molecules are shown in Fig. 1. 15-Epi-LXA4 was purchased from Cayman (Ann Arbor, MI, USA). The concentration of 15-epi-LXA4 was determined accurately immediately before starting any biochemical or cellular experimental work by measuring ultraviolet (UV) absorbance by spectrophotometry at the UV spectrum of lipoxins (lambda max at 301 nm) to confirm that the material has not been degraded. In addition, 15-epi-LXA4 stability was monitored by liquid chromatography-mass spectrometry (LC-MS). Chromatographic separation was carried out on a Acquity ultra-performance liquid chromatograph (UPLC) from Waters (Milford, MA, USA) with a BEH C18 column (50 mm × 2 1 internal diameter, particle size 1·7 μm) at a constant flow rate of 0·4 ml/min. The mobile phase consisted of 10 mM formic acid (pH 2·8) (A) and acetonitrile (B), linear gradient from 30 to 55% B within 1·8 min. The mobile phase was then returned to the starting solvent mixture in 0·1 min and the system equilibrated for 0·4 min between runs. UPLC was coupled to an Applied Biosystems API 4000 QTrap hybrid triple quadrupole linear ion trap mass spectrometer (Applied Biosystems, Foster City, CA, USA). Samples were analysed using negative electrospray ionization (ESI). The ion spray voltage was set at −4500 V. The source temperature was set at 400°C. Nitrogen was used as the nebulizer and auxiliary gas and was set at 20, 50 and 50 arbitrary units for the curtain gas, the ion source gas 1 and the ion source gas 2, respectively. MS/MS spectra of 15-epi-LXA4 showed the same fragmentation pattern as the published 31 and commercial source (data not shown) spectra. Moreover, LC-MS/MS analysis confirmed 15-epi-LXA4 stability and no changes in height peak and area were observed during the time of the in-vitro assay conditions and using the 15-epi-LXA4 concentration reported to show biological activity (data not shown). The synthetic peptide WKYMVm (Trp-Lys-Tyr-Met-Val-D-Met-NH2) was purchased from Tocris Bioscience (Bristol, UK). IL-8 was purchased from Peprotech (Rocky Hill, NJ, USA). Montelukast, MK-571, compound 43 and SCH527123 were synthesized at the Medicinal Chemistry Department in Almirall R&D Centre (Sant Feliu de Llobregat, Barcelona, Spain).
Fig. 1.
Chemical structure of reference compounds. The chemical structures of the FPR2/ALX natural ligand 15-epi-lipoxin A4 (LXA4), the FPR2/ALX agonist compound 43, two cysteinyl leukotriene receptor 1 (CysLT1) antagonists montelukast (MK-476) and MK-571, and the synthetic peptide WKYMVm are detailed.
Cell culture and maintenance
Human Chinese hamster ovary (CHO)-FPR2/ALX (ES-610-C) and human CHO-CysLT1 (ES-470-C) cell lines were purchased from Perkin Elmer (Waltham, MA, USA). Surface expression of the receptor FPR2/ALX was monitored by flow cytometry using a commercial monoclonal antibody against the receptor. Results clearly show high levels of receptor expression in FPR2/ALX-recombinant CHO cells compared to non-transfected CHO cells (increased 40-fold in mean expression). In addition, information on Bmax of recombinant cell lines by a radioligand saturation binding assay was provided by Perkin Elmer and confirmed activity of both receptors in the recombinant cells. Ham's F12 culture medium supplemented with 100 IU/ml penicillin and 400 μg/ml G418 was used to grow the cells.
FPR2/ALX-CHO cell membrane preparation
FPR2/ALX cell membrane preparation was performed from FPR2/ALX stable transfected CHO cells purchased from Perkin-Elmer. Adherent-growing CHO-h FPR2/ALX cells were washed in cold phosphate-buffered saline (PBS), harvested by scraping and collected by centrifugation at 1500 g for 5 min. The cell pellet was washed twice with cold PBS and resuspended in homogenization buffer [15 mM Tris-HCl, pH 7·5, 2 mM MgCl2, 0·3 mM ethylenediamine teraacetic acid (EDTA), 1 mM ethylene glycol tetraacetic acid (EGTA)]. The cells were then lysed with an Ultraturrax homogenizer. Intact cells and nuclei were removed by centrifugation at 1000 g for 5 min. The cell membranes in the supernatant were then pelleted by centrifugation at 40 000 g for 25 min and resuspended in storage buffer (50 mM Tris-HCl pH 7·4, 0·5 mM EDTA, 10 mM MgCl2, 10% sucrose), aliquoted, quick-frozen in liquid N2 and stored at −80°C. Protein concentration in membrane preparations was determined using the DC Protein Assay kit (Bio-Rad, Hercules, CA, USA).
Cyclic adenosine monophosphate (cAMP) assay in CHO-FPR2/ALX cell line
CHO-FPR2/ALX cells (1 × 104 per well) were seeded in 96 half-area plates using Optimem with phenol red (Invitrogen, Carlsbad, CA, USA) overnight at 37°C in 5% CO2. For the agonist mode, CHO cells were incubated with reference compounds at 0·01 pM–100 μM final concentration with 10 μM forskolin for 30 min. After incubation, detection mixture (cAMP-D2 and cAMP-antibody-Europium) was added following the time-resolved fluorescence resonance energy transfer (TR-FRET) dynamic-2 cAMP kit (Cisbio, Bagnols-sur-Cèze, France) instructions. After 1 h incubation, cAMP levels were read on Envision (Perkin Elmer). For the antagonist mode, CHO-FPR2/ALX cells were preincubated with reference compounds at 0·01 pM–100 μM final concentration 1 h prior to adding 10 μM forskolin and the agonist at the effective dose (EC80) (20 nM and 0·05 nM for compound 43 and WKYMVm peptide, respectively). After 30 min of incubation, cAMP levels were measured as in the agonist mode. All incubations were performed at room temperature.
[35S]-guanosine triphosphate-gamma (GTPγ)S binding assay in CHO-FPR2/ALX cell membranes
FPR2/ALX cell membranes (2 μg) were incubated in a 200 μl total volume containing 20 mM HEPES pH 7·4, 100 mM NaCl, 10 mM MgCl2, 10 μM GDP, 50 μg/ml saponin, 0·2% BSA (Sigma, Saint Louis, MI, USA) and 0·1 nM [35S]-GTPγS (NEN; specific activity 1250 Ci/mmol). For agonist mode, reference compounds were incubated with the membranes for 90 min with gentle mixing. Briefly, the reaction mixture was filtrated through GF/C filter plates (Millipore, Billerica, MA, USA) using the Manifold Filtration System (Millipore). The filters were washed immediately six times with 200 μl of sodium phosphate buffer pH 7·4. After drying the filter plates for 20 min at 65°C, 30 μl of Optiphase Hisafe II scintillant liquid were added to each well and [35S]-GTPγS were measured on a Trilux Scintillation Counter. For antagonist mode, reference compounds were preincubated with membranes for 1 h before addition of the agonist compound 43 at the EC80 (716 nM). After 90 min incubation, the same protocol as in the agonist mode was used for [35S]-GTPγS detection. All incubations were performed at room temperature.
[3H]-LTD4 binding to CHO-CysLT1 cell membranes
Competition binding experiments were conducted in 96-well polypropylene plates in a total volume of 200 μl using 0·62 nM of [3H]-LTD4 and 7·5 μg/well of CHO-CysLT1 membranes (ES-470-M, Euroscreen; Perkin Elmer, Waltham, MA, USA). All reagents were prepared in the binding assay buffer (20 mM Tris pH 7·4, 5 mM MgCl2), except for compounds that were dissolved in 100% dimethylsulphoxide (DMSO). Non-specific binding (NSB) was measured in the presence of 10 μM zafirlukast. After an incubation period of 30 min with gentle agitation, 150 μl of the reaction mix was transferred to 96-well GF/C filter plates (Millipore) treated previously for 1 h with binding assay buffer plus 0·05% Brij 35. Bound and free [3H]-LTD4 were separated by rapid vacuum filtration in a manifold and washed four times with ice-cold washing buffer. After drying for 30 min, 30 μl of OPTIPHASE Hisafe II were added to each well and radioactivity was measured using a Microbeta microplate scintillation counter.
Calcium flux assay in CHO-CysLT1 cell line
The calcium flux assay was performed using Flexstation fluorescence spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). The day before the assay, 4 × 104 CHO-CysLT1 cells per well were seeded in a 96-well dark-walled plate (Costar, Corning, NY, USA) in 50 μl of Ham's F12 medium,10% FBS and 1% L-glutamine. After overnight incubation at 37°C in 5% CO2 cells were washed four times with buffer [Hanks's balanced salt solution (HBSS) ×1 with calcium and magnesium and 20 mM HEPES, pH 7·4], resuspended in 50 μl of buffer and loaded using the Calcium 5 kit dye (Molecular Devices) for 1 h at room temperature. For the agonist mode, reference compounds dissolved with buffer plus 2·5 mM probenecid were added to CHO-CysLT1-loaded cells at 0·01 pM–100 μM final concentration and kinetic measurement of cytoplasmic calcium was determined in the Flexstation at an extinction wavelength of 485 nm and an emission wavelength of 525 nm. For antagonist mode, CHO-CysLT1 cells were preincubated for 1 h with reference compounds dissolved with buffer plus 2·5 mM probenecid at 0·01 pM–100 μM final concentration in addition to the calcium dye. The CysLT1 agonist (LTD4, 1 nM) was added and cytoplasmic calcium kinetics were measured.
Human neutrophil chemotaxis
Polymorphonuclear leukocytes (PMNs) were isolated from peripheral blood of normal volunteers by dextran sedimentation and centrifugation through PolymorphoPrep (Axis-Shield, Dundee, Scotland, UK). After erythrocyte lysis, PMNs were washed and resuspended at a concentration of 1 × 106 cells/ml in Dulbecco's phosphate-buffered saline (DPBS) with Ca2+ and Mg2+ containing BSA 0·1%, Hepes 10 mM (Invitrogen), glucose 10 mM at pH 7·4 (DPBS++). Before starting the chemotaxis assay, 24-transwell plates (Corning Inc., Corning, NY, USA) were equilibrated with DPBS+/+ (100 μl upper well and 600 μl lower well) for at least 1 h. Human recombinant IL-8 (600 μl) at 1·25 nM or vehicle (DPBS+/+) were added to the lower wells of the chemotaxis chamber. The wells were overlaid with a 5-μm pore size polycarbonate filter. PMNs (100 μl) were placed in the upper wells, and the transwell plate was incubated (37°C, 5% CO2) for 30 min. Following incubation, media from the lower wells were placed into a clean tube. Each condition was run in duplicate, and cells that migrated across the filter towards the lower well were enumerated by fluorescence activated cell sorter (FACS). To assess inhibition, PMNs were suspended in DPBS++ with vehicle (ethanol or DMSO < 0·1%), increasing concentrations (1 μM–0·1 nM) of the FPR2/ALX agonists (15-epi-LXA4 or compound 43) or CysTL1 antagonists (montelukast or MK-571) and incubated for 30 min at 37°C before their placement into the upper wells. The chemotactic properties of FPR2/ALX agonists and CysLT antagonists by themselves were studied by adding the compounds (100 nM) alone in the lower compartment of the migration chambers. Compound 43 was tested at three concentrations (0·01, 0·1 and 1 μM). IL-8 (1·25 nM) was used as positive control of neutrophil migration. A control of neutrophil inhibition was assessed by incubating compound 43 and SCH527123 for 30 min with human neutrophils prior to placing them into the upper compartment.
Human neutrophil apoptosis: caspase 3/7 activity
PMNs, 2 × 104 cells/ml in PBS+/+, were incubated with increasing concentrations (1 μM–0·1 nM) of the FPR2/ALX agonists (15-epi-LXA4 or compound 43) or CysTL1 antagonists (montelukast or MK-571) for 30 min at 37°C or vehicle (DMSO < 0·1%) in 96-well plates. Human recombinant IL-8 (100 nM) was added to the wells and incubated for 4 h. After covering the bottom of the plate with the adhesive non-translucid paper, the caspase 3/7 reagent was added and incubated for 30 min. Caspase 3/7 activity was measured by luminometry using a Luminoskan Ascent (Thermo Labsystems, Bar Hill, Cambridge, UK). Caspase inhibitor I (5 μM) was used as a control of apoptosis inhibition and staurosporine (1 μg/ml) as a control of apoptosis induction. In order to avoid LPS contamination, fresh buffers were prepared using sterile and filtered solutions on the same day of the apoptosis assay.
Human neutrophil apoptosis: annexin V staining
PMNs at 1 × 106 cells/ml in PBS+/+ were incubated with the FPR2/ALX agonists (15-epi-LXA4 and compound 43) and CysTL1 antagonists (montelukast and MK-571) (100 nM) for 30 min at 37°C or vehicle (DMSO < 0·1%) in 24-well plates. Human recombinant IL-8 (100 nM) was added to the wells and incubated for 4 h. After incubation cells were transferred to a clean FACS tube and washed with PBS (×2). Briefly, cells were resuspended with ×1 binding buffer (500 μl) and 5 μl of annexin V-FITC (Sigma, Saint Louis, MO, USA) and 10 μl of propidium iodide were added. Cells were incubated at room temperature for 10 min and fluorescence was measured immediately by flow cytometry using a FACSCanto (BD Biosciences, Franklin Lakes, NJ, USA).
Calculations and statistical analysis
Dose–response curves were set up in duplicate. Half maximal inhibitory concentration (IC50) and Half maximal effective concentration (EC50) calculations were performed using the four-parameter logistic (4PL) non-linear regression [log (inhibitor) versus response with variable slope equation] using GraphPad Prism software. IC50 values are reported as geometric mean (GeoMean) ± standard error of the mean. Values for chemotaxis and apoptosis assessment were analysed by Student's t-test.
Results
Effects of FPR2/ALX agonists and CysLT1 antagonists on cAMP and GTPγ pathways in hFPR2/ALX-expressing cells and binding and calcium flux in hCysLT1-expressing cells
In order to study the signalling pathway triggered by activation of FPR2/ALX and CysLT1 by each reference compound, cAMP and GTPγ binding assays in FPR2/ALX recombinant cells and membranes and binding and calcium flux assays in CysLT1 recombinant cells were performed. IC50 and percentage of inhibition of the reference compounds in agonist and antagonist mode in FPR2/ALX and CysLT1 are shown in Table 1 and Fig. 2, respectively.
Table 1.
Characterization of reference compounds behaviour, agonism and antagonism in FPR2/ALX and CysLT1-related assays
| FPR2/ALX receptor | CysLT1 receptor | |||||||
|---|---|---|---|---|---|---|---|---|
| Inhibition cAMP levels (IC50, nM) | GTPγ binding (IC50, nM) | [3H]-LTD4 binding (IC50, nM) | Calcium flux (IC50, nM) | |||||
| Mechanism of action | Reference compound | Agonist mode | Antagonist mode | Agonist mode | Antagonist mode | Agonist mode | Antagonist mode | |
| FPR2/ALX agonist and/or CysLT1 antagonist? | 15-epi-LXA4 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| FPR2/ALX agonist | Compound 43 | 11·6 ± 1·9 | n.t. | 207 ± 51 | n.t. | Inactive | Inactive | Inactive |
| CysLT1 antagonist | Montelukast (MK-476) | Inactive | Inactive | Inactive | Inactive | 1·9 ± 1·1 | Inactive | 16·1 ± 3·3 |
| CysLT1 antagonist | MK-571 | Inactive | Inactive | Inactive | Inactive | 11·5 ± 11 | Inactive | 13·9 ± 1·0 |
Half maximal inhibitory concentrations (IC50) of reference compounds in FPR2/ALX [cyclic adenosine monophosphate (cAMP) and guanosine triphosphate-gamma (GTPγ) binding assays] and CysLT1 [leukotriene D4 (LTD4) binding and Ca2+ flux assays] are detailed in an agonist and antagonist mode. IC50 values are reported as the geometric mean (GeoMean) ± standard error of the mean (s.e.m.) of more than three independent assays. cAMP and GTPγ binding assays in FPR2/ALX recombinant cells and membranes were evaluated in the antagonist mode using compound 43 and the peptide WKYMVm as agonists. Calcium flux assay in CysLT1 recombinant cells was evaluated in an antagonist mode using LTD4 as agonist. A compound is defined as inactive when 0% of inhibition is observed at a concentration of 100 μM; n.t.: not tested. 15-epi-LXA4 is inactive in all the assays tested (FPR2/ALX or CysLT1 assays), whereas compound 43 behaves as an agonist in FPR2/ALX cAMP and GTPγ binding assays, being inactive in CysLT1 assays similar to the proinflammatory FPR2/ALX agonist peptide WKYMVm. Montelukast (MK-476) and MK-571 bind to CysLT1 and behave as CysLT1 antagonists, with no effect on FPR2/ALX signalling.
Fig. 2.
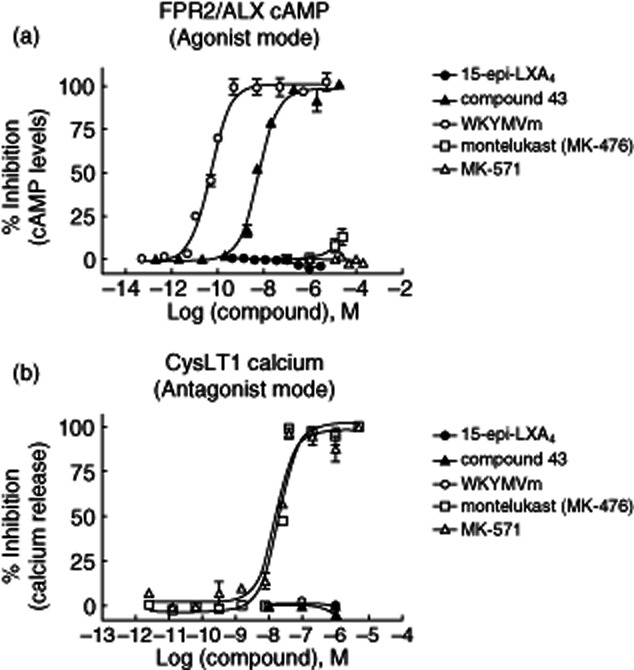
FPR2/ALX agonists and CysLT1 antagonist behaviour in cyclic adenosine monophosphate (cAMP) and CysLT1 Ca2+ flux signalling assays in Chinese hamster ovary (CHO) over-expressing human FPR2/ALX and CysLT1 receptors, respectively. (a) Effect of the reference compounds on cAMP in human FPR2/ALX recombinant cells. The agonist behaviour was measured as the percentage of inhibition after preincubation of the compounds (100 μM–0·1 pM) with human FPR2/ALX recombinant cells for 1 h prior to adding 10 μM forskolin. Fluorescence signal was detected after 1 h incubation with time-resolved fluorescence resonance energy transfer (TR-FRET) dynamic-2 cAMP kit (further information detailed in Material and methods). Compound 43 and the proinflammatory peptide (WKYMVm) inhibited cAMP in recombinant cells, whereas 15-epi-lipoxin (LX)A4, montelukast (MK-476) or MK-571 did not exert any effect. (b) Effect of the reference compounds on calcium release in human CysLT1 recombinant cells. The antagonist behaviour was measured as the percentage of inhibition after compound preincubation (0·01 pM–100 μM) and agonist addition [leukotriene D4 (LTD4), 1 nM]. Cytoplasmic calcium concentration was determined by fluorescence signal in the Flexstation (further information detailed in Material and methods). In the CysLT1 calcium flux assay only montelukast and MK-571 inhibited the signal potently.
15-Epi-LXA4 was inactive (0% of inhibition at 100 μM) in either GTPγ binding or cAMP assays in both agonist or antagonist mode in FPR2/ALX-expressing cells (Table 1 and Fig. 2a). Calcium release was not increased after stimulation of FPR2/ALX recombinant cells by 15-epi-LXA4 (data not shown). Similarly, 15-epi-LXA4 did not block the binding of LTD4 to CysLT1 and, consistent with the binding data, was inactive in the calcium influx assay in both agonist or antagonist mode in CysLT1-expressing cells (Table 1 and Fig. 2b). Conversely, compound 43 and the peptide WKYMVm were actively potent in the cAMP assay in FPR2/ALX over-expressing CHO cells (IC50 = 11·6 ± 1·9 nM and 0·14 ± 0·11 nM, respectively) (Table 1 and Fig. 2a); compound 43 was also active in the GTPγ binding assay (IC50 = 207 ± 51 nM) (Table 1), confirming that FPR2/ALX is the functional receptor for this small molecular weight compound. Furthermore, compound 43 and WKYMVm were not acting as agonists or antagonists of the CysLT1 receptor.
The CysLT1 antagonists montelukast (MK-476) and MK-571 were inactive in GTPγ binding (Table 1), cAMP (Table 1 and Fig. 2a) and intracellular calcium release (data not shown) assays in FPR2/ALX recombinant cells, whereas they exerted potent inhibition of [3H]-LTD4 binding to CysLT1-expressing cell membranes (IC50 = 1·9 ± 1·1 nM and 11·5 ± 11 nM, respectively) and, as expected, inhibited LTD4-induced calcium influx in CysLT1-expressing cells (IC50 = 16·1 ± 3·3 nM and 13·9 ± 1·0 nM, respectively) (Table 1 and Fig. 2b). Taken together, our initial hypothesis was not confirmed, as 15-epi-LXA4 did not function either as an FPR2/ALX agonist or CysLT1 antagonist, whereas compound 43 and WKYMVm peptide behaved as FPR2/ALX agonists and montelukast and MK571 exerted the expected antagonist properties on CysLT1.
Effect of reference compounds on IL-8-induced neutrophil chemotaxis
Because no data have been reported so far regarding the effect of LXs in IL-8-mediated neutrophil function, we evaluated the effect of 15-epi-LXA4 on the induction of chemotaxis induced by IL-8 in freshly isolated peripheral blood human neutrophils. 15-epi-LXA4 showed partial blockage of IL-8-induced neutrophil chemotaxis with a maximum inhibition of 40% at 10 nM (Fig. 3a). However, neutrophil migration was reduced significantly by 15-epi-LXA4 at a concentration ≥ 10 nM (P < 0·05). In contrast, compound 43 inhibited IL-8-induced neutrophil migration potently (IC50 = 67 nM) at the same extension as the CXCR2 antagonist SCH527123 (IC50 = 9·3 nM) (Fig. 3a). Conversely, no inhibition of IL-8-induced neutrophil chemotaxis was observed with the CysLT1 antagonists montelukast or MK-571 at the nanomolar range (data not shown). 15-epi-LXA4, montelukast, MK-571 and SCH527123 at 100 nM did not evoke neutrophil chemotaxis by themselves (Fig. 3b). However, compound 43 induced a concentration-dependent increase of neutrophil migration.
Fig. 3.
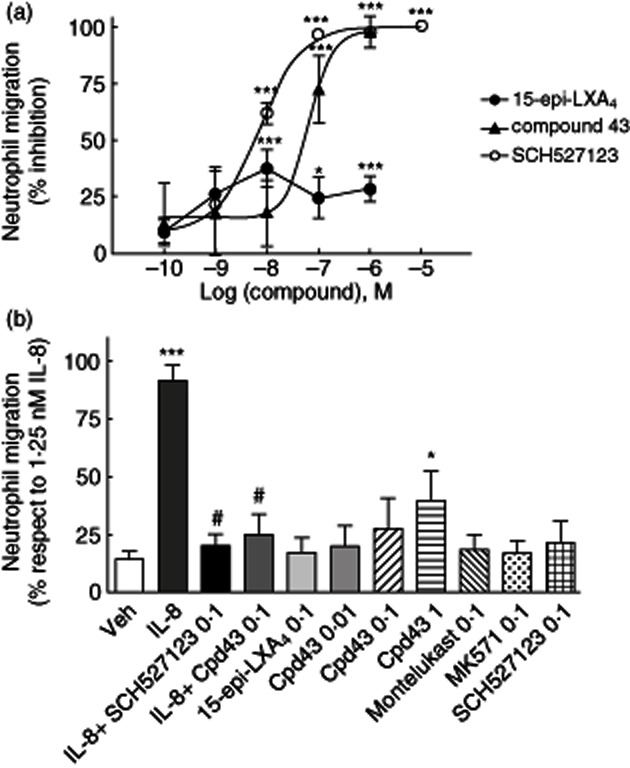
Effect of 15-epi-lipoxin (LX)A4 and compound 43 on interleukin (IL)-8-induced neutrophil chemotaxis. (a) The percentage of inhibition of neutrophil chemotaxis was calculated after preincubation of the compounds (0·1 nM–1 μM) with human peripheral blood isolated neutrophils for 30 min. Briefly, chemotaxis was run for additional 30 min against an IL-8 (1·25 nM) gradient, as described in Material and methods. IL-8-induced neutrophil chemotaxis was dampened by 15-epi-LXA4 (maximum inhibition 40% at 10 nM) and compound 43 (IC50 = 67 nM). SCH527123, a CXCR2 antagonist, was used as a positive control of inhibition of neutrophil migration (IC50 = 9·3 nM). *P < 0·05; ***P < 0·001. (b) Direct chemotactic effect of the reference compounds on neutrophils was measured by incubating 15-epi-LXA4, montelukast, MK-571, SCH527123 (0·1 μM) and compound 43 (0·01, 0·1 and 1 μM) alone in the lower chemotactic chamber compartment without IL-8. Neutrophils were added on top of the filter that separated the upper compartment containing cells, but no agonist, from the lower compartment containing the reference compounds. IL-8 (1·25 nM) was used as a positive control of neutrophil migration and controls of inhibition of IL-8-induced chemotaxis were assessed by incubating compound 43 (0·1 μM) and SCH527123 (0·1 μM) with neutrophils prior to placing them in the upper compartment. ***P < 0·001 IL-8 versus vehicle; *P < 0·05 compound 43 (1 μM) versus vehicle; #P < 0·05 compound 43 and SCH527123 versus IL-8.
Evaluation of reference compounds on IL-8-induced neutrophil apoptosis arrest
One of the important reported functions for LXs in neutrophils is their role in inducing apoptosis of activated cells 23,24. It is suggested that FPR2/ALX plays a major role in the resolution of inflammation by inducing apoptosis of activated neutrophils. For this reason, we evaluated the effect of 15-epi-LXA4, montelukast, MK-571 and compound 43 on neutrophil survival activated by IL-8 by measuring caspase 3/7 activity; 100% apoptosis is the basal apoptosis achieved by neutrophils during the time without stimulation (under vehicle conditions). Results of the apoptosis percentage are referred to this basal value. In our study, neither FPR2/ALX agonists nor CysLT1 antagonists exerted any effect on the inhibition of neutrophil survival induced by IL-8 (100 nM) at the concentrations tested (0·1 nM–1 μM) (Fig. 4). Caspase inhibitor I was used as a control of apoptosis inhibition, resulting in a complete blockade of caspase 3/7 activity.
Fig. 4.
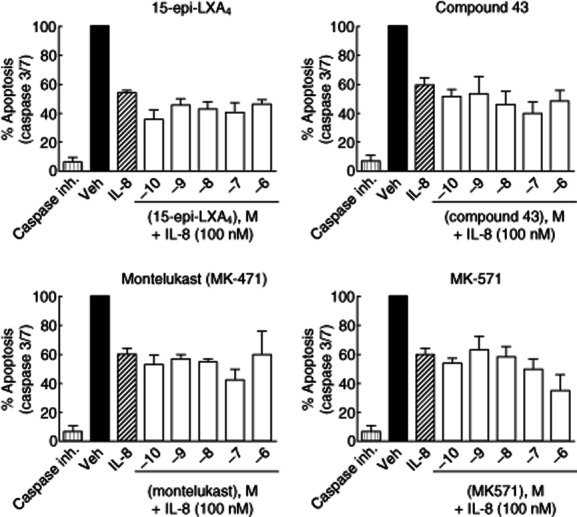
Effect of 15-epi-lipoxin (LX)A4, compound 43, montelukast and MK-571 on interleukin (IL)-8-induced neutrophil apoptosis arrest by measuring caspase 3/7 activity. Human neutrophil survival was induced by incubation with IL-8 (100 nM), and the apoptosis produced by the reference compounds (0·1 nM–1 μM) was measured by the percentage of induction of caspase 3/7 activity after 4 h of IL-8 addition. Caspase inhibitor I (Caspase inh.) at 5 μM was used as a control of apoptosis inhibition. None of the reference compounds reversed IL-8-induced neutrophil apoptosis arrest at the doses tested.
Similar results were observed using annexin V staining as a marker of apoptotic cells and propidium iodide as a control of the number of necrotic cells (Figs 5 and 5). 15-epi-LXA4 (100 nM) could not reverse the percentage of neutrophil apoptosis arrest induced by IL-8 stimulation (21% and 23% of apoptotic cells in IL-8 alone and IL-8 plus 15-epi-LXA4, respectively). As expected, the CXCR2 antagonist SCH527123 reversed IL-8-induced apoptosis arrest and returned the apoptotic cell index to the basal conditions (Fig. 6). Of interest, compound 43 (100 nM) by itself increased neutrophil survival in the absence of IL-8, confirming the recent published data regarding the inflammatory actions associated with this small molecule FPR2/ALX agonist 28,32. All the other reference compounds tested showed no effect on neutrophil survival by themselves (Fig. 6). Overall, these results indicate that 15-epi-LXA4 is inactive in reversing the survival signal induced by proinflammatory chemokines such as IL-8 in human neutrophils, and compound 43 by itself induces proinflammatory signals in neutrophils.
Fig. 5.
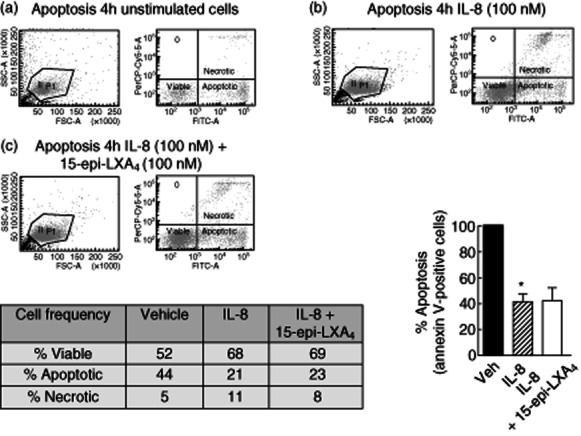
Measurement of interleukin (IL)-8-induced neutrophil survival by annexin V staining: effect of 15-epi-lipoxin (LX)A4. Cell apoptosis was measured in unstimulated human neutrophils (a) stimulated with IL-8 (100 nM) for 4 h (b) or preincubated with 15-epi-LXA4 (100 nM) for 30 min before addition of IL-8 (100 nM) for 4 h (c). Apoptosis induction was measured by annexin V staining. Propidium iodide staining was included for necrotic cell detection. Total neutrophils were gathered (left panel) and the number of viable neutrophils (double-negative cells), apoptotic neutrophils (annexin V-positive cells) and necrotic neutrophils (IP-positive cells) was detected using the proper probes by flow cytometry. Percentage of viable, apoptotic and necrotic cells are shown in the insert table. The percentage of apoptotic neutrophils after IL-8 and IL-8 in addition to 15-epi-LXA4 treatment compared to vehicle is represented in the bottom right graph. 15-epi-LXA4 did not increase annexin V-positive cells and did not reverse IL-8-induced neutrophil apoptosis arrest. *P < 0·05 IL-8 versus Vehicle.
Fig. 6.
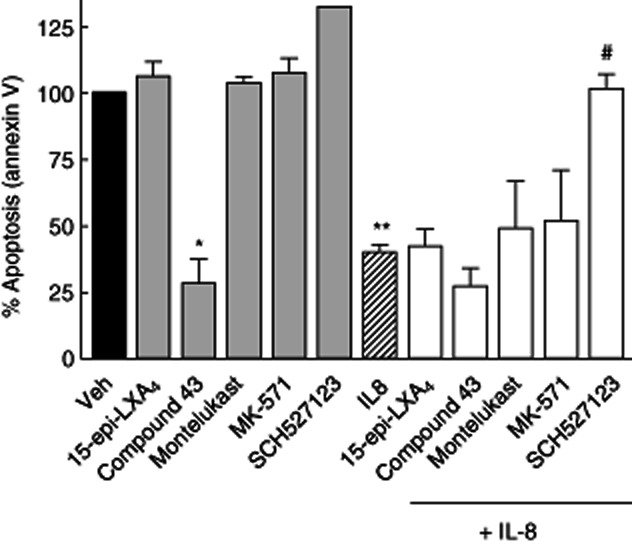
Effect of 15-epi-lipoxin (LX)A4, compound 43, montelukast, MK-571 and SCH527123 alone or in the presence of interleukin (IL)-8 on neutrophil apoptosis by annexin V staining assessment. 15-epi-LXA4, montelukast and MK-571 did not produce any effect by themselves on neutrophil survival, whereas compound 43 alone arrested the neutrophils to enter apoptosis. Similar to the caspase 3/7 activity results, none of the compounds reversed IL-8 induced neutrophil apoptosis arrest, except for the CXCR2 antagonist SCH527123 that restores the normal levels of apoptosis. *P < 0·05 compound 43 versus Veh; **P < 0·001 IL8 versus Veh; #P < 0·001 SCH527123 versus IL-8 (Student's t-test).
Discussion
LXs and 15-epi-LXs are arachidonic acid-derived metabolites suggested to play an important role as novel anti-inflammatory and pro-resolution agents. LX stable analogues display potent bioactivity in vivo in several murine model systems of acute inflammation 25 and block airway hyper-responsiveness and allergic inflammation in ovalbumin and cockroach allergen-induced airway inflammation models 26. In addition, transgenic over-expressing mice of human FPR2/ALX receptor show shorter resolution times and doses required in response to lipoxin stable analogues 16, and are protected from acid-induced acute lung injury 33 and allergen-induced pulmonary inflammation 34. FPR2 knock-down cell lines no longer signal in response to LXA4 and deficiency of FPR2 in mice decreases the ability of lipoxin A4 and annexin peptide to reduce inflammation in vivo 14,15. Nevertheless, all the in-vivo data supporting the role of FPR2/ALX mediating the anti-inflammatory actions of LXs has been generated in mice and differences in FPR2/ALX signalling between species cannot be discarded. Moreover, no FPR2/ALX knock-out or transgenic mice studies have been addressed to study in particular the relevance of the LX–FPR2/ALX axis in neutrophil migration in vivo.
In humans, differences in FPR2/ALX expression have been observed in acute and chronic inflammatory responses. Whereas in acute inflammation FPR2/ALX expression is up-regulated on macrophages from 24 h-inflamed skin blisters 18, and the same over-expression is observed in colonic mucosal biopsies of ulcerative colitis patients 35, FPR2/ALX mRNA expression is down-regulated in chronic severe asthmatics compared to healthy subjects 36,37, suggesting a deficit in the LX FPR2/ALX axis in chronic respiratory diseases. However, no significant changes have been detected in LX biosynthesis in other chronic inflammatory diseases such as COPD 38,39; thus, general conclusions cannot be drawn and lipoxin receptor levels may be specific for each disease condition.
Although the well-documented beneficial actions reported for LXs are suggested to involve FPR2/ALX-triggered signalling, the specific associated pathways responsible for in-vivo lipoxin activity remain to be elucidated. In addition, data supporting a role for LXs in modulating human neutrophil function in an IL-8 environment is missing, although moderate efficacy has been shown on human neutrophil transmigration across the intestinal epithelium and on the blockade of the release of human neutrophil azurophilic granules 40,41. The reported binding data indicate that FPR2/ALX is a high-affinity receptor for LXs and its analogues 12, but in our study the signalling activated by LXs– FPR2/ALX interactions are not the classical G-protein-activated pathways involving an increase in GTPγ binding response, a decrease in cAMP or enhancement of the intracellular calcium flux. However, in the same FPR2/ALX recombinant cells the peptide ligand WKYMVm and the small molecule FPR2/ALX agonist compound 43 induced GTPγ binding and calcium influx, suggesting that proinflammatory peptides and synthetic FPR2/ALX compounds present agonist properties whereas, in principle, 15-epi-LXA4 binds but not acts as an FPR2/ALX agonist. Similarly, recent work from an independent group has shown lack of signalling induced by 15-epi-LXA4 through enhancement in intracellular calcium in FPR2/ALX over-expressing cells 32. Conversely, a novel lipid-mediated downstream FPR2/ALX signalling has been described, involving intracellular polyisoprenyl phosphate remodelling. Interaction of these endogenous lipids with FPR2/ALX block agonist-induced presqualene diphosphate (PSDP) turnover to presqualene monophosphate (PSMP) and an increase in PSDP accentuates anti-inflammatory actions through inhibition of PLD and PI3K in human neutrophils 42,43. Nevertheless, the role for these pathways in FPR2/ALX-associated functions in vivo remains to be elucidated.
In addition to reducing acute inflammation induced by the potent neutrophil chemoattractant LTB4, LXs are able to modulate neutrophil functions induced by proinflammatory FPR2/ALX peptides. It has been reported that LXs reverse both neutrophil chemotaxis induced by MHC- and MMK-1-derived peptides 44 and neutrophil apoptosis arrest mediated by SAA 23. Due to the importance of IL-8 in recruiting neutrophils to the site of inflammation, it is clue to understand the potential role of LXs and FPR2/ALX signalling in the resolution process of inflammation mediated by IL-8 in chronic inflammatory diseases such as COPD. To our knowledge, the effect of LXs on IL-8-mediated neutrophil function has not been described in the literature. In our study, 15-epi-LXA4 could exert only a mild inhibition of IL-8-mediated neutrophil migration (40% at 10 nM), consistent with the findings reported in the literature by LXA4, 15-epi-LXA4 and their stable analogues in LTB4-induced neutrophil migration 22. In contrast, compound 43, a known synthetic agonist for FPR2/ALX, blocked IL-8-induced neutrophil chemotaxis potently, consistent with previous data published by Amgen, describing this small molecule as an anti-inflammatory FPR2/ALX agonist able to block neutrophil migration and reduce ear swelling in vivo 29,30. However, recent publications suggest that compound 43 is a dual fMLF receptor (FPR1) and FPR2/ALX agonist, because calcium mobilization increases not only in FPR2/ALX over-expressing cells but also in FPR1 recombinant cells 32, being FPR1 the suggested receptor preferred for compound 43 in neutrophils. In this sense, the inhibition of IL-8-mediated chemotaxis in the presence of compound 43 could be explained by the reported FPR2/ALX cross-desensitization of other chemoattractant receptors on the neutrophil surface, such as FPR1 or IL-8 receptor (CXCR2) 32. Similar to neutrophil migration, 15-epi-LXA4 was unable to restore apoptosis levels to normal after IL-8-induced cell survival, discarding other potential anti-inflammatory actions in an IL-8 inflammation environment. None of the reference compounds enhanced neutrophil migration or arrested neutrophils to enter into apoptosis by themselves, with the exception of compound 43, confirming the proinflammatory actions associated to the Amgen molecule 28.
It is interesting to note that recent work published by Bozinovski and colleagues 45 indicates that LXA4 directs allosteric inhibition of SAA-initiated epithelial cell proinflammatory responses such as release of IL-8. In line with this, LXs would behave as non-competitive negative modulators on SAA-mediated actions. Although their conclusion was that LXs act as allosteric inhibitors for FPR2/ALX, no experimental data were presented showing a direct role for the LX–FPR2/ALX interaction in this modulation. It is possible that LXs interact with other receptor or cell surface molecules on human cells to modulate neutrophil chemotaxis or survival induced by multiple proinflammatory ligands, including LTB4, IL-8 or FPR2/ALX peptides. To establish if LXs could reverse FPR2/ALX peptide agonist-induced proinflammatory actions, we investigated the effects of 15-epi-LXA4 as an antagonist in FPR2/ALX-expressing cells. Both FPR2/ALX proinflammatory agonists tested, the peptide WKYMVm and the synthetic small molecule compound 43, induced GTPγ binding or decreased cAMP in FPR2/ALX-expressing cells by themselves, but 15-epi-LXA4 could not antagonize the activity of any of them. The lack of signalling of the endogenous lipid mediator through its receptor, despite the well-documented binding data, and the absence of antagonism of LXs in peptide-induced inflammation raises concern for the direct role of LX–FPR2/ALX-mediated anti-inflammatory actions.
Conversely, and because LX analogues have been shown to bind with high affinity to the CysLT1, we explored if LXs could exert their actions modulating other receptors involved in inflammatory responses. In our study, 15-epi-LXA4 did not show any binding affinity for CysLT1 or any cellular signalling induction in CysLT1 over-expressing cells, whereas the described CysLT1 antagonists montelukast and MK-571 inhibited potently both LTD4-binding and calcium release 12,46. Moreover, our data indicate that MK-571 did not signal through FPR2/ALX because no effect on cAMP and GTPγ binding assays was observed. Differences between our data and the published literature results may be due to the use of different types of assay (GTPγ binding or cAMP versus radioligand binding assays), different classes of over-expressing cell lines (CHO versus HEK over-expressing cells) and discrepancies between binding and functional assays 12. The data generated in cell functional systems (human neutrophil chemotaxis and apoptosis assays) are of great value, and closer to a physiological condition compared to the limited binding results derived from over-expressing cell lines. In our study, the initial working hypothesis of cross-talk between FPR2/ALX and CysLT1 ligands is discarded, ruling out the potentially beneficial dual role of 15-epi-LXA4 on CysLT1 signalling as well as on FPR2/ALX-regulated neutrophil activation and migration. These results, together with the lack of activity observed by 15-epi-LXA4 on FPR2/ALX in cAMP and GTPγ binding assays, indicate that FPR2/ALX over-expressing cells do not respond to the described anti-inflammatory mediators (15-epi-LXA4 and MK-571), whereas they respond to proinflammatory ligands (compound 43 and WKYMVm).
Our data suggest that with current knowledge of the LX–FPR2/ALX-mediated signalling pathway, it would be difficult to identify potential non-lipid small molecule agonists to mimic LX function in vivo. IL-8 is considered to be an important chemokine for inflammatory diseases where neutrophils play a crucial role, such as COPD and cystic fibrosis, and no significant evidence for LXs or other FPR2/ALX agonists has been described in reversing IL-8-mediated in-vitro functions. Species differences could explain the discrepancy in efficacy of LXs in inflammatory preclinical models in rodents and in human cellular assays. Nevertheless, the recent published findings describing the antagonist behaviour of LXs on peptide-mediated inflammation opens a new field of investigation for LX-mediated actions in vivo. Overall, our data do not support a relevant direct role of 15-epi-LXA4–FPR2/ALX interaction in inflammatory diseases, and further work is required to identify other potential LX-binding molecules on the surface of innate cells with signalling capacity to establish a role for FPR2/ALX modulation in health and disease.
Acknowledgments
The authors thank Rosario Cerrato for her excellent technical assistance in running the cAMP and GTPγ binding assays in FPR2/ALX recombinant cells and membranes. The authors also thank Sonia Pascual and Vicente García for technical and scientific support.
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol. 2011;32:452–460. doi: 10.1016/j.it.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–416. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 3.Nathan C. Points of control of inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 4.Stockley RA. Neutrophils and the pathogenesis of COPD. Chest. 2002;121:151S–155S. doi: 10.1378/chest.121.5_suppl.151s. [DOI] [PubMed] [Google Scholar]
- 5.Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153:530–534. doi: 10.1164/ajrccm.153.2.8564092. [DOI] [PubMed] [Google Scholar]
- 6.Barnes PJ. Mechanisms in COPD: differences from asthma. Chest. 2000;117:10S–14S. doi: 10.1378/chest.117.2_suppl.10s. [DOI] [PubMed] [Google Scholar]
- 7.Stănescu D, Sanna A, Veriter C, et al. Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax. 1996;51:267–271. doi: 10.1136/thx.51.3.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto C, Yoneda T, Yoshikawa M, et al. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest. 1997;112:505–510. doi: 10.1378/chest.112.2.505. [DOI] [PubMed] [Google Scholar]
- 9.Stillie R, Farooq SM, Gordon JR, Stadnyk AW. The functional significance behind expressing two IL-8 receptor types on PMN. J Leukoc Biol. 2009;86:529–543. doi: 10.1189/jlb.0208125. [DOI] [PubMed] [Google Scholar]
- 10.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fiore S, Ryeom SW, Weller PF, Serhan CN. Lipoxin recognition sites. Specific binding of labeled lipoxin A4 with human neutrophils. J Biol Chem. 1992;267:16168–16176. [PubMed] [Google Scholar]
- 12.Chiang N, Serhan CN, Dahlén SE, et al. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev. 2006;58:463–487. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- 13.Fiore S, Maddox JF, Perez HD, Serhan CN. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J Exp Med. 1994;180:253–256. doi: 10.1084/jem.180.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dufton N, Hannon R, Brancaleone V, et al. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–2619. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Cai L, Wang H, et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene. 2011;30:3887–3899. doi: 10.1038/onc.2011.112. [DOI] [PubMed] [Google Scholar]
- 16.Devchand PR, Arita M, Hong S, et al. Human ALX receptor regulates neutrophil recruitment in transgenic mice: roles in inflammation and host defense. FASEB J. 2003;17:652–659. doi: 10.1096/fj.02-0770com. [DOI] [PubMed] [Google Scholar]
- 17.Damian M, Mary S, Martin A, Pin JP, Banères JL. G protein activation by the leukotriene B4 receptor dimer. Evidence for an absence of trans-activation. J Biol Chem. 2008;283:21084–21092. doi: 10.1074/jbc.M710419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris T, Stables M, Hobbs A, et al. Effects of low-dose aspirin on acute inflammatory responses in humans. J Immunol. 2009;183:2089–2096. doi: 10.4049/jimmunol.0900477. [DOI] [PubMed] [Google Scholar]
- 19.Simiele F, Recchiuti A, Mattoscio D, et al. Transcriptional regulation of the human FPR2/ALX gene: evidence of a heritable genetic variant that impairs promoter activity. FASEB J. 2012;26:1323–1333. doi: 10.1096/fj.11-198069. [DOI] [PubMed] [Google Scholar]
- 20.Gronert K, Martinsson-Niskanen T, Ravasi S, Chiang N, Serhan CN. Selectivity of recombinant human leukotriene D(4), leukotriene B(4), and lipoxin A(4) receptors with aspirin-triggered 15-epi-LXA(4) and regulation of vascular and inflammatory responses. Am J Pathol. 2001;158:3–9. doi: 10.1016/S0002-9440(10)63937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diamant Z, Mantzouranis E, Bjermer L. Montelukast in the treatment of asthma and beyond. Exp Rev Clin Immunol. 2009;5:639–658. doi: 10.1586/eci.09.62. [DOI] [PubMed] [Google Scholar]
- 22.Fierro IM, Colgan SP, Bernasconi G, et al. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit human neutrophil migration: comparisons between synthetic 15 epimers in chemotaxis and transmigration with microvessel endothelial cells and epithelial cells. J Immunol. 2003;170:2688–2694. doi: 10.4049/jimmunol.170.5.2688. [DOI] [PubMed] [Google Scholar]
- 23.El Kebir D, József L, Khreiss T, et al. Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: a novel mechanism for resolution of inflammation. J Immunol. 2007;179:616–622. doi: 10.4049/jimmunol.179.1.616. [DOI] [PubMed] [Google Scholar]
- 24.El Kebir D, József L, Pan W, et al. 15-Epi-lipoxin A4 inhibits myeloperoxidase signalling and enhances resolution of acute lung injury. Am J Respir Crit Care Med. 2009;180:311–319. doi: 10.1164/rccm.200810-1601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clish CB, O'Brien JA, Gronert K, Stahl GL, Petasis NA, Serhan CN. Local and systemic delivery of a stable aspirin-triggered lipoxin prevents neutrophil recruitment in vivo. Proc Natl Acad Sci USA. 1999;96:8247–8252. doi: 10.1073/pnas.96.14.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levy BD, Lukacs NW, Berlin AA, et al. Lipoxin A4 stable analogs reduce allergic airway responses via mechanisms distinct from CysLT1 receptor antagonism. FASEB J. 2007;21:3877–3884. doi: 10.1096/fj.07-8653com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahlgren C, Christophe T, Boulay F, Madianos PN, Rabiet MJ, Karlsson A. The synthetic chemoattractant Trp-Lys-Tyr-Met-Val-DMet activates neutrophils preferentially through the lipoxin A(4) receptor. Blood. 2000;95:1810–1818. [PubMed] [Google Scholar]
- 28.Forsman H, Kalderén C, Nordin A, Nordling E, Jensen AJ, Dahlgren C. Stable formyl peptide receptor agonists that activate the neutrophil NADPH-oxidase identified through screening of a compound library. Biochem Pharmacol. 2011;81:402–411. doi: 10.1016/j.bcp.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 29.Bürli RW, Xu H, Zou X, et al. Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorg Med Chem Lett. 2006;16:3713–3718. doi: 10.1016/j.bmcl.2006.04.068. [DOI] [PubMed] [Google Scholar]
- 30.Frohn M, Xu H, Zou X, et al. New ‘chemical probes’ to examine the role of the hFPRL1 (or ALXR) receptor in inflammation. Bioorg Med Chem Lett. 2007;17:6633–6637. doi: 10.1016/j.bmcl.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 31.Chiang N, Takano T, Clish CB, Petasis NA, Tai HH, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (ATL) generation by human leukocytes and murine peritonitis exudates: development of a specific 15-epi-LXA4 ELISA. J Pharmacol Exp Ther. 1998;287:779–790. [PubMed] [Google Scholar]
- 32.Forsman H, Onnheim K, Andreasson E, Dahlgren C. What formyl peptide receptors, if any, are triggered by compound 43 and lipoxin A4? Scand J Immunol. 2011;74:227–234. doi: 10.1111/j.1365-3083.2011.02570.x. [DOI] [PubMed] [Google Scholar]
- 33.Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol. 2005;174:5033–5039. doi: 10.4049/jimmunol.174.8.5033. [DOI] [PubMed] [Google Scholar]
- 34.Levy BD, De Sanctis GT, Devchand PR, et al. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4) Nat Med. 2002;8:1018–1023. doi: 10.1038/nm748. [DOI] [PubMed] [Google Scholar]
- 35.Vong L, Ferraz JG, Dufton N, et al. Up-regulation of Annexin-A1 and lipoxin A(4) in individuals with ulcerative colitis may promote mucosal homeostasis. PLoS ONE. 2012;7:e39244. doi: 10.1371/journal.pone.0039244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levy BD, Bonnans C, Silverman ES, Palmer LJ, Marigowda G, Israel E Severe Asthma Research Program, National Heart, Lung, and Blood Institute. Diminished lipoxin biosynthesis in severe asthma. Am J Respir Crit Care Med. 2005;172:824–830. doi: 10.1164/rccm.200410-1413OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Planagumà A, Kazani S, Marigowda G, et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am J Respir Crit Care Med. 2008;178:574–582. doi: 10.1164/rccm.200801-061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noguera A, Gomez C, Faner R, et al. An investigation of the resolution of inflammation (catabasis) in COPD. Respir Res. 2012;13:101–110. doi: 10.1186/1465-9921-13-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vachier I, Bonnans C, Chavis C, et al. Severe asthma is associated with a loss of LX4, an endogenous anti-inflammatory compound. J Allergy Clin Immunol. 2005;115:55–60. doi: 10.1016/j.jaci.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 40.Colgan SP, Serhan CN, Parkos CA, Delp-Archer C, Madara JL. Lipoxin A4 modulates transmigration of human neutrophils across intestinal epithelial monolayers. J Clin Invest. 1993;92:75–82. doi: 10.1172/JCI116601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gewirtz AT, Fokin VV, Petasis NA, Serhan CN, Madara JL. LXA4, aspirin-triggered 15-epi-LXA4, and their analogs selectively downregulate PMN azurophilic degranulation. Am J Physiol. 1999;276:C988–994. doi: 10.1152/ajpcell.1999.276.4.C988. [DOI] [PubMed] [Google Scholar]
- 42.Levy BD, Petasis NA, Serhan CN. Polyisoprenyl phosphates in intracellular signalling. Nature. 1997;389:985–990. doi: 10.1038/40180. [DOI] [PubMed] [Google Scholar]
- 43.Levy BD, Fokin VV, Clark JM, Wakelam MJ, Petasis NA, Serhan CN. Polyisoprenyl phosphate (PIPP) signalling regulates phospholipase D activity: a ‘stop’ signalling switch for aspirin-triggered lipoxin A4. FASEB J. 1999;13:903–911. doi: 10.1096/fasebj.13.8.903. [DOI] [PubMed] [Google Scholar]
- 44.Chiang N, Fierro IM, Gronert K, Serhan CN. Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J Exp Med. 2000;191:1197–1208. doi: 10.1084/jem.191.7.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bozinovski S, Uddin M, Vlahos R, et al. Serum amyloid A opposes lipoxin A4 to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc Natl Acad Sci USA. 2012;109:935–940. doi: 10.1073/pnas.1109382109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lynch KR, O'Neill GP, Liu Q, et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399:789–793. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]